

Abstract
Neuronal ceroid lipofuscinosis (NCL), also known as Batten disease, is a debilitating neurological disorder that affects both children and adults. Thirteen genetically distinct genes have been identified that when mutated, result in abnormal lysosomal function and an excessive accumulation of ceroid lipofuscin in neurons, as well as other cell types outside of the central nervous system. The NCL family of proteins is comprised of lysosomal enzymes (PPT1/CLN1, TPP1/CLN2, CTSD/CLN10, CTSF/CLN13), proteins that peripherally associate with membranes (DNAJC5/CLN4, KCTD7/CLN14), a soluble lysosomal protein (CLN5), a protein present in the secretory pathway (PGRN/CLN11), and several proteins that display different subcellular localizations (CLN3, CLN6, MFSD8/CLN7, CLN8, ATP13A2/CLN12). Unfortunately, the precise functions of many of the NCL proteins are still unclear, which has made targeted therapy development challenging. The social amoeba Dictyostelium discoideum has emerged as an excellent model system for studying the normal functions of proteins linked to human neurological disorders. Intriguingly, the genome of this eukaryotic soil microbe encodes homologs of 11 of the 13 known genes linked to NCL. The genetic tractability of the organism, combined with its unique life cycle, makes Dictyostelium an attractive model system for studying the functions of NCL proteins. Moreover, the ability of human NCL proteins to rescue gene-deficiency phenotypes in Dictyostelium suggests that the biological pathways regulating NCL protein function are likely conserved from Dictyostelium to human. In this review, I will discuss each of the NCL homologs in Dictyostelium in turn and describe how future studies can exploit the advantages of the system by testing new hypotheses that may ultimately lead to effective therapy options for this devastating and currently untreatable neurological disorder.
Keywords: Dictyostelium discoideum, Neuronal ceroid lipofuscinosis, Batten disease, Growth, Development, Calcium, Lysosome, Model organism
Background
Dictyostelium as a model system for studying human neurological disorders
The social amoeba Dictyostelium discoideum is a fascinating microbe that has emerged as a valuable model organism for biomedical and human disease research. This model eukaryote, which has historically been used to study basic cell function and multicellular development, undergoes a 24-h asexual life cycle comprised of both single-cell and multicellular phases [1] (Fig. 1). As a result, it is an excellent system for studying a variety of cellular and developmental processes, including lysosome function and intracellular trafficking and signalling [2, 3]. In nature, Dictyostelium feeds and grows as single cells (Fig. 1). When prompted by starvation, cells undergo chemotactic aggregation towards cAMP to form a multicellular aggregate (i.e., a mound), which then undergoes a series of morphological changes to form a motile multicellular pseudoplasmodium, also referred to as a slug (Fig. 1). Cells within the slug then terminally differentiate into either stalk or spore to form a fruiting body [4] (Fig. 1). Unlike immortalized mammalian cells that have been removed from their respective tissues, Dictyostelium represents a true organism in the cellular state that retains all of its dynamic physiological processes. Moreover, the cellular processes and signalling pathways that regulate the behaviour of Dictyostelium cells are remarkably similar to those observed in metazoan cells, indicating that findings from Dictyostelium are highly likely to be translatable to more complex eukaryotic systems [5].
Fig. 1.
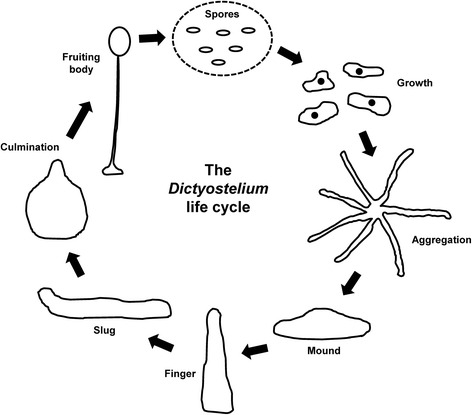
The life cycle of Dictyostelium. During growth, single cells feed on bacteria. Upon starvation, cells undergo chemotactic aggregation towards cAMP to form a multicellular mound. The mound then forms a finger, which falls on the surface to generate a motile pseudoplasmodium, or slug. During culmination, terminal differentiation of pre-stalk and pre-spore cells forms a fruiting body composed of a mass of viable spores supported atop a slender stalk. When a food source becomes available, the spores germinate allowing the cells to restart the life cycle
Dictyostelium is recognized as an excellent model system for studying human neurological disorders, including epilepsy, lissencephaly, Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease [6–10]. Dictyostelium also contains the highest content of prion-like proteins of all organisms investigated to date [11, 12]. Intriguingly, prion-like proteins known to generate insoluble deposits in other eukaryotic cells, do not aggregate in Dictyostelium [11, 12]. Instead these proteins accumulate in the nucleus and are broken down by the ubiquitin–proteasome system suggesting that Dictyostelium has undergone specific adaptations that allow it to efficiently regulate its prion-like proteome [11, 12]. Thus, understanding the pathways involved in preventing protein aggregation in Dictyostelium could provide novel routes to tackling neurological disorders characterized by protein aggregation (e.g., Alzheimer’s, Parkinson’s, and Huntington’s disease).
Dictyostelium as a model system for studying neuronal ceroid lipofuscinosis
Recently, Dictyostelium has emerged as a significant model system for studying neuronal ceroid lipofuscinosis (NCL) [13–15]. NCL, also known as Batten disease, is a neurological disorder that affects both children and adults [16]. The disease, which is the most common form of neurodegeneration in children, affects all ethnicities worldwide. The highest rates of incidence are observed in Northern European populations and Newfoundland, Canada [17–20]. Over 430 mutations have been documented in thirteen genetically distinct NCL genes, which are categorized based on the age of onset and pathological features [16, 21, 22] (Table 1). However, more genes likely remained to be identified as some patients who present with NCL-like symptoms do not carry mutations in any of the known NCL genes. Clinical manifestations of the disease include the progressive loss of vision, mental ability, and motor function, as well as epileptic seizures and a reduced lifespan [23]. At the cellular level, NCL disorders characteristically display aberrant lysosomal function and an excessive accumulation of ceroid lipofuscin in neurons and other cell types, with pathology often seen outside of the central nervous system [22].
Table 1.
List of genes linked to NCL in humans
| Protein | Disease |
|---|---|
| PPT1/CLN1 | Infantile NCL |
| TPP1/CLN2 | Late-infantile NCL |
| CLN3 | Juvenile NCL |
| DNAJC5/CLN4 | Adult-onset NCL, Kufs disease, Parry disease |
| CLN5 | Late-infantile NCL (Finnish variant) |
| CLN6 | Variant late-infantile NCL |
| MFSD8/CLN7 | Late-infantile NCL (Turkish variant) |
| CLN8 | Northern epilepsy (epilespy, progressive with mental retardation, EPMR) |
| CTSD/CLN10 | Congenital, neonatal and late infantile NCL |
| PGRN/CLN11 | Adult-onset NCL, frontotemporal dementia in heterozygotes |
| ATP13A2/CLN12 | Juvenile-onset NCL |
| CTSF/CLN13 | Adult-onset NCL |
| KCTD7/CLN14 | Infantile NCL |
Substantial molecular advances have been made in the past decade, which has made a significant impact on genetic diagnosis and our understanding of the biology underlying the NCL disorders. The NCL family of proteins is comprised of lysosomal enzymes (PPT1/CLN1, TPP1/CLN2, CTSD/CLN10, CTSF/CLN13), proteins that peripherally associate with membranes (DNAJC5/CLN4, KCTD7/CLN14), a soluble lysosomal protein (CLN5), a protein that is present in the secretory pathway (PGRN/CLN11), and several transmembrane proteins that display different subcellular localizations (CLN3, CLN6, MFSD8/CLN7, CLN8, ATP13A2/CLN12) [16, 21]. Unfortunately, the precise functions of many of the NCL proteins are still unknown [21]. As a result, new innovative approaches are critically needed to identify the primary functions of these proteins, which will hopefully lead to targeted therapy development for this devastating and currently untreatable disease. Interestingly, recent genetic advances now clearly support a strong overlap of NCL phenotypes with later onset neurodegenerative diseases, including Parkinson’s and frontotemporal dementia [24, 25]. Therefore, studying the cellular mechanisms underlying NCL protein function may provide fresh new insight into these and other related neurological disorders.
Homologs of NCL proteins have been identified and studied in several model organisms and this work has significantly enhanced our understanding of the localization and functions of NCL proteins in humans [26–28]. The Dictyostelium genome contains homologs of 11 of the 13 known NCL genes, but does not contain homologs of CLN6 or CLN8 (Table 2). Of all the NCL proteins, these are the only ER-resident membrane proteins [29, 30]. By comparison, Dictyostelium contains more homologs of NCL proteins than other model organisms including budding and fission yeast, the nematode Caenorhabditis elegans, and the fruit fly Drosophila melanogaster, but fewer than zebrafish (Danio rerio) [26–28] (Table 3). While the functions of some of the homologs in Dictyostelium have been investigated using gene-deficiency models, knockout mutants for several of the genes have yet to be generated (e.g., ppt1/cln1, ddj1/cln4, cln5, mfsd8/cln7, grn/cln11, cprA/cln13). Moreover, of the genes that have been studied, many have not been investigated in relation to NCL (e.g., ctsD/cln10, kil2/cln12, kctd9/cln14). The mRNA expression profiles of the Dictyostelium NCL homologs suggest different roles during the life cycle, with some being required for growth, and others for development [31] (Fig. 2). Moreover, many of the NCL homologs were detected in a proteomic profile of the macropinocytic pathway suggesting that the proteins share common functions or participate in the same biological pathway or process [32]. In this review, I will discuss each of the NCL homologs in Dictyostelium in turn, and highlight how we can use this model organism to provide fresh new insight into NCL protein function. While the focus of this review will be on the Dictyostelium homologs of human TPP1/CLN2, CLN3, CTSD/CLN10, ATP13A2/CLN12, CTSF/CLN13, and KCTD7/CLN14, the potential to use Dictyostelium to elucidate the functions of the other NCL proteins will also be discussed.
Table 2.
Dictyostelium proteins homologous to human NCL proteins
| Human protein | Size (aa) | Dictyostelium homolog (dictyBase ID) | Size (aa) | Region of similarity (aa) | Identities (%) | Positives (%) | E-value |
|---|---|---|---|---|---|---|---|
| PPT1/CLN1 Accession: NP_000301 |
306 | Ppt1 (DDB0233890) | 303 | 273 | 46 | 69 | 8.00E-80 |
| TPP1/CLN2 Accession: NP_000382 |
563 | Tpp1 (DDB0234303) | 600 | 552 | 36 | 52 | 4.00E-91 |
| CLN3 Accession: NP_000077 |
438 | Cln3 (DDB0233983) | 421 | 429 | 27 | 45 | 3.00E-31 |
| DNAJC5/CLN4 Accession: NP_079495 |
198 | Ddj1 (DDB0215016) | 411 | 69 | 57 | 76 | 2.00E-18 |
| CLN5 Accession: NP_006484 |
407 | Cln5 (DDB0234077) | 322 | 301 | 30 | 47 | 2.00E-27 |
| CLN6 Accession: NP_060352 |
311 | No homolog | |||||
| MFSD8/CLN7 Accession: NP_689991 |
518 | Mfsd8 (DDB0307149) | 498 | 530 | 29 | 47 | 1.00E-41 |
| CLN8 Accession: NP_061764 |
286 | No homolog | |||||
| CTSD/CLN10 Accession: NP_001900 |
412 | CtsD (DDB0215012) | 383 | 359 | 47 | 66 | 9.00E-91 |
| PGRN/CLN11 Accession: NP_002078 |
593 | Grn (DDB0238428) | 130 | 47 | 55 | 68 | 2.00E-08 |
| ATP13A2/CLN12 Accession: NP_071372 |
1180 | Kil2 (DDB0237611) | 1158 | 1089 | 35 | 53 | 1.00E-175 |
| CTSF/CLN13 Accession: NP_003784 |
484 | CprA (DDB0201647) | 343 | 340 | 37 | 55 | 6.00E-64 |
| KCTD7/CLN14 Accession: NP_694578 |
289 | Kctd9 (DDB0231824) | 488 | 96 | 38 | 58 | 2.00E-11 |
The amino acid sequences of human NCL proteins were inputted into the BLASTp server of dictyBase (http://www.dictybase.org). The following parameters were set: E-value, 1000; Matrix, BLOSUM62; Filter, no
Table 3.
Established and putative homologs of NCL proteins in other model organisms
| Human Protein |
S. cerevisiae (budding yeast) S. pombe (fission yeast) |
C. elegans (nematode) | D. melanogaster (fruit fly) | D. rerio (zebrafish) |
|---|---|---|---|---|
| PPT1/CLN1 Accession: NP_000301 |
Yes (S. pombe) [26–28] | Yes [26] | Yes [26–28] | Yes [27, 28] |
| TPP1/CLN2 Accession: NP_000382 |
No | No | No | Yes [27, 28] |
| CLN3 Accession: NP_000077 |
Yes [26–28] | Yes [26–28] | Yes [26–28] | Yes [27, 28] |
| DNAJC5/CLN4 Accession: NP_079495 |
Yes [a] | Yes [28] | Yes [c] | Yes [27, 28] |
| CLN5 Accession: NP_006484 |
No | No | No | Yes [27, 28] |
| CLN6 Accession: NP_060352 |
No | No | No | Yes [27, 28] |
| MFSD8/CLN7 Accession: NP_689991 |
No | Yes [b] | Yes [27, 28] | Yes [27, 28] |
| CLN8 Accession: NP_061764 |
No | No | No | Yes [27, 28] |
| CTSD/CLN10 Accession: NP_001900 |
Yes [26–28] | Yes [26, 28] | Yes [26–28] | Yes [27, 28] |
| PGRN/CLN11 Accession: NP_002078 |
No | Yes [b] | No | Yes [27, 28] |
| ATP13A2/CLN12 Accession: NP_071372 |
Yes [28],[a] | Yes [b] | Yes [c] | Yes [27, 28] |
| CTSF/CLN13 Accession: NP_003784 |
No | Yes [b] | Yes [c] | Yes [27, 28] |
| KCTD7/CLN14 Accession: NP_694578 |
No | Yes [b] | Yes [c] | Yes [27, 28] |
Fig. 2.
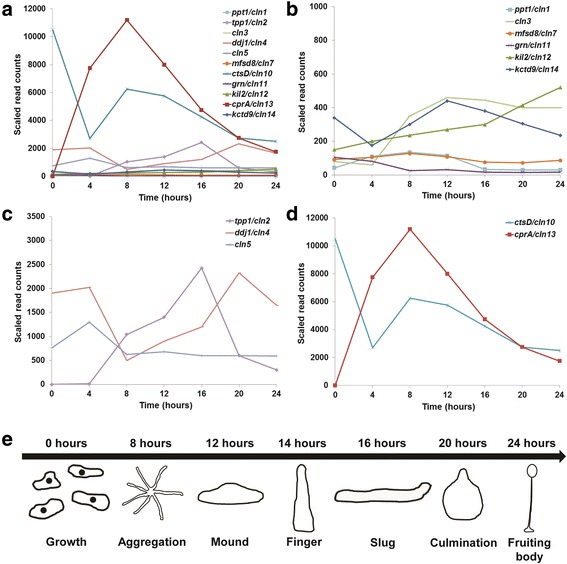
Gene expression analysis of NCL homologs in Dictyostelium. RNA-Seq data was obtained from dictyExpress (http://www.dictyexpress.biolab.si) [31] and re-plotted using Microsoft Excel. Genes with similar magnitudes of scaled read counts were grouped together on the same plot. a Expression profiles of all NCL homologs. b Expression profiles of ppt1, cln3, mfsd8, grn, kil2, kctd9. c Expression profiles of tpp1, ddj1, and cln5. d Expression profiles of ctsD and cprA. e Timeline of the various stages of Dictyostelium development. Images are not drawn to scale
Ppt1, the Dictyostelium homolog of human PPT1/CLN1
Mutations in palmitoyl-protein thioesterase 1 (PPT1/CLN1) cause one of the two infantile forms of NCL and the most severe subtype of the disease [21, 23] (Table 1). Clinical symptoms present between the ages of 1 and 2, and usually results in death between the ages of 8 and 11 [33]. However late infantile, juvenile, and adult forms of this NCL subtype have also been reported [34–38]. The PPT1 gene is conserved from yeast to human [26–28] (Table 3). PPT1 localizes to the lysosomal matrix, cytoplasmic vesicles, the endoplasmic reticulum (ER), lipid rafts, and presynaptic areas in neurons [21] (Table 4). The targets of PPT1-mediated de-palmitoylation and the precise function of the protein are still not known, however the protein is thought to be involved in apoptosis, cholesterol metabolism, and the recycling of synaptic vesicles [21] (Table 5).
Table 4.
Localization of NCL homologs in Dictyostelium and other systems
| Dictyostelium homolog | dictyBase ID | Localization in Dictyostelium | Localization of human protein (or homologs in other systems) |
|---|---|---|---|
| Ppt1 | DDB0233890 | Extracellular space [40] Macropinocytic pathway [32] |
Cytoplasmic vesicles [21] Endoplasmic reticulum [21] Lipid rafts [21] Lysosomal matrix [21] Presynaptic areas in neurons [21] |
| Tpp1 | DDB0234303 | Lysosome [14] | Endoplasmic reticulum [21] Lysosomal matrix [21] |
| Cln3 | DDB0233983 | CV system [13, 15] Endocytic pathway [13, 15] Macropinocytic pathway [32] |
Late endosomal membrane [21] Lysosomal membrane [21] |
| Ddj1 | DDB0215016 | Centrosome [69] Macropinocytic pathway [32] Phagosome [70] |
Cell membrane [68] Cytoplasm [21] Melanosomes [67] Synaptic vesicles in neurons [21] Secretory granules in endocrine, neurocrine and exocrine cells [21] Vesicular membranes [21] |
| Cln5 | DDB0234077 | Macropinocytic pathway [32] | Extracellular space [76] Lysosomal matrix [21, 75] |
| Mfsd8 | DDB0307149 | Macropinocytic pathway [32] | Lysosomal membrane [21] |
| CtsD | DDB0215012 | Endocytic pathway (e.g., phagosome, lysosome) [81–83] Macropinocytic pathway [32] |
Extracellular space [21, 79, 80] Lysosomal matrix [21] |
| Grn | DDB0238428 | Not known | Extracellular space [21, 97] |
| Kil2 | DDB0237611 | Macropinocytic pathway [32] Phagosomal membrane [102] |
Lysosomal membrane [21, 99, 100] Multi-vesicular bodies [21, 99, 100] |
| CprA | DDB0201647 | Not known | Lysosomal matrix [21] |
| Kctd9 | DDB0231824 | Not known | Cell membrane [21, 114] Punctate cytoplasmic distributions [21, 114] |
Table 5.
Cellular processes modulated by NCL homologs in Dictyostelium and other systems
| Dictyostelium homolog | dictyBase ID | Processes modulated in Dictyostelium | Knockout mutant generated | Processes modulated by human protein (or homologs in other systems) |
|---|---|---|---|---|
| Ppt1 | DDB0233890 | Phagocytosis [41] | No | Apoptosis [21] Cholesterol metabolism [21] Phagocytosis [42] Recycling of synaptic vesicles [21] |
| Tpp1 | DDB0234303 | Autophagy [14] Pre-spore cell differentiation [14] Timing of mid-stage development [14] |
Yes [14] | Endocytosis [21] Macroautophagy [21] TNF-α-induced apoptosis [21] |
| Cln3 | DDB0233983 | Cell proliferation [13] Pinocytosis [13] Protein secretion and cleavage [13] Cell adhesion [15] Timing of mid-stage development [15] Timing of late-stage development [15] Response to bacterial pathogens [66] |
Yes [13] | Apoptosis [21] Autophagy [21] Calcium homeostasis [21] Cell cycle control [21] Endocytosis [21] Intracellular trafficking [21] Lysosomal pH homeostasis [21] Osmoregulation [21] |
| Ddj1 | DDB0215016 | Phagocytosis [70] | No | Presynaptic endocytosis and exocytosis [21] |
| Cln5 | DDB0234077 | Not known | No | Apoptosis [21] Cell growth [21] Myelination [21] Sorting and recycling of lysosomal receptors [21] Sphingolipid transport and synthesis [21] |
| Mfsd8 | DDB0307149 | Not known | No | Predicted to have a role in: Transport of small substrates across the lysosomal membrane [21] |
| CtsD | DDB0215012 | Bacterial killing [86] Cell death [88, 89] Timing of early-stage development [81] |
Yes [79] | Apoptosis [21, 90] Autophagy [21, 91] Cell migration [85] Innate immune response [87] |
| Grn | DDB0238428 | Not known | No | Autophagy [21, 97] Cell migration [21, 97] Embryogenesis [21, 97] Inflammation [21, 97] Tumorigenesis [21, 97] |
| Kil2 | DDB0237611 | Bacterial killing [102, 105] Predation on yeast [104] |
Yes [100] | Predicted to have a role in: Lysosomal degradation [21] pH and biometal homeostasis within lysosomes [21] Transport across cellular membranes [21] |
| CprA | DDB0201647 | Early-stage development [108] Osmoregulation [110] |
No | Autophagy [21] Cell immunity [21, 113] Lipoprotein degradation [21] Proteasome degradation [21] |
| Kctd9 | DDB0231824 | Not known | Yes [118] | Predicted to have a role in: Hyperpolarization of the neuronal cell membrane [21] Proteasome degradation [21] |
The Dictyostelium homolog of human PPT1, ppt1, is composed of three exons that encode a 303 amino acid, 34 kDa protein (Ppt1; DDB0233890) (Table 2). Like PPT1, the Dictyostelium homolog contains a signal peptide for secretion suggesting that it may function extracellularly (SignalP 4.0) [39]. Not surprisingly, the enzyme has been detected in the macropinocytic pathway of Dictyostelium as well as in conditioned media from developing cells [32, 40] (Table 4). ppt1 expression increases during the early stages of development and reaches its peak level after 8 h [31] (Fig. 2b). Expression then decreases dramatically during mid-stage development and remains low during terminal differentiation and fruiting body formation (Fig. 2b). These results suggest that Ppt1 may be important for processes that occur during the early stages of development, specifically cAMP chemotaxis and aggregation. A knockout mutant has not yet been generated to study the function of Ppt1 in Dictyostelium. However, a previous study that analyzed genome-wide transcriptional changes during bacterial feeding and growth suggests that Ppt1 may function during phagocytosis, which supports data from mice linking PPT1 function to phagocyte infiltration following neuronal cell death [41, 42] (Table 5).
Tpp1, the Dictyostelium homolog of human TPP1/CLN2
Mutations in tri-peptidyl peptidase 1 (TPP1/CLN2) cause a late infantile form of NCL [21, 23] (Table 1). Clinical symptoms present between the ages of 2 and 4, with death typically occurring between the ages of 6 and 15 [43]. In addition to NCL, mutations in TPP1 have also been linked to autosomal recessive spinocerebellar ataxia 7 (SCAR7) [44]. The TPP1 protein localizes to the lysosomal matrix and the ER, functions as a serine protease, and has been linked to endocytosis, macroautophagy, and TNF-α-induced apoptosis [21] (Tables 4 and 5). While TPP1 is highly conserved in vertebrates, homologs are not present in classical model organisms such as yeast, the nematode, and the fruit fly [45] (Table 3). As a result, the NCL research community has been unable to exploit the relative advantages of these systems to study TPP1 function. Fortunately, the Dictyostelium genome encodes a homolog of TPP1 which was recently characterized [14]. The Dictyostelium homolog, tpp1, is composed of one exon that encodes a 600 amino acid, 67 kDa protein (Tpp1; DDB0234303) (Table 2). Tpp1 contains peptidase S8 and S53 domains as well as a putative signal peptide for secretion (SignalP 4.0) [39]. However the protein has not been detected in conditioned media from developing Dictyostelium cells [40]. tpp1 expression increases during the early-to-mid stages of development reaching peak levels after 16 h [31] (Fig. 2c). Expression then decreases dramatically during the later stages of development (Fig. 2c). The function of the protein, which is enriched in pre-spore cells, was recently characterized [14, 46]. Importantly, Dictyostelium Tpp1 shares many attributes with human TPP1. As in human cells, Dictyostelium Tpp1 localizes to the lysosome [14, 22] (Table 4). Auto-fluorescent bodies are observed in Dictyostelium tpp1 − cells during mid-stage development, which is similar to the auto-fluorescent material observed in human cells expressing mutated TPP1 [14, 22]. However the composition of the aggregated material in Dictyostelium cells remains to be identified. tpp1 − cells develop precociously and show a reduced ability to cleave target substrates and form spores [14] (Table 5). Similar to phenotypes observed in mammalian cell models, Tpp1-deficient cells also show defects in autophagy [14] (Table 5). More specifically, in response to starvation, tpp1 − cells display reduced cell size and viability when compared to wild-type [14]. In support of its localization to the lysosome, Tpp1-deficienct cells display strongly impaired development when treated with the lysosome-perturbing drug chloroquine [14]. Importantly, Phillips and Gomer [14] showed that this phenotype can be suppressed by a secondary mutation in stpA (suppressor of Tpp1, DDB_G0282973), which encodes a protein that shares some sequence similarity to mammalian oxysterol-binding proteins. This finding indicates that Dictyostelium has the potential to make substantial contributions to NCL protein research, specifically by exploiting the genetic tractability of the organism to identify other secondary mutations that suppress NCL phenotypes.
Cln3, the Dictyostelium homolog of human CLN3
The most common subtype of NCL is caused by loss-of-function mutations in ceroid lipofuscinosis neuronal 3 (CLN3), which is one of the two juvenile forms of the disease [21, 23] (Table 1). Clinical symptoms present between the ages of 5 and 10 and result in premature death usually in the late teenage years or early 20s [22, 33]. The human CLN3 gene encodes a 438 amino acid multi-pass transmembrane protein that is predicted to contain 6 transmembrane domains [47]. The gene is conserved from yeast to human and has been studied using a number of systems and cell models [26–28] (Table 3). The CLN3 protein, which localizes to late endosomes and the lysosomal membrane, has been linked to autophagy, apoptosis, intracellular trafficking, lysosomal pH homeostasis, cell cycle control, osmoregulation, and calcium homeostasis [21] (Tables 4 and 5). However, the precise function of CLN3 and the mechanism by which the protein affects these processes remains unknown [26–28].
The Dictyostelium homolog, cln3, is composed of three exons that encode a 421 amino acid, 47 kDa protein (Cln3; DDB0233983) predicted to contain 10–11 transmembrane domains (Table 2). Importantly, amino acid residues that are post-translationally modified in human CLN3 are conserved in the Dictyostelium homolog, as are residues mutated in patients with the disease [13]. cln3 expression increases dramatically during the early stages of development reaching peak levels after 12 h [31] (Fig. 2b). Expression then remains high during the mid-to-late stages of development (Fig. 2b). Loss of Cln3 results in aberrant secretion and cleavage of autocrine proliferation repressor A (AprA) and counting factor-associated protein A (CfaD) during growth [13]. As a consequence, Cln3-deficient cells display an increased rate of proliferation compared to wild-type [13] (Table 5). During the early stages of development, the aggregation of cln3 − cells is delayed and aberrant, but this phenotype is not due to reduced chemotaxis, alterations in the expression or localization of the cAMP receptor (CarA), or aberrant expression of genes linked to cAMP signal transduction [15] (Table 5). However, cln3 − cells do display reduced cell-substrate and cell-cell adhesion, which correlates with a decreased intracellular amount of the cell adhesion protein contact site A (CsaA), and an increased amount of soluble calcium-dependent cell adhesion molecule (CadA) in conditioned media [15] (Table 5). These results suggest that the reduced adhesion of cln3 − cells to the substrate and to each other causes the delayed and aberrant aggregation. As proof-of-concept, aberrant adhesion has also been observed in mammalian cells lacking functional CLN3 (e.g., mouse cerebellar cells, human neuronal progenitor cells derived from induced pluripotent stem cells generated from patient fibroblasts) (Huber, Atwal, Chandrachud, Cotman, unpublished observations). Finally, during the mid-to-late stages of development, cln3 − cells develop precociously and cln3 − slugs display enhanced migration [13] (Table 5).
In Dictyostelium, Cln3 localizes to both the contractile vacuole (CV) system and the endocytic pathway (late endosomes/lysosomes), and was detected in a proteomic profile of the macropinocytic pathway [13, 15, 32] (Table 4). Like other protists, the Dictyostelium CV system functions primarily as an osmoregulatory organelle [48]. However roles in ion homeostasis, vesicular trafficking, and unconventional protein secretion have also been reported [48–50]. Importantly, defects in osmoregulation have been reported in mammalian cell models lacking functional CLN3 [51–53]. In Dictyostelium, the CV system is a major intracellular store of ions, particularly calcium, and is required for cAMP-mediated calcium influx during chemotaxis [48, 54–56]. Calcium has also been shown to function as a chemoattractant during the early stages of Dictyostelium development [57]. Moreover, the primary sensor of intracellular calcium, calmodulin, localizes predominantly on the CV system [50, 58]. Previous work has shown that Cln3-deficiency phenotypes during development can be rescued by chelating calcium with EGTA [13, 15]. More specifically, calcium chelation rescues the aberrant streaming and aggregation of cln3 − cells, the precocious formation and enhanced migration of cln3 − slugs, and the precocious formation of cln3 − fruiting bodies [13, 15]. These findings suggest that cln3 − cells may inappropriately buffer calcium from the environment, and are consistent with studies in mammalian systems that have reported altered ion homeostasis, most notably calcium, in the absence of functional CLN3 [59–64]. Given its localization to the CV system, Dictyostelium presents an excellent system for studying the possible function of Cln3 in calcium homeostasis. As discussed above, Cln3-deficiency alters the extracellular levels of the cell proliferation regulators AprA and CfaD during growth, and CadA during the early stages of development [13, 15]. Intriguingly, CadA is secreted via an unconventional pathway involving the CV system [65]. Coupled with the localization of GFP-Cln3 to the CV system, these observations suggest that Cln3 may function to regulate unconventional protein secretion via the CV system during Dictyostelium growth and development. Together these results indicate that further study of Cln3 localization to the CV system could provide valuable new insight into the primary function of CLN3 in human cells.
In Dictyostelium, Cln3-deficiency causes pleiotropic effects that can be rescued through the expression of either Dictyostelium Cln3 or human CLN3, thus confirming that the function of CLN3 is evolutionarily conserved [13, 15]. Moreover, the localization of Cln3 to the CV system in Dictyostelium suggests that this model organism could provide an excellent system for assessing the role of the protein in osmotic stress, vesicular trafficking, ion homeostasis, and unconventional protein secretion. On a final note, Dictyostelium has been used as a model system to study the virulence of Pseudomonas aeruginosa, which is one of the most relevant human opportunistic bacterial pathogens. Dictyostelium cells infected with one of two strains of P. aeruginosa (PAO1 and PA14) cause a decrease in the expression of cln3 suggesting that Cln3 is also involved in the response of Dictyostelium cells to bacterial pathogens [66] (Table 5).
Ddj1, the Dictyostelium homolog of DNAJC5/CLN4
Mutations in DNAJC5/CLN4 cause an adult-onset form of NCL that is also known as Kufs disease or Parry disease [21, 23] (Table 1). DNAJC5 is conserved from yeast to human and encodes cysteine-string protein alpha (CSPα), which is a protein chaperone that localizes to the cytoplasm and associates with vesicular membranes, synaptic vesicles in neurons, and secretory granules in endocrine, neurocrine, and exocrine cells [21, 27, 28] (Tables 3 and 4). It has also been detected at the cell membrane and in melanosomes [67, 68] (Table 4). The disease-causing mutations in CSPα occur within the highly conserved cysteine-string region that is responsible for membrane binding and oligomerization [21].
The Dictyostelium homolog of DNAJC5, Ddj1 (DnaJ homolog 1, DDB0215016), localizes to both the cytoplasm and cell cortex, and was detected in proteomic profiles of the centrosome and macropinocytic pathway [32, 69] (Tables 2 and 4). The ddj1 gene is composed of two exons that encode a 411 amino acid, 46 kDa protein. ddj1 expression is highest during growth, the first 4 h of development, and fruiting body formation [31] (Fig. 2c). Expression decreases substantially during aggregation reaching its lowest level after 8 h of development, after which time the expression begins to increase (Fig. 2c). The peak expression level occurs after 20 h of development (Fig. 2c). Together, these observations suggest that the protein likely functions during growth and terminal differentiation. A Dictyostelium ddj1 knockout mutant has not yet been generated, however the function of the protein has been linked to phagocytosis in Dictyostelium [70] (Table 5).
Cln5, the Dictyostelium homolog of human CLN5
Mutations in ceroid lipofuscinosis neuronal 5 (CLN5) cause a late-infantile form of NCL [21, 23]. However, early juvenile and adult cases have also been reported [71–73] (Table 1). While this subtype had previously been referred to as the Finnish variant, it now appears in other regions of world [74]. The CLN5 protein localizes to the lysosomal matrix, but has also been detected in conditioned media from cultured cells [21, 75, 76] (Table 4). While the precise function of CLN5 in human cells is not known, it is proposed to function in sphingolipid transport and synthesis, myelination, cell growth, apoptosis, and the sorting and recycling of lysosomal receptors [21] (Table 5). Like CLN2, homologs of CLN5 do not exist in yeast, the nematode, or the fruit fly (Table 3).
The Dictyostelium homolog of human CLN5, cln5, is composed of three exons that encode a 322 amino acid, 37 kDa protein (Cln5; DDB0234077) (Table 2). While the localization of Cln5 in Dictyostelium has not yet been revealed, the protein was detected in a proteomic analysis of the macropinocytic pathway [32] (Table 4). Interestingly, like human CLN5, the Dictyostelium homolog contains a signal peptide for secretion (SignalP 4.0) [39] and has been detected in conditioned media from developing cells (Huber, manuscript in preparation). cln5 expression levels indicate that the protein may play an important role during the early stages of development [31] (Fig. 2c). Expression increases dramatically during the first 4 h of development, decreases significantly during the next four hours, and then remains low during the remaining stages of development (Fig. 2c). Future work to generate and characterize a cln5 knockout mutant should reveal Cln5-dependent processes in Dictyostelium that may provide insight into the precise function of the protein in human cells. This is especially important considering that classical model organisms such as yeast, the nematode, and the fruit fly lack homologs of CLN5 (Table 3).
Mfsd8, the Dictyostelium homolog of human MFSD8/CLN7
Mutations in major facilitator superfamily domain-containing protein 8 (MFSD8), also known as CLN7, cause a late-infantile form of NCL that was previously referred to as the Turkish variant [21, 23] (Table 1). The protein, which contains 12 transmembrane domains, localizes to the lysosomal membrane and is predicted to play a role in the transport of small substrates across the lysosomal membrane [21] (Tables 4 and 5). However, the precise function of the protein and its substrates are still not known. Homologs of MFSD8 are present in the nematode, the fruit fly, and zebrafish, but absent in yeast [27, 28] (Table 3).
The Dictyostelium homolog of human MFSD8, mfsd8, is composed of one exon that encodes a 498 amino acid, 55 kDa protein (Mfsd8; DDB0307149) (Table 2). Proteomic analyses have localized the protein to the macropinocytic pathway [32] (Table 4). Although a knockout mutant has not yet been generated in Dictyostelium, RNA-seq studies have provided some insight into when during the life cycle the protein may function. mfsd8 expression increases during the first 8 h of development [31] (Fig. 2b) suggesting that the protein functions during cAMP chemotaxis and aggregation. Expression then decreases between 8 and 20 h before rising slightly during fruiting body formation (Fig. 2b). By generating and characterizing a Dictyostelium mfsd8 knockout mutant, future research may be able to provide insight into the primary function of MFSD8 that is lost in NCL patients.
CtsD, the Dictyostelium homolog of human CTSD/CLN10
Mutations in cathepsin D (CTSD), also known as CLN10, have been linked to aging and neurodegeneration, including congenital, neonatal, and late infantile forms of NCL [21, 23, 77] (Table 1). Juvenile forms of this NCL subtype have also been reported [78]. CTSD is an aspartic protease that is conserved from yeast to human [26–28] (Table 3). It localizes to the lysosomal matrix and is linked to aberrant apoptosis and autophagy [21] (Tables 4 and 5). The enzyme, which contains a peptidase A1 domain and a signal peptide for secretion, has also been detected extracellularly, where its function is linked to a number of human cancers (SignalP 4.0) [39, 79] (Table 4). Proteomic profiling has also detected extracellular CTSD in aortic samples, where it was found to be loosely bound to the matrix [80] (Table 4).
The Dictyostelium homolog of human CTSD, ctsD, is composed of two exons that encode a 383 amino acid, 41 kDa protein (CtsD; DDB0215012) that is present throughout the endocytic pathway, including maturing phagosomes and lysosomes [32, 81–83] (Tables 2 and 4). Like human CTSD, the Dictyostelium homolog contains a signal peptide for secretion (SignalP 4.0) [39] and has been detected in conditioned media from developing cells (Huber, manuscript in preparation). The mRNA expression profile of ctsD indicates that the protein functions primarily during growth, where its expression in the highest [31] (Fig. 2d). ctsD expression decreases dramatically during the first four hours of development before rising slightly between 4 and 8 h (Fig. 2d). Expression then steadily decreases during the remaining stages of development (Fig. 2d). ctsD − cells display no overt phenotypes during growth or development, except for a slight delay in aggregation [81] (Table 5). Interestingly, ctsD expression is downregulated 10-fold in srfB − cells [84]. SrfB (serum response factor B) is a DNA-binding protein that similar to ctsD, is expressed in growth-phase cells as well as during the later stages of aggregation [84]. srfB − cells display decreased proliferation, aberrant cytokinesis, increased pinocytosis, abolished streaming, and precocious aggregation [84]. The defects in streaming and aggregation can be rescued by manually stimulating cells with pulses of cAMP suggesting that cAMP signaling is aberrant in srfB − cells [84]. These phenotypes, coupled with the similar expression profiles of srfB and ctsD, the dramatic reduction in the expression of ctsD in srfB − cells, and the delayed aggregation of ctsD − cells suggest that CtsD is required for optimal migration and aggregation in Dictyostelium. This is supported by studies in mammalian cells that have reported reduced migration of CTSD-deficient cells due to aberrant organization of cytoskeletal components, which if present in neurons, could have negative effects on neurogenesis, maintenance of neuronal polarity and shape, and migration [85] (Table 5).
In addition to studies on SrfB function, other work in Dictyostelium has also provided insight into CtsD function, specifically its role in bacterial killing and cell death. During infection with Salmonella typhimurium, the agent of food-borne gastroenteritis, Dictyostelium cells activate a defense response involving the upregulation of a number of cysteine proteases and cathepsins, including CtsD [86] (Table 5). Interestingly, studies in mice have linked CTSD function to the innate immune response against Listeria monocytogenes [87]. In Dictyostelium, research suggests that CtsD is involved in staurosporine-induced cell death and functions with calpains to facilitate cell dismantling during oxidative stress-induced cell death [88, 89] (Table 5). Intriguingly, CTSD is linked to staurosporine-induced apoptosis in human fibroblasts and activates autophagy to inhibit oxidative stress-induced cell death in human cancer cells [90, 91]. Staurosporine induces apoptosis in a number of mammalian cell lines, activating both caspase-dependent and caspase-independent types of cell death [92]. Caspase-independent cell death occurs through the release of lysosomal enzymes into the cytosol [93]. This is significant since the Dictyostelium genome does not encode caspases and instead undergoes caspase-independent cell death, which results in the release of lysosomal enzymes such as CtsD into the cytoplasm [94, 95]. Together, these results have provided insight into the functions of CtsD during the Dictyostelium life cycle. Future research using this model organism may be able to assess the effects of NCL-causing CTSD mutations on CtsD-dependent processes in Dictyostelium.
Grn, the Dictyostelium homolog of human PGRN/CLN11
Mutations in PGRN (progranulin), also referred to as CLN11, cause an adult-onset form of NCL in humans [21, 23] (Table 1). Interestingly, heterozygote mutations in PGRN also cause frontotemporal dementia [96]. PGRN is a multi-domain protein that is secreted and proteolytically cleaved to generate 6-kDa cleavage products called granulins (GRNs) (1–7) [21, 97] (Table 4). The primary functions of GRNs are still not known, however they are proposed to function in autophagy, embryogenesis, cell motility, inflammation, and tumorigenesis [21, 97] (Table 5). Homologs of GRNs are present in the nematode and zebrafish, but absent in yeast and the fruit fly [27, 28] (Table 3).
The Dictyostelium homolog of human GRN, Grn, is highly similar in sequence to human GRNs 1–7 (Table 2). The grn gene is composed of two exons that encode a 130 amino acid, 14 kDa protein (Grn; DDB0238428). The mRNA expression profile of grn indicates that the protein likely functions during growth where its expression is the highest [31] (Fig. 2b). Expression then decreases dramatically during development reaching its lowest level between 16 and 24 h suggesting that it may not be required for processes that occur during the mid-to-late stages of Dictyostelium development (e.g., terminal differentiation) (Fig. 2b). Although a grn knockout mutant has not yet been generated, future work aimed at creating a Dictyostelium Grn-deficiency model may be able to provide insight into the primary function of GRNs, and how mutations in PGRN cause NCL and frontotemporal dementia in humans.
Kil2, the Dictyostelium homolog of human ATP13A2/CLN12
Another juvenile-onset form of NCL is caused by mutations in ATP13A2, which is also known as CLN12 [21, 23] (Table 1). ATP13A2 contains 10 transmembrane domains and belongs to a family of P-type ATPases that are involved in the active transport of cations, heavy metals, and lipids across cellular membranes [98]. ATP13A2 is conserved from yeast to human and localizes to the lysosomal membrane and multi-vesicular bodies [21, 27, 28, 99, 100] (Tables 3 and 4). The precise function of the protein is not yet known however it is predicted to transport heavy metals, cations, and lipids across cellular membranes, and to be involved in degrading material and maintaining pH and biometal homeostasis within lysosomes [21] (Table 5). Interestingly, ATP13A2 also protects against α-synuclein toxicity in several models of Parkinson’s disease [101].
The Dictyostelium homolog of ATP13A2 is Kil2 (DDB0237611), which is a V-type P-ATPase that contains 10 transmembrane domains [102] (Table 2). The kil2 gene is composed of two exons that encode an 1158 amino acid, 131 kDa protein that localizes to the phagosomal membrane and is present in the macropinocytic pathway [32, 102] (Table 4). The mRNA expression profile of kil2 suggests that the protein functions primarily during development, specifically during terminal differentiation. Expression increases steadily during development reaching peak levels during fruiting body formation [31] (Fig. 2b).
Dictyostelium serves as an excellent model system for studying the cellular mechanisms involved in the degradation of ingested bacteria and yeast [103, 104], and previous work investigating this process has provided some insight into the function of Kil2. In humans, the efficient ingestion and killing of bacteria by phagocytic cells is necessary to protect the human body from infectious microorganisms. Kil2-deficient cells show reduced phagosomal activity compared to wild-type cells and are not able to kill ingested Klebsiella pneumoniae bacteria [102, 105] (Table 5). However these defects can be restored by supplementing the medium with magnesium suggesting that Kil2 functions as a magnesium pump to maintain the optimal concentration of magnesium in phagosomes, and to ensure the activity of phagosomal proteases [102]. Intriguingly, kil2 − cells are still able to kill several other species of bacteria (e.g., B. subtilis and P. aeruginosa) suggesting that the response of cells to ingested bacteria likely involves a diversity of proteins with different specificities [102]. More recently, Kil2 function has also been linked to the predation of Dictyostelium amoeba on yeast [104]. Since Kil2 appears to function as a magnesium pump in Dictyostelium, and ATP13A2 is proposed to function in maintaining biometal homeostasis in human lysosomes, these results support the use of Dictyostelium as a model system for studying the function of this protein. Thus, future work in Dictyostelium may be able to provide new insight into the function of ATP13A2 in humans and how mutations in this gene cause NCL.
CprA, the Dictyostelium homolog of human CTSF/CLN13
Mutations in cathepsin F (CTSF), also known as CLN13, have been linked to another adult-onset form of NCL [21, 23] (Table 1). CTSF is a cysteine protease that belongs to the peptidase C1 family of proteases. The protein localizes to the lysosomal matrix, and has been proposed to function in autophagy, proteasome degradation, cell immunity, and lipoprotein degradation [21] (Tables 4 and 5). CTSF contains a signal peptide for secretion and has been detected extracellularly (SignalP 4.0) [39, 106] (Table 4). Homologs of CTSF are present in the nematode, the fruit fly, and zebrafish, but absent in yeast [27, 28] (Table 3).
CprA (cysteine proteinase A; DDB0201647) is the Dictyostelium homolog of human CTSF (Table 2). The gene, which shares a high degree of homology to plant and animal sulphydryl proteinases, is composed of six exons that encode a 343 amino acid, 39 kDa protein [107]. cprA is not expressed during the growth phase of the life cycle [31]. However, its expression, which is inducible by extracellular cAMP, increases dramatically during the early stages of development [31, 108] (Fig. 2d). By 10–12 h, cprA mRNA comprises approximately 1% of the total cellular mRNA [109]. While the expression of cprA steadily decreases during the remaining stages of development, cprA has been identified as a pre-stalk specific gene [31, 46, 109] (Fig. 2d). It has been suggested that CprA functions to digest proteins during development to provide amino acids and a source of energy for developing cells [109]. A cprA knockout mutant has not yet been generated however previous studies have provided some insight into the function of the protein. cprA expression was shown to be induced during hyperosmotic stress with 200 mM sorbitol suggesting that protein functions during the osmotic stress response [110] (Table 5). cprA expression is also upregulated in rpkA − cells during starvation [111]. RpkA (receptor phosphatidylinositol kinase A) is seven-transmembrane G-protein coupled receptor (GPCR) that contains a C-terminal phosphatidylinositol-4-phosphate 5-kinase (PIP5K) domain (http://www.dictybase.org). Importantly, RpkA localizes to late phagosomes and is involved in the phagocytosis of infectious bacteria [112], which adheres to the proposed function of CTSF in cell immunity in mammalian cells [113] (Table 5). At low density, rpkA − cells fail to aggregate due to their inability to respond to cAMP and conditioned media factor (CMF) [111]. Given the increased expression of cprA in rpkA − cells, and the dramatic increase in expression of cprA during the early stages of development, these results suggest that CprA is required for cAMP chemotaxis and aggregation in Dictyostelium. Finally, like human CTSF, CprA contains a signal peptide for secretion and has been detected in conditioned media from developing cells (SignalP 4.0) [39] (Huber, manuscript in preparation). Together, these results have provided insight into the function of CprA during the Dictyostelium life cycle. Further examination of its localization in Dictyostelium, and the creation of a CprA-deficiency model, may be able to provide additional insight into the role of CTSF in human disease.
Kctd9, the Dictyostelium homolog of human KCTD7/CLN14
Mutations in potassium channel tetramerization-domain 7 (KCTD7), also known as CLN14, have been linked to another infantile form of NCL [21, 23, 114] (Table 1). In humans, the KCTD family of proteins consists of 26 members with mostly unknown functions [115]. Mutations in KCTD7 cause vision loss, progressive myoclonic epilepsy, progressive decline in cognition and motor skills, and a reduced lifespan [114]. NCL-type storage material is also observed in patient samples [114]. In a wild-type mouse cerebellar cell line, KCTD7 fused to GFP localizes to the cell membrane, as well as punctate distributions within the cytoplasm [114] (Table 4). The precise function of the protein is not yet known however it is predicted to be involved in proteasome degradation and hyperpolarization of the neuronal cell membrane [21] (Table 5). Homologs of KCTD7 are present in the nematode, the fruit fly, and zebrafish, but absent in yeast [27, 28] (Table 3).
Kctd9 (DDB0231824) is the Dictyostelium homolog of human KCTD7 (Table 2). The protein contains four penta-peptide repeats, a double-courtin domain, and is highly similar to vertebrate potassium channel tetramerization domain-containing proteins. The gene is composed of one exon that encodes a 488 amino acid, 53 kDa protein. The mRNA expression profile of kctd9 suggests that the protein functions during the mid-stages of development. kctd9 expression decreases during the early stages of development reaching its lowest level after 4 h [31] (Fig. 2b). Expression then increases significantly to reach its peak level after 12 h, and is followed by a steady decrease in expression between 12 and 24 h of development (Fig. 2b). Although the effect of Kctd9-deficiency during growth and development has not yet been studied, gene expression studies have provided some insight into its function during sexual development. In addition to the asexual life cycle, Dictyostelium can also undergo sexual reproduction to form macrocysts [116]. During this alternative life cycle, phermonal interactions generate fusion-competent cells, which then undergo cell and pronuclear fusion through a process involving cAMP chemotaxis. kctd9 − cells show no obvious phenotypes during macrocyst formation, however kctd9 expression is enriched in gametes (e.g., sexually mature, fusion-competent cells) [117, 118] suggesting that the protein may be involved in membrane fusion during sexual development. The research community would benefit from future work examining the precise function of Kctd9 during membrane fusion as well as examining the effect of Kctd9 loss on processes that occur during the Dictyostelium asexual life cycle.
Conclusions
Accumulated evidence indicates that Dictyostelium could provide unprecedented opportunities to elucidate the normal functions of NCL proteins and the signal transduction regulating their activities. This is especially true for those proteins that have no clear homologs in other classical model organisms. Many of the NCL homologs in Dictyostelium were detected in a proteomic profile of the macropinocytic pathway, which supports the suggestion that NCL proteins share common functions or participate in the same biological pathway or process [32, 119] (Table 4). While the functions of some of the NCL homologs in Dictyostelium have been previously studied, many have not been investigated in relation to human disease. A major benefit of using Dictyostelium to study NCL protein function is the ability to knockout multiple genes in a single cell line using homologous recombination [120]. Importantly, this allows researchers to study epistatic relationships between genes and will allow future studies to assess potential functional redundancies between the NCL homologs in Dictyostelium. However, one must acknowledge that using Dictyostelium as model system for studying NCL protein function does present some limitations. For instance, while Dictyostelium does develop into a true multicellular organism, it contains a limited number of cell types that may present challenges when attempting to relate gene-deficiency phenotypes in Dictyostelium to pathologies in specific tissues or organs in humans. Since Dictyostelium lacks a central nervous system, discoveries made in Dictyostelium must be evaluated in the relevant mammalian cell type. Nonetheless, the ability of human NCL proteins to rescue deficiency phenotypes in Dictyostelium [13–15] suggests that the biological pathways regulating the functions of NCL proteins are likely conserved from Dictyostelium to human. In addition to normal function models, future studies can use Dictyostelium to assess the effects of newly discovered mutations on NCL protein function, which will greatly improve our understanding of the molecular basis of the NCL disorders, and will hopefully aid in the development of therapies that rescue the molecular defects in NCL patients.
Acknowledgements
The author would like to thank the anonymous reviewers for their helpful and constructive comments.
Funding
The author is supported by internal funds provided by Trent University.
Availability of data and materials
Not applicable.
Author’s contributions
RJH contributed to the conception, design, and drafting of this manuscript.
Author’s information
RJH is an Assistant Professor at Trent University and uses Dictyostelium as a model system to study the functions of NCL proteins.
Competing interests
The author declares that he has no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AprA
Autocrine proliferation repressor A
- cAMP
cyclic AMP
- CfaD
Counting factor-associated protein A
- CLN
Ceroid lipofuscinosis neuronal
- CMF
Conditioned media factor
- CprA
Cysteine proteinase A
- CSPα
Cysteine-string protein alpha
- CTSD
Cathepsin D
- CTSF
Cathepsin F
- CV
Contractile vacuole
- Ddj1
DnaJ homolog 1
- ER
Endoplasmic reticulum
- GPCR
G protein-coupled receptor
- KCTD
Potassium channel tetramerization-domain
- MFSD8
Major facilitator superfamily domain-containing protein 8
- NCL
Neuronal ceroid lipofuscinosis
- PGRN
Progranulin
- PIP5K
Phosphatidylinositol-4-phosphate 5-kinase
- PPT1
Palmitoyl-protein thioesterase 1
- RpkA
Receptor phosphatidylinositol kinase A
- SCAR7
Spinocerebellar ataxia 7
- SrfB
Serum response factor B
- StpA
Suppressor of Tpp1 A
- TPP1
Tri-peptidyl peptidase 1
References
- 1.Fey P, Kowal AS, Gaudet P, Pilcher KE, Chisholm RL. Protocols for growth and development of Dictyostelium discoideum. Nat Protoc. 2007;2:1307–1316. doi: 10.1038/nprot.2007.178. [DOI] [PubMed] [Google Scholar]
- 2.Simon M, Plattner H. Unicellular eukaryotes as models in cell and molecular biology: critical appraisal of their past and future value. Int Rev Cell Mol Biol. 2014;309:141–198. doi: 10.1016/B978-0-12-800255-1.00003-X. [DOI] [PubMed] [Google Scholar]
- 3.Maniak M. Dictyostelium as a model for human lysosomal and trafficking diseases. Semin Cell Dev Biol. 2011;22:114–119. doi: 10.1016/j.semcdb.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Du Q, Kawabe Y, Schilde C, Chen ZH, Schaap P. The evolution of aggregative multicellularity and cell-cell communication in the Dictyostelia. J Mol Biol. 2015;427:3722–3733. doi: 10.1016/j.jmb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller-Taubenberger A, Kortholt A, Eichinger L. Simple system—substantial share: the use of Dictyostelium in cell biology and molecular medicine. Eur J Cell Biol. 2013;92:45–53. doi: 10.1016/j.ejcb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Walker MC, Williams RS. The search for better epilepsy treatments: from slime mould to coconuts. Biochem Soc Trans. 2013;41:1625–1628. doi: 10.1042/BST20130183. [DOI] [PubMed] [Google Scholar]
- 7.Meyer I, Kuhnert O, Gräf R. Functional analyses of lissencephaly-related proteins in Dictyostelium. Semin Cell Dev Biol. 2011;22:89–96. doi: 10.1016/j.semcdb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Kortholt A, Gilsbach B, van Haastert, PJM. Dictyostelium discoideum: A Model System to Study LRRK2-Mediated Parkinson Disease. In: Dushanova J, editor. Mechanisms in Parkinson’s Disease–Models and Treatments. Croatia: InTech; 2012. p. 293–310.
- 9.McMains VC, Myre M, Kreppel L, Kimmel AR. Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: a system for functional studies of the presenilin/gamma-secretase complex. Dis Model Mech. 2010;3:581–594. doi: 10.1242/dmm.004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myre MA, Lumsden AL, Thompson MN, Wasco W, MacDonald ME, Gusella JF. Deficiency of huntingtin has pleiotropic effects in the social amoeba Dictyostelium discoideum. PLoS Genet. 2011;7:e1002052. doi: 10.1371/journal.pgen.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santarriaga S, Petersen A, Ndukwe K, Brandt A, Gerges N, Bruns Scaglione J, Scaglione KM. The social amoeba Dictyostelium discoideum is highly resistant to polyglutamine aggregation. J Biol Chem. 2015;290:25571–25578. doi: 10.1074/jbc.M115.676247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malinovska L, Palm S, Gibson K, Verbavatz JM, Alberti S. Dictyostelium discoideum has a highly Q/N-rich proteome and shows an unusual resilience to protein aggregation. Proc Natl Acad Sci U S A. 2015;112:E2620–E2629. doi: 10.1073/pnas.1504459112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber RJ, Myre MA, Cotman SL. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS One. 2014;9:e110544. doi: 10.1371/journal.pone.0110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips JE, Gomer RH. Partial genetic suppression of a loss-of-function mutant of the neuronal ceroid lipofuscinosis-associated protease TPP1 in Dictyostelium discoideum. Dis Model Mech. 2015;8:147–156. doi: 10.1242/dmm.018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber RJ, Myre MA, Cotman SL. Aberrant adhesion impacts early development in a Dictyostelium model for juvenile neuronal ceroid lipofuscinosis. Cell Adh Migr. in press. [DOI] [PMC free article] [PubMed]
- 16.Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease) Biochim Biophys Acta. 2015;1852:2237–2241. doi: 10.1016/j.bbadis.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andermann E, Jacob JC, Andermann F, Carpenter S, Wolfe L, Berkovic S. The Newfoundland aggregate of neuronal ceroid-lipofuscinosis. Am J Med Genet. 1988;5:111–116. doi: 10.1002/ajmg.1320310615. [DOI] [PubMed] [Google Scholar]
- 18.Ju W, Zhong R, Moore S, Moroziewicz D, Currie JR, Parfrey P, Brown WT, Zhong N. Identification of novel CLN2 mutations shows Canadian specific NCL2 alleles. J Med Genet. 2002;39:822–825. doi: 10.1136/jmg.39.11.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore SJ, Buckley DJ, MacMillan A, Marshall HD, Steele L, Ray PN, Nawaz Z, Baskin B, Frecker M, Carr SM, Ives E, Parfrey PS. The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clin Genet. 2008;74:213–222. doi: 10.1111/j.1399-0004.2008.01054.x. [DOI] [PubMed] [Google Scholar]
- 20.Williams RE. Appendix 1: NCL incidence and prevalence data. In: Mole SE, Williams RE, Goebel HH, editors. The Neuronal Ceroid Lipofuscinoses (Batten Disease) 2. Oxford: Oxford University Press; 2011. pp. 361–365. [Google Scholar]
- 21.Cárcel-Trullols J, Kovács AD, Pearce DA. Cell biology of the NCL proteins: What they do and don’t do. Biochim Biophys Acta. 2015;1852:2242–2255. doi: 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 22.Radke J, Stenzel W, Goebel HH. Human NCL Neuropathology. Biochim Biophys Acta. 2015;1852:2262–2266. doi: 10.1016/j.bbadis.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases–clinical perspectives. Biochim Biophys Acta. 2013;1832:1801–1806. doi: 10.1016/j.bbadis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng H, Xiu X, Jankovic J. Genetic convergence of Parkinson’s disease and lysosomal storage disorders. Mol Neurobiol. 2015;51:1554–1568. doi: 10.1007/s12035-014-8832-4. [DOI] [PubMed] [Google Scholar]
- 25.Dearborn JT, Harmon SK, Fowler SC, O’Malley KL, Taylor GT, Sands MS, Wozniak DF. Comprehensive functional characterization of murine infantile Batten disease including Parkinson-like behavior and dopaminergic markers. Sci Rep. 2015;5:12752. doi: 10.1038/srep12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips SN, Muzaffar N, Codlin S, Korey CA, Taschner PE, de Voer G, Mole SE, Pearce DA. Characterizing pathogenic processes in Batten disease: use of small eukaryotic model systems. Biochim Biophys Acta. 2006;1762:906–919. doi: 10.1016/j.bbadis.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Bond M, Holthaus SM, Tammen I, Tear G, Russell C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim Biophys Acta. 2013;1832:1842–1865. doi: 10.1016/j.bbadis.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Faller KM, Gutierrez-Quintana R, Mohammed A, Rahim AA, Tuxworth RI, Wager K, Bond M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim Biophys Acta. 2015;1852:2267–2278. doi: 10.1016/j.bbadis.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 29.Heine C, Koch B, Storch S, Kohlschütter A, Palmer DN, Braulke T. Defective endoplasmic reticulum-resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J Biol Chem. 2004;279:22347–22352. doi: 10.1074/jbc.M400643200. [DOI] [PubMed] [Google Scholar]
- 30.Lonka L, Kyttälä A, Ranta S, Jalanko A, Lehesjoki AE. The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Hum Mol Genet. 2000;9:1691–1697. doi: 10.1093/hmg/9.11.1691. [DOI] [PubMed] [Google Scholar]
- 31.Rot G, Parikh A, Curk T, Kuspa A, Shaulsky G, Zupan B. dictyExpress: a Dictyostelium discoideum gene expression database with an explorative data analysis web-based interface. BMC Bioinformatics. 2009;10:265. doi: 10.1186/1471-2105-10-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Journet A, Klein G, Brugière S, Vandenbrouck Y, Chapel A, Kieffer S, Bruley C, Masselon C, Aubry L. Investigating the macropinocytic proteome of Dictyostelium amoebae by high-resolution mass spectrometry. Proteomics. 2012;12:241–245. doi: 10.1002/pmic.201100313. [DOI] [PubMed] [Google Scholar]
- 33.Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta. 2013;1832:1795–1800. doi: 10.1016/j.bbadis.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Bonsignore M, Tessa A, Di Rosa G, Piemonte F, Dionisi-Vici C, Simonati A, Calamoneri F, Tortorella G, Santorelli FM. Novel CLN1 mutation in two Italian sibs with late infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol. 2006;10:154–156. doi: 10.1016/j.ejpn.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Mitchison HM, Hofmann SL, Becerra CH, Munroe PB, Lake BD, Crow YJ, Stephenson JB, Williams RE, Hofman IL, Taschner PE, Martin JJ, Philippart M, Andermann E, Andermann F, Mole SE, Gardiner RM, O’Rawe AM. Mutations in the palmitoyl-protein thioesterase gene (PPT; CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum Mol Genet. 1998;7:291–297. doi: 10.1093/hmg/7.2.291. [DOI] [PubMed] [Google Scholar]
- 36.Khan A, Chieng KS, Baheerathan A, Hussain N, Gosalakkal J. Novel CLN1 mutation with atypical juvenile neuronal ceroid lipofuscinosis. J Pediatr Neurosci. 2013;8:49–51. doi: 10.4103/1817-1745.111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalviainen R, Eriksson K, Losekoot M, Sorri I, Harvima I, Santavuori P, Jarvela I, Autti T, Vanninen R, Salmenpera T, van Diggelen OP. Juvenile-onset neuronal ceroid lipofuscinosis with infantile CLN1 mutation and palmitoyl-protein thioesterase deficiency. Eur J Neurol. 2007;14:369–372. doi: 10.1111/j.1468-1331.2007.01668.x. [DOI] [PubMed] [Google Scholar]
- 38.Ramadan H, Al-Din AS, Ismail A, Balen F, Varma A, Twomey A, Watts R, Jackson M, Anderson G, Green E, Mole SE. Adult neuronal ceroid lipofuscinosis caused by deficiency in palmitoyl protein thioesterase 1. Neurology. 2007;68:387–388. doi: 10.1212/01.wnl.0000252825.85947.2f. [DOI] [PubMed] [Google Scholar]
- 39.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 40.Bakthavatsalam D, Gomer RH. The secreted proteome profile of developing Dictyostelium discoideum cells. Proteomics. 2010;10:2556–2559. doi: 10.1002/pmic.200900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sillo A, Bloomfield G, Balest A, Balbo A, Pergolizzi B, Peracino B, Skelton J, Ivens A, Bozzaro S. Genome-wide transcriptional changes induced by phagocytosis or growth on bacteria in Dictyostelium. BMC Genomics. 2008;9:291. doi: 10.1186/1471-2164-9-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Lee YC, Kim SJ, Choi MS, Tsai PC, Saha A, Wei H, Xu Y, Xiao YJ, Zhang P, Heffer A, Mukherjee AB. Production of lysophosphatidylcholine by cPLA2 in the brain of mice lacking PPT1 is a signal for phagocyte infiltration. Hum Mol Genet. 2007;16:837–847. doi: 10.1093/hmg/ddm029. [DOI] [PubMed] [Google Scholar]
- 43.Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–83. doi: 10.1016/S0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- 44.Sun Y, Almomani R, Breedveld GJ, Santen GWE, Aten E, Lefeber DJ, Hoff JI, Brusse E, Verheijen FW, Verdijk RM, Kriek M, Oostra B, Breuning MH, Losekoot M, den Dunnen JT, van de Warrenburg BP, Maat-Kievit AJA. Autosomal recessive spinocerebellar ataxia 7 (SCAR7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN2 disease) Hum Mutat. 2013;34:706–713. doi: 10.1002/humu.22292. [DOI] [PubMed] [Google Scholar]
- 45.Wlodawer A, Li M, Gustchina A, Oyama H, Dunn BM, Oda K. Structural and enzymatic properties of the sedolisin family of serine-carboxyl peptidases. Acta Biochim Pol. 2003;50:81–102. [PubMed] [Google Scholar]
- 46.Iranfar N, Fuller D, Sasik R, Hwa T, Laub M, Loomis WF. Expression patterns of cell-type-specific genes in Dictyostelium. Mol Biol Cell. 2001;12:2590–2600. doi: 10.1091/mbc.12.9.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The International Batten Disease Consortium Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82:949–957. doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 48.Plattner H. Contractile vacuole complex—its expanding protein inventory. Int Rev Cell Mol Biol. 2013;306:371–416. doi: 10.1016/B978-0-12-407694-5.00009-2. [DOI] [PubMed] [Google Scholar]
- 49.Kinseth MA, Anjard C, Fuller D, Guizzunti G, Loomis WF, Malhotra V. The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell. 2007;130:524–534. doi: 10.1016/j.cell.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 50.Sriskanthadevan S, Brar SK, Manoharan K, Siu CH. Ca(2+) -calmodulin interacts with DdCAD-1 and promotes DdCAD-1 transport by contractile vacuoles in Dictyostelium cells. FEBS J. 2013;280:1795–1806. doi: 10.1111/febs.12203. [DOI] [PubMed] [Google Scholar]
- 51.Stein CS, Yancey PH, Martins I, Sigmund RD, Stokes JB, Davidson BL. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am J Physiol Cell Physiol. 2010;298:C1388–C1400. doi: 10.1152/ajpcell.00272.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Getty A, Kovács AD, Lengyel-Nelson T, Cardillo A, Hof C, Chan CH, Pearce DA. Osmotic stress changes the expression and subcellular localization of the Batten disease protein CLN3. PLoS One. 2013;8:e66203. doi: 10.1371/journal.pone.0066203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, Davidson BL. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci. 2013;33:18065–18079. doi: 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moniakis J, Coukell MB, Janiec A. Involvement of the Ca2 +-ATPase PAT1 and the contractile vacuole in calcium regulation in Dictyostelium discoideum. J Cell Sci. 1999;112:405–414. doi: 10.1242/jcs.112.3.405. [DOI] [PubMed] [Google Scholar]
- 55.Malchow D, Lusche DF, Schlatterer C, De Lozanne A, Müller-Taubenberger A. The contractile vacuole in Ca2+-regulation in Dictyostelium: its essential function for cAMP-induced Ca2+-influx. BMC Dev Biol. 2006;6:31. doi: 10.1186/1471-213X-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bozzaro S, Buracco S, Peracino B. Iron metabolism and resistance to infection by invasive bacteria in the social amoeba Dictyostelium discoideum. Front Cell Infect Microbiol. 2013;3:50. doi: 10.3389/fcimb.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scherer A, Kuhl S, Wessels D, Lusche DF, Raisley B, Soll DR. Ca2+ chemotaxis in Dictyostelium discoideum. J Cell Sci. 2010;123:3756–3767. doi: 10.1242/jcs.068619. [DOI] [PubMed] [Google Scholar]
- 58.Zhu Q, Clarke M. Association of calmodulin and an unconventional myosin with the contractile vacuole complex of Dictyostelium discoideum. J Cell Biol. 1992;118:347–358. doi: 10.1083/jcb.118.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boriack RL, Bennett MJ. CLN-3 protein is expressed in the pancreatic somatostatin-secreting delta cells. Eur J Paediatr Neurol. 2001;5(Suppl A):99–102. doi: 10.1053/ejpn.2000.0443. [DOI] [PubMed] [Google Scholar]
- 60.An Haack K, Narayan SB, Li H, Warnock A, Tan L, Bennett MJ. Screening for calcium channel modulators in CLN3 siRNA knock down SH-SY5Y neuroblastoma cells reveals a significant decrease of intracellular calcium levels by selected L-type calcium channel blockers. Biochim Biophys Acta. 2011;1810:186–191. doi: 10.1016/j.bbagen.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warnock A, Tan L, Li C, An Haack K, Narayan SB, Bennett MJ. Amlodipine prevents apoptotic cell death by correction of elevated intracellular calcium in a primary neuronal model of Batten disease (CLN3 disease) Biochem Biophys Res Commun. 2013;436:645–649. doi: 10.1016/j.bbrc.2013.04.113. [DOI] [PubMed] [Google Scholar]
- 62.Chandrachud U, Walker MW, Simas AM, Heetveld S, Petcherski A, Klein M, Oh H, Wolf P, Zhao WN, Norton S, Haggarty SJ, Lloyd-Evans E, Cotman SL. Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J Biol Chem. 2015;290:14361–14380. doi: 10.1074/jbc.M114.621706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang JW, Choi H, Kim HJ, Jo DG, Jeon YJ, Noh JY, Park WJ, Jung YK. Neuronal vulnerability of CLN3 deletion to calcium-induced cytotoxicity is mediated by calsenilin. Hum Mol Genet. 2007;16:317–326. doi: 10.1093/hmg/ddl466. [DOI] [PubMed] [Google Scholar]
- 64.Grubman A, Pollari E, Duncan C, Caragounis A, Blom T, Volitakis I, Wong A, Cooper J, Crouch PJ, Koistinaho J, Jalanko A, White AR, Kanninen KM. Deregulation of biometal homeostasis: the missing link for neuronal ceroid lipofuscinoses? Metallomics. 2014;6:932–943. doi: 10.1039/c4mt00032c. [DOI] [PubMed] [Google Scholar]
- 65.Sesaki H, Wong EF, Siu CH. The cell adhesion molecule DdCAD-1 in Dictyostelium is targeted to the cell surface by a nonclassical transport pathway involving contractile vacuoles. J Cell Biol. 1997;138:939–951. doi: 10.1083/jcb.138.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carilla-Latorre S, Calvo-Garrido J, Bloomfield G, Skelton J, Kay RR, Ivens A, Martinez JL, Escalante R. Dictyostelium transcriptional responses to Pseudomonas aeruginosa: common and specific effects from PAO1 and PA14 strains. BMC Microbiol. 2008;8:109. doi: 10.1186/1471-2180-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chi A, Valencia JC, Hu Z-Z, Watabe H, Yamaguchi H, Mangini NJ, Huang H, Canfield VA, Cheng KC, Yang F, Abe R, Yamagishi S, Shabanowitz J, Hearing VJ, Wu C, Appella E, Hunt DF. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res. 2006;5:3135–3144. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- 68.Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R, Hulkova H, Jahnova H, van der Zee J, Staropoli JF, Sims KB, Tyynela J, Van Broeckhoven C, Nijssen PC, Mole SE, Elleder M, Kmoch S. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reinders Y, Schulz I, Gräf R, Sickmann A. Identification of novel centrosomal proteins in Dictyostelium discoideum by comparative proteomic approaches. J Proteome Res. 2006;5:589–598. doi: 10.1021/pr050350q. [DOI] [PubMed] [Google Scholar]
- 70.Gotthardt D, Blancheteau V, Bosserhoff A, Ruppert T, Delorenzi M, Soldati T. Proteomics fingerprinting of phagosome maturation and evidence for the role of a Galpha during uptake. Mol Cell Proteomics. 2006;5:2228–2243. doi: 10.1074/mcp.M600113-MCP200. [DOI] [PubMed] [Google Scholar]
- 71.Cannelli N, Nardocci N, Cassandrini D, Morbin M, Aiello C, Bugiani M, Criscuolo L, Zara F, Striano P, Granata T, Bertini E, Simonati A, Santorelli FM. Revelation of a novel CLN5 mutation in early juvenile neuronal ceroid lipofuscinosis. Neuropediatrics. 2007;38:46–49. doi: 10.1055/s-2007-981449. [DOI] [PubMed] [Google Scholar]
- 72.Pineda-Trujillo N, Cornejo W, Carrizosa J, Wheeler RB, Munera S, Valencia A, Agudelo-Arango J, Cogollo A, Anderson G, Bedoya G, Mole SE, Ruíz-Linares A. A CLN5 mutation causing an atypical neuronal ceroid lipofuscinosis of juvenile onset. Neurology. 2005;64:740–742. doi: 10.1212/01.WNL.0000151974.44980.F1. [DOI] [PubMed] [Google Scholar]
- 73.Xin W, Mullen TE, Kiely R, Min J, Feng X, Cao Y, O’Malley L, Shen Y, Chu-Shore C, Mole SE, Goebel HH, Sims K. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology. 2010;74:565–571. doi: 10.1212/WNL.0b013e3181cff70d. [DOI] [PubMed] [Google Scholar]
- 74.Lebrun AH, Storch S, Ruschendorf F, Schmiedt ML, Kyttala A, Mole SE, Kitzmuller C, Saar K, Mewasingh LD, Boda V, Kohlschutter A, Ullrich K, Braulke T, Schulz A. Retention of lysosomal protein CLN5 in the endoplasmic reticulum causes neuronal ceroid lipofuscinosis in Asian sibship. Hum Mutat. 2009;30:E651–E661. doi: 10.1002/humu.21010. [DOI] [PubMed] [Google Scholar]
- 75.Isosomppi J, Vesa J, Jalanko A, Peltonen L. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum Mol Genet. 2002;11:885–891. doi: 10.1093/hmg/11.8.885. [DOI] [PubMed] [Google Scholar]
- 76.Moharir A, Peck SH, Budden T, Lee SY. The role of N-Glycosylation in folding, trafficking, and functionality of lysosomal protein CLN5. PLoS One. 2013;8:e74299. doi: 10.1371/journal.pone.0074299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;S1568-1637(16)30067-8. [DOI] [PubMed]
- 78.Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W, Saftig P, Gartner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. 2006;78:988–998. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vetvicka V, Fusek M. Procathepsin D as a tumor marker, anti-cancer drug or screening agent. Anticancer Agents Med Chem. 2012;12:172–175. doi: 10.2174/187152012799014904. [DOI] [PubMed] [Google Scholar]
- 80.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Journet A, Chapel A, Jehan S, Adessi C, Freeze H, Klein G, Garin J. Characterization of Dictyostelium discoideum cathepsin D. J Cell Sci. 1999;112:3833–3843. doi: 10.1242/jcs.112.21.3833. [DOI] [PubMed] [Google Scholar]
- 82.Harris E, Wang N, Wu Wl WL, Weatherford A, De Lozanne A, Cardelli J. Dictyostelium LvsB mutants model the lysosomal defects associated with Chediak-Higashi syndrome. Mol Biol Cell. 2002;13:656–669. doi: 10.1091/mbc.01-09-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagedorn M, Soldati T. Flotillin and RacH modulate the intracellular immunity of Dictyostelium to Mycobacterium marinum infection. Cell Microbiol. 2007;9:2716–2733. doi: 10.1111/j.1462-5822.2007.00993.x. [DOI] [PubMed] [Google Scholar]
- 84.Galardi-Castilla M, Pergolizzi B, Bloomfield G, Skelton J, Ivens A, Kay RR, Bozzaro S, Sastre L. SrfB, a member of the Serum Response Factor family of transcription factors, regulates starvation response and early development in Dictyostelium. Dev Biol. 2008;316:260–274. doi: 10.1016/j.ydbio.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koch S, Scifo E, Rokka A, Trippner P, Lindfors M, Korhonen R, Corthals GL, Virtanen I, Lalowski M, Tyynelä J. Cathepsin D deficiency induces cytoskeletal changes and affects cell migration pathways in the brain. Neurobiol Dis. 2013;50:107–119. doi: 10.1016/j.nbd.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 86.Sillo A, Matthias J, Konertz R, Bozzaro S, Eichinger L. Salmonella typhimurium is pathogenic for Dictyostelium cells and subverts the starvation response. Cell Microbiol. 2011;13:1793–1811. doi: 10.1111/j.1462-5822.2011.01662.x. [DOI] [PubMed] [Google Scholar]
- 87.Carrasco-Marín E, Madrazo-Toca F, de los Toyos JR, Cacho-Alonso E, Tobes R, Pareja E, Paradela A, Albar JP, Chen W, Gomez-Lopez MT, Alvarez-Dominguez C. The innate immunity role of cathepsin-D is linked to Trp-491 and Trp-492 residues of listeriolysin O. Mol Microbiol. 2009;72:668–682. doi: 10.1111/j.1365-2958.2009.06673.x. [DOI] [PubMed] [Google Scholar]
- 88.Mir H, Rajawat J, Begum R. Staurosporine induced poly (ADP-ribose) polymerase independent cell death in Dictyostelium discoideum. Indian J Exp Biol. 2012;50:80–86. [PubMed] [Google Scholar]
- 89.Rajawat J, Alex T, Mir H, Kadam A, Begum R. Proteases involved during oxidative stress-induced poly(ADP-ribose) polymerase-mediated cell death in Dictyostelium discoideum. Microbiology. 2014;160:1101–1111. doi: 10.1099/mic.0.076620-0. [DOI] [PubMed] [Google Scholar]
- 90.Johansson AC, Steen H, Ollinger K, Roberg K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003;10:1253–1259. doi: 10.1038/sj.cdd.4401290. [DOI] [PubMed] [Google Scholar]
- 91.Hah YS, Noh HS, Ha JH, Ahn JS, Hahm JR, Cho HY, Kim DR. Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 2012;323:208–214. doi: 10.1016/j.canlet.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 92.Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res. 1998;51:293–308. doi: 10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 93.Zhang XD, Gillespie SK, Hersey P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol Cancer Ther. 2004;3:187–197. [PubMed] [Google Scholar]
- 94.Olie RA, Durrieu F, Cornillon S, Loughran G, Gross J, Earnshaw WC, Golstein P. Apparent caspase independence of programmed cell death in Dictyostelium. Curr Biol. 1998;8:955–958. doi: 10.1016/S0960-9822(98)70395-1. [DOI] [PubMed] [Google Scholar]
- 95.Roisin-Bouffay C, Luciani MF, Klein G, Levraud JP, Adam M, Golstein P. Developmental cell death in Dictyostelium does not require paracaspase. J Biol Chem. 2004;279:11489–11494. doi: 10.1074/jbc.M312741200. [DOI] [PubMed] [Google Scholar]
- 96.Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:171. doi: 10.3389/fnagi.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petkau TL, Leavitt BR. Progranulin in neurodegenerative disease. Trends Neurosci. 2014;37:388–398. doi: 10.1016/j.tins.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 98.Bublitz MJ, Morth JP, Nissen P. P-type ATPases at a glance. J Cell Sci. 2011;124:2515–2519. doi: 10.1242/jcs.088716. [DOI] [PubMed] [Google Scholar]
- 99.Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 100.Covy JP, Waxman EA, Giasson BI. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J Neurosci Res. 2012;90:2306–2316. doi: 10.1002/jnr.23112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord. 2015;30:770–779. doi: 10.1002/mds.26243. [DOI] [PubMed] [Google Scholar]
- 102.Lelong E, Marchetti A, Guého A, Lima WC, Sattler N, Molmeret M, Hagedorn M, Soldati T, Cosson P. Role of magnesium and a phagosomal P-type ATPase in intracellular bacterial killing. Cell Microbiol. 2011;13:246–258. doi: 10.1111/j.1462-5822.2010.01532.x. [DOI] [PubMed] [Google Scholar]
- 103.Cosson P, Lima WC. Intracellular killing of bacteria: is Dictyostelium a model macrophage or an alien? Cell Microbiol. 2014;16:816–823. doi: 10.1111/cmi.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koller B, Schramm C, Siebert S, Triebel J, Deland E, Pfefferkorn AM, Rickerts V, Thewes S. Dictyostelium discoideum as a novel host system to study the interaction between phagocytes and yeasts. Front Microbiol. 2016;7:1665. doi: 10.3389/fmicb.2016.01665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Le Coadic M, Froquet R, Lima WC, Dias M, Marchetti A, Cosson P. Phg1/TM9 proteins control intracellular killing of bacteria by determining cellular levels of the Kil1 sulfotransferase in Dictyostelium. PLoS One. 2013;8:e53259. doi: 10.1371/journal.pone.0053259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaakinen R, Lindstedt KA, Sneck M, Kovanen PT, Oörni K. Angiotensin II increases expression and secretion of cathepsin F in cultured human monocyte-derived macrophages: an angiotensin II type 2 receptor-mediated effect. Atherosclerosis. 2007;192:323–327. doi: 10.1016/j.atherosclerosis.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 107.Pears CJ, Mahbubani HM, Williams JG. Characterization of two highly diverged but developmentally co-regulated cysteine proteinase genes in Dictyostelium discoideum. Nucleic Acids Res. 1985;13:8853–8866. doi: 10.1093/nar/13.24.8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Driscoll DM, Williams JG. Two divergently transcribed genes of Dictyostelium discoideum are cyclic AMP-inducible and coregulated during development. Mol Cell Biol. 1987;7:4482–4489. doi: 10.1128/MCB.7.12.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Williams JG, North MJ, Mahbubani H. A developmentally regulated cysteine proteinase in Dictyostelium discoideum. EMBO J. 1985;4:999–1006. doi: 10.1002/j.1460-2075.1985.tb03730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Na J, Tunggal B, Eichinger L. STATc is a key regulator of the transcriptional response to hyperosmotic shock. BMC Genomics. 2007;8:123. doi: 10.1186/1471-2164-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bakthavatsalam D, Brazill D, Gomer RH, Eichinger L, Rivero F, Noegel AA. A G protein-coupled receptor with a lipid kinase domain is involved in cell-density sensing. Curr Biol. 2007;17:892–897. doi: 10.1016/j.cub.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 112.Riyahi TY, Frese F, Steinert M, Omosigho NN, Glöckner G, Eichinger L, Orabi B, Williams RS, Noegel AA. RpkA, a highly conserved GPCR with a lipid kinase domain, has a role in phagocytosis and anti-bacterial defense. PLoS One. 2011;6:e27311. doi: 10.1371/journal.pone.0027311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi GP, Bryant RA, Riese R, Verhelst S, Driessen C, Li Z, Bromme D, Ploegh HL, Chapman HA. Role for cathepsin F in invariant chain processing and major histocompatibility complex class II peptide loading by macrophages. J Exp Med. 2000;191:1177–1186. doi: 10.1084/jem.191.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Staropoli JF, Karaa A, Lim ET, Kirby A, Elbalalesy N, Romansky SG, Leydiker KB, Coppel SH, Barone R, Xin W, MacDonald ME, Abdenur JE, Daly MJ, Sims KB, Cotman SL. A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Am J Hum Genet. 2012;91:202–208. doi: 10.1016/j.ajhg.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Z, Xiang Y, Sun G. The KCTD family of proteins: structure, function, disease relevance. Cell Biosci. 2013;3:45. doi: 10.1186/2045-3701-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Day DH, Keszei A. Signalling and sex in the social amoebozoans. Biol Rev Camb Philos Soc. 2012;87:313–329. doi: 10.1111/j.1469-185X.2011.00200.x. [DOI] [PubMed] [Google Scholar]
- 117.Muramoto T, Suzuki K, Shimizu H, Kohara Y, Kohriki E, Obara S, Tanaka Y, Urushihara H. Construction of a gamete-enriched gene pool and RNAi-mediated functional analysis in Dictyostelium discoideum. Mech Dev. 2003;120:965–975. doi: 10.1016/S0925-4773(03)00136-9. [DOI] [PubMed] [Google Scholar]
- 118.Muramoto T, Takeda S, Furuya Y, Urushihara H. Reverse genetic analyses of gamete-enriched genes revealed a novel regulator of the cAMP signaling pathway in Dictyostelium discoideum. Mech Dev. 2005;122:733–743. doi: 10.1016/j.mod.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 119.Persaud-Sawin DA, Mousallem T, Wang C, Zucker A, Kominami E, Boustany RM. Neuronal ceroid lipofuscinosis: a common pathway? Pediatr Res. 2007;61:146–152. doi: 10.1203/pdr.0b013e31802d8a4a. [DOI] [PubMed] [Google Scholar]
- 120.Faix J, Linkner J, Nordholz B, Platt JL, Liao XH, Kimmel AR. The application of the Cre-loxP system for generating multiple knock-out and knock-in targeted loci. Methods Mol Biol. 2013;983:249–267. doi: 10.1007/978-1-62703-302-2_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.