

Abstract
Background
Neuroinflammation plays a key role in intracerebral hemorrhage (ICH)–induced secondary brain injury, but the specific roles of peripheral inflammatory cells such as macrophages and lymphocytes remain unknown. The purpose of this study was to explore the roles of macrophages, T lymphocytes, and the cytokines they secrete as potential targets for treating secondary brain injury after ICH.
Methods and Results
Our results showed that peripheral macrophages and T lymphocytes successively infiltrated the brain, with macrophage counts peaking 1 day after ICH and T‐lymphocyte counts peaking after 4 days. These peaks in cellular infiltration corresponded to increases in interleukin (IL)‐23 and IL‐17 expression, respectively. We found that hemoglobin from the hematoma activated IL‐23 secretion by infiltrating macrophages by inducing the formation of toll‐like receptor (TLR) 2/4 heterodimer. This increased IL‐23 expression stimulated γδT‐cell production of IL‐17, which increased brain edema and neurologic deficits in the model mice as a proinflammatory factor. Finally, we found that sparstolonin B (SsnB) could ameliorate brain edema and neurologic deficits in ICH model mice via inhibition of TLR2/TLR4 heterodimer formation, and notably, SsnB interacted with myeloid differentiation factor 88 Arg196.
Conclusions
Together, our results reveal the importance of the IL‐23/IL‐17 inflammatory axis in secondary brain injury after ICH and thus provide a new therapeutic target for ICH treatment.
Keywords: IL‐17, IL‐23, intracerebral hemorrhage, lymphocyte, macrophage, TLR2, TLR4
Subject Categories: Cerebrovascular Disease/Stroke, Blood-Brain Barrier, Intracranial Hemorrhage
Introduction
Intracerebral hemorrhage (ICH) accounts for 20% to 30% of all cases of cerebrovascular disease in China and remains the leading cause of mortality, largely because there currently is no specific treatment.1, 2, 3ICH is associated with both primary and secondary injury. The physical effects of the hematoma (mass effect) and disturbance of the adjacent tissue that occur within hours of the onset of bleeding represent the primary insult.4 The potential benefit of clot removal has been investigated in multiple clinical trials but not confirmed.4, 5, 6 Instead, most recent attempts to identify novel therapeutic targets have focused on the mechanisms responsible for ICH‐induced secondary injury, which is caused by the development of edema, free radicals, inflammation, and direct cellular toxicity of the degradation by products of the hematoma.4 Increasing evidence indicates that inflammation plays a critical role in ICH‐induced secondary injury.7, 8, 9 Within minutes after ICH, microglia are activated and begin to release large amounts of cytokines and chemokines to destroy the blood‐brain barrier (BBB).8, 10 In the later phase, peripheral inflammatory cells such as macrophages and lymphocytes are mobilized to the perihematoma brain tissue through the damaged BBB.11, 12, 13 The infiltrating macrophages and T lymphocytes further produce proinflammatory factors to exacerbate brain edema and neurologic deficits after ICH.12, 14 However, little is known about the mechanisms by which macrophages and lymphocytes are activated after ICH as well as the proinflammatory factors they release to induce the later brain injury.
Among the proinflammatory factors released within the injured brain, interleukin (IL)‐17 generated by IL‐17–expressing helper T (Th17) cells has been shown to induce inflammation in experimental autoimmune encephalomyelitis (EAE), which is a mouse model of multiple sclerosis.15, 16 Research has also shown that IL‐23, a heterodimer of the IL‐23p19 and Il‐12p40 subunits, released by activated macrophages and dendritic cells (DCs) can induce differentiation of CD4+ T cells into Th17 cells to generate a major inflammatory reaction.17, 18 This IL‐23/IL‐17 inflammatory axis has been shown to be essential for the onset and progression of autoimmune inflammatory diseases in multiple mouse models, including EAE18 and collagen‐induced arthritis.19 Moreover, the T‐lymphocyte production of IL‐17 in response to macrophage‐derived IL‐23 was found to play a critical role in the later stage of brain infarction.20, 21 These data highlight an important role of the IL‐23/IL‐17 inflammatory axis in the acute neuroinflammatory response. However, the roles of macrophages and T lymphocytes as well as the IL‐23/IL‐17 inflammatory axis within the neuroinflammation that occurs after ICH remain to be determined.
The purpose of this study was to explore the roles of macrophages, T lymphocytes, and the cytokines they secrete in the secondary brain injury caused by ICH. ICH was induced using an established model in various lines of knockout mice, and the molecular events underlying the resulting neuroinflammatory responses were studied. The results of these experiments provide insight into the mechanisms of the neuroinflammation responsible for secondary brain injury after ICH and provide new targets for the development of therapies for ICH patients.
Materials and Methods
Animals
Male C57BL/6 mice, weighing 20 to 24 g, were obtained from the Animal Center of the Third Military Medical University (Chongqing, China). IL‐17−/−, IL‐23p19−/−, T‐cell receptor (TCR) γδ−/− (γδT−/−), and TIR domain‐containing adaptor‐inducing interferon‐β (TRIF)−/− mice (male, 20–24 g) were purchased from the Jackson Laboratory (Bar Harbor, ME). TLR2−/−, TLR4−/−, myeloid differentiation factor 88 (MyD88)−/−, and Rag1−/− mice (20–24 g) were obtained from the Model Animal Research Institute of Nanjing University (Nanjing Biomedical Research Institute, Nanjing, China). Each knockout mouse model was backcrossed onto the C57BL/6 background. TLR2−/− and TLR4−/− mice were bred to obtain TLR2/TLR4 double‐knockout mice (TLR2−/−/TLR4−/−), and TLR2 and TLR4 expression in mouse brain was assessed by Western blotting to identify the genotype of TLR2−/−/TLR4−/− mice, as previously reported.22 All animal experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals, 8th edition (2011) and ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
ICH Model
A previously reported model of ICH employing autologous blood was used in this study.23, 24 After mice were anesthetized by intraperitoneal (i.p.) injection of 80 mg/kg phenobarbital, they were fixed on a mouse stereotaxic frame (Stoelting, Wood Dale, IL). Then 20 μL of autologous blood collected from the tail vein and not treated with an anticoagulant was injected into the caudate nucleus using a syringe pump (KD Scientific, Holliston, MA) at 0.8 mm anterior, 2 mm right lateral, and 3.5 mm deep from the bregma. During the procedure, body temperature was kept at 37°C, and the mice had free access to food and water once awake. Mice that died were excluded from the study.
Cell Isolation and Adoptive Transfer in Mice
Based on previously reported methods,25 CD3+ T lymphocytes (with a purity >90%) were isolated from the spleen of wild‐type (WT) mice by following the manufacturer's instructions for the mouse CD3ε microbead kit (Miltenyi Biotec, Cologne, Germany). The isolated CD3+ T lymphocytes (1×107) were transfused back into the Rag1−/− mice intravenously 5 days before ICH.
BM‐Chimeric Mice
According to previously reported methods,20 recipient mice were given two doses (5‐Gy) of total body radiation 4 hours apart. After irradiation, recipients received an injection of 1×107 donor bone marrow (BM) cells in the tail vein. Ly5.1 and Ly5.2 mice were used to confirm the success rate of chimerism. At 6 weeks after irradiation and BM transplantation, >99% of CD11b+ cells in the blood of irradiated Ly5.2 mice were of the donor (Ly5.1+) origin. ICH was induced in the BM‐chimeric mice according to the model described above at 7 weeks after irradiation and transplantation.
Macrophage Depletion and CLP Administration
Clodronate liposomes (CLPs) and control liposomes were purchased from FormuMax Scientific, Inc. (Palo Alto, CA) and injected intraperitoneally 1 day before and 1 day after induction of ICH according to the dosing recommended by the manufacturer (0.2 mL/20–25 g). Four days after induction of ICH, the number of F4/80+ macrophages in the spleen was examined by flow cytometry to determine the efficiency of macrophage depletion.
Fingolimod Administration
According to previously reported methods,12, 13 fingolimod (Gilenya; Novartis Pharmaceuticals Corporation, East Hanover, NJ) was dissolved in 2% dimethyl sulfoxide (DMSO). Mice were divided into 4 groups: sham‐operated, ICH+vehicle (vehicle), ICH+fingolimod (1 mg/kg, i.p.) 5 minutes before ICH (fingolimod), and ICH+repeated fingolimod injection (1 mg/kg, i.p; fingolimod+R, once 5 minutes before ICH and once daily for the next 3 consecutive days after ICH). Both the sham‐ and vehicle‐treated groups received the same volume of 2% DMSO i.p.
SsnB Administration
Our previous research revealed that sparstolonin B (SsnB) reduces lipopolysaccharide‐induced Nuclear factor kappa B (NF‐κB) activity in a dose‐dependent manner, and the half‐maximal inhibitory concentration of SsnB is ≈10 μmol/L.26 For in vitro experiments, SsnB (1, 10, or 50 μmol/L) was dissolved in polyethylene glycol 400 and added to the cell culture medium for 3 hours. For in vivo experiments, SsnB was injected intravenously into mice at a dose of 1, 5, or 10 mg/kg at 2 hours after induction of ICH with different administration protocols (twice a day, once daily, or once every 2 days for 3 successive days). Because a dose of 5 mg/kg once daily achieved the maximum inhibitory effect on neurologic deficit score (NDS) after ICH (data not shown), the protocol (5 mg/kg SsnB injected 2 hours after ICH and once daily for the next 3 consecutive days) was used to test the therapeutic effects of SsnB on ICH mice. Mice in the control group received the same volume of vehicle.
Cell Culture
Bone marrow DCs
As previously described,27 BM was collected from the tibias and femurs of C57BL/6, TLR2−/−, TLR4−/−, TLR2−/−/TLR4−/−, MyD88−/−, and TRIF−/− mice. BM cells were passed through a nylon mesh and then cultured for 8 days in RPMI‐1640 medium with 10% fetal calf serum and 1 μg/mL mouse granulocyte‐macrophage colony‐stimulating factor Recombinant Protein (eBioscience, San Diego, CA).
HEK‐293 cells
Human embryonic kidney (HEK)‐293 cells for use in transfection experiments were obtained from ATCC (American Type Culture Collection, Manassas, VA) and cultured in high glucose–containing Dulbecco's modified Eagle medium (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Life Technologies).
Real‐Time PCR Analysis
Real‐time polymerase chain reaction (PCR) analysis was performed with the primers listed in Table S1, according to the manufacturer's instructions (Takara Biotechnology, Dalian, China). The reaction conditions were as follows: 95°C for 30 seconds; 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 1 minute; followed by 72°C for 5 minutes. The 2−ΔΔCT method was applied to calculate levels of mRNA expression relative to that of β‐actin.
Western Blot Analysis
According to previously reported methods,22 proteins from perihematoma tissues were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto polyvinylidene fluoride membranes via electroblotting. Membranes were incubated with IL‐23p19 antibody (rabbit polyclonal antibody to IL‐23p19, 1:200, Abcam, Cambridge, UK) or IL‐17 antibody (rabbit polyclonal antibody to IL‐17, 1:400; Abcam) at 4°C overnight, and the next day membranes were incubated with horseradish peroxidase–conjugated goat antimouse secondary antibody (1:300; Santa Cruz Biotechnology, Santa Cruz, CA) at 25°C for 1.5 hour. A chemiluminescence detection system was used to observe the resulting protein bands.
Immunoprecipitation
Cultured cells or infiltrating inflammatory cells sorted by fluorescence‐activated cell sorting (FACS) were lysed in radio immunoprecipitation assay (RIPA) lysis buffer as previously described.22 The extracted protein was then incubated with antibody to mouse TLR2 (1:100, Abcam), TLR4 (1:100, Abcam), or IgG as a control (1:100, Abcam) at 4°C overnight, followed by addition of unblocked protein A beads (1:400, Abcam) for 2 hours. After centrifugation, the immunoprecipitates were resuspended in SDS‐PAGE buffer and subjected to Western blot analysis to quantify protein expression.
Immunofluorescence and TUNEL Staining
Immunofluorescence staining was performed on brain tissue sections (thickness of 25 μm) and cultured BM–derived DCs (BMDCs) using the primary and secondary antibodies listed in Table S2 as well as 4′‐6‐diamidino‐2‐phenylindole (DAPI; 0.5 μg/mL; Roche, Indianapolis, IN). For staining, samples were blocked with goat serum, incubated with the primary antibodies at 4°C overnight, washed, and then exposed to secondary antibodies at 25°C for 1 hour.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the In Situ Cell Death Detection Kit of Roche Molecular Biochemicals (Mannheim, Germany), according to the manufacturer's instructions.
All stained sections were examined using a confocal laser microscope (Leica, Hamburg, Germany). Positively stained cells were counted on the cortex around the hematoma. The number of positive cells within 3 regions of interest, each 0.1 mm2 was expressed as an averaged value. Photomicrograph overlay and mild adjustment of contrast and brightness were achieved using Adobe Photoshop CS3 (Adobe Systems, San Jose, CA). Three trained researchers who were blinded to the grouping information performed the counting.
Flow Cytometry
Preparation of inflammatory cells in perihematomal tissues
Single‐cell suspensions of brain cells were prepared as previously described.28, 29 Briefly, at the designated time points, mice (n=5–8) were perfused transcardially using ice‐cold PBS to clear blood cells before dissection of the forebrain from the cerebellum. The harvested brain tissue was suspended in RPMI‐1640 medium and then digested in a 37°C shaker rotating at 180 rpm for 45 minutes upon addition of type IV collagenase (1 mg/mL; Sigma‐Aldrich, St. Louis, MO) and DNase I (50 mg/mL, Roche). From the brain tissue, inflammatory cells were isolated by 37% to 70% Percoll (GE Healthcare) density gradient centrifugation as previously described30 and then washed with RPMI‐1640 prior to flow cytometric analysis.
Surface marker and intracellular cytokine staining
For surface and intracellular cytokine staining, infiltrating inflammatory cells isolated by Percoll density gradient centrifugation as described above were stimulated for 5 hours with 50 nmol/L phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich), 1 μg/mL ionomycin (Sigma‐Aldrich), and 1 μmol/L brefeldin A (eBioscience). Surface staining of infiltrating inflammatory cells from ICH brain tissue was performed with fluorescently labeled antibodies to the surface markers CD45‐APC, CD3‐percpcy5.5, CD4‐PE/cy7, CD11b‐FITC, γδT‐PE, and Gr‐1‐PE (all from Biolegend, San Diego, CA) according to the manufacturer's protocol (Biolegend) for 15 minutes. Different types of inflammatory cells were identified according to expression of specific markers as follows: macrophages (CD45high CD11bhigh), microglia (CD45int CD11bint); neutrophils (CD45high, CD11bhigh, Gr‐1+); T lymphocytes (CD45high CD3+); Th lymphocytes (CD45high CD3+ CD4+); and γδT lymphocytes (CD45high CD3+ CD4− TCRγδ+). After surface staining, the cells were resuspended in Fixation Buffer (eBioscience), and intracellular cytokine staining was performed using FITC‐conjugated IL‐17 (Biolegend) according to the manufacturer's protocol.
FACS analysis
FACS analysis was performed using a FACSVerse analyzer (BD Biosciences, San Jose, CA), and the data were analyzed using FlowJo software 7.6.1 (FlowJo, LLC, Ashland, OR).
NF‐κB Activity
According to a previously reported method,22 HEK‐293 cells (ATCC, 1×107) were transfected with both NF‐κB‐luciferase reporter gene (Addgen.org, plasmids 13642) and pcDNA3.1 vectors containing TLR2‐YFP, TLR4‐YFP, or MyD88 (Addgen.org, plasmids 13016, 13018, and 13026, respectively) and fused to yellow fluorescent protein (YFP). Mutation of the Arg196 of MyD88 was conducted as previously reported.22 Forty‐eight hours after cotransfection, cells were stimulated under the specified conditions for 12 hours. Cells were subsequently lysed, and reporter gene activity was measured using a Dual Luciferase Assay Reporter System (E1910, Promega, Madison, WI).
Liquid Chromatography‐Mass Spectrometry
Four days after ICH, WT mice were intravenously injected with SsnB at a dose of 5 mg/kg in polyethylene glycol 400, and mice in the control group received the same volume of vehicle intravenously. Cerebral tissues were sampled at 10, 20, and 30 minutes after injection. SsnB levels in the harvested samples were measured using liquid chromatography‐mass spectrometry (LC‐MS) (6410B Triple Quad LC/MS, Agilent Technologies, Palo Alto, CA).
Brain Water Content
Brain water content (BWC) was measured in mouse cerebral tissues after ICH as previously described.22 Briefly, the surface water on harvested cerebral tissue samples was blotted with filter paper, and the cerebellum and contralateral cerebral hemisphere were removed. Brain samples were immediately weighed on an electric analytic balance to obtain the wet weight and then dried at 100°C for 24 hours before measurement of the dry weight. BWC was calculated using the following formula: BWC (%)=(wet weight–dry weight)/wet weight×100%.
Neurologic Deficit Score
Multiple behavioral tests were applied to characterize neurological deficits as previously reported.22, 31 Two trained investigators who were blinded to animal grouping performed the scoring, and the final score for each mouse was obtained by calculating the mean scores for the subscales.
Statistical Analysis
All statistical tests were performed using SPSS 18.0 software (IBM, Armonk, NY), and all data are presented as the mean±standard error. The significance of differences between 2 and ≥3 groups was determined using nonparametric tests, Student t test, and 1‐way ANOVA followed by Scheffe post hoc test, respectively. The effects of multiple factors were studied via 2‐way ANOVA with repeated measures. For all tests, P<0.05 was considered statistically significant.
Results
Infiltration of Peripheral Inflammatory Cells and Their Involvement in Secondary Injury After ICH
After induction of ICH in WT C57BL/6 mice, the temporal changes in the numbers of different types of inflammatory cells in the hemorrhagic hemisphere were analyzed by FACS. Microglia and macrophages were gated according to the fluorescence intensity of CD45 and CD11b staining. The CD45high group was gated and further analyzed for the expression of CD3 and CD4. The CD45high CD3+ cells were gated and further analyzed for the expression of γδT (Figure S1). We observed the highest absolute numbers of CD45+ cells (Figure 1A), including T lymphocytes (CD3+ cells; Figure 1B), CD4+ T lymphocytes (Figure 1C), and γδT lymphocytes (Figure 1D), on day 4 after ICH, while the number of macrophages peaked at day 1 (Figure 1E). In contrast, no changes in the numbers of microglia were observed after ICH (Figure 1F). In confirmation of these FACS results, immunofluorescence staining showed that F4/80+ (macrophages/microglia; Figure 1G) and CD3+ cells (T lymphocyte; Figure 1H) were located along the perihematoma area after ICH.
Figure 1.
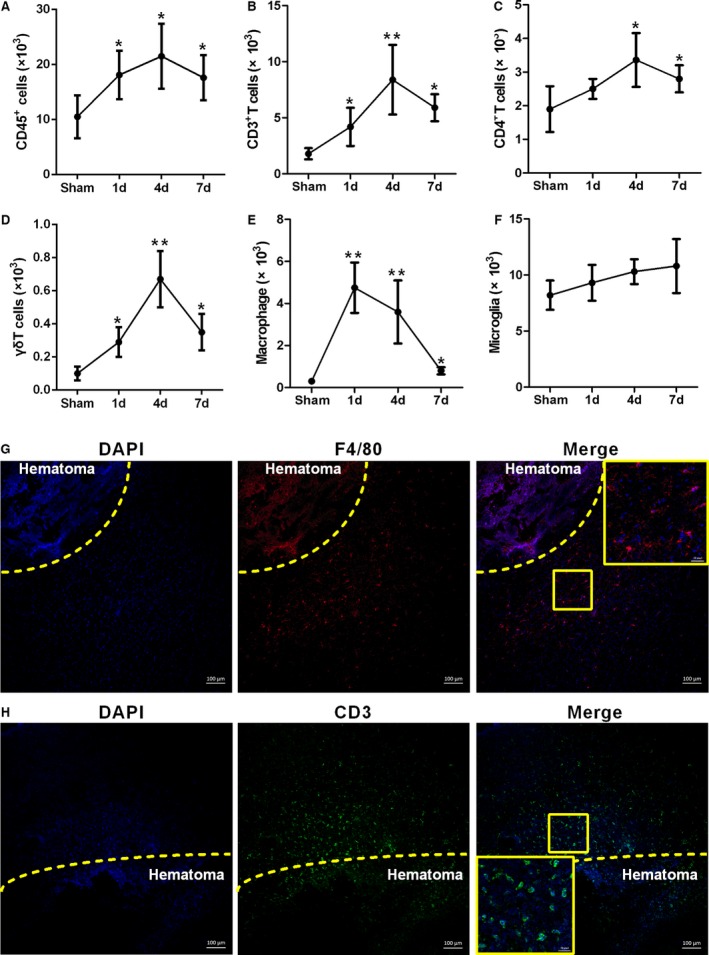
Macrophage and T‐lymphocyte infiltration into mouse brain after intracerebral hemorrhage (ICH). A through F, Temporal changes in absolute numbers of different inflammatory cell types in hemorrhagic hemispheres at 1, 4, and 7 days after ICH. Data were obtained for cells pooled from 6 mice, and the experiments were repeated 3 times. *P<0.05, **P<0.01 vs sham. G, Representative fluorescence microscopy images showing infiltrating F4/80+ cells in the perihematoma area at 1 day after ICH (blue=4′‐6‐diamidino‐2‐phenylindole [DAPI], red=F4/80, scale bars=100 μm). H, Representative fluorescence microscopy images showing CD3+ cell infiltration into the perihematoma area at 4 days after ICH (blue=DAPI, green=CD3, scale bars=100 μm).
Because the absolute numbers of macrophages peaked on day 1 after ICH, we first explored whether macrophage infiltration of the brain is required for ICH‐induced injury by depleting peripheral macrophages using CLPs. In these liposomes, clodronate is encapsulated at a concentration of 7 mg/mL, and systemic administration with a dose of 0.2 mL/20 to 25 g has been demonstrated to achieve efficient depletion of macrophages within 24 to 36 hours.32, 33 We confirmed depletion of 76.2% of F4/80+ macrophages in the spleen of ICH mice at 4 days after the first CLP injection (Figure S2). Intraperitoneal injection of CLPs also significantly reduced the number of infiltrating macrophages in the brain at 1 day and 4 days after ICH (Figure 2A), without influencing the infiltration of T lymphocytes (Figure 2B). Moreover, we found that CLP injection significantly reduced the NDS (Figure 2C) and BWC (Figure 2D) of WT mice with ICH. These findings that macrophage depletion alleviated ICH‐induced brain damage in mice suggest that macrophage infiltration plays a key role in ICH‐induced brain injury.
Figure 2.
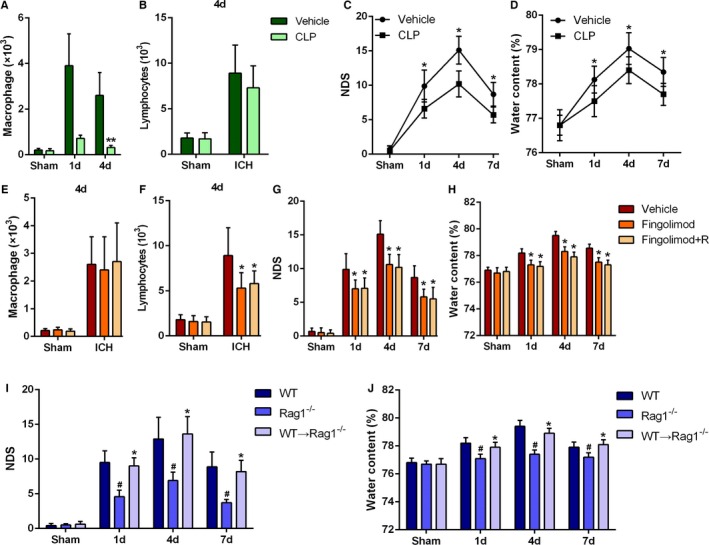
Roles of macrophages and T lymphocytes in intracerebral hemorrhage (ICH)–induced inflammation. A, Absolute numbers of infiltrating macrophages on day 1 and day 4 after ICH. B, Absolute numbers of infiltrating T lymphocytes on day 4 after ICH in clodronate liposomes (CLP)–treated or liposome (vehicle)‐treated mice. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times. **P<0.01 vs vehicle. C, Neurologic deficit score (NDS) at 1, 4, and 7 days after ICH in the CLP‐ and vehicle‐treated mice. *P<0.05 vs vehicle, n=6 per group. D, Brain water content (BWC) at 1, 4, and 7 days after ICH in the CLP‐ and vehicle‐treated mice. *P<0.05 vs vehicle, n=4 per group. Two‐way ANOVA reported a significant difference in main effects of all treatment groups (P<0.05) but not of time points (P>0.05), there was no interaction between treatments and time points (P>0.05). E and F, Absolute numbers of infiltrating macrophages and T lymphocytes on day 4 after ICH in fingolimod‐treated or untreated mice. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times. *P<0.05 vs vehicle. G, NDS for vehicle (dimethyl sulfoxide [DMSO])‐, fingolimod‐, and fingolimod+R‐treated mice at 1, 4, and 7 days after ICH. *P<0.05 vs vehicle, n=6 per group. Two‐ANOVA reported a significant difference in main effects of all treatment groups (P<0.05) but not of time points (P>0.05), there was no interaction between treatments and time points (P>0.05). H, BWC for vehicle (DMSO)‐, fingolimod‐, and fingolimod+R‐treated mice at 1, 4, and 7 days after ICH. *P<0.05 vs vehicle, n=4 per group. I, NDS for wild‐type (WT), Rag1−/−, and Rag1−/− mice that received CD3+ T lymphocytes from WT mice (WT CD3→Rag1−/−) at 1, 4, and 7 days after ICH. # P<0.05 vs WT mice, *P<0.05 vs Rag1−/− mice, n=5. Two‐way ANOVA reported significant difference in main effects of genotype (P<0.05) but not of time points (P>0.05), there was no interaction between genotypes and time points (P>0.05). J, BWC for WT, Rag1−/−, and WT CD3→Rag1−/− mice at 1, 4, and 7 days after ICH. # P<0.05 vs WT mice, *P<0.05 vs Rag1−/− mice, n=5.
We next investigated the role of T lymphocytes in ICH‐induced secondary brain injury by using fingolimod to inhibit T‐lymphocyte migration. At day 4 after the ICH in mice treated with fingolimod, no change was observed in macrophage infiltration (Figure 2E), but T‐lymphocyte infiltration was significantly reduced (Figure 2F). In addition, we also found that fingolimod treatment ameliorated the neurologic deficit (Figure 2G) and BWC (Figure 2H) changes induced by ICH in WT mice over 7 days. To confirm that these protective effects of fingolimod were related to reduced infiltration of peripheral T cells, we performed adoptive transfer of T lymphocytes into mice deficient in recombination activating gene‐1 (Rag1−/− mice). First, we found that Rag1−/− mice exhibited a lower NDS (Figure 2I) and BWC (Figure 2J) than WT mice after ICH, which was consistent with previous results for experimentally induced ischemic stroke.34 Moreover, the NDS (Figure 2I) and BWC (Figure 2J) were increased when WT T lymphocytes were transferred into Rag1−/− mice. These results suggest that infiltrating peripheral T lymphocytes can aggravate secondary brain injury in mice after ICH.
Hb‐Induced TLR2/TLR4 Heterodimer Formation on Infiltrating Macrophages Aggravated Secondary Brain Injury After ICH
Our previous research showed that hemoglobin (Hb) promotes microglial activation by inducing the formation of TLR2/TLR4 heterodimers and then exacerbates brain injury following ICH by amplifying the production of inflammatory cytokines.22 To determine whether Hb has a similar effect on infiltrating macrophages, we observed the colocalization of TLR2 and TLR4 on F4/80+ cells perihematomal in ICH mouse brain tissue by immunohistochemical staining (Figure 3A). However, F4/80+ cells included not only macrophages but also microglia. To clarify whether TLR2 and TLR4 colocalized on macrophages, we stimulated BMDCs with Hb (5 μmol/L, 3 hours) and observed colocalization of TLR2 and TLR4 on these cells (Figure 3B). Furthermore, to investigate the link between TLR2 and TLR4 on macrophages, we cultured BMDCs from WT mice and separated macrophages from the brain or spleen of WT mice at day 1 after ICH. Then immunoprecipitation assays were performed to detect the coprecipitation of TLR2 and TLR4 in those cells. Among the red blood cell components Hb, hemin, bilirubin, Fe2+, and Fe3+, only Hb was found to induce coprecipitation of TLR2 and TLR4 (Figure 3C). Immunoprecipitation assays also revealed that Hb induced coprecipitation of TLR2 and TLR4 on brain‐derived macrophages of ICH mice, but not on spleen‐derived macrophages (Figure 3D). These findings suggest that Hb induces TLR2/TLR4 heterodimer formation on macrophages located specifically in the brain but not in the peripheral region.
Figure 3.
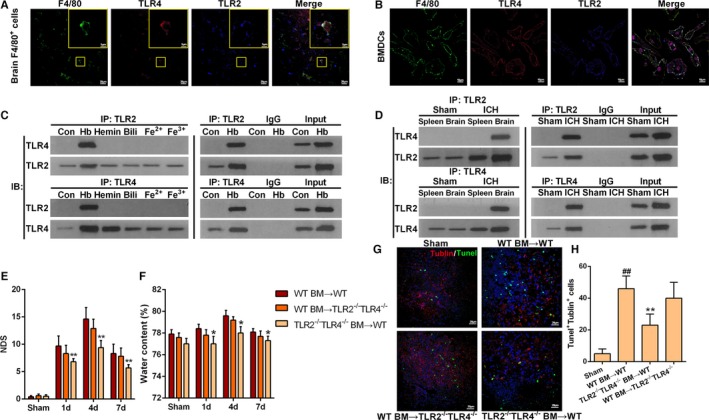
Hemoglobin (Hb)‐induced toll‐like receptor (TLR) 2/4 heterodimer formation on infiltrating macrophages. A, Representative immunofluorescence staining images showing colocalization of TLR2 and TLR4 in F4/80+ cells of perihematomal tissues at 1 day after intracerebral hemorrhage (ICH; scale bars=20 μm). B, Representative immunofluorescence staining images showing colocalization of TLR2 and TLR4 in cultured bone marrow (BM)–derived dendritic cells (BMDCs) after stimulation with Hb (5 μmol/L) for 3 hours (scale bars=10 μm). C, Coprecipitation of TLR2 and TLR4 on cultured BMDCs stimulated with components of red blood cells including Hb, hemin, bilirubin (Bili), Fe2+, and Fe3+, each at a concentration of 5 μmol/L for 3 hours (n=3). D, Coprecipitation of TLR2 and TLR4 on brain infiltrating macrophages or macrophages separated from spleen at 1 day after ICH. Data were obtained for samples pooled from 10 mice, and the experiments were repeated 3 times. E, Neurologic deficit score (NDS) for BM‐chimeric mice analyzed at 1, 4, and 7 days after ICH. TLR2−/−/TLR4−/− BM→WT represents transfer of TLR2/TLR4 double‐knockout BM cells into wild‐type (WT) mice. *P<0.05 vs WT BM→TLR2−/−/TLR4−/− mice, n=4. Two‐way ANOVA reported significant difference in main effects of BM‐chimeric (P<0.05) but not of time points (P>0.05), there was no interaction between BM‐chiemeric and time points (P>0.05). F, Brain water content for BM‐chimeric mice analyzed at 1, 4, and 7 days after ICH. *P<0.05 vs WT BM→TLR2−/−/TLR4−/− mice, n=4. G, Representative immunofluorescence staining images showing βIII tubulin and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) double‐positive cells in the perihematoma area. Brain sections obtained on day 4 after ICH (red=tubulin, green=TUNEL, blue=4′‐6‐diamidino‐2‐phenylindole [DAPI], scale bars=20 μm). H, Quantification of βIII tubulin and TUNEL double‐positive cells at 4 days after ICH. *P<0.05 vs WT, ## P<0.01 vs sham, n=5.
To test whether TLR2/TLR4 heterodimer on hematopoietic cells rather than resident brain cells plays a key role in ICH‐induced secondary injury, we used BM‐chimeric mice. The NDS (Figure 3E), BWC (Figure 3F), and number of apoptotic neurons (Figure 3G and 3H) were lower in WT mice that received TLR2−/−/TLR4−/− BM cells compared with those in TLR2−/−/TLR4−/− mice that received WT BM cells.
IL‐23p19 Produced by Infiltrating Macrophages Exacerbated Secondary Brain Injury Following ICH
IL‐23p19 is known to play key roles in the establishment and maintenance of organ‐specific inflammatory autoimmune diseases.18, 19, 35, 36 To study IL‐23p19 involvement in ICH‐induced brain injury, Western blot and real‐time PCR analyses were performed to detect IL‐23p19 expression at days 1, 4, and 7 after ICH. Compared with those in sham controls, IL‐23p19 protein (Figure 4A) and mRNA (Figure 4B) levels were significantly higher in the perihematoma tissues of ICH mice, peaking on day 1 after ICH (Figure 4A and 4B). To further elucidate the role of IL‐23p19, we compared ICH injury levels between IL‐23p19−/− mice and WT mice. IL‐23p19−/− mice showed a lower NDS (Figure 4C) and BWC (Figure 4D) and fewer apoptotic neurons after ICH (Figure 4E and 4F) than WT mice. In addition, IL‐1β and tumor necrosis factor‐α (TNF‐α) gene expression in infiltrating CD45+ cells was significantly lower in IL‐23p19−/− mice than in WT mice at day 4 after ICH (Figure 4G). These results suggest that IL‐23p19 contributed to brain injury after ICH, which prompted us to next investigate the source of IL‐23p19 in the injured brain.
Figure 4.
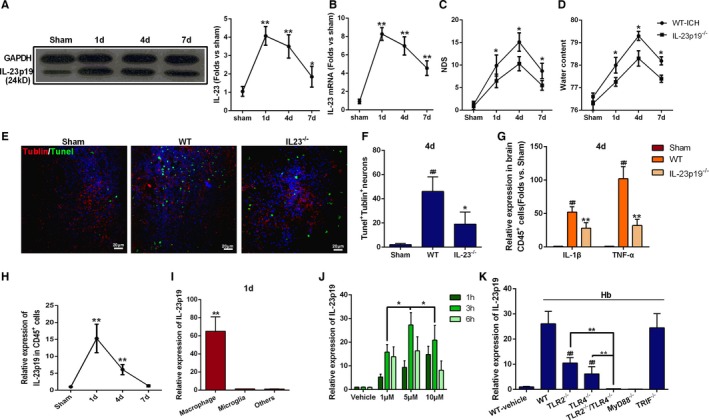
Infiltrating macrophage production of interleukin (IL)‐23p19 in brain injury following intracerebral hemorrhage (ICH). A and B, Protein and mRNA expression of IL‐23p19 in perihematoma tissues were obtained from wild‐type (WT) mice on days 1, 4, and 7 after ICH. *P<0.05 vs sham, **P<0.01 vs sham, n=6. C, Neurologic deficit score (NDS) for WT and IL‐23p19−/− mice at 1, 4, and 7 days after ICH. *P<0.05 vs WT, n=6. Two‐way ANOVA reported significant difference in main effects of genotype (P<0.05) but not of time points (P>0.05), there was no interaction between genotype and time points (P>0.05). D, Brain water content (BWC) for WT and IL‐23p19−/− mice at 1, 4, and 7 days after ICH. *P<0.05 vs WT, n=6. E, Representative immunofluorescence staining images showing βIII tubulin and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) double‐positive cells in the perihematoma area of WT and IL‐23−/− mice. Brain sections were obtained on day 4 after ICH (scale bars=20 μm). F, Quantification of βIII tubulin and TUNEL double‐positive cells on day 4 after ICH. *P<0.05 vs WT, ## P<0.01 vs sham, n=5. G, mRNA levels of IL‐1β and tumor necrosis factor‐α (TNF‐α) in infiltrating CD45+ cells of WT and IL‐23p19−/− mice on day 4 after ICH. *P<0.05, **P<0.01 vs WT, ## P<0.01 vs sham, n=4. H, Time course of IL‐23p19 mRNA expression in infiltrating CD45+ cells of WT mice after ICH. Expression was normalized to that in the sham group. **P<0.01 vs sham, n=5. I, IL‐23p19 mRNA expression in macrophages, microglia, and other cells. Data were obtained for samples pooled from 5 mice and normalized to expression in microglia, and the experiments were repeated 3 times. **P<0.01 vs microglia. J, IL‐23p19 mRNA expression in bone marrow–derived dendritic cells (BMDCs) from WT mice cultured with various concentrations of hemoglobin (Hb; 1, 5, and 10 μmol/L) for 1, 3, and 6 hours. Relative expression to that of the vehicle‐treated group. *P<0.05 vs 1 μmol/L and 10 μmol/L for 3 hours, n=5. K, IL‐23p19 mRNA levels in BMDCs from WT, toll‐like receptors (TLRs) 2−/−, TLR4−/−, TLR2−/−/TLR4−/−, myeloid differentiation factor 88 (Myd88)−/−, and TIR domain‐containing adaptor‐inducing interferon‐β (TRIF)−/− mice detected 3 hours after stimulation with 5 μmol/L Hb. Relative expression to that of the vehicle‐treated WT group. ## P<0.01 vs WT, **P<0.05 vs TLR2−/− and TLR4−/−, n=4).
IL‐23p19 mRNA expression on infiltrating inflammatory (CD45+) cells was highest at day 1 after ICH (Figure 4H), corresponding to the peak in macrophage infiltration after ICH. Upon sorting of the infiltrating inflammatory cells into macrophages, microglia, and “other” cells (Figure S3), IL‐23p19 mRNA was found to be expressed dominantly in the macrophages, with little expression in microglia and other cells (Figure 4I). Additionally, IL‐23p19 mRNA expression in perihematoma tissues was markedly decreased in the CLP mice compared with the vehicle‐treated groups (Figure S2C). These data indicate that infiltrating macrophages were the major source of IL‐23p19 in the mouse brain after ICH.
To verify whether Hb is essential for IL‐23p19 production, we cultured BMDCs with different concentrations of Hb for 1, 3, and 6 hours. Stimulation with 5 μmol/L Hb for 3 hours led to the greatest IL‐23p19 production (Figure 4J), and, thus, this concentration and stimulation duration were used in subsequent experiments. Next, we investigated the relationship between TLR2/TLR4 heterodimer formation and IL‐23 production. Hb‐mediated induction of IL‐23p19 expression was lower in TLR2−/− and TLR4−/− BMDCs than in WT BMDCs and lower still in TLR2−/−/TLR4−/− BMDCs (Figure 4K).
Given that MyD88 and TRIF are independent adaptor proteins in the TLR signaling pathway, they also may be involved in the Hb‐mediated expression of IL‐23p19 in BMDCs. We found that MyD88−/− BMDCs produced less IL‐23p19 than WT BMDCs (Figure 4K), whereas IL‐23p19 production was comparable between TRIF−/− BMDCs and WT BMDCs. These results suggest that IL‐23p19 expression relies on TLR2/TLR4 heterodimer formation in a MyD88‐dependent manner.
γδT+ T Lymphocyte Produced IL‐17 in Brain Injured by ICH
IL‐23 has been shown to induce CD4+ T‐cell differentiation into Th17 cells, and this IL‐23/IL‐17 axis plays a central role in several inflammatory diseases.17, 18, 19, 35, 36 Thus, we examined the role of IL‐17 in ICH‐induced brain injury by intracellular cytokine staining and found enrichment of IL‐17+ cells in the perihematoma tissue at day 4 after ICH (Figure 5A), which corresponds to the peak in T‐lymphocyte infiltration. To examine the source of IL‐17 in the injured brain, we performed immunostaining for various surface markers along with intracellular cytokine staining and found that IL‐17 was colocalized with CD3 in the perihematoma area at 4 days after ICH (Figure 5B), but absent in CD11b+, βIII tubulin+, and GFAP+ cells (data were not shown), indicating that T lymphocytes were likely the source of IL‐17 in ICH brain, which was further confirmed by FACS data (Figure S4). Flow cytometric analysis of γδT and CD4 expression on IL‐17+ CD3+ T cells indicated that most IL‐17–producing T lymphocytes were CD4− γδT+ cells (Figure 5C).
Figure 5.
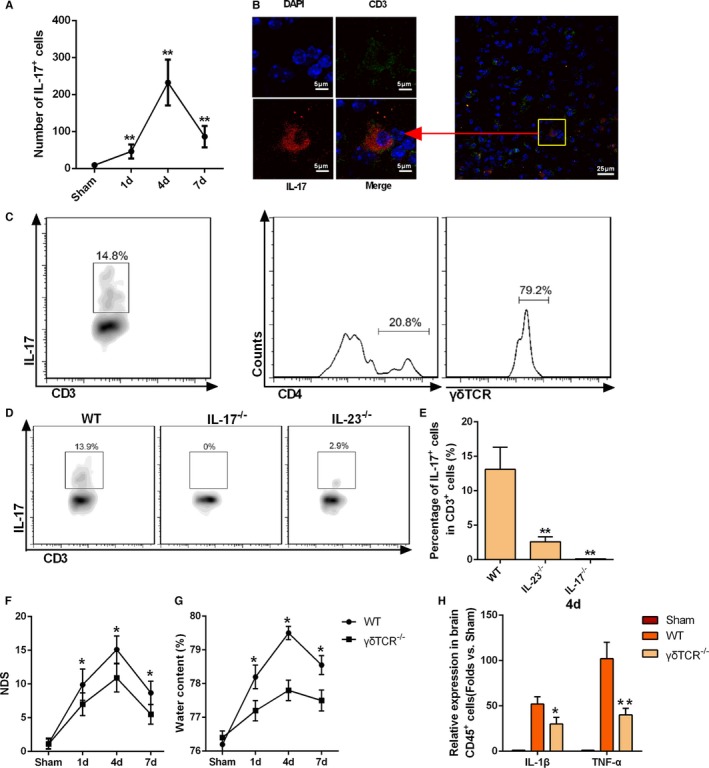
γδT‐cell production of interleukin (IL)‐17 after intracerebral hemorrhage (ICH). A, IL‐17+ T lymphocytes identified by fluorescence‐activated cell sorting (FACS) staining at 1, 4, and 7 days after ICH. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times, *P<0.05 vs sham. B, Immunofluorescence staining of CD3+ and IL‐17+‐positive cells in the perihematoma area of wild‐type (WT) mice at 4 days after ICH (scale bars=20 μm). C, Flow cytometric analysis of γδT cell receptor (γδT) and CD4 expression on the CD3+ IL‐17+ T cells. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times. D and E, Percentages of IL‐17+ cells among infiltrating T lymphocytes of WT, IL‐17−/−, and IL‐23−/− mice. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times. **P<0.01 vs WT. F, Neurologic deficit score (NDS) for WT and γδT −/− mice as analyzed at 1, 4, and 7 days after ICH. *P<0.05 vs WT, n=5. Two‐way ANOVA reported significant difference in main effects of genotype (P<0.05) but not of time points (P>0.05), there was no interaction between genotype and time points (P>0.05). G, Brain water content for WT and γδT −/− mice as analyzed at 1, 4, and 7 days after ICH. *P<0.05 vs WT, n=5. H, IL‐1β and tumor necrosis factor‐α (TNF‐α) mRNA levels in infiltrating CD45+ cells of WT and γδT −/− mice at day 4 after ICH. *P<0.05, **P<0.01 vs WT, n=4.
To confirm the relationship between IL‐23 expression and IL‐17 production in the ICH brain, we examined the percentage of IL‐17+ cells among infiltrating CD3+ T lymphocytes in IL‐23p19−/− mice. The lower percentage of IL‐17+ CD3+ T lymphocytes in these mice (Figure 5D and 5E) suggests that IL‐23p19 is essential for production of IL‐17 after ICH.
Finally, to verify the contribution of γδT lymphocytes in ICH‐induced brain injury, γδT−/− mice were used. The NDS (Figure 5F), BWC (Figure 5G), and mRNA expression of IL‐1β and TNF‐α (Figure 5H) were significantly lower in γδT−/− mice than in WT mice after ICH.
IL‐17 Played a Critical Role in ICH‐Induced Secondary Brain Injury
To confirm the importance of IL‐17 expression in ICH‐induced secondary brain injury, we detected the dynamic expression of IL‐17 in the mouse brain after ICH. Significant increases in both IL‐17 protein (Figure 6A) and mRNA (Figure 6B) were observed from day 4 after ICH. We next compared the severity of ICH‐induced injury between IL‐17−/− mice and WT mice. IL‐17−/− mice had significantly lower NDS (Figure 6C) and BWC (Figure 6D), and fewer apoptotic neurons were detected in IL‐17−/− mice compared with WT mice (Figure 6E and 6F). However, no differences in macrophage and T‐lymphocyte infiltration in response to ICH were observed between IL‐17−/− mice and WT mice (Figure 6G). Still, IL‐1β and TNF‐α mRNA expression in perihematoma tissues was significantly lower in IL‐17−/− mice than in WT mice on day 4 after ICH (Figure 6H). CD45+ inflammatory cells in IL‐17−/− mice also showed lower IL‐1β and TNF‐α mRNA expression on day 4 after ICH compared with these cells in WT mice (Figure 6I). Together, these findings indicate that elevated IL‐17 expression aggravated secondary brain injury after ICH, and although IL‐17 deficiency was not associated with decreased infiltration of inflammatory cells, increased expression of inflammatory factors including IL‐1β and TNF‐α may contribute to alleviation of ICH‐induced injury.
Figure 6.
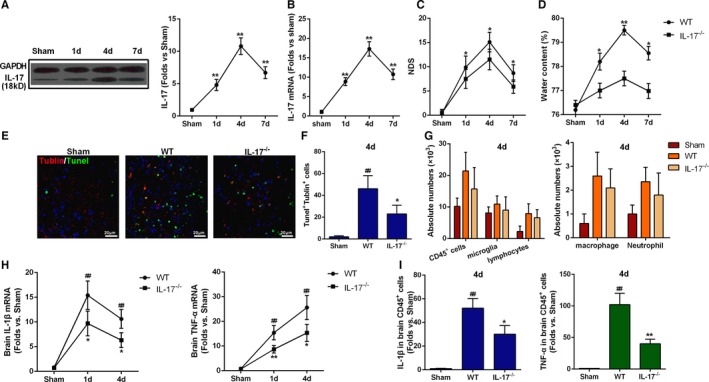
Interleukin (IL)‐17 aggravated brain injury induced by intracerebral hemorrhage (ICH). A and B, IL‐17 protein and mRNA expression in the perihematoma tissues at days 1, 4, and 7 after ICH. **P<0.01 vs sham, n=5. C, Neurologic deficit score (NDS) for wild‐type (WT) and IL‐17−/− mice at 1, 4, and 7 days after ICH. **P<0.01 vs sham, n=5. Two‐way ANOVA reported significant difference in main effects of genotype (P<0.05) but not of time points (P>0.05), there was no interaction between genotype and time points (P>0.05). D, Brain water content for WT and IL‐17−/− mice at 1, 4, and 7 days after ICH. **P<0.01 vs sham, n=5. E, Representative immunofluorescence staining images showing βIII tubulin and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) double‐positive cells around the perihematoma of WT and IL‐17−/− mice at day 4 after ICH. Scale bars=20 μm,*P<0.05 vs WT, ## P<0.01 vs sham, n=5. F, Quantification of the βIII tubulin and TUNEL double‐positive cells in the perihematoma of WT and IL‐17−/− mice at 4 days after ICH. *P<0.05 vs WT, ## P<0.01 vs sham, n=5. G, Absolute numbers of cells (CD45+ cells, microglia, lymphocytes, macrophages, and neutrophils) in sham‐treated, WT, and IL‐17−/− mice. Data were obtained for samples pooled from 5 mice, and the experiments were repeated 3 times. H, mRNA levels of IL‐1β and tumor necrosis factor‐α (TNF‐α) in brain tissue at 1, 4, and 7 days after ICH. I, mRNA levels of IL‐1β and TNF‐α in infiltrating CD45+ cells at day 4 after ICH. *P<0.05, **P<0.01 vs WT, ## P<0.01 vs sham, n=5.
SsnB Ameliorated Secondary Brain Injury After ICH by Disrupting TLR2/TLR4 Heterodimer Formation
Because our results indicate that TLR2/TLR4 heterodimer formation is essential for secondary injury following ICH, disruption of TLR2/TLR4 heterodimer formation may be a potential therapeutic strategy for ICH. We previously found that SsnB, a monomer component of Chinese traditional herbs (Figure S5A), can reduce lipopolysaccharide (LPS)‐induced inflammatory injury, which suggested that SsnB may inhibit the TLR pathway.26, 37 In the present study, immunoprecipitation indicated that SsnB inhibited TLR2/TLR4 heterodimer formation on cultured BMDCs (Figure 7A). We further examined the inhibitory activity of derivatives of SsnB (Figure S5B). SA3 treatment inhibited formation of TLR2/TLR4 heterodimer, whereas SsnB exhibited the greatest potential to inhibit TLR2/TLR4 heterodimer formation (Figure 7A). Furthermore, TLR2/TLR4 heterodimer formation was inhibited by SsnB in a dose‐dependent manner (Figure 7B), with a high dose (50 μmol/L) of SsnB completely abolished Hb‐induced TLR2/TLR4 heterodimer formation (Figure 7B). Based on the involvement of TLR2/TLR4 heterodimer in Hb‐mediated expression of IL‐23p19, we detected IL‐23p19 in SsnB‐treated BMDCs and observed a reduction in IL‐23p19 expression (Figure 7C).
Figure 7.
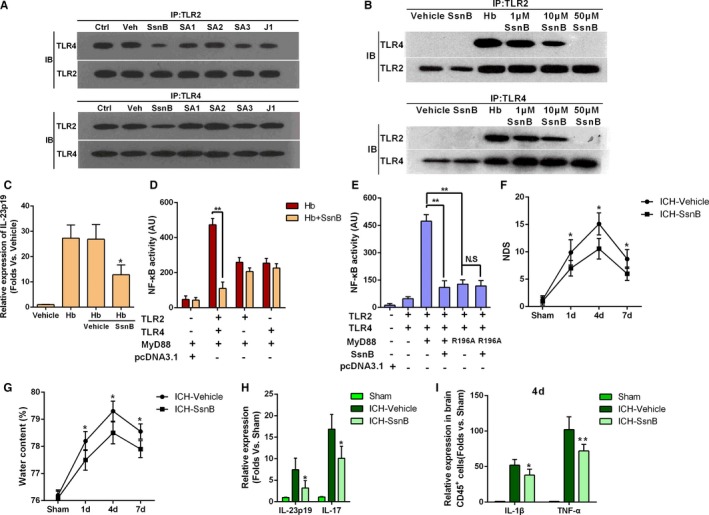
Sparstolonin B (SsnB) ameliorated intracerebral hemorrhage (ICH)–induced secondary injury by disrupting toll‐like receptor (TLR) 2/4 heterodimer formation. A, Coimmunoprecipitation of compounds derived from SsnB/J1 in cultured bone marrow–derived dendritic cells (BMDCs) exposed to 5 μmol/L hemoglobin (Hb; Ctrl) or Hb+vehicle/SsnB/one (all at 10 μmol/L) (with Ctrl indicating control only treated with Hb; Veh, Hb+ vehicle; IP, immunoprecipitation; IB, immunoblotting; n=5). B, IP of TLR2/TLR4 heterodimer in cultured BMDCs treated with Hb (5 μmol/L), SsnB (10 μmol/L), and Hb (5 μmol/L)+SsnB (1, 10, 20 μmol/L) (n=4). C, Interleukin (IL)‐23p19 mRNA expression in cultured Hb‐activated BMDCs treated with vehicle and SsnB (10 μmol/L). *P<0.05 vs Hb+vehicle, n=3. D, Nuclear factor‐κB (NF‐κB) activity in human embryonic kidney (HEK)‐293 cells treated with Hb and SsnB with or without TLR2 and TLR4 expression. **P<0.01, n=5. E, Effect of SsnB on Hb‐induced NF‐κB activity in TLR2‐TLR4‐myeloid differentiation factor 88 (MyD88)–transfected HEK‐293 cells (R196A, N‐terminal domain mutation of MyD88). **P<0.01, NS=no significant difference, n=5. F, Neurologic deficit score (NDS) for SsnB‐treated and untreated WT mice after ICH at 1, 4, and 7 days after ICH. *P<0.05 vs vehicle, n=5. Two‐way ANOVA reported significant difference in main effects of treatments (P<0.05) but not of time points (P>0.05), there was no interaction between treatments and time points (P>0.05). G, Brain water content for SsnB‐treated and untreated WT mice after ICH at 1, 4, and 7 days after ICH. *P<0.05 vs vehicle, n=5. H, IL‐23p19 and IL‐17 mRNA expression in the hemorrhagic hemisphere of SsnB‐treated or untreated WT mice on day 4 after ICH (*P<0.05 vs ICH‐vehicle, n=5). I, IL‐1β and tumor necrosis factor‐α (TNF‐α) mRNA levels in infiltrating CD45+ cells on day 4 after ICH. *P<0.05, **P<0.01 vs vehicle, n=4.
NF‐κB activity assays were performed to test the ability of SsnB to inhibit the inflammatory reaction. Before the test, HEK‐293 cells were transfected with TLR2, TLR4, and MyD88 containing pcDNA3.1. SsnB significantly inhibited NF‐κB activity in HEK‐293 cells transfected with both TLR2 and TLR4, but the activity remained unchanged in HEK‐293 cells expressing only TLR2 or TLR4 (Figure 7D). These results indicate that SsnB inhibited NF‐κB activity by interrupting TLR2/TLR4 heterodimer formation. Furthermore, compared with that in control MyD88‐expressing HEK‐293 cells, NF‐κB activity was significantly reduced in MyD88 nonexpressing HEK‐293 cells (Figure 7E). This finding confirmed that MyD88 was essential for TLR2/TLR4 heterodimer formation, and the inhibitory effect of SsnB may be related to MyD88. Our previous research demonstrated that mutation of residue Arg196 of MyD88 can interrupt TLR2/TLR4 dimerization,22 which together with our present findings suggests that residue Arg196 on MyD88 may be the active site of SsnB. In support of this notion, a mutation assay showed that NF‐κB activity was significantly inhibited when SsnB was added to HEK‐293 cells expressing normal MyD88, while the NF‐κB activity was both significantly inhibited in cells expressing mutant MyD88 and showed no significant difference with the absence or presence of SsnB (Figure 7E).
To further investigate whether SsnB treatment benefits ICH model mice, we first examined the ability of SsnB to cross the BBB. Indeed, SsnB was detected in the brains of ICH mice by LC‐MS (Figure S6). Then, the effect of SsnB on ICH‐induced injury in mice was evaluated. SsnB (5 mg/kg) was injected intravenously into mice 2 hours after ICH and then once daily for the next 3 days. Compared with the vehicle treatment, SsnB treatment led to a decreased NDS (Figure 7F), BWC (Figure 7G), and expression of proinflammatory factors including IL‐23p19, IL‐17 (Figure 7H), IL‐1β, and TNF‐α (Figure 7I). In addition, SsnB had no effect on the rectal temperature and peripheral white cell counts before and after ICH (Table S3), which suggests that SsnB treatment did not increase the risk of secondary infection. Together, our data indicate that SsnB significantly inhibited TLR2/TLR4 heterodimer formation and ameliorated secondary brain injury after ICH.
Discussion
The peripheral inflammatory cells responsible for neuroinflammation play important roles in the secondary brain injury that occurs after ICH, but the precise roles and mechanisms remain to be elucidated. The results of the present study revealed that infiltrating macrophages and γδT cells could worsen the secondary injury after ICH. We found that: (1) Hb from the hematoma could activate infiltrating macrophages to secrete IL‐23 by inducing formation of TLR2/TLR4 heterodimer; (2) IL‐23 derived from activated macrophages could drive IL‐17 production by γδT cells and further increase neuroinflammation and secondary injury after ICH; and (3) SsnB could ameliorate secondary brain injury of ICH by disrupting TLR2/TLR4 heterodimer formation. Together, these findings reveal the mechanisms of neuroinflammation in ICH‐induced secondary brain injury and provide new insight for the development of a novel targeted ICH therapy.
Our previous research revealed that during the hyperacute phase (<6 hours) of ICH, Hb from the hematoma can induce TLR2/TLR4 heterodimer formation on microglia. In the present study, we also found that only Hb from the hematoma could induce heterodimer formation on BMDCs, whereas other components including hemin, bilirubin, Fe2+, and Fe3+ have little effect on formation of the heterodimer. Furthermore, the TLR2/TLR4 heterodimer could only be detected on macrophages that had infiltrated the central nervous system, but not on the peripheral macrophages in the spleen. These results suggest that formation of the heterodimer may be closely related to the CNS microenvironment. Finally, compared with those of WT BM‐transplanted TLR2−/−/TLR4−/− mice, the NDS, BWC, and percentage of apoptotic neurons were all significantly decreased in TLR2−/−/TLR4−/− BM‐transplanted WT mice. Thus, the heterodimer on peripheral inflammatory cells plays a more important role than the heterodimer on the resident microglia. In conclusion, Hb from the hematoma could induce activation of both microglia and macrophages by inducing TLR2/TLR4 heterodimer formation, but the infiltrating macrophages may be more important in ICH‐induced inflammatory injury. Accordingly, another study also indicated that macrophages have higher phagocytic and inflammatory potential than the microglia at the onset of neuroinflammation.38
Peroxiredoxin originating from degenerating neurons was shown to induce macrophage release of IL‐23 through TLR2 and TLR4 signaling, which is an initiator of postischemic inflammation in the brain.21 Our present data first indicated that hematoma‐derived Hb activated macrophage release of IL‐23 by inducing the formation of TLR2/TLR4 heterodimer after ICH. This finding suggests that the Hb‐induced TLR2/TLR4 heterodimer initiates neuroinflammation following ICH. Therefore, disruption of TLR2/TLR4 heterodimer formation may represent a new treatment strategy for ICH.
Our results indicating that macrophages are the main source of IL‐23 and γδT cells are the main source of IL‐17 in the hemorrhagic hemisphere are consistent with the findings of several previous studies involving inflammatory diseases. Shichita et al20 showed that IL‐23 is expressed by infiltrating macrophages from the initiation of stroke and is an essential inducing factor for IL‐17 production by γδT cells in the delayed phase of cerebral ischemia in mice as well as that IL‐17–producing γδT cells have an important function during late‐stage brain infarction and are the main source of IL‐17, rather than Th17 cells. Another recent study demonstrated that γδT cells are an important source of IL‐17 and can act in an amplification loop to promote IL‐17 production by Th17 cells in EAE.17 Accordingly, in our study, IL‐17– and γδT‐deficient mice showed marked reductions in brain edema and neurologic deficits compared with WT mice. In addition, the number of macrophages and IL‐23 expression in perihematomal tissues peaked at 1 day after ICH, whereas the number of γδT cells and IL‐17 expression in these tissues peaked at 4 days after ICH. Together, these findings suggest that macrophages infiltrated the tissue immediately after ICH (acute phase, 6–24 hours post‐ICH) and secreted elevated levels of IL‐23, whereas γδT cells infiltrated the brain tissue in the delayed phase (>24 hours) of ICH and released IL‐17 to exert a detrimental function. Furthermore, we observed that the number of IL‐17+ cells was significantly lower in IL‐23−/− mice after ICH, further indicating that IL‐17 production is dependent on IL‐23 expression. Although our results indicate that IL‐17 production by γδT cells is essential in secondary brain injury of ICH, the precise role of IL‐17 in inducing brain damage is not well understood and the target cells of IL‐17 in the central nervous system remain unclear. Moreover, several cell types beyond only T‐helper cells are capable of producing IL‐17, including natural Th17 cells and innate lymphoid cells.39, 40, 41
Administration of SsnB 2 hours after ICH dramatically reduced neuroinflammation corresponding to the secondary injury. Because microglia are activated within minutes after ICH,42, 43 inhibition of TLR2/TLR4 heterodimer formation on microglia seems difficult. Macrophages instead accumulate and become activated during the first 24 hours after ICH, which offers a prolonged therapeutic window. We propose that the clinical benefit of SsnB administrated 2 hours after ICH may be due to inhibition of heterodimer formation on macrophages. Furthermore, we demonstrated that SsnB can penetrate the BBB, which is important for inhibition of neuroinflammation in ICH. Nonetheless, inflammation is generally considered a “double‐edged sword,” because the use of anti‐inflammatory drugs may increase the risk of secondary infection. Our findings that SsnB had no effect on rectal temperature and peripheral white cell counts before and after ICH suggest that SsnB did not increase the chance of secondary infection after ICH in our model. Therefore, SsnB warrants further testing as a potentially safe and efficient agent in ICH therapy.
Conclusions
The present study showed that Hb from the hematoma formed upon ICH induced the formation of TLR2/TLR4 heterodimer to activate IL‐23 secretion by infiltrating macrophages. This macrophage‐derived IL‐23 induced γδT cell production of IL‐17, which exacerbated the secondary brain injury after ICH in mice by amplifying the secretion of proinflammatory factors (Figure 8). Finally, SsnB may be able to ameliorate secondary brain injury in ICH by disrupting the formation of TLR2/TLR4 heterodimer.
Figure 8.
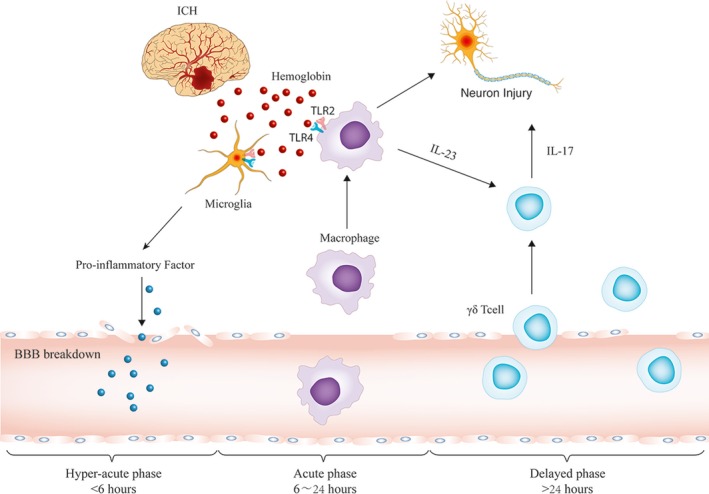
Schematic model of the roles of inflammatory cells and cytokines in intracerebral hemorrhage (ICH)–induced secondary brain injury. In the hyperacute phase of ICH (within 6 hours after ICH onset), hemoglobin (Hb) is released from red blood cells and activates microglia by inducing the formation of toll‐like receptors (TLRs) 2/4 heterodimer. Activated microglia release cytokines and chemokines to promote blood‐brain barrier (BBB) breakdown. Thereafter, peripheral blood cells begin to infiltrate into the perihematoma brain during the acute phase of ICH (6–24 hours after ICH onset). In this phase, Hb activates the infiltrating macrophages via TLR2/TLR4 heterodimer. Activated macrophages produce interleukin (IL)‐23. In the delayed phase of ICH (>24 hours after ICH onset), IL‐23 induces IL‐17 production from γδT cell receptor cells and causes further brain injury.
Sources of Funding
This work was supported by grants from the National Natural Science Fund for Distinguished Young Scholars (81525008), National Science Foundation of China (81400974), and the National Basic Research Program of China (973 Program) (2014CB541605).
Disclosures
None.
Supporting information
Table S1. Primers Used for Real‐Time PCR
Table S2. Primary and Secondary Antibodies Used for Immunofluorescence Staining
Table S3. Physiological Parameters of Mice Before and After ICH
Figure S1. Macrophages, microglia, and other cell types were identified by FACS from pooled infiltrating inflammatory cells collected from 8 mice on day 4 after induction of ICH, according to the following markers: CD45high CD11bhigh cell were macrophages; CD45int CD11bint were microglia; CD45high CD3+CD4+ cells were defined as brain‐invading T helper cells; and CD45high CD3+ cells were further analyzed to identify γδT lymphocytes. FSC‐A, forward scatter channel area; SSC‐A, side scatter channel area.
Figure S2. Depletion of macrophages in the spleen upon clodronate liposome (CLP) treatment. A, Percentages of F4/80+macrophages in spleens of vehicle‐treated and CLP‐treated mice at 1 day after ICH determined by flow cytometric analysis. B, Quantified percentages of F4/80+ macrophages in spleens of vehicle‐treated and CLP‐treated groups (**P<0.01 vs vehicle, n=5). C, mRNA expression of IL‐23p19 in perihematoma tissues at 1, 4, and 7 days after ICH in the CLP‐and vehicle‐treated mice (**P<0.01 vs vehicle, n=4).
Figure S3. Sorting of macrophages, microglia, and other cells from pooled infiltrating inflammatory cells collected from 7 mice at day 4 by FACS for subsequent evaluation of IL‐23p19 expression. CD45high CD11bhigh cell were macrophages; CD45int CD11bint were microglia; and CD45+, CD11b− were grouped as “other” cells.
Figure S4. CD3+T lymphocytes were the main source of IL‐17. The indicated surface markers were used to identify IL‐17–producing cells among pooled infiltrating inflammatory cells collected from 8 mice on day 4 after induction of ICH and sorted by FACS.
Figure S5. A, Structures of SsnB and the compounds derived from SsnB. B, Structure of J1, the isomorphic isomer of SsnB.
Figure S6. SsnB crosses the mouse blood‐brain barrier. A, Standard curve for SsnB. B, SsnB concentrations in cerebral tissues of mice subjected to ICH and in the vehicle‐treated group. C, SsnB concentrations in cerebral tissues of ICH model mice at 10, 20, and 30 minutes after intravenous injection of SsnB.
(J Am Heart Assoc. 2016;5:e004340 doi: 10.1161/JAHA.116.004340)
References
- 1. Cheung RT. Update on medical and surgical management of intracerebral hemorrhage. Rev Recent Clin Trials. 2007;2:174–181. [DOI] [PubMed] [Google Scholar]
- 2. Zhou M, Wang H, Zhu J, Chen W, Wang L, Liu S, Li Y, Liu Y, Yin P, Liu J, Yu S, Tan F, Barber RM, Coates MM, Dicker D, Fraser M, Gonzalez‐Medina D, Hamavid H, Hao Y, Hu G, Jiang G, Kan H, Lopez AD, Phillips MR, She J, Vos T, Wan X, Xu G, Yan LL, Yu C, Zhao Y, Zheng Y, Zou X, Naghavi M, Wang Y, Murray CJ, Yang G, Liang X. Cause‐specific mortality for 240 causes in China during 1990–2013: a systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet. 2016;387:251–272. [DOI] [PubMed] [Google Scholar]
- 3. Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. [DOI] [PubMed] [Google Scholar]
- 5. Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, Karimi A, Shaw MD, Barer DH. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387–397. [DOI] [PubMed] [Google Scholar]
- 6. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, Skolnick BE, Steiner T. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127–2137. [DOI] [PubMed] [Google Scholar]
- 7. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hwang BY, Appelboom G, Ayer A, Kellner CP, Kotchetkov IS, Gigante PR, Haque R, Kellner M, Connolly ES. Advances in neuroprotective strategies: potential therapies for intracerebral hemorrhage. Cerebrovasc Dis. 2011;31:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Dore S. Heme oxygenase‐1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rolland WB II, Manaenko A, Lekic T, Hasegawa Y, Ostrowski R, Tang J, Zhang JH. FTY720 is neuroprotective and improves functional outcomes after intracerebral hemorrhage in mice. Acta Neurochir Suppl. 2011;111:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ, Yan Y, Huang D, Yu C, Shi FD. Fingolimod for the treatment of intracerebral hemorrhage: a 2‐arm proof‐of‐concept study. JAMA Neurol. 2014;71:1092–1101. [DOI] [PubMed] [Google Scholar]
- 14. Sansing LH, Harris TH, Kasner SE, Hunter CA, Kariko K. Neutrophil depletion diminishes monocyte infiltration and improves functional outcome after experimental intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer‐Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene‐microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. [DOI] [PubMed] [Google Scholar]
- 16. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)‐1 in the induction of IL‐17‐producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. [DOI] [PubMed] [Google Scholar]
- 18. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corneth OB, Mus AM, Asmawidjaja PS, Klein Wolterink RG, van Nimwegen M, Brem MD, Hofman Y, Hendriks RW, Lubberts E. Absence of interleukin‐17 receptor a signaling prevents autoimmune inflammation of the joint and leads to a Th2‐like phenotype in collagen‐induced arthritis. Arthritis Rheumatol. 2014;66:340–349. [DOI] [PubMed] [Google Scholar]
- 20. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin‐17‐producing gammadelta T cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. [DOI] [PubMed] [Google Scholar]
- 21. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post‐ischemic inflammation in the brain. Nat Med. 2012;18:911–917. [DOI] [PubMed] [Google Scholar]
- 22. Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, Chen J, Xiong XY, Lv FL, Liang QL, Yang QW. Toll‐like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. 2014;75:876–889. [DOI] [PubMed] [Google Scholar]
- 23. Wang YC, Wang PF, Fang H, Chen J, Xiong XY, Yang QW. Toll‐like receptor 4 antagonist attenuates intracerebral hemorrhage‐induced brain injury. Stroke. 2013;44:2545–2552. [DOI] [PubMed] [Google Scholar]
- 24. MacLellan CL, Silasi G, Poon CC, Edmundson CL, Buist R, Peeling J, Colbourne F. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008;28:516–525. [DOI] [PubMed] [Google Scholar]
- 25. Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, Langhauser F, Helluy X, Schwarz T, Bittner S, Mayer CT, Brede M, Varallyay C, Pham M, Bendszus M, Jakob P, Magnus T, Meuth SG, Iwakura Y, Zernecke A, Sparwasser T, Nieswandt B, Stoll G, Wiendl H. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, Lu H, Wang Q, Nagarkatti P, Fan D. Characterization of sparstolonin B, a Chinese herb‐derived compound, as a selective Toll‐like receptor antagonist with potent anti‐inflammatory properties. J Biol Chem. 2011;286:26470–26479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weischenfeldt J, Porse B. Bone marrow‐derived macrophages (BMM): isolation and applications. CSH Protoc. 2008;2008:pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 28. Mracsko E, Javidi E, Na SY, Kahn A, Liesz A, Veltkamp R. Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke. 2014;45:2107–2114. [DOI] [PubMed] [Google Scholar]
- 29. Loftspring MC, McDole J, Lu A, Clark JF, Johnson AJ. Intracerebral hemorrhage leads to infiltration of several leukocyte populations with concomitant pathophysiological changes. J Cereb Blood Flow Metab. 2009;29:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. [DOI] [PubMed] [Google Scholar]
- 31. Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d‐Prostaglandin J2 activates peroxisome proliferator‐activated receptor‐gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006;26:811–820. [DOI] [PubMed] [Google Scholar]
- 32. Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71:743–752. [DOI] [PubMed] [Google Scholar]
- 33. Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, Schwendener RA. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol. 1997;27:2626–2633. [DOI] [PubMed] [Google Scholar]
- 34. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon‐gamma in ischemic stroke. Circulation. 2006;113:2105–2112. [DOI] [PubMed] [Google Scholar]
- 35. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin‐23 drives innate and T cell‐mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL‐23 is essential for T cell‐mediated colitis and promotes inflammation via IL‐17 and IL‐6. J Clin Invest. 2006;116:1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liang QL, Lei LL, Cui X, Zou NS, Duan JA. Bioactive cis‐stilbenoids from the tubers of Scirpus yagara. Fitoterapia. 2013;84:170–173. [DOI] [PubMed] [Google Scholar]
- 38. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conti HR, Peterson AC, Brane L, Huppler AR, Hernandez‐Santos N, Whibley N, Garg AV, Simpson‐Abelson MR, Gibson GA, Mamo AJ, Osborne LC, Bishu S, Ghilardi N, Siebenlist U, Watkins SC, Artis D, McGeachy MJ, Gaffen SL. Oral‐resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J Exp Med. 2014;211:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marks BR, Nowyhed HN, Choi JY, Poholek AC, Odegard JM, Flavell RA, Craft J. Thymic self‐reactivity selects natural interleukin 17‐producing T cells that can regulate peripheral inflammation. Nat Immunol. 2009;10:1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sutton CE, Mielke LA, Mills KH. IL‐17‐producing gammadelta T cells and innate lymphoid cells. Eur J Immunol. 2012;42:2221–2231. [DOI] [PubMed] [Google Scholar]
- 42. Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–279. [DOI] [PubMed] [Google Scholar]
- 43. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183:25–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Used for Real‐Time PCR
Table S2. Primary and Secondary Antibodies Used for Immunofluorescence Staining
Table S3. Physiological Parameters of Mice Before and After ICH
Figure S1. Macrophages, microglia, and other cell types were identified by FACS from pooled infiltrating inflammatory cells collected from 8 mice on day 4 after induction of ICH, according to the following markers: CD45high CD11bhigh cell were macrophages; CD45int CD11bint were microglia; CD45high CD3+CD4+ cells were defined as brain‐invading T helper cells; and CD45high CD3+ cells were further analyzed to identify γδT lymphocytes. FSC‐A, forward scatter channel area; SSC‐A, side scatter channel area.
Figure S2. Depletion of macrophages in the spleen upon clodronate liposome (CLP) treatment. A, Percentages of F4/80+macrophages in spleens of vehicle‐treated and CLP‐treated mice at 1 day after ICH determined by flow cytometric analysis. B, Quantified percentages of F4/80+ macrophages in spleens of vehicle‐treated and CLP‐treated groups (**P<0.01 vs vehicle, n=5). C, mRNA expression of IL‐23p19 in perihematoma tissues at 1, 4, and 7 days after ICH in the CLP‐and vehicle‐treated mice (**P<0.01 vs vehicle, n=4).
Figure S3. Sorting of macrophages, microglia, and other cells from pooled infiltrating inflammatory cells collected from 7 mice at day 4 by FACS for subsequent evaluation of IL‐23p19 expression. CD45high CD11bhigh cell were macrophages; CD45int CD11bint were microglia; and CD45+, CD11b− were grouped as “other” cells.
Figure S4. CD3+T lymphocytes were the main source of IL‐17. The indicated surface markers were used to identify IL‐17–producing cells among pooled infiltrating inflammatory cells collected from 8 mice on day 4 after induction of ICH and sorted by FACS.
Figure S5. A, Structures of SsnB and the compounds derived from SsnB. B, Structure of J1, the isomorphic isomer of SsnB.
Figure S6. SsnB crosses the mouse blood‐brain barrier. A, Standard curve for SsnB. B, SsnB concentrations in cerebral tissues of mice subjected to ICH and in the vehicle‐treated group. C, SsnB concentrations in cerebral tissues of ICH model mice at 10, 20, and 30 minutes after intravenous injection of SsnB.