

Abstract
Background
Production of the proatherogenic metabolite, trimethylamine N‐oxide (TMAO), from dietary nutrients by intestinal microbiota enhances atherosclerosis development in animal models and is associated with atherosclerotic coronary artery disease in humans. The utility of studying plasma levels of TMAO to risk stratify in patients with peripheral artery disease (PAD) has not been reported.
Methods and Results
We examined the relationship between fasting plasma TMAO and all‐cause mortality (5‐year), stratified by subtypes of PAD and presence of coronary artery disease in 935 patients with PAD who underwent elective angiography for cardiac evaluation at a tertiary care hospital. Median plasma TMAO was 4.8 μmol/L (interquartile range, 2.9–8.0 μmol/L). Elevated TMAO levels were associated with 2.7‐fold increased mortality risk (fourth versus first quartiles, hazard ratio 2.86, 95% CI 1.82–3.97, P<0.001). Following adjustments for traditional risk factors, inflammatory biomarkers, and history of coronary artery disease, the highest TMAO quartile remained predictive of 5‐year mortality (adjusted hazard ratio 2.06, 95% CI 1.36–3.11, P<0.001). Similar prognostic value for elevated TMAO was seen for subjects with carotid artery, non–carotid artery, or lower extremity PAD. TMAO provided incremental prognostic value for all‐cause mortality (net reclassification index, 40.22%; P<0.001) and improvement in area under receiver operator characteristic curve (65.7% versus 69.4%; P=0.013).
Conclusions
TMAO, a pro‐atherogenic metabolite formed by gut microbes, predicts long‐term adverse event risk and incremental prognostic value in patients with PAD. These findings point to the potential for TMAO to help improve selection of high‐risk PAD patients with or without significant coronary artery disease, who likely need more aggressive and specific dietary and pharmacologic therapy.
Keywords: intestinal microbiota, peripheral vascular disease, trimethylamine N‐oxide, vascular biology
Subject Categories: Peripheral Vascular Disease, Metabolism, Mortality/Survival
Introduction
Increasing data support a role of gut microbiota and dietary ingestion of phosphatidylcholine (PC or lecithin) in the pathogenesis of atherosclerosis.1, 2 PC is the major dietary source of choline, which often is found in the Western diet, such as red meat, eggs, and meat products. Production of trimethylamine N‐oxide (TMAO) by gut microbiota metabolism of dietary PC has been associated with the development of atherosclerosis in animals and in humans.1, 2, 3 Increased levels of plasma TMAO are associated with an enhanced number of diseased major coronary vessels,1 as well as increased risk of major adverse cardiac events in patients undergoing elective coronary angiography.3, 4
Peripheral artery disease (PAD) is a common manifestation of systemic atherosclerosis and is associated with significantly increased cardiovascular death and mortality.5, 6 It affects over 27 million people across Europe and North America.7 An estimated 8.5 million Americans aged ≥40 years carry the diagnosis of PAD.8 Despite its association with high prevalence and extremely poor prognosis, PAD remains undiagnosed and rarely receives attention in the scientific literature compared to patients with coronary artery disease (CAD) and other atherosclerotic conditions.7, 9 To date, predictors of mortality and underlying pathophysiology in patients with PAD are not well defined, which may be a contributing factor to why the mortality risk and ischemic amputations in PAD patients remained high.10 Therefore, there is a need to find new prognostic markers that may provide insight into underlying pathophysiology, improve long‐term clinical risk prediction incremental to traditional risk factors, and suggest avenues for therapeutic development in PAD. The primary objective of this study was to determine the clinical prognostic value of plasma TMAO levels in a contemporary cohort of patients with established but stable PAD.
Methods
Study Design and Study Population
This is a single‐center prospective cohort study approved by the Cleveland Clinic Institutional Review Board, and all participants gave written informed consent. We enrolled sequential consenting patients who underwent elective, nonurgent coronary angiographic evaluation at the Cleveland Clinic between 2001 and 2007, as previously described. All patients were seen by a cardiologist at Cleveland Clinic before the left heart catheterization. Enrolled subjects were directly asked by research personal about past history of non‐CAD cardiovascular disease and/or history of or repair of aortic dissection/aneurysm. We carefully reviewed the electronic medical record (including angiographic data) for validation of diagnosis subtypes of PAD. A confirmed diagnosis of PAD was based primarily on the type of PAD, based on reporting evidence of stenosis at the corresponding vasculature (Table 1).11, 12, 13 All‐cause mortality was prospectively tracked over 5 years for all participants with medical chart review and confirmed by follow‐up contact and the Social Security Death Index. In this analysis, we included those with a documented history of PAD.
Table 1.
Criteria Diagnosis of PAD Based Primarily on the Type of PAD
| Diagnosis | Criteria Diagnosis |
|---|---|
| Carotid artery stenosis (CAS)12 |
At least 1 of the following:
|
| Lower extremity peripheral artery disease (LEAD)12 |
At least 1 of the following:
|
| Aortic abdominal aneurysm (AAA), or dissection12 |
At least 1 of the following:
|
| Renal artery stenosis (RAS)12 |
At least 1 of the following:
|
| Upper extremity artery stenosis (defined as stenotic of subclavian or brachial artery)13 |
At least 1 of the following:
|
ABI indicates ankle–brachial index; CTA, computed tomography angiography; DUS, duplex ultrasound; MRA, magnetic resonance angiography; NASCET, North American Symptomatic Carotid Endarterectomy Trial; PAD, peripheral artery disease.
*Determining percentage stenosis based on internally developed diagnosis criteria in the noninvasive vascular laboratory of the Cleveland Clinic.
Diagnosis Validation of PAD Subtypes
Peripheral artery disease is defined by American College of Cardiology/American Heart Association as a group of clinical disorders that includes abdominal aortic aneurysm, renal artery stenosis (RAS), mesenteric artery stenosis (MAS), and lower extremity peripheral artery disease (LEAD).6, 12 However, The European Society of Cardiology uses the term PAD to include several vascular sites, including extracranial carotid artery stenosis (CAS) and vertebral artery diseases, upper extremity artery stenosis, MAS, RAS, and LEAD, but diseases of the aorta are not covered.13 In our cohort, the term PAD is used to indicate the majority of noncoronary arterial territories including CAS, upper extremity artery stenosis, MAS, RAS, and LEAD, while diseases of the aorta were not included. In this analysis, we defined non‐CAS if the primary diagnosis was not CAS (which included LEAD, RAS, upper extremity artery stenosis, and MAS).
Laboratory Testing
After informed consent was obtained from all patients, fasting blood samples were collected using EDTA tubes at the time of cardiac catheterization, immediately prior to heparin injection. Samples were maintained on ice or at 4°C, immediately processed within 3 hours of collection, and frozen at −80°C until analysis. Creatinine clearance was estimated using the Cockcroft‐Gault equation. Myeloperoxidase was determined by the CardioMPO assay (Cleveland Heart Labs, Cleveland, OH). TMAO, choline, and betaine levels in plasma were determined using stable isotope dilution high‐performance liquid chromatography with online electrospray ionization tandem mass spectrometry on Shimadzu LCMS‐8050 CL Triple Quadrupole mass spectrometer (Shimadzu Corp, Kyoto, Japan) using d9‐(trimethyl)‐labeled internal standards as described previously.1, 14
Statistical Analysis
Continuous data are presented as mean (±SD) or median (interquartile range) and compared with Student t test and ANOVA or nonparametric alternatives (Kruskal–Wallis test) when appropriate. Categorical variables are presented as number (%) and compared between groups with χ2 tests. We divided plasma TMAO levels into quartiles. Kaplan–Meier analysis with Cox proportional hazards regression was used for the time‐to‐event analysis to determine hazard ratios (HR) and 95% CI for 5‐year all‐cause mortality stratified according to TMAO as a continuous variable (log‐transformed per SD increase), as well as according to quartiles. Adjustments were made for individual traditional cardiovascular risk factors (age, sex, systolic blood pressure, diabetes mellitus, low‐density and high‐density lipoprotein cholesterol levels, triglyceride levels, and smoking status) and for log‐transformed high‐sensitivity C‐reactive protein (hsCRP), log‐transformed estimated glomerular filtration rate (eGFR), log‐transformed myeloperoxidase, apolipoprotein A‐1 and apolipoprotein B, history of CAD, and statin use, to predict all‐cause mortality. Category‐free net reclassification improvement, Integrated Discrimination Improvement, and area under the receiver‐operating characteristic curve were calculated to quantify improvement in model performance according to mortality risk estimated using Cox models adjusted for the abovementioned traditional risk factors with versus without TMAO, as previously described.15 Subgroups were stratified according to diagnosis subtypes of PAD (LEAD, CAS, and non‐CAS) as well as other baseline clinical and laboratory subgroups that might be affecting mortality risks. All analyses were performed used R 2.15.1 (R Foundation for Statistical Computing, Vienna, Austria). A P value <0.05 was considered to indicate statistically significant.
Results
Baseline Characteristics
In our study cohort, a total of 935 subjects had reported a history of PAD. Of these, confirmed diagnosis data were not available for 14 patients, and 100 patients with a diagnosis of aortic aneurysm were excluded. Therefore, 821 consecutive patients were included in this study. Baseline characteristics of the 821 participants are shown in Table 2. In our study cohort, the median TMAO level was 4.8 μmol/L (interquartile range 2.9–8.0 μmol/L), which was similar between patients with CAS and non‐CAS (Figure 1). Subjects with elevated plasma TMAO levels were more likely to be older, with diabetes, with elevated hsCRP, as well as with lower eGFR. In contrast, prior history of CAD, smoking status, myeloperoxidase levels, apolipoprotein B levels, and medication use, were similar across TMAO levels.
Table 2.
Baseline Characteristics of PAD Cohort
| Overall (n=821) | Quartile 1 (n=205) | Quartile 2 (n=205) | Quartile 3 (n=205) | Quartile 4 (n=206) | P Value | |
|---|---|---|---|---|---|---|
| Age, y | 66±10 | 63±12 | 66±10 | 69±9 | 68±10 | <0.001 |
| Male, % | 66 | 65 | 68 | 68 | 61 | 0.346 |
| Diabetes mellitus, % | 43 | 29 | 40 | 44 | 60 | <0.001 |
| Hypertension, % | 83 | 80 | 82 | 81 | 87 | 0.186 |
| Former/current smokers, % | 74 | 77 | 73 | 74 | 74 | 0.864 |
| Prior CAD, % | 90 | 89 | 91 | 90 | 92 | 0.717 |
| LDL cholesterol, mg/dL | 92 (74–110) | 96 (77–115) | 90 (74–107) | 93 (79–111) | 91 (70–110) | 0.079 |
| HDL cholesterol, mg/dL | 33 (27–39) | 34 (28–40) | 34 (28–40) | 32 (26–38) | 31 (26–38) | 0.004 |
| hsCRP, mg/L | 3.2 (1.3–8.3) | 3 (1–8.4) | 2.5 (1.2–6.2) | 3.1 (1.5–7.2) | 4.5 (1.6–9.4) | 0.006 |
| MPO, mg/L | 118.6 (80.3–261.8) | 127.8 (84.3–263.8) | 114.4 (79.9–246) | 121 (83.5–279.5) | 115.7 (78.8–277.8) | 0.795 |
| ApoA1 | 112 (100–128) | 113 (101–128) | 114 (101–131) | 111 (99–129) | 109 (97.2–123.8) | 0.142 |
| ApoB | 80 (68–93) | 82 (70–97) | 79 (66–91) | 81 (72–92) | 79 (67–92.8) | 0.167 |
| WBC, ×109 | 6.4 (5.2–7.9) | 6.5 (5.5–8.2) | 6.3 (5.2–7.7) | 6.4 (5.2–7.8) | 6.5 (5.2–8) | 0.439 |
| eGFR, mL/min per 1.73 m2 | 78.8 (59.4–90.9) | 90.8 (81.9–98.5) | 80.6 (67.3–92) | 74.8 (57.9–86.2) | 56.5 (39–78.1) | <0.001 |
| Medication | ||||||
| ACE inhibitor or ARB, % | 60 | 56 | 59 | 67 | 59 | 0.088 |
| β‐Blocker, % | 69 | 70 | 69 | 68 | 67 | 0.895 |
| Statin, % | 70 | 73 | 74 | 69 | 66 | 0.249 |
| Aspirin, % | 76 | 78 | 78 | 76 | 73 | 0.632 |
| TMAO, μmol/L | 4.8 (2.9–8) | 2.2 (1.7–2.6) | 3.7 (3.3–4.3) | 6.1 (5.4–7) | 13 (9.8–21.9) | <0.001 |
| Choline, μmol/L | 10.7 (8.7–13.8) | 8.7 (7.1–10.7) | 10 (8.1–12.4) | 11.7 (9.7–14.7) | 13.2 (10.4–17.6) | <0.001 |
| Betaine, μmol/L | 41.9 (32.6–53.4) | 38.6 (30.7–49.5) | 42.9 (33.6–53.9) | 42.1 (32.2–53.5) | 44.2 (34.7–53.9) | 0.004 |
Values expressed in mean±SD, %, or median (interquartile range). ACE indicates angiotensin‐converting enzyme; ApoA1, apolipoprotein A1; ApoB, apolipoprotein B; ARB, angiotensin‐receptor blocker; CAD, coronary artery disease; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; hsCRP, high‐sensitivity C‐reactive protein; LDL, low‐density lipoprotein; MPO, myeloperoxidase; PAD, peripheral artery disease; TMAO, trimethylamine N‐oxide; WBC, total leukocyte count.
Figure 1.
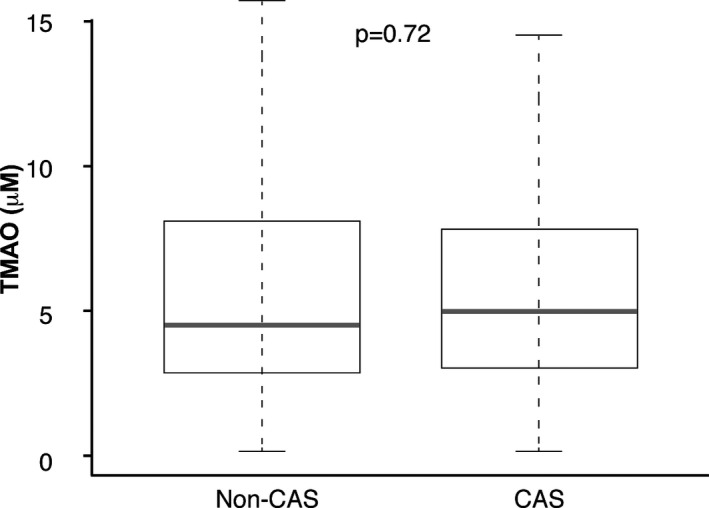
Comparison of fasting trimethylamine‐N‐oxide (TMAO) levels between patients with carotid artery stenosis and non–carotid artery stenosis. TMAO concentration was not significantly different between patients with carotid artery stenosis (CAS) and non–carotid artery stenosis (Non‐CAS).
Of the 821 subjects included, 371 had diagnosis of CAS confirmed by duplex ultrasonography (83%), magnetic resonance angiography (2.7%), catheter‐based radiocontrast angiography (4.0%), prior endovascular intervention (3.8%), and open carotid endarterectomy (6.5%); 421 patients had LEAD confirmed by the following: ankle–brachial index <0.9 (65.5%), duplex ultrasonography (7.0%), computed tomography angiography (1.5%), magnetic resonance angiography (0.5%), catheter‐based radiocontrast angiography (7.7%), prior endovascular intervention (8.8%), and prior open surgical procedure (9.0%); 15 patients had RAS confirmed by magnetic resonance angiography (20%), catheter‐based radiocontrast angiography (53%), and prior endovascular intervention (27%); 13 patients had upper extremity artery stenosis confirmed by catheter‐based radiocontrast angiography (38.5%), and prior endovascular intervention (61.5%), and 1 patient had MAS confirmed by prior endovascular intervention (Figure 2).
Figure 2.
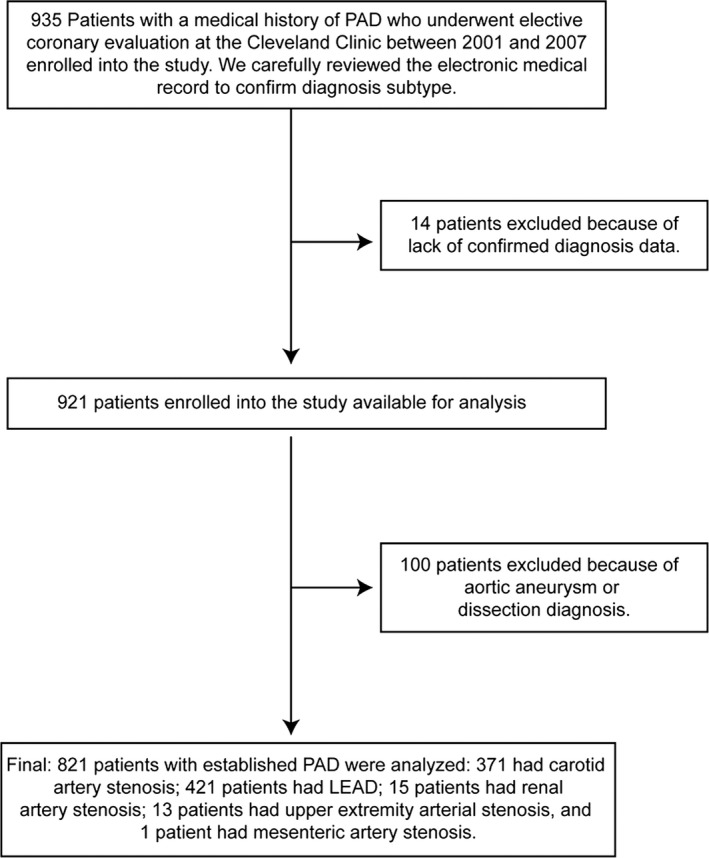
Study design. LEAD indicates lower extremity peripheral artery disease; PAD, peripheral artery disease.
TMAO Levels and Mortality Risks
Over the 5‐year follow‐up, 222 (27%) deaths occurred in our cohort. Nineteen patients (19/821=2%) were not reached at 5‐year follow‐up. As illustrated in the Kaplan–Meier analyses shown in Figure 3, a graded increase in risk for all‐cause mortality was observed with increasing plasma levels of TMAO. Elevated plasma TMAO levels were associated with a 2.7‐fold increased risk for all‐cause mortality (unadjusted HR=2.69, 95% CI 1.82–3.97, P<0.001) (Table 3). Following adjustments for traditional risk factors, history of CAD, statin use, and inflammatory marker including log‐transformed myeloperoxidase, log‐transformed hsCRP and apolipoprotein A‐1 and apolipoprotein B,, elevated plasma TMAO levels remained a significant predictor of the risk of 5‐year all‐cause mortality (adjusted HR 1.88, 95% CI 1.21–2.92, P=0.002), as well as after adjusting for traditional risk factor, log‐transformed hsCRP, and log‐transformed eGFR (adjusted HR 1.59, 95% CI 1.03–2.45, P=0.038) (Table 3). A similar graded increase in risk was observed when levels of TMAO were analyzed as a continuous variable in increments of 1 SD (unadjusted HR 1.53, 95% CI 1.35–1.74, P<0.001) and remained significant after adjusting for traditional risk factor, log‐transformed hsCRP and log‐transformed eGFR (adjusted HR 1.26, 95% CI 1.03–1.53, P=0.016). The inclusion of TMAO as a covariate resulted in a significant improvement in risk estimation over traditional risk factors (net reclassification improvement, 40.22%, [P<0.001]; Integrated Discrimination Improvement, 10.0%, [P<0.001]; and differences in area under the receiver‐operating characteristic curve 65.69 versus 69.42, P=0.013). Interestingly, mortality risks were similar between PAD diagnosis subgroups (between CAS, non‐CAS, and LEAD), as well as other clinical and laboratory subgroups that might be affected for mortality risks (Figure 4).
Figure 3.
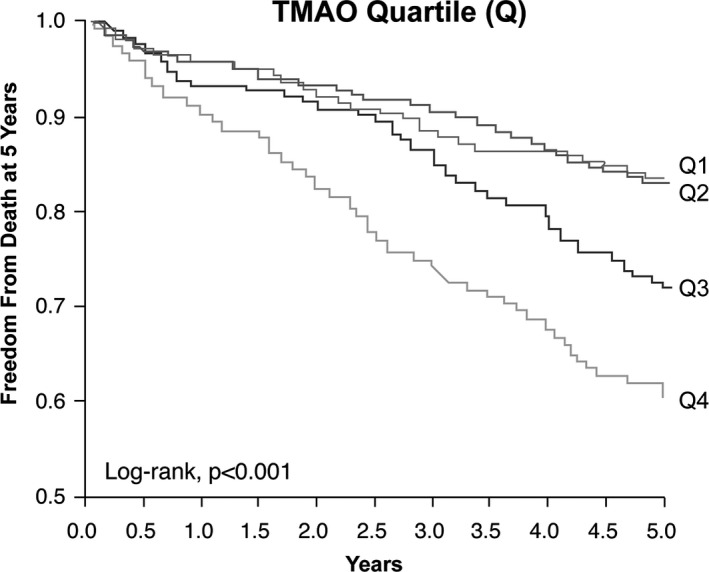
Kaplan–Meier estimates of risk of all‐cause mortality according to the quartiles of plasma levels of trimethylamine‐N‐oxide (TMAO). Kaplan–Meier curves for 5‐year all‐cause mortality with TMAO stratified as quartiles. The P value is for all comparisons.
Table 3.
Hazard Ratio of Fasting Plasma TMAO Levels for 5‐Year All‐Cause Mortality Stratified According to All Subjects and Each Diagnosis Subtype of PAD
| TMAO | ||||
|---|---|---|---|---|
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | |
| Range, μmol/L | <2.94 | 2.94 to 4.81 | 4.81 to 8.01 | ≥8.01 |
| Unadjusted | 1 | 0.99 (0.62–1.56) | 1.75 (1.17–2.64)** | 2.69 (1.82–3.97)*** |
| Adjusted model 1 | 1 | 0.99 (0.62–1.56) | 1.36 (0.89–2.08) | 2.06 (1.36–3.11)*** |
| Adjusted model 2 | 1 | 0.86 (0.54–1.38) | 1.25 (0.82–1.92) | 1.59 (1.03–2.45)* |
| Adjusted model 3 | 1 | 0.9 (0.55–1.47) | 1.34 (0.85–2.11) | 1.88 (1.21–2.92)** |
| Event rate | 37/205=18.1% | 37/205=18.1% | 62/205=30.2% | 86/206=41.8% |
Adjusted model 1: adjusted for traditional risk factors including age, sex, systolic blood pressure, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, smoking, and diabetes mellitus; Adjusted model 2: adjusted for model 1 plus high‐sensitivity C‐reactive protein (log‐transformed) and estimated glomerular filtration rate (log‐transformed); Adjusted model 3: adjusted for model 1 plus history of coronary artery disease, statin use, apolipoprotein A1, apolipoprotein B, myeloperoxidase (log‐transformed), and high‐sensitivity C‐reactive protein (log‐transformed). PAD indicates peripheral artery disease; TMAO, trimethylamine N‐oxide. ***P<0.001; **P<0.01; *P<0.05.
Figure 4.
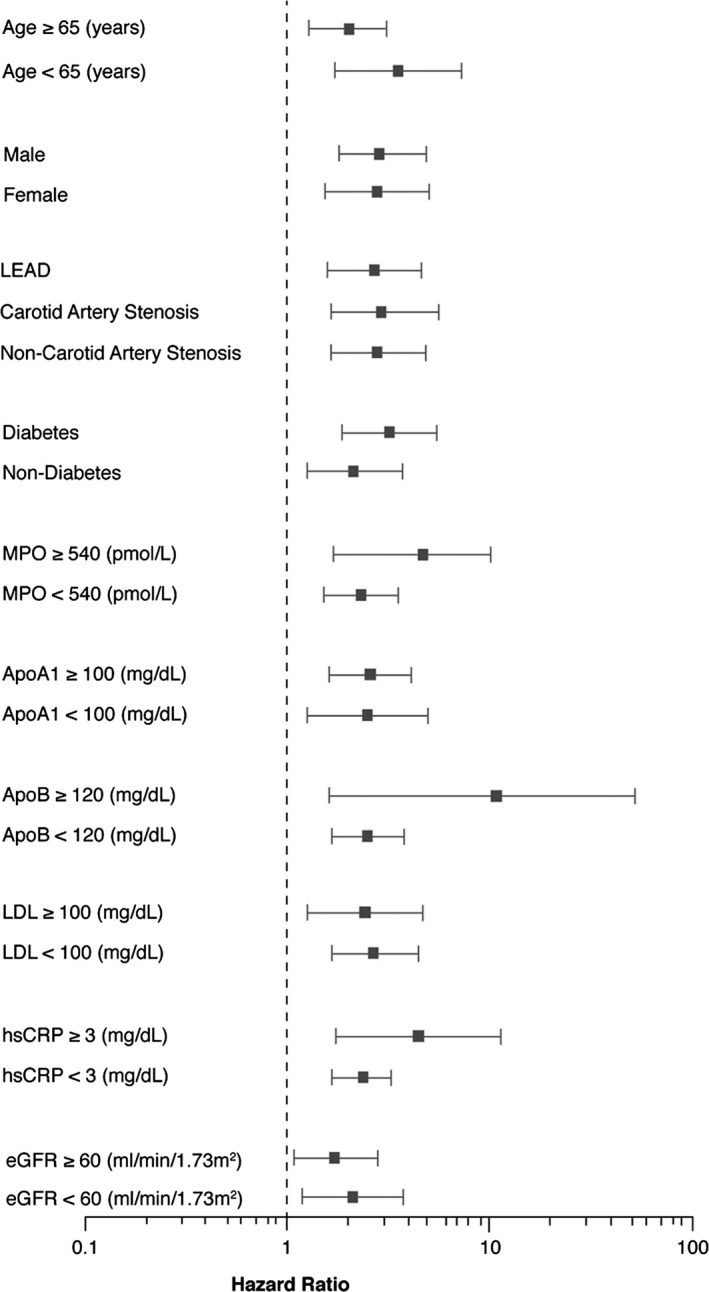
Relationship between plasma trimethylamine‐N‐oxide (TMAO) concentration and mortality risk stratified according to baseline characteristics. Forest plot of hazard ratio (squares) of 5‐year all‐cause mortality comparing first and fourth quartiles of plasma TMAO levels. Bars represent 95% CI. The wide confidence intervals in some subgroups are in part due to their small sample sizes and event rate. ApoA1 indicates apolipoprotein A1; ApoB, apolipoprotein B; eGFR, estimated glomerular filtration rate; hsCRP, high‐sensitivity C‐reactive protein; LDL, low‐density lipoprotein; LEAD, lower extremity peripheral artery disease; MPO, myeloperoxidase.
Discussion
To our knowledge, this represents the first report of elevated plasma TMAO levels as a significant prognostic marker in patients with a PAD. Our major finding is that elevated plasma TMAO is a significant predictor of 5‐year all‐cause mortality risk among stable patients with a PAD. Moreover, mortality risks were not significantly different among all different subtypes of diagnosis of PAD, presence or absence of CAD, as well as other clinical and laboratory subgroups.
The role of gut microbiota is increasingly being accepted as an environmental factor that affects global metabolism of their host and contributes to associated pathological conditions, such as obesity, insulin resistance, chronic kidney disease, and cardiovascular disease.1, 2, 3, 16, 17 Our recent studies in animals and in humans suggested a role for gut microbiota in the pathogenesis of atherosclerosis.1, 2 Trimethylamine‐containing nutrients (eg, PC, choline, and l‐carnitine) are metabolized by the gut microbiota to trimethylamine (TMA) and converted to TMAO.1 PC is a major dietary source of choline in omnivores, which often is found in the Western diet, such as red meat, eggs, and meat products. Direct ingestion of PC was shown to result in a rise in choline, betaine, and TMAO levels.1, 3 Moreover, previous studies have report that elevated plasma TMAO levels predicted future risk of major adverse cardiovascular events and have a direct mechanistic link to development of cardiovascular disease.1, 2, 3, 4 Extending these observations, in the present prospective study focused exclusively on patients with adjudicated diagnosis of PAD, an upper quartile for TMAO levels (compared with the lowest quartile) had a significant, 1.6‐fold increased risk for future all‐cause mortality, independent of traditional cardiovascular risk factors, inflammation markers, and eGFR.
Additional studies demonstrated that the mechanism through which dietary PC or choline enhances atherosclerosis in mouse models requires the presence of intact gut microbiota and formation of TMAO.1, 2 Furthermore, in a study examining plasma carnitine, choline, or betaine levels in sequential subjects undergoing elective coronary angiography, each was associated with future risk of major adverse cardiovascular events independent of traditional risk factors. However, their prognostic values were restricted only to those with concomitant increased levels of TMAO, consistent with this gut microbe metabolite being the proximate and mechanistically linked metabolite in the pathway.4 In several recent studies genetically repressing hepatic flavin monooxygenase 3 expression, the host enzyme primarily responsible for converting gut microbe–generated TMA into TMAO, choline diet–enhanced atherosclerosis was inhibited, further implicating the TMAO pathway in atherosclerosis development.18, 19 These and other studies have also implicated FMO3 as an important regulator of body cholesterol and bile acid metabolism, suggesting mechanistic links between the gut microbe–TMAO pathway and atherosclerosis pathogenesis.18, 19, 20 Interestingly, TMAO can promote atherosclerosis by inhibiting reverse cholesterol transport and enhanced macrophage foam cell formation in both the artery wall and peritoneal cavity, as well as promoting aortic root atherosclerotic plaque development.1, 2 Recently, exciting new human data reported by Randrianarisoa et al have found that increased fasting serum TMAO levels were associated with increased carotid intima–media thickness, which is a marker of atherosclerosis. Furthermore, after the lifestyle intervention, carotid intima–media thickness decreased significantly (P=0.0056) specifically in subjects in the largest decrease (>20%) of TMAO levels.21 Thus, gut microbial generation of TMAO from dietary nutrients such as PC may represent an attractive target for intervention in subjects with PAD.
Patients with PAD show a 2‐ to 6‐fold increased risk of death from cardiovascular causes and mortality relative to those without PAD.6, 8 Data from the Reduction of Atherothrombosis for Continued Health (REACH) registry have shown that PAD is strongly associated with concomitant CAD and atherosclerosis in multiple vascular beds, and indicated that these patients have poorer prognosis than patients with just 1 territory affected.22 It is also recognized that the majority of patients with PAD die from CAD.5 There are a number of proven therapies to reduce mortality among patients with PAD including antiplatelet agents, lipid‐lowering therapy with the statins, and aggressive cardiovascular risk factor modification.6 Unfortunately, lifesaving medications are underprescribed among patients with PAD, particularly in comparison with patients with CAD,23 and also the true atherosclerosis burden and mortality risk of PAD are underestimated.9, 10 In contrast, the overall rate of use of cardioprotective medication for secondary prevention in our study cohort was relatively high; at baseline 70% were on statins, 76% on aspirin, 69% on β‐blockers, and 60% on angiogensin‐converting enzyme inhibitor/angiotensin‐receptor blockers. Yet, despite patients having received optimal cardioprotective medication, the 5‐year all‐cause mortality rate in our study cohort remained high, with 27% deaths. These higher event rates may be driven by a large proportion of patients with CAD (90%). Interestingly, these data are consistent with findings from the REACH registry, which has a large percentage of patients with PAD combined with CAD and is associated with high mortality rates.22 Therefore, accurate risk prediction and identification for underlying pathophysiology in patients with PAD is critically meaningful. Our findings suggest a link between plasma TMAO, a metabolite formed by gut microbiota metabolism of the choline group of PC, and prognosis risk prediction. They may be a new prognostic marker for risk stratification and suggest avenues for therapeutic development to reduce future all‐cause mortality in patients with PAD.
Our findings may be important to understand a potential pathophysiological contribution of intestinal microbiota in the development of atherosclerosis and adverse prognosis in patients with PAD, including the majority of noncoronary territories. Since occult atherosclerotic plaque burden in extracoronary territories is a common issue hindering PAD diagnosis, and given the mechanistic links between TMAO and atherosclerosis development and progression, the present studies suggest that plasma TMAO level measurements could be used to identify patients in whom more detailed PAD surveillance efforts are needed given the heightened adverse prognosis risk associated with higher TMAO levels. An improvement in understanding of the pathophysiology linking gut microbes, TMAO, and PAD development may serve to help improve selection of high‐risk PAD patients who need more aggressive and specific dietary and pharmacologic therapy.
Study Limitations
This was a single, tertiary care center study that recruited patients at the point of cardiac evaluation for coronary angiography; therefore, there was a higher proportion of patients with CAD. Our population consisted of a heterogeneous subgroup of patients with PAD, yet we believe that the confirmed diagnoses were carefully captured individually. Also, we lacked complete information regarding New York Heart Association functional class, claudication symptom, and disease severity, but we addressed this issue by enrolling patients in stable condition, confirmed by careful review of the electronic medical record individually. We also acknowledge that many factors may have influenced TMAO levels, including increased age, diabetes, elevated hsCRP, or low eGFR. All of these factors can contribute to increased mortality in this population, and we do not have an external validation cohort to confirm our findings. Despite these limitations, our findings are the first report to point to novel insights that provide a mechanistic link between gut microbiota–associated metabolism involved in TMAO formation and PAD, and substantially improved risk stratification for adverse prognosis in patients with PAD.
Conclusions
Elevated plasma TMAO levels, a pro‐atherogenic metabolite formed by gut microbiota metabolism of the choline group of PC, portend stronger incident risk of major adverse cardiovascular events and all‐cause death in patients with PAD, independent of traditional risk factors.
Sources of Funding
This research was supported by grants from the National Institutes of Health (NIH) and the Office of Dietary Supplements (R01HL103866, P20HL113452, R01DK106000). The GeneBank study has been supported by NIH grants P01HL076491, P01HL098055, R01HL103931, and the Cleveland Clinic Clinical Research Unit of the Case Western Reserve University CTSA (UL1TR000439). Dr Wang was partially supported by NIH grant R01HL130819. Dr Hazen was partially supported by a gift from the Leonard Krieger Endowment. Mass spectrometry studies were performed on instruments housed in a facility supported in part by a Center of Innovations Award by Shimadzu. High‐sensitivity cardiac troponin T testing reagents were provided by Roche Diagnostics.
Disclosures
Drs Wang and Hazen are named as co‐inventors on pending patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr Wang has received royalty payment for inventions or discoveries related to cardiovascular diagnostics from Cleveland HeartLab. Dr Hazen is a paid consultant for Esperion and P&G. Dr Hazen has received research funds from Abbott, P&G, Pfizer Inc, Roche Diagnostics, and Takeda. Dr Hazen has received royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Siemens, Esperion, and Frantz Biomarkers, LLC. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
(J Am Heart Assoc. 2016;5:e004237 doi: 10.1161/JAHA.116.004237)
References
- 1. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of l‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota‐generated metabolite trimethylamine‐n‐oxide. Eur Heart J. 2014;35:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Criqui MH, Langer RD, Fronek A, Feigelson HS, Klauber MR, McCann TJ, Browner D. Mortality over a period of 10 years in patients with peripheral arterial disease. N Engl J Med. 1992;326:381–386. [DOI] [PubMed] [Google Scholar]
- 6. Rooke TW, Hirsch AT, Misra S, Sidawy AN, Beckman JA, Findeiss LK, Golzarian J, Gornik HL, Halperin JL, Jaff MR, Moneta GL, Olin JW, Stanley JC, White CJ, White JV, Zierler RE; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society for Vascular Medicine; Society for Vascular Surgery . ACCF/AHA focused update of the guideline for the management of patients with peripheral artery disease (updating the guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2020–2045.21959305 [Google Scholar]
- 7. Belch JJ, Topol EJ, Agnelli G, Bertrand M, Califf RM, Clement DL, Creager MA, Easton JD, Gavin JR III, Greenland P, Hankey G, Hanrath P, Hirsch AT, Meyer J, Smith SC, Sullivan F, Weber MA. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med. 2003;163:884–892. [DOI] [PubMed] [Google Scholar]
- 8. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirsch AT, Criqui MH, Treat‐Jacobson D, Regensteiner JG, Creager MA, Olin JW, Krook SH, Hunninghake DB, Comerota AJ, Walsh ME, McDermott MM, Hiatt WR. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001;286:1317–1324. [DOI] [PubMed] [Google Scholar]
- 10. Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, Criqui MH. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329–1340. [DOI] [PubMed] [Google Scholar]
- 11. North American Symptomatic Carotid Endarterectomy Trial Collaborators . Beneficial effect of carotid endarterectomy in symptomatic patients with high‐grade carotid stenosis. N Engl J Med. 1991;325:445–453. [DOI] [PubMed] [Google Scholar]
- 12. Creager MA, Belkin M, Bluth EI, Casey DE Jr, Chaturvedi S, Dake MD, Fleg JL, Hirsch AT, Jaff MR, Kern JA, Malenka DJ, Martin ET, Mohler ER III, Murphy T, Olin JW, Regensteiner JG, Rosenwasser RH, Sheehan P, Stewart KJ, Treat‐Jacobson D, Upchurch GR Jr, White CJ, Ziffer JA, Hendel RC, Bozkurt B, Fonarow GC, Jacobs JP, Peterson PN, Roger VL, Smith EE, Tcheng JE, Wang T, Weintraub WS. 2012 ACCF/AHA/ACR/SCAI/SIR/STS/SVM/SVN/SVS key data elements and definitions for peripheral atherosclerotic vascular disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Clinical Data Standards for Peripheral Atherosclerotic Vascular Disease). Circulation. 2012;125:395–467. [DOI] [PubMed] [Google Scholar]
- 13. Tendera M, Aboyans V, Bartelink ML, Baumgartner I, Clement D, Collet JP, Cremonesi A, De Carlo M, Erbel R, Fowkes FG, Heras M, Kownator S, Minar E, Ostergren J, Poldermans D, Riambau V, Roffi M, Rother J, Sievert H, van Sambeek M, Zeller T. ESC guidelines on the diagnosis and treatment of peripheral artery diseases: document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries: the task force on the diagnosis and treatment of peripheral artery diseases of the European Society of Cardiology (ESC). Eur Heart J. 2011;32:2851–2906. [DOI] [PubMed] [Google Scholar]
- 14. Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, Hazen SL. Measurement of trimethylamine‐n‐oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem. 2014;455:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the roc curve to reclassification and beyond. Stat Med. 2008;27:157–172. [DOI] [PubMed] [Google Scholar]
- 16. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. [DOI] [PubMed] [Google Scholar]
- 17. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa‐Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shih DM, Wang Z, Lee R, Meng Y, Che N, Charugundla S, Qi H, Wu J, Pan C, Brown JM, Vallim T, Bennett BJ, Graham M, Hazen SL, Lusis AJ. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miao J, Ling AV, Manthena PV, Gearing ME, Graham MJ, Crooke RM, Croce KJ, Esquejo RM, Clish CB, Vicent D, Biddinger SB. Flavin‐containing monooxygenase 3 as a potential player in diabetes‐associated atherosclerosis. Nat Commun. 2015;6:6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, Sacks J, Rong X, Vallim TA, Chou J, Ivanova PT, Myers DS, Brown HA, Lee RG, Crooke RM, Graham MJ, Liu X, Parini P, Tontonoz P, Lusis AJ, Hazen SL, Temel RE, Brown JM. The TMAO‐generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Randrianarisoa E, Lehn‐Stefan A, Wang X, Hoene M, Peter A, Heinzmann SS, Zhao X, Konigsrainer I, Konigsrainer A, Balletshofer B, Machann J, Schick F, Fritsche A, Haring HU, Xu G, Lehmann R, Stefan N. Relationship of serum trimethylamine N‐oxide (TMAO) levels with early atherosclerosis in humans. Sci Rep. 2016;6:26745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhatt DL, Steg PG, Ohman EM, Hirsch AT, Ikeda Y, Mas JL, Goto S, Liau CS, Richard AJ, Rother J, Wilson PW. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA. 2006;295:180–189. [DOI] [PubMed] [Google Scholar]
- 23. Subherwal S, Patel MR, Kober L, Peterson ED, Jones WS, Gislason GH, Berger J, Torp‐Pedersen C, Fosbol EL. Missed opportunities: despite improvement in use of cardioprotective medications among patients with lower‐extremity peripheral artery disease, underuse remains. Circulation. 2012;126:1345–1354. [DOI] [PubMed] [Google Scholar]