

Abstract
At present, when a clinical diagnosis of Parkinson's disease (PD) is made, serious damage has already been done to nerve cells of the substantia nigra pars compacta. The diagnosis of PD in its earlier stages, before this irreversible damage, would be of enormous benefit for future treatment strategies designed to slow or halt the progression of this disease that possibly prevents accumulation of toxic aggregates. As a molecular biomarker for the detection of PD in its earlier stages, alpha-synuclein (α-syn), which is a key component of Lewy bodies, in which it is found in an aggregated and fibrillar form, has attracted considerable attention. Here, α-syn is reviewed in details.
Keywords: Alpha-synuclein, biomarker, Parkinson's disease
INTRODUCTION
Human alpha-synuclein (α-syn) is predominantly expressed in the brain, especially in the neocortex, hippocampus, substantia nigra (SN), thalamus, and cerebellum, and is found in Lewy bodies (LBs). It is encoded by the SNCA that consists of 6 exons ranging in size from 42 to 1110 bp.[1,2] Translation of SNCA starts from an autophagy-related start codon that is encoded by exon 2 and stops at a TAA stop codon that is encoded by exon 6. As noted above, the predominant form of α-syn is the full-length protein, but other shorter isoforms have also been described. In addition, C-terminal truncations of α-syn induce aggregation, suggesting that C-terminal modifications might be involved in the pathology of α-syn.[3] Changes in the levels of α-syn have been reported in cerebrospinal fluid and plasma of Parkinson's disease (PD) patients compared to control individuals.[4] Therefore, α-syn can be considered a potential biomarker for PD.
ALPHA-SYNUCLEIN IDENTIFICATION AND MAPPING
In 1985, a neuron-specific protein of 143 amino acids (AA) was identified using an antibody against Torpedo californica purified cholinergic synaptic vesicles. Moreover, a 140-AA protein was also identified in a rat brain cDNA library with a high homology with the torpedo electric organ protein. The researchers concerned named the protein synuclein as they found it in both cytoplasmic and nuclear regions of neurons although later studies have never confirmed the nuclear localization of synuclein. Torpedo synucleins based on their molecular weights were classified in three major subclasses as 17.5 kDa, 18.5 kDa, and 20 kDa synucleins. The 17.5 kDa synuclein was predominantly expressed among other species.[5]
In a later study, amyloid protein from senile plaques of Alzheimer disease (AD) brain was analyzed, and this led to the identification of two other unknown peptides in addition to Aβ. Structural analysis of these proteins showed a high tendency for them to form β-structures. Therefore, it was hypothesized that these proteins, based on their hydrophobic identity and their ability to form β-structures, may act as a “seed” for Aβ aggregation. Therefore, Aβ protein as a consequence of interaction with these proteins might be converted into insoluble Aβ and trigger later neurodegenerative processes. These two unknown proteins were named non-Aβ components of AD amyloid (Non-Aβ component of AD plaques [NAC]). The concentration of both NAC proteins was the same in the amyloid fractions, and thus, it was suggested that they can both be cleaved from a single larger precursor. Later on, a 140-AA protein from a full-length encoding cDNA was identified as the precursor of NAC (NACP). NACP was detected in all tissues except liver with the highest concentration in the brain. These findings suggest a brain-specific role for NACP although it is expressed ubiquitously.[6]
Shortly afterward, 140-AA and 134-AA proteins that were present in the human brain in high amounts were purified and sequenced. Interestingly, the 140-AA protein was exactly the same as NACP, and it showed a high homology with torpedo and rat synuclein. Moreover, the 134-AA protein was identified as the human homolog of bovine phosphoneuroprotein 14 and showed 61% homology with the 140-AA protein. These findings suggested the existence of a family of human brain proteins, and the 140-AA protein was named α-syn while the 134-AA protein was named β-synuclein (β-syn). It was also shown that α-syn and β-syn are expressed predominantly in the brain and are concentrated in the presynaptic terminals of nerve cells.[7] Soon after, using FISH on human metaphase chromosomes, the chromosomal locations of SNCA and SNCB were identified. The results showed that α-syn is encoded by a gene on chromosome 4q21 and β-syn is encoded by a different gene located on chromosome 5q35.[8] Later, another type of synuclein was found to be expressed in high levels in ovarian and breast carcinomas. This was named γ-syn, and its gene was reported as breast cancer-specific gene 1.[9] Although γ-syn is also found in the peripheral nervous system,[10] its functional role is not completely understood.
ALPHA-SYNUCLEIN STRUCTURE
α-Syn is a small acidic protein with three domains namely N-terminal lipid-binding α-helix, amyloid-binding central domain (NAC), and C-terminal acidic tail. α-Syn can be present as an α-helix structure in association with phospholipids or an unfolded conformation in the cytosol, suggesting that it plays specific roles in different cellular locations based on its dynamic structure.[11]
The N-terminal domain of α-syn (residues 1–87) is a positively charged region, including seven series of 11-AA repeats. Each 11-AA repeat contains a highly conserved KTKEGV hexameric motif that is also present in the α-helical domain of apolipoproteins. Moreover, the ability of α-syn to disrupt lipid bilayers is related to these repeat sequences. These repeats, via their ability to induce α-syn helical structure and subsequently reduce the tendency of α-syn to form β-structures, are important in α-syn-lipid interactions.[12]
The core region of α-syn (residues 61–95), also known as NAC, is involved in fibril formation and aggregation[13] as it can to form cross β-structures.[14]
The C-terminal domain of α-syn (residues 96–140) is an acidic tail of 43-AA residues, containing 10 Glu and 5 Asp residues. Structurally, the C-terminal domain of α-syn is present in a random coil structure due to its low hydrophobicity and high net negative charge. In vitro studies have revealed that α-syn aggregation can be induced by reduction of pH which neutralizes these negative charges.[15,16] An interaction between the C-terminal domain and the NAC region of α-syn is thought to be responsible for inhibition of α-syn aggregation. Moreover, in the presence of Al3+, the C-terminal domain of α-syn binds to this metal ion and thus the ruined inhibitory effect of the C-terminal on NAC leads to α-syn aggregation.[17] The phosphorylation of residue serine 129 is important in the inhibitory property of the C-terminal region as dephosphorylation of serine 129 causes α-syn aggregation.[18] Moreover, the C-terminal of α-syn is homologous with small heat shock proteins (HSPs), suggesting a protective role for α-syn in keeping proteins out of the degradation process.[19]
ALPHA-SYNUCLEIN STRUCTURE AND LIPID INTERACTION
Discovering the fact that α-syn has the ability to interact with membrane lipids encouraged scientists to find out how this interaction is related to the biophysical features of lipids and α-syn. Therefore, in vitro studies were established using membrane mimics, with different acyl head groups, different fatty acid chains, and various degrees of saturation of fatty acyl chains.[20] Liposomes are spherical lipid bilayers with the head groups located either inside or outside of the sphere and the fatty acid tails concealed within the space between these two layers. Liposomes differ in size and lipid composition, and they usually are classified as small unilamellar vesicles (SUVs) with a diameter 10–100 nm, large unilamellar vesicles (LUVs) with a diameter 100 nm - 1 μm, and giant unilamellar vesicles (GUVs) with a diameter >1 μm. SUVs are good mimics for small secretory vesicles while GUVs can be good models for cellular membranes.[21]
α-Syn preferably binds to SUVs or LUVs containing negatively charged head groups. This interaction is mediated by the α-syn N-terminal region that is rich in lysine residues, suggesting that the α-syn N-terminal region preferentially binds to anionic lipids electrostatically. α-Syn also prefers some acidic head groups more than the others. For example, phosphatidylethanolamine, phosphatidic acid (PA), phosphatidylinositol, and ganglioside attract α-syn more than phosphatidylserine or phosphatidylglycerol.[21]
Moreover, polyunsaturated acyl chains can increase the ability of α-syn to interact with the membrane. This could be due to the larger space between poorly packed unsaturated lipids than compact saturated forms. In addition, the higher binding affinity of α-syn to SUVs and LUVs than GUVs may be related to the ability of α-syn to sense the membrane curvature. Disordered α-syn folds into an α-helical structure when it interacts with the membrane. This folding happens when α-syn inserts into hydrophobic acyl chains. Thus, α-syn has an elevated affinity for SUVs with a loose force between acyl chains due to high curvature. Taken together, these data indicate that α-syn has a preference for interaction with lipids and membranes containing unsaturated fatty acids with small anionic head groups and high curvature.[20]
Membrane-bound α-syn forms a helical structure that spans eleven residues in three distinct regions. A break at the middle of this structure divides α-syn into two helical zones, increasing its ability to bind to highly curved membranes. The α-syn A30P mutation shifts two of these turns and reduces the α-syn-lipid interaction strength. However, the α-syn A53T mutation seems not to affect α-syn conformation and interaction affinity[22] whereas E46K may increase the tendency of α-syn to interact with the membrane.[23]
Although the first 100 residues of α-syn are important for lipid interactions, different N-terminal regions of α-syn bind to membranes based on the lipid to protein ratio. Typically, α-syn binds to membranes with its first 25 AA residues (known as α-syn SL1 mode) when its ratio is high enough. However, reduction in the lipid to protein ratio causes α-syn to interact with the membrane with its SL2 mode which involves binding of the first 97 AA residues.[21]
The interaction of α-syn with the membrane may initiate its aggregation. Cytosolic α-syn does not aggregate unless membrane fractions are added to the experiment. Moreover, this aggregation produces protofibrillar α-syn that causes formation of open holes in the membrane. These open channels lead to leakage of neurotransmitter from synaptic vesicles and apoptosis due to mitochondrial membrane depolarization. Interestingly, deletion of α-syn residues 74-84 can stop aggregation. Thus, the central NAC, region of α-syn, with the ability to form β-sheet aggregates, seems to be responsible for membrane disruption.[24]
Thus, α-syn can interact with the negatively charged head groups of synthetic phospholipid vesicles. This interaction causes an alternation in the conformation of α-syn from a random coil to an α-helical pattern. The Asp98–Ala140 (C-terminal region) of α-syn that is rich in glutamate residues does not form the α-helical structure on interaction with vesicles and remains unstructured. Moreover, some studies suggest a protective role for α-syn on lipid membranes to prevent lipid oxidation. In addition, it has been found that α-syn interacts with multilamellar vesicles to produce of nonbilayer or small vesicles. Thus, α-syn may play a role in the organization of membrane lipid molecules.[21]
ALPHA-SYNUCLEIN STRUCTURE AND DYNAMICS
The dynamics of α-syn in cells can be modulated in different ways. A relationship between the N-terminal region of α-syn and α-syn cellular trafficking has been demonstrated by tracking α-syn within cells. Both A30P and A53T mutant α-syn increase its transition speed between the cytoplasm and the nucleus, suggesting that mutant α-syn shows enhanced transit to the nucleus compared to normal protein. Therefore, an elevated inhibitory effect of α-syn on chromatin acetylation may lead to cell death. The way that mutations alter α-syn dynamics is based on changes in α-syn secondary structure due to the mutations. For example, A30P mutant α-syn loses its membrane binding ability and thus locates to the nucleus in higher amounts than normal α-syn.[25] Phosphorylation of α-syn at S129 is another method for cells to control α-syn dynamics. Blocking of phosphorylation at S129 residue leads to the movement of nuclear α-syn to the cytoplasm, potentially followed by the formation of LB.[25]
ALPHA-SYNUCLEIN FUNCTIONS
The function of α-syn is not entirely understood, but the following functions appear to be consistent with some of this protein's activities. These functions are also summarized in Table 1.
Table 1.
Alpha-Synuclein functions
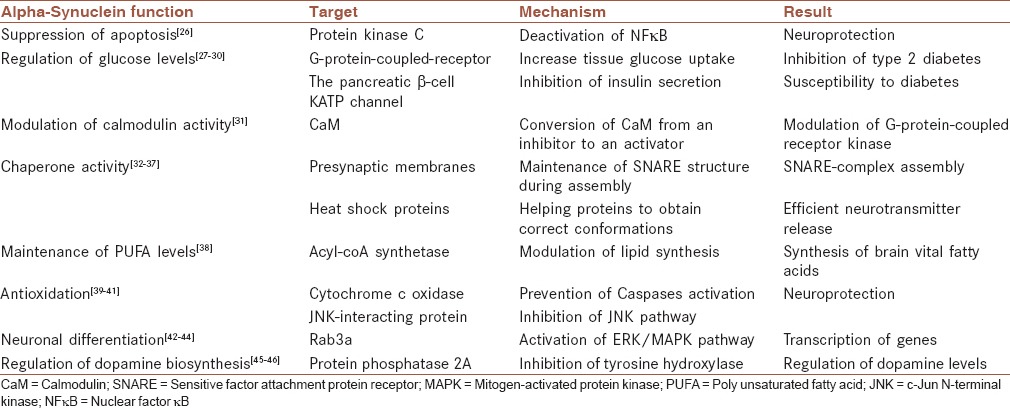
Suppression of apoptosis in dopaminergic neurons by reducing protein kinase C activity
Protein kinase C (PKC) is a serine-threonine kinase that phosphorylates different target proteins and therefore controls many mechanisms, such as apoptosis. This kinase is very sensitive to oxidative stress and triggers an apoptotic cascade in dopaminergic cells. α-Syn seems to be a PKC downregulator that can protect dopaminergic cells against apoptosis. Particularly, it has been shown that α-syn can switch off the proteolytic cascade by downregulation of PKCd expression. In dopaminergic cells, α-syn by deactivation of nuclear factor-κB reduces transcription of PKCd. Therefore, α-syn can be considered to be a neuroprotective protein in dopaminergic cells.[26]
Regulation of glucose levels
In addition to neurons, α-syn has also been identified in many other cell types, especially those involved in secretory processes. α-Syn decreases insulin secretion as a consequence of interaction with insulin-containing secretory granules. α-Syn interaction with KATP channels of insulin-secretory granules leads to inhibition of insulin secretion triggered by glucose stimulation. These findings suggest a role for α-syn in diabetes that, as other degenerative disorders such as PD, is caused by a problem in the modulation of secretory processes.[27] Moreover, it has been shown that in type 2 diabetes (T2Ds), an amyloidogenic protein deposits in pancreatic β-cells and these patients are most likely to develop PD as α-syn may combine to amyloid fibrils and form irreversible damaging complexes in dopaminergic cells.[28] On the other hand, in a recent study, α-syn is shown to increase tissue glucose uptake and thus diminish insulin resistance and possibly T2D. This study shows that in both humans and mice, the higher levels of serum α-syn is directly associated with glucose metabolism.[29] It seems that α-syn can facilitate the entrance of glucose into adipose tissues and skeletal muscle through a G-protein-coupled receptor known as LPAR2-CD90.[30]
Modulation of calmodulin activity
Calcium-modulated protein, also known as calmodulin (CaM), is a messenger protein that can be activated through binding to Ca2+ ions and triggers various mechanisms such as those involved in short- and long-term memory. It has been shown that both wild-type (WT) and mutant α-syn can interact with CaM not only in vitro but also in vivo. This interaction, which is dependent on calcium ions, causes α-syn fibrillization. Therefore, calcium ions can modulate α-syn function by inducing an interaction between α-syn and CaM. Moreover, CaM has an inhibitory effect on G-protein-coupled receptor kinase 5 (GRK5) in the absence of α-syn. On the other hand, CaM stimulates GRK5 activity in the presence of α-syn. Taken together, α-syn by controlling the conversion of CaM from an inhibitor to an activator, and vice versa, is involved in the modulation of GRK indirectly.[31]
Promotion of sensitive factor attachment protein receptor-complex assembly in vivo and in vitro
Neurotransmitters are secreted several times from presynaptic vesicles within 1 min. The sequential secretion pathway of the neurotransmitter is strongly related to the organized activity of membrane fusion proteins. Assembly and disassembly of the N-ethylmaleimide – sensitive factor attachment protein receptor (SNARE) complex is one of the reactions, which has to be repeated for each single neurotransmitter release cycle. However, assembly and disassembly of SNARE proteins are accompanied by the formation of intermediate SNARE protein structures that are unfolded and vulnerable to degeneration. α-Syn, by its chaperone activity, is involved in the maintenance of SNARE structure during these assembly/disassembly cycles. Specifically, direct interaction of α-syn with the SNARE -protein synaptobrevin-2/vesicle-associated membrane protein 2 causes the inhibition of SNARE complex assembly. Thus, α-syn dysfunction may lead to neurological problems because its deficiency reduces SNARE complex assembly.[32] During the SNARE complex assembly, unfolded cytosolic α-syn monomers bind to presynaptic membranes and form a complex of α-helically folded α-syn homomers, that is, chaperones assembly of SNARE complex and lead to neuroprotection in presynaptic terminals.[33] Moreover, large oligomers of α-syn unlike monomers are harmful, and bind to an N-terminal domain of a specific SNARE protein called synaptobrevin-2 and prevent assembly of SNARE complex leading to neurodegeneration.[34]
Acting as a molecular chaperone
α-Syn is considered to be a chaperone protein because it contains regions that are homologous with 14-3-3 proteins that interact with many cellular proteins. 14-3-3 proteins are members of the cytoplasmic chaperone family that phosphorylate the binding site of their targets, including PKC, Bcl-2-associated death promoter (BAD), and Extracellular-signal-regulated kinases (ERK). It has been shown that α-syn can also bind to the targets of 14-3-3. In addition, overexpression of α-syn under thermal and chemical conditions that prevent aggregation of target proteins is another reason for suggesting α-syn as a chaperone. α-Syn chaperone activity is dependent on both its N- and C-terminal regions. The N-terminal domain is responsible for α-syn interaction with substrate proteins, leading to the formation of a large complex while the C-terminal domain is responsible for the solubilization of that complex.[35]
The HSPs are also a family of chaperone proteins that are expressed in response to cellular stresses. HSPs are involved in stabilizing proteins to obtain correct conformations, refolding of incorrect protein structures, and degradation of misfolded proteins. It has been shown that α-syn is part of a chaperone complex containing Hsc70/Hsp70 and two co-chaperones that are responsible for efficient neurotransmitter release.[36] The functional link between α-syn and HSPs is also examined in an experiment that the csp gene was deleted in knockout (KO) mice. CSPα as an HSP is recruited into the outer membrane of presynaptic vesicles and plays role in neurotransmitter release. Deletion of this protein is associated with the reduction of α-syn levels and lethal neurodegeneration. Interestingly, CSPα KO mice can be rescued from death if human WT α-syn was overexpressed that shows how α-syn can recover a chaperone activity.[37]
Maintenance of polyunsaturated fatty acids levels
The brain as the second main source of lipid content after adipose tissue is enriched in two polyunsaturated fatty acids (PUFAs), arachidonic acid and docosahexaenoic acid, which constitute about 20% of the brain fatty acids. Acyl-coA synthetase (ACSL) by adding acyl groups to a fatty acid backbone synthesizes these fatty acids from other available fatty acid stocks. The ACSL6 isoform that incorporates arachidonic acid into phospholipids is modulated by α-syn. Therefore, α-syn may be involved in the presentation of substrates to ACSL and consequently, in control of the brain's PUFAs levels.[38]
Acting as an antioxidant and preventing oxidation of unsaturated lipids in vesicles
Oxidative stress in dopaminergic neurons could initiate the pathological damage leading to PD. This damage is triggered by oxidation of unsaturated phospholipids that consequently damage both cellular and intracellular membranes. The sensitivity of dopaminergic neurons to such damage is caused by the oxidative metabolism of dopamine within these cells. Therefore, dopaminergic cell membranes that are rich in unsaturated fatty acids are more vulnerable to this oxidation and damage. The monomeric form of α-syn by interaction with lipid membranes can protect lipids from oxidation, but the fibrillar form of α-syn does not have this capability. Therefore, it seems that monomeric α-syn could act as an antioxidant that plays an important role in dopaminergic neurons to protect them against oxidative damage.[39] Another study suggests that maybe α-syn is not an antioxidant but can prevent apoptosis by binding to cytochrome c oxidase in mitochondrial membrane and inhibits release of cytochrome c from mitochondria to cytosol. Consequently, caspases cannot be activated and thus cannot trigger apoptosis.[40] Moreover, hydrogen peroxide cannot damage α-syn expressing neural cells while it is toxic in control cells. Hydrogen peroxide in the absence of α-syn activates c-Jun N-terminal kinase (JNK), a member of mitogen-activated protein kinase (MAPK) family that promotes apoptosis under oxidative stress condition. α-Syn by activation of JNK-interacting protein/islet-brain (JIP-1b/IB1) as an inhibitor of JNK pathway may protect cells from injury.[41]
Neuronal differentiation
As α-syn is both functionally and structurally homologous with chaperone protein 14-3-3,[42] it can interact not only with 14-3-3 itself but also with its ligands, such as kinase suppressor of Ras. Therefore, α-syn is considered to be involved in different cellular functions via activation of Ras. The activated form of Ras can activate a chain of events including the ERK/MAPK pathway that is involved in sending a growth factor signal from the cell receptor to transcription factors in the nucleus.[43] Moreover, α-syn binds to membrane via interaction by Ras-related GTPase Rab3a. This molecular association is evidenced using antibody against Rab3a that dissociates bound α-syn from membrane signifying that Rab3a is involved in stabilizing α-syn on synaptosomes.[44]
Regulation of dopamine biosynthesis
α-Syn as a downregulator of tyrosine hydroxylase (TH) activity can modulate production of dopamine and control its cellular levels. Therefore, α-syn-reduced expression or α-syn aggregation leads to increased dopamine synthesis, leading to oxidative stress caused by dopamine metabolism. The inhibitory effect of α-syn on TH activity is not direct and is dependent on the interaction between α-syn and protein phosphatase 2A (PP2A). This α-syn PP2A interaction activates PP2A phosphatase function, leading to dephosphorylation of TH-Ser 40 residue, and therefore TH inhibition. Both α-syn overexpression and mutations have been shown to upregulate the inhibitory effect of α-syn on TH and dopamine levels, leading to downregulation of dopamine synthesis and release.[45] On the other hand, it has been shown that overexpression of α-syn increases activity of src, a member of nonreceptor tyrosine kinases family, leading to hyperphosphorylation of proteins and neurodegeneration. Scr can phosphorylate PP2A as a substrate at Tyr 307 and consequently inhibit its phosphates activity.[46]
Modulating vesicle trafficking
The synaptic vesicle formation pathway starts with the curvation of the membrane into a bud with the help of coat proteins such as clathrin which act as a spherical frame. This process is followed by the targeting of vesicles to a specific secretory point with the help of SNARE proteins. In this pathway, α-syn binds to the curved membrane selectively to modulate vesicle trafficking in synapses.[47] α-Syn possibly reduces both the amount and the speed of vesicle recycling from synapses to the presynaptic area. This reduction in vesicular dynamics is important for synaptic homeostasis as the absence of α-syn disturbs; this equilibrium by increased vesicle trafficking.[48]
This modulating role of α-syn is because of its specific structure. The 11 residue repeats of α-syn in the presence of liposomes can fold to a membrane binding helix that is divided by a mid-unstructured fragment.[49] α-Syn can detect the vesicle pool as it has an affinity for lipid bilayers. This means that α-syn is more likely to be present at high levels in vesicle pools containing lots of small liposomes than in other parts of the cell since it can sense the membrane curvature of the vesicles.[50]
ALPHA-SYNUCLEIN-PROTEIN INTERACTIONS AND PARKINSON DISEASE
It is possible that α-syn could damage cells by interrupting the function of other proteins since it can interact with many proteins, including tubulin,[51] parkin,[52] phospholipases D (PLD),[53] tau protein, 14-3-3, and the dopamine receptor.[54]
Alpha-synuclein interaction with tubulin
α-Syn has been identified in combination with tubulin in PD brain extracts including LBs and Lewy neuritis. Moreover, in vitro studies have revealed that adding a small amount of tubulin in physiological media containing α-syn can be sufficient not only to start but also to accelerate α-syn aggregation into fibrils.[55] Furthermore, it has been shown that extracellular oligomeric α-syn can reduce polymerization of tubulins in dopaminergic neurons leading to cell death.[51] These findings suggest that in the brain, a similar interaction between tubulin and α-syn may be responsible for the formation of LBs. Therefore, it seems possible that environmental toxins could initiate α-syn aggregation by disrupting the assembly of tubulins into microtubules, leading to increased free cytosolic tubulins that can interact with α-syn.
Alpha-synuclein interaction with parkin
Parkin as the ubiquitin ligase member of the proteasome system is involved in the degradation of redundant and disordered proteins. Therefore, mutations in parkin by affecting its ligase activity could lead to the formation of insoluble aggregates. Parkin mutations that damage parkin function are the most common causes of familial PD in the form of autosomal recessive juvenile PD. It also seems possible that the interaction between α-syn and parkin in stress conditions forms a site for deposition of α-tubulin, and as a result, triggers alteration of the neuronal cytoskeleton, causing neuronal dysfunction.[52] Moreover, parkin and Leucine-rich repeat kinase2 (LRRK2) are two modulators of the secretory pathway, and they are assumed to be involved in the development of synapses. As regulation of synaptic vesicle dynamics is important for proper neuronal activity, both protein mutations could be related to synucleinopathy.[56]
Alpha-synuclein interaction with LRRK2
α-Syn as a presynaptic protein may accumulate in the cytosol when parkin and LRRK2 mutations destroy synaptic assemblies, leading to the unwanted distribution of α-syn in the cytosol. It has been shown that LRRK2 can affect α-syn trafficking as its overexpression destroys polymerization of tubulin. In addition, mutant LRRK2 may induce phosphorylation of α-syn at S129, propagating oligomerization. However, phosphorylation of α-syn is not due directly to LRRK2 activity or its interaction with α-syn. Possibly, 14-3-3 is a mediator between α-syn and LRRK2. Mitochondrial improper function and imperfect biogenesis are associated with mutant LRRK2, leading to the production of superoxide and subsequently aggregation of α-syn. Moreover, the ubiquitin-proteasome system activity and chaperone-mediated autophagy can be destroyed by either LRRK2 mutations or α-syn A53T mutation. Therefore, mutation of both proteins at the same time can even worsen the situation, leading to very high accumulation of proteins in neurons.[57]
Alpha-synuclein interaction with dopamine receptor
The dopamine transporter (DAT) is responsible for the reuptake of dopamine from the synapse and its delivery back to the presynaptic terminal of neurosecretory cells. It has been reported that, in PD brains, α-syn binds to the C-terminal region of DAT via its NAC sequence. This interaction increases neuronal levels of dopamine by facilitating the membrane clustering of DAT and enhancing its activity. Increased levels of dopamine in neurons can damage cells as a consequence of enhanced dopamine metabolism reactions leading to oxidative stress.[54] However, in normal brains, α-syn controls the neuronal levels of dopamine by decreasing DAT activity. These findings suggest that aggregated α-syn could be neurotoxic in dopaminergic cells by initiating exposure to free radical products of dopamine oxidative metabolism.[58]
Alpha-synuclein interaction with synphilin-1
An interaction between synphilin-1 and α-syn has been reported in vivo using the yeast two-hybrid system.[59] Moreover, α-syn and synphilin-1 aggregate into an eosinophilic cytosolic complex similar to LB in α-syn and synphilin-1 co-transfected mammalian cells. In normal conditions, the N-terminal domain of WT α-syn interacts with the central region (ankyrin and coiled-coil domains) of synphilin-1. The sufficient regions for this interaction are α-syn AA 1–65 and synphilin-1 AA 349–555. This interaction protects α-syn from degradation by inhibition of 20S proteasome and thus formation of harmful aggregates in PD.[60] However, the α-syn A53T mutant interacts with synphilin-1 via its C-terminal domain, while the α-syn A30P mutant increases the number of α-syn synphilin-1 compounds.[61] Taken together, α-syn mutations could alter the site and efficiency of α-syn synphilin-1 interactions.
Alpha-Synuclein interaction with phospholipases
PLD is a hydrolytic enzyme that is located in the plasma membrane and hydrolyzes phosphatidylcholine to PA. PA, as a signaling lipid, modulates different processes, especially those involved in cellular morphology and vesicular transport. In neurons, PA may be involved in the growth, differentiation, and releases of neurotransmitters. α-Syn inhibits PLD activity. Therefore, α-syn, by controlling the production of PA, modulates cell signaling.[11] It has been shown that overexpression of PLD2 in rat brain leads to severe damage of dopaminergic neurons while either coexpression of human WT α-syn or reduction of PLD2 activity by siRNA inhibits the lethal effect of PLD2. Moreover, coimmunocapture of α-syn with PLD2 in the extract of striatal tissues revealed that α-syn interacts with PLD2 in vivo and downregulates its activity.[53]
Alpha-Synuclein interaction with small ubiquitin-related modifiers
Small ubiquitin-related modifiers usually attach to proteins and modify their structures after translation. This process, known as sumoylation, may help proteins to remain soluble. A study has shown that sumoylated α-syn does not aggregate, suggesting that sumoylation deficits could lead to α-syn accumulation. α-Syn residue lysine 96 and lysine 102 seem to be sites of sumoylation of α-syn.[62]
CONCLUSION
In PD brains, α-syn accumulates in nerve cells of the SN, pons, medulla, and gut leading to inflammation and cell death and subsequently difficulties in movement, digestion, circulation, and sleep. It seems that PD may be cured by stop α-syn from aggregation. Some pharmaceutical companies are focusing on compounds that prevent α-syn from aggregation such as NPT200-11 that is produced in Neuropore Company, and its human trial is begun. As α-syn is the most potential target for treatment of PD, many scientists around the world work on α-syn to know much more about its structure, functions, and interactions.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
I would like to acknowledge Professor David Allsop. This work is supported by an EC Framework 7 Marie Curie Fellowship Training Network Grant (NEURASYNC) on “α-synuclein-related brain diseases.”
REFERENCES
- 1.McLean PJ, Kawamata H, Ribich S, Hyman BT. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson's disease-linked mutations. J Biol Chem. 2000;275:8812–6. doi: 10.1074/jbc.275.12.8812. [DOI] [PubMed] [Google Scholar]
- 2.Yu S, Li X, Liu G, Han J, Zhang C, Li Y, et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience. 2007;145:539–55. doi: 10.1016/j.neuroscience.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 3.Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. α-synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 2010;33:559–68. doi: 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Agnaf OM, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson's disease. FASEB J. 2006;20:419–25. doi: 10.1096/fj.03-1449com. [DOI] [PubMed] [Google Scholar]
- 5.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–15. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–6. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 8.Spillantini MG, Divane A, Goedert M. Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics. 1995;27:379–81. doi: 10.1006/geno.1995.1063. [DOI] [PubMed] [Google Scholar]
- 9.Bruening W, Giasson BI, Klein-Szanto AJ, Lee VM, Trojanowski JQ, Godwin AK. Synucleins are expressed in the majority of breast and ovarian carcinomas and in preneoplastic lesions of the ovary. Cancer. 2000;88:2154–63. [PubMed] [Google Scholar]
- 10.George JM. The synucleins. Genome Biol. 2001 doi: 10.1186/gb-2001-3-1-reviews3002. reviews3002.1-reviews3002.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn BH, Rhim H, Kim SY, Sung YM, Lee MY, Choi JY, et al. Alpha-synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J Biol Chem. 2002;277:12334–42. doi: 10.1074/jbc.M110414200. [DOI] [PubMed] [Google Scholar]
- 12.Sode K, Ochiai S, Kobayashi N, Usuzaka E. Effect of reparation of repeat sequences in the human α-synuclein on fibrillation ability. Biol Sci. 2007;3:1–7. doi: 10.7150/ijbs.3.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajagopalan S, Andersen JK. Alpha synuclein aggregation: Is it the toxic gain of function responsible for neurodegeneration in Parkinson's disease? Mech Ageing Dev. 2001;122:1499–510. doi: 10.1016/s0047-6374(01)00283-4. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525:486–90. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn M, Kim S, Kang M, Ryu Y, Kim TD. Chaperone-like activities of alpha-synuclein: Alpha-synuclein assists enzyme activities of esterases. Biochem Biophys Res Commun. 2006;346:1142–9. doi: 10.1016/j.bbrc.2006.05.213. [DOI] [PubMed] [Google Scholar]
- 16.Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J Mol Biol. 2002;322:383–93. doi: 10.1016/s0022-2836(02)00775-1. [DOI] [PubMed] [Google Scholar]
- 17.Ly T, Julian RR. Protein-metal interactions of calmodulin and alpha-synuclein monitored by selective noncovalent adduct protein probing mass spectrometry. J Am Soc Mass Spectrom. 2008;19:1663–72. doi: 10.1016/j.jasms.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Esposito A, Dohm CP, Kermer P, Bähr M, Wouters FS. Alpha-synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis. 2007;26:521–31. doi: 10.1016/j.nbd.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Kim TD, Choi E, Rhim H, Paik SR, Yang CH. Alpha-synuclein has structural and functional similarities to small heat shock proteins. Biochem Biophys Res Commun. 2004;324:1352–9. doi: 10.1016/j.bbrc.2004.09.208. [DOI] [PubMed] [Google Scholar]
- 20.Wang GF, Li C, Pielak GJ. 19F NMR studies of α-synuclein-membrane interactions. Protein Sci. 2010;19:1686–91. doi: 10.1002/pro.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim Biophys Acta. 2012;1818:162–71. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh D, Sahay S, Ranjan P, Salot S, Mohite GM, Singh PK, et al. The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry. 2014;53:6419–21. doi: 10.1021/bi5010365. [DOI] [PubMed] [Google Scholar]
- 23.Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, et al. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry. 2007;46:7107–18. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- 24.Högen T, Levin J, Schmidt F, Caruana M, Vassallo N, Kretzschmar H, et al. Two different binding modes of α-synuclein to lipid vesicles depending on its aggregation state. Biophys J. 2012;102:1646–55. doi: 10.1016/j.bpj.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonçalves S, Outeiro TF. Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol Neurobiol. 2013;47:1081–92. doi: 10.1007/s12035-013-8406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. α-synuclein negatively regulates protein kinase Cd expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011;31:2035–51. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng X, Lou H, Wang J, Li L, Swanson AL, Sun M, et al. α-synuclein binds the K (ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am J Physiol Endocrinol Metab. 2011;300:E276–86. doi: 10.1152/ajpendo.00262.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma SK, Chorell E, Steneberg P, Vernersson-Lindahl E, Edlund H, Wittung-Stafshede P. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci Rep. 2015;5:12531. doi: 10.1038/srep12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Araujo G, Nakagami H, Takami Y, Katsuya T, Akasaka H, Saitoh S, et al. Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci Rep. 2015;5:12081. doi: 10.1038/srep12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Araujo G, Nakagami H, Hayashi H, Mori M, Shiuchi T, Minokoshi Y, et al. Alpha-synuclein elicits glucose uptake and utilization in adipocytes through the Gab1/PI3K/Akt transduction pathway. Cell Mol Life Sci. 2013;70:1123–33. doi: 10.1007/s00018-012-1198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez J, Moeller I, Erdjument-Bromage H, Tempst P, Lauring B. Parkinson's disease-associated alpha-synuclein is a calmodulin substrate. J Biol Chem. 2003;278:17379–87. doi: 10.1074/jbc.M209020200. [DOI] [PubMed] [Google Scholar]
- 32.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burré J, Sharma M, Südhof TC. α-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–83. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, et al. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A. 2013;110:4087–92. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SM, Jung HY, Kim TD, Park JH, Yang CH, Kim J. Distinct Roles of the N-terminal-binding Domain and the C-terminal-solubilizing Domain of á-Synuclein, a Molecular Chaperone. Biological Chemistry. 2002;277:28512–20. doi: 10.1074/jbc.M111971200. [DOI] [PubMed] [Google Scholar]
- 36.Witt SN. Molecular chaperones, α-synuclein, and neurodegeneration. Mol Neurobiol. 2013;47:552–60. doi: 10.1007/s12035-012-8325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Ruipérez V, Darios F, Davletov B. Alpha-synuclein, lipids and Parkinson's disease. Prog Lipid Res. 2010;49:420–8. doi: 10.1016/j.plipres.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Zhu M, Qin ZJ, Hu D, Munishkina LA, Fink AL. Alpha-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry. 2006;45:8135–42. doi: 10.1021/bi052584t. [DOI] [PubMed] [Google Scholar]
- 40.Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther. 2005;313:46–55. doi: 10.1124/jpet.104.078469. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto M, Hsu LJ, Rockenstein E, Takenouchi T, Mallory M, Masliah E. Alpha-synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J Biol Chem. 2002;277:11465–72. doi: 10.1074/jbc.M111428200. [DOI] [PubMed] [Google Scholar]
- 42.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, et al. Alpha-synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19:5782–91. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: Structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–47. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 44.Chen RH, Wislet-Gendebien S, Samuel F, Visanji NP, Zhang G, Marsilio D, et al. α-synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J Biol Chem. 2013;288:7438–49. doi: 10.1074/jbc.M112.439497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng X, Tehranian R, Dietrich P, Stefanis L, Perez RG. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J Cell Sci. 2005;118(Pt 15):3523–30. doi: 10.1242/jcs.02481. [DOI] [PubMed] [Google Scholar]
- 46.Yang W, Wang X, Duan C, Lu L, Yang H. Alpha-synuclein overexpression increases phospho-protein phosphatase 2A levels via formation of calmodulin/Src complex. Neurochem Int. 2013;63:180–94. doi: 10.1016/j.neuint.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 47.Pranke IM, Morello V, Bigay J, Gibson K, Verbavatz JM, Antonny B, et al. α-synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol. 2011;194:89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott D, Roy S. α-synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–35. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–93. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drin G, Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584:1840–7. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Jin J, Davis J, Zhou Y, Wang Y, Liu J, et al. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem Biophys Res Commun. 2007;356:548–53. doi: 10.1016/j.bbrc.2007.02.163. [DOI] [PubMed] [Google Scholar]
- 52.Kawahara K, Hashimoto M, Bar-On P, Ho GJ, Crews L, Mizuno H, et al. Alpha-synuclein aggregates interfere with Parkin solubility and distribution: Role in the pathogenesis of Parkinson disease. J Biol Chem. 2008;283:6979–87. doi: 10.1074/jbc.M710418200. [DOI] [PubMed] [Google Scholar]
- 53.Gorbatyuk OS, Li S, Nguyen FN, Manfredsson FP, Kondrikova G, Sullivan LF, et al. α-synuclein expression in rat substantia nigra suppresses phospholipase D2 toxicity and nigral neurodegeneration. Mol Ther. 2010;18:1758–68. doi: 10.1038/mt.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–26. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 55.Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, et al. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem. 2002;277:2112–7. doi: 10.1074/jbc.M102981200. [DOI] [PubMed] [Google Scholar]
- 56.Plowey ED, Chu CT. Synaptic dysfunction in genetic models of Parkinson's disease: A role for autophagy? Neurobiol Dis. 2011;43:60–7. doi: 10.1016/j.nbd.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu G, Aliaga L, Cai H. α-synuclein, LRRK2 and their interplay in Parkinson's disease. Future Neurol. 2012;7:145–153. doi: 10.2217/fnl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wersinger C, Sidhu A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci Lett. 2003;340:189–92. doi: 10.1016/s0304-3940(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 59.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–4. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 60.Alvarez-Castelao B, Castaño JG. Synphilin-1 inhibits alpha-synuclein degradation by the proteasome. Cell Mol Life Sci. 2011;68:2643–54. doi: 10.1007/s00018-010-0592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neystat M, Rzhetskaya M, Kholodilov N, Burke RE. Analysis of synphilin-1 and synuclein interactions by yeast two-hybrid beta-galactosidase liquid assay. Neurosci Lett. 2002;325:119–23. doi: 10.1016/s0304-3940(02)00253-7. [DOI] [PubMed] [Google Scholar]
- 62.Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol. 2011;194:49–60. doi: 10.1083/jcb.201010117. [DOI] [PMC free article] [PubMed] [Google Scholar]