

Abstract
Neuroligins are postsynaptic cell-adhesion molecules implicated in autism and other neuropsychiatric disorders. Despite extensive work, the role of neuroligins in synapse function and plasticity, especially NMDA receptor (NMDAR)-dependent LTP, remains unclear. To establish which synaptic functions unequivocally require neuroligins, we analyzed single and triple conditional knockout (cKO) mice for all three major neuroligin isoforms (NL1-NL3). We inactivated neuroligins by stereotactic viral expression of Cre-recombinase in hippocampal CA1 region pyramidal neurons at postnatal day 0 (P0) or day 21 (P21), and measured synaptic function, synaptic plasticity, and spine numbers in acute hippocampal slices 2–3 weeks later. Surprisingly, we find that ablation of neuroligins in newborn or juvenile mice only modestly impaired basal synaptic function in hippocampus, and caused no alteration in postsynaptic spine numbers. However, triple cKO of NL1-NL3 or single cKO of NL1 impaired NMDAR-mediated excitatory postsynaptic currents (NMDAR EPSCs), and abolished NMDAR-dependent LTP. Strikingly, the NL1 cKO also abolished LTP elicited by activation of L-type Ca2+-channels during blockade of NMDARs. These findings demonstrate that neuroligins are generally not essential for synapse formation in CA1 pyramidal neurons but shape synaptic properties and that NL1 specifically is required for LTP induced by postsynaptic Ca2+-elevations, a function which may contribute to the pathophysiological role of neuroligins in brain disorders.
Keywords: neuroligin, synapse, NMDA receptor, long-term potentiation (LTP)
INTRODUCTION
Neuroligins are postsynaptic cell-adhesion molecules that interact with presynaptic neurexins to shape synaptic properties in robust and complex ways1–3. Interest in elucidating the specific functions of neuroligins has been stimulated by their genetic association with neuropsychiatric diseases, most notably autism spectrum disorders (ASD)1,4–6. Despite decades of work, however, controversies remain about specific functions of neuroligins (reviewed in 7). The inconsistencies in results from experiments that study neuroligins have legitimate biological causes (e.g. studying different synapses in different preparations; performing neuroligin manipulations at different ages), but may also partly be due to the methods used. Knockdown approaches using shRNAs or microRNAs as well as overexpression approaches provide valuable information but suffer from inherent limitations. By definition, knockdown approaches do not eliminate all of the targeted proteins and often have off-target effects. Overexpression of proteins may cause targeting of proteins inappropriately such that the observed effects do not necessarily reflect the functions of the endogenous protein. Similarly, constitutive knockouts (KOs) of neuroligins provide information about their absolute necessity but are potentially compromised by developmental compensation, which may hinder functional analyses. Finally, many studies on neuroligins used dissociated cultured neurons or slice cultures, preparations that are generated from developing brains and exhibit on-going synaptogenesis with altered target specificity and thus may not precisely mimic properties of synapses in the intact mammalian brain.
To address these issues, we generated single and triple conditional knockout (cKO) lines of the three major neuroligins found in mouse brain8–10 (NL1, NL2, and NL3). NL1 is exclusively localized at excitatory synapses11; NL2 is selectively localized at inhibitory and cholinergic synapses12,13; while NL3 is localized at both excitatory and inhibitory synapses1,14. We did not examine NL4 because it is found at very low levels in mouse brain, and predominantly localizes to glycinergic synapses15. We chose to genetically delete neuroligins in vivo at two different developmental stages in hippocampal CA1 pyramidal neurons by stereotactically injecting viruses expressing Cre-recombinase. By deleting one or more neuroligins at P0 and analyzing acute slices ~3 weeks later, we could define the role of neuroligins in synapse formation and synapse maturation since P0-P14 is a time of robust synaptogenesis in CA116,17. By performing the same experiments after deleting one or more neuroligins at P21, a time at which synapse formation is largely complete, we could assess the role of neuroligins in mature synapses. These experimental procedures provide, arguably, the most biologically relevant and rigorous test of the role of neuroligins in synapse function since conditional genetic deletion in vivo minimizes the possibility of compensatory adaptations while performing the deletion in the intact brain. Our results demonstrate that neuroligins perform an essential role in long-term synaptic plasticity in the hippocampus and additionally contribute to shaping synapse properties and strength, but are expendable for synapse formation and maintenance both in developing and in mature neurons.
MATERIALS AND METHODS
Mouse lines
All experiments were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. All experiments used homozygous NL1, NL2, and NL3 single and triple cKO mice8–10 that were maintained on a mixed CD1/C57BL6 background except for the NL3 cKO line (pure C57BL6 background) and NL2 cKO line (pure CD1 background).
Stereotactic injections of lentiviruses
Lentiviruses expressing eGFP-tagged Cre-recombinase driven by a ubiquitin promoter were procured from the Stanford Gene Vector and Virus Core and stereotactically injected into the CA1 region of the hippocampus as described18,19. For P0 injections (coordinates: ~1.0 mm anterior to interaural; ~0.7 mm lateral to midline; depth of ~1.15 mm from the surface of the skull, with 0.5 μl lentivirus injected at 0.8 μl/min with a microinjection pump), pups were anesthetized on ice for 5 min and immobilized with tape on a mold that maintained the top surface of the skull horizontally. Procedures for virus injections at P21 were as previously described20. For vGAT quantification experiments 1.5 μl of lentivirus was injected unilaterally.
Electrophysiology
Mice were analyzed at P18-25 (P0 injections) or at P35-42 (P21 injections). Transverse hippocampal slices from dorsal hippocampus were cut20,21 in a solution containing (in mM): 228 sucrose, 26 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 7 MgSO4 and 0.5 CaCl2, and recovered in artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 26 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4 and 2.5 CaCl2. Cre-recombinase expressing cells were identified by eGFP epifluorescence; uninfected cells (eGFP negative) from the same slices were used as controls. The identity of recorded infected versus uninfected cells was confirmed by the eGFP signal in the nucleus which was pulled into the recording electrode after the recording. Whole-cell recordings were made using an internal solution containing (in mM): 140 CsMeSO4, 8 CsCl, 10 HEPES, 0.25 EGTA, 2 Mg2ATP, 0.3 Na3GTP, 0.1 spermine, 7 phosphocreatine (pH 7.25–7.3; osmolarity 294–298). For morphological reconstruction experiments, the internal solution additionally contained 0.2% biocytin. AMPAR-mediated mEPSCs were recorded with tetrodotoxin (TTX, 1 μM), D-APV (50 μM), and picrotoxin (PTX, 50 μM) in the ACSF. For GABAAR-mediated mIPSCs, CsMeSO4 was replaced by 140 mM CsCl in the internal soultion and TTX, CNQX (20 μM), and D-APV (50 μM) were added to the ACSF. A theta glass pipette filled with ACSF was placed in the stratum radiatum to evoke EPSCs in CA1 pyramidal cells. PTX was included in extracellular ACSF in all experiments examining excitatory synaptic transmission. Stimulation pulses were delivered every 10 s. NMDAR/AMPAR ratios were calculated as the averaged NMDAR-mediated EPSC (20 trials, measured at 50 ms after the onset of EPSCs at +40 mV) divided by the averaged AMPAR-mediated EPSC (20 trials, measured as the peak amplitude of EPSCs at −70 mV). Two pulses at different intervals (20, 50,100, 200, 500 ms) were delivered to calculate paired pulse ratios (PPR). To induce NMDAR-dependent LTP, after collecting a 5–10 min baseline 2 trains of high frequency stimulation (100 Hz, 1s) separated by 20 s were delivered while clamping the postsynaptic cell at 0 mV20. To induce NMDAR-independent LTP22, twenty postsynaptic depolarization pulses (80 mV, 1 s separated by 6 s) were applied to cells clamped at −70 mV. 5 μM Bay K8644 (an L-type calcium channel activator) and 50 μM APV were included in the ACSF. All drugs were obtained from Tocris except tetanus-toxin light chain (List Biological Laboratories).
Immunohistochemistry
Immunohistochemical experiments were performed as previously described10. Briefly, triple NL123 floxed mice (~P21) were anesthetized with isoflurane, perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in 0.1 M PBS via a perfusion pump (2 ml/min). Perfused brains were post-fixed in 4% PFA for 2 h at room temperature (RT) then cryoprotected in 30% sucrose (in 1× PBS) for 24 h at 4°C. Coronal brain sections (30 μm) were cryo-sectioned at −20°C (Leica CM1050). Sections were serially washed with PBS and incubated in blocking solution (0.3 % Triton X-100 and 5 % goat serum in PBS) for 1 h at RT, and incubated for 24 h at 4 °C with primary antibodies diluted in PBS (anti-vGAT, 1:500, rabbit, Synaptic Systems). Sections were washed 4 times (15 min each time) in PBS, treated with secondary antibodies (1:1000, Alexa 555, Invitrogen) at 4°C overnight, and washed 4 times (15 min each time) again in PBS. Sections were then mounted on superfrost slides and covered with mounting media (Vectashield, Vector Labs). For triple NL123 cKO group, only slices with robust virus infection were selected for imaging. Single plane images (at 1024 x 1024 resolution) from hippocampal CA1 region were acquired using a Nikon confocal microscope (A1Rsi) with a 60x oil objective (PlanApo, NA1.4). All acquisition parameters were kept constant among different conditions within experiments. Image backgrounds were normalized, and immunoreactive elements were analyzed with Nikon analysis software. The density of vGAT positive puncta in striatum pyramidale (40 μm X 40 μm) and striatum radiatum (40 μm X 60 μm) were automatically analyzed using the same setting for both control and cKO images.
For spine imaging experiments, slices were fixed after electrophysiology experiments with 4% PFA in PBS for 2 h, washed 3 times (15 min each time) with PBS, and sequentially incubated at room temperature with PBS containing 10% goat serum/0.5% Triton X-100 for 1 h, and with PBS containing streptavidin (Alexa Fluor 594 conjugate, 1:1000, Invitrogen) for 2 h. Slices were washed 3 times (15 min each time) in PBS, mounted, and analyzed by 3D-imaging of dendrites using the same confocal microscope. For every labeled neuron, 5–8 regions of interest (10 μm diameter) from different secondary/tertiary dendrites were selected for manual spine counting on anonymized images.
RESULTS
Conditional deletion of neuroligins at P0 reduces inhibitory but not excitatory synapse formation
To define the requisite roles of NL1-3 in synapse formation, maturation and function, we used mice containing cKO alleles of NL1, NL2 and NL3 that were generated using standard approaches8–10 (Fig. 1A). Lentiviruses expressing eGFP-tagged cre-recombinase were stereotactically injected into the CA1 region of the hippocampus at P0, and whole-cell recordings from infected and uninfected cells were made from acute slices prepared at P18-25 (Fig. 1B). Using modest titers of lentivirus, we obtained sparse infectivity (Fig. 1C) that allowed recordings in the same slice from control and neuroligin-deficient neurons.
Figure 1. Sparse deletion of Neuroligins from hippocampal CA1 pyramidal neurons during postnatal development produces impairments in inhibitory but not fast excitatory synaptic transmission.
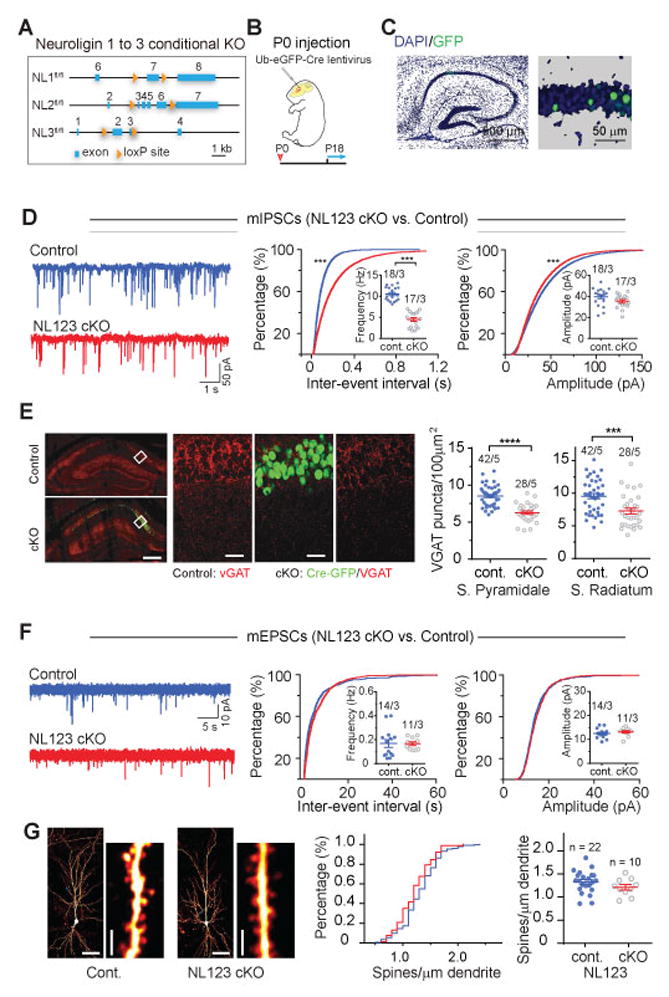
(A) Schematic diagram of the NL cKO alleles.
(B) Experimental design for injections at P0, followed by experimental analyses at postnatal P18–25.
(C) Representative images of the hippocampus from a P0 injected mouse (left) and of sparsely infected CA1-region neurons (right; blue, DAPI; green, GFP). In all experiments, non-infected neurons served as controls for adjacent infected neurons in the same slices.
(D) Analysis of mIPSCs in triple NL123 cKO neurons. Left, representative traces; middle, cumulative distribution of the mIPSC inter-event interval (inset: data points from individual cells and means of mIPSC frequency); right, cumulative distribution of mIPSC amplitudes (inset: data points from individual cells and mean mIPSC amplitudes).
(E) vGAT staining in stratum pyramidale and stratum radiatum of hippocampal CA1 region. Left, low resolution images showing without (top, control) and with (bottom, cKO) robust infection of Ub-eGFP-Cre lentivirus, scale bar: 200 μm; Middle, high resolution images from the white box in control and cKO slices used for analysis, scale bar: 50 μm; Group data showing that the density of inhibitory synapses in stratum pyramidale and stratum radiatum of CA1 region was decreased in triple NL123 cKO.
(F) Same as D, but for mEPSCs in triple NL123 cKO neurons.
(G) NL123 deletion in newborn mice does not change the spine density of CA1 pyramidal cells. Left, representative images of biocytin-labeled CA1 pyramidal neurons after patch-clamp recording, with a lower and higher magnification images shown side by side (calibration bars: 50 μm and 2 μm, respectively); middle, cumulative distribution of spine density (control: 173 dendrites/22 neurons; cKO: 77 dendrites/10 NL123 cKO pyramidal neurons); right, summary graph of mean spine densities with data points from individual neurons.
Data in summary graphs are means ± SEM; statistical comparisons were performed with the Kolmogorov-Smirnov test (cumulative distributions) or student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001; non-significant comparisons are not labeled). Numbers indicate number of cells/mice examined.
The triple NL123 cKO at PO caused ~50% reduction in mean mIPSC frequency with a clear right-shift in the cumulative distribution of mIPSC inter-event intervals and only a minor decrease in mIPSC amplitudes (Fig. 1D). Accordingly, immunostaining for GABAergic synapses showed a ~30% decreased number of vGAT puncta in the CA1 pyramidal cell layer and stratum radiatum in NL123 cKO mice (Fig 1E). To assess the relative contributions of NL2 and NL3, which are both expressed at inhibitory synapses13,14, we repeated this analysis in single NL2 and NL3 cKOs. The NL2 cKO phenocopied the NL123 cKO (Fig. S1A). Surprisingly, NL3 cKO at P0 caused a modest increase in mIPSC frequency but no change in mIPSC amplitudes (Fig. S1B).
In marked contrast to the effects on mIPSCs, the triple NL123 cKO had no detectable effect on the frequency or amplitude of AMPA receptor (AMPAR)-mediated mEPSCs (Fig. 1F). Similarly, the triple NL123 cKO at P0 had no effect on the density of dendritic spines on secondary and tertiary dendritic branches of CA1 pyramidal neurons (Fig. 1G). Thus, neuroligins are not required for the formation of excitatory synapses or function of AMPARs on CA1 pyramidal neurons during early postnatal development, but are required for the development of normal inhibitory synaptic function.
Neuroligins play a modest role in mature inhibitory and excitatory synapse function
To evaluate whether neuroligins play a requisite role in the maintenance of function at mature inhibitory and excitatory synapses on CA1 pyramidal neurons, we injected Cre-expressing lentivirus at P21 into triple NL123 cKO mice and prepared acute slices 2–3 weeks later (Fig 2A). Unexpectedly, this manipulation caused only very small decreases in both mIPSC frequency and mIPSC amplitudes (Fig. 2B). To assess the relative contributions of NL2 and NL3 in the maintenance of inhibitory synapse function, we repeated this analysis in single NL2 and NL3 cKOs. The NL2 cKO showed a small decrease in the amplitudes of mIPSCs without any change in their frequency (Fig. S2A) while NL3 cKO at P21 caused no changes in either mIPSC amplitude or frequency (Fig. S2B) thus specifying a redundant role for NL2 and NL3 in inhibitory synapse maintenance and a role for NL2 alone in the functioning of these synapses in adult mice. The genetic deletion of NL123 or NL1 alone at P21 had no detectable effects on mEPSC frequency or amplitude (Fig. 2C, S3A). Morphological analysis revealed that spine density was also not altered by NL123 KO at P21 (Fig. 2D) Thus, neuroligins are not necessary for maintaining a normal complement of excitatory synapses on CA1 pyramidal cells and play only a minor role in maintaining inhibitory synaptic function.
Figure 2. Sparse deletion of Neuroligins from mature hippocampal CA1 pyramidal neurons in juvenile mice causes similar but less severe phenotypes as deletions in developing neurons.
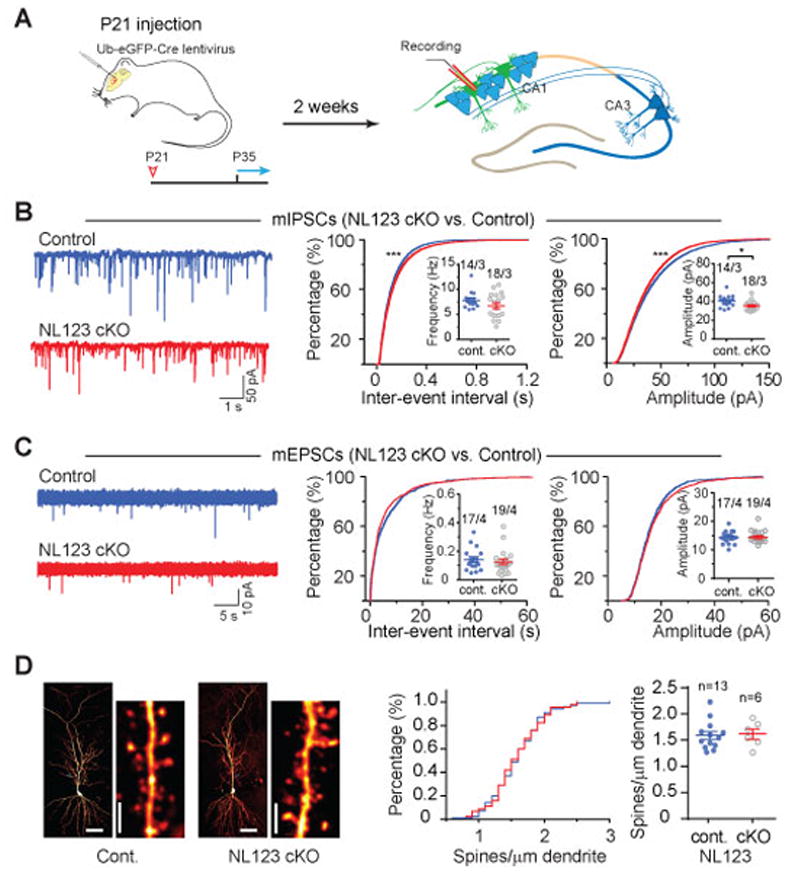
(A) Experimental design for injections at P21, followed by experimental analyses from P35 onwards.
(B) Analysis of mIPSCs in triple NL123 cKO neurons. Left, representative traces; middle, cumulative distribution of the mIPSC inter-event interval (inset: data points from individual cells and means of mIPSC frequency); right, cumulative distribution of mIPSC amplitudes (inset: data points from individual cells and mean mIPSC amplitudes).
(C) Same as B, but for mEPSCs.
(D) NL123 deletion in juvenile mice also does not change the spine density of CA1 pyramidal cells. Left, representative images of biocytin-stained CA1 pyramidal neurons after patch-clamp recording, with a lower and higher magnification images shown side by side (calibration bars: 50 μm and 2 μm, respectively); middle, cumulative distribution of spine density (control: 100 dendrites/13 neurons; cKO: 45 dendrites/6 NL123 cKO pyramidal neurons); right, summary graph of mean spine densities with data points from individual neurons.
Data in summary graphs are means ± SEM; statistical comparisons were performed with the Kolmogorov-Smirnov test (cumulative distributions) or student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001; non-significant comparisons are not labeled). Numbers indicate number of cells/mice examined.
NL1 cKO at P0 and P21 reduces NMDAR-mediated synaptic transmission
Previous work using constitutive NL1 KO mice or NL1 KD approaches demonstrated a critical role for NL1 in maintaining a normal complement of NMDARs, as measured by changes in NMDAR EPSCs7, 23–27. Consistent with these findings, the triple NL123 cKO at P0 caused ~40% decrease in the ratio of NMDAR- to AMPAR EPSCs (NMDAR:AMPAR ratio; Fig. 3A, B). This decrease was entirely due to the loss of NL1 as the NL1 cKO phenocopied the decrease in the NMDAR:AMPAR ratio, whereas the NL3 cKO did not (Fig. 3B). This finding was further validated by a ~35% decrease in the input-output relationship for NMDAR EPSCs without any change in the input-output relationship for AMPAR EPSCs in the NL1 cKO (Fig. S3B). The lack of an effect in the NL3 cKO is consistent with previous results obtained in NL3 constitutive KO mice28, demonstrating that NL3 cannot compensate for the loss of NL1 in CA1 pyramidal cells.
Figure 3. Single NL1 but not NL3 deletion significantly reduces NMDAR-mediated synaptic transmission in developing and juvenile CA1 pyramidal neurons.
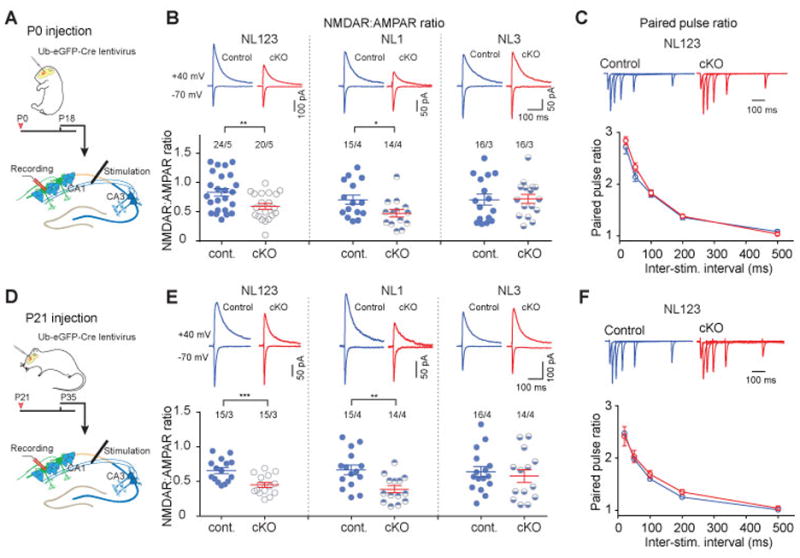
(A) Experimental design for conditional NL deletions in developing mice using P0 injections.
(B) Measurements of the ratio of NMDAR- to AMPAR-mediated EPSCs in CA1 pyramidal neurons in triple NL123 cKO (left) and NL1 (middle) and NL3 single cKO mice (right). AMPAR-mediated EPSCs were quantified as peak EPSC amplitude monitored at -70 mV; NMDAR-mediated EPSCs were quantified as the EPSC amplitude at 50 ms after presynaptic stimulation monitored at +40 mV. Top, representative traces; bottom, summary plots of mean NMDAR/AMPAR ratios and the values from individual neurons
(C) Paired pulse ratio (PPR) of EPSCs was not changed in triple NL123 cKO neurons. Top, representative traces; bottom, summary plot of mean PPRs as a function of the inter-stimulus interval (n = 16 control neurons/5 mice and 14 cKO neurons/5 mice, respectively).
(D) Experimental design for conditional NL deletions in juvenile mice using P21 injections.
(E) Same as B, but for juvenile mice.
(F) Same as C, but for juvenile mice (n = 10 control neurons/3 mice and 10 cKO neurons/3 mice, respectively).
Data in summary graphs are means ± SEM; statistical comparisons were performed with student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001; non-significant comparisons are not labeled). Numbers indicate number of cells/mice examined.
To examine whether the triple NL123 cKO influences presynaptic function via trans-synaptic interactions, we measured paired-pulse ratios (PPRs) of AMPAR EPSCs, which inversely correlate with presynaptic release probability29. The NL123 cKO had no effect on PPRs examined at multiple inter-stimulus intervals (Fig. 3C). Essentially identical results were obtained when the triple NL123 cKO and the single NL1 and NL3 cKOs were analyzed after injections of Cre lentivirus at P21 (Fig. 3D, E). Neither NL1 nor NL3 cKO at P21 showed any changes in PPRs, confirming that deletion of neuroligins has no effect on presynaptic release probability (Fig. S4). Thus, at excitatory synapses on CA1 pyramidal cells NL1 is required for the development and maintenance of normal NMDAR-mediated synaptic transmission.
NL1 cKO blocks long-term potentiation (LTP)
Constitutive KO of NL1 impairs LTP in CA1 pyramidal neurons30, 31, yet microRNA mediated KD of NL1 in mature hippocampus had no effect on LTP at these same CA1 synapses27,30. To address whether neuroligins are in fact required for this classic form of synaptic plasticity, we genetically deleted NL123 at P0 (Fig. 4A–D) and P21 (Fig. 4E–H). Strikingly, we found that at both ages, LTP was, on average, completely blocked. This block was entirely due to the loss of NL1 as the single NL1 but not NL3 cKO at P21 phenocopied the block of LTP (Fig. 4I–P).
Figure 4. NL123 triple and NL1 single deletion but not NL3 deletion abolishes NMDAR-dependent LTP in CA1 pyramidal neurons.
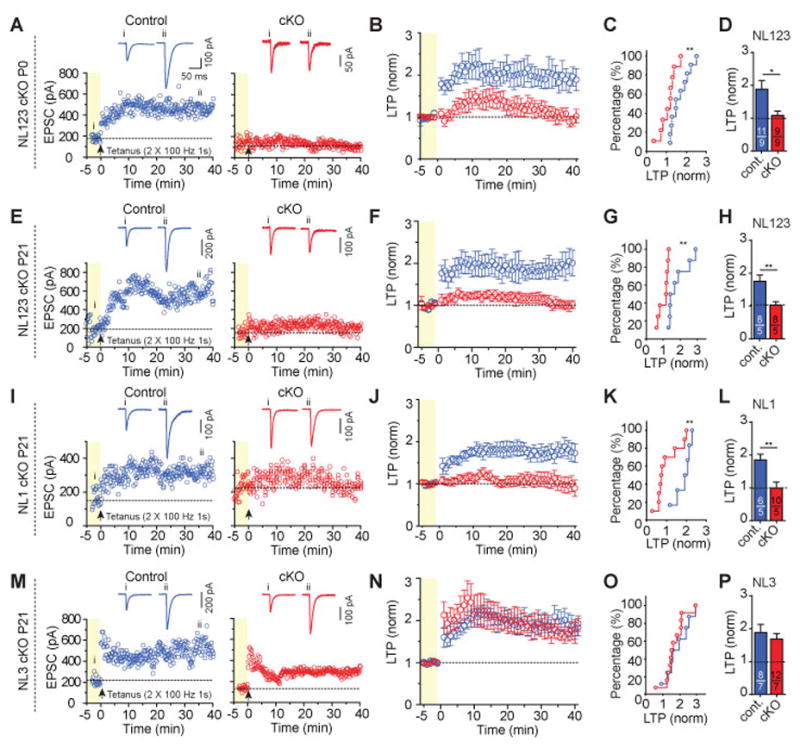
(A) Representative LTP experiments in a control (left) and NL123 cKO neuron (right) at P18 after lentiviral deletion of Neuroligins at P0. Top, sample traces; bottom, plots of the EPSC amplitude as a function of time before (yellow background) and after the LTP induction stimulus (2 trains of 100 Hz for 1 s with the cell depolarized to 0mV, separated by 20 s).
(B) Summary plot of LTP shows LTP was completely blocked in triple NL123 cKO following P0 lentiviral injection.
(C) Cumulative distribution of normalized LTP magnitud at 35-40 min after LTP induction.
(D) Summary graph of the mean LTP magnitude at 35-40 min after LTP induction.
(E–H), same as (A–D), except for the triple NL123 cKO was induced by lentiviral infection of CA1 pyramidal neurons in juvenile mice at P21.
(I–L), same as (E–H), except for the NL1 single cKO at P21.
(M–P), same as (E–H), except for the NL3 single cKO at P21.
Data in summary graphs are means ± SEM; statistical comparisons were performed with the Kolmogorov-Smirnov test (cumulative distributions) or student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001; non-significant comparisons are not labeled). Numbers indicate number of cells/mice examined.
Given that the genetic deletions of neuroligins, which blocked LTP, also caused a decrease in NMDAR EPSCs, an obvious possibility is that the block of LTP is due to the ~40% decrease in NMDAR-mediated synaptic transmission. To address this possibility, we first identified a concentration of D-APV (2 μM) that reduced isolated NMDAR-EPSCs and NMDAR charge transfer during high frequency stimulation by ~50% (Fig. S5A, B). At this concentration of D-APV, LTP was not blocked, although its magnitude was reduced (Fig. 5A–D). Thus, this experiment suggested that the block in LTP was independent of the effect of the neuroligin ablation on NMDARs, but is not conclusive given the partial effect.
Figure 5. NL1 cKO abolishes NMDAR-independent LTP in juvenile mice.
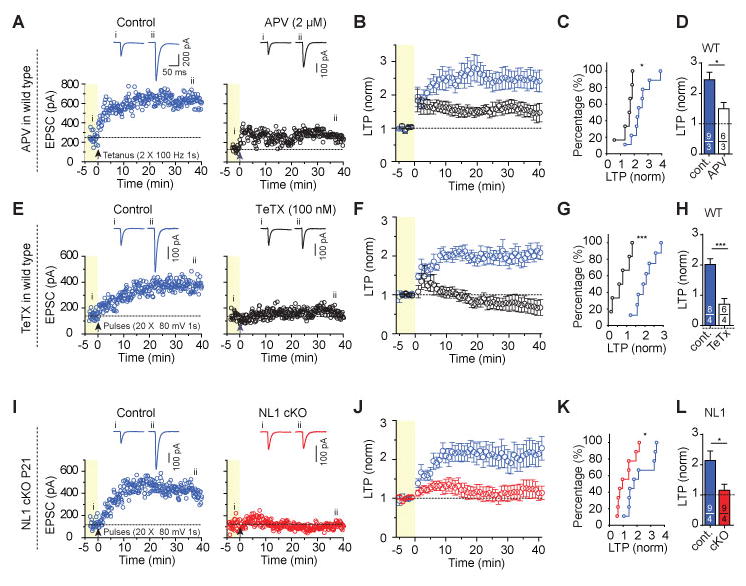
(A) Representative LTP experiments in wild-type mice showing that partial inhibition of NMDARs with AP5 (2 μM) impairs, but does not block, LTP induced by a standard induction protocol (2 trains of 100 Hz for 1 s separated by 20 s). Top, sample traces; bottom, plots of the EPSC amplitude as a function of time before (yellow background) and after the LTP induction stimulus.
(B) Summary plot of LTP in wild-type mice showing that this low dose of AP5 impaired, but did not block, LTP induced by a standard induction protocol.
(C) Cumulative distribution of normalized LTP magnitude at 35–40 min after LTP induction for B.
(D) Summary graph of the mean LTP magnitude at 35–40 min after LTP induction for B.
(E) Representative LTP experiments in wild-type mice using an NMDAR-independent induction protocol and demonstrating that similar to NMDAR-dependent LTP, NMDAR-independent LTP is also inhibited by postsynaptic tetanus-toxin (TeTx) light chain (right, 100 nM tetanus-toxin in the pipette solution). LTP was induced by 20 postsynaptic depolarizations (80 mV, 1 s separated by 6 s) in the presence of 50 μM AP5 and 5 μM Bay K 8644 Top, sample traces; bottom, plots of the EPSC amplitude as a function of time before (yellow background) and after the LTP induction stimulus.
(F) Summary plot of LTP induced by L-type Ca2+-channel mediated Ca2+-influx under continuous NMDAR-inhibition in control cells from wild-type mice, and its inhibition by tetanus toxin light chain.
(G) Cumulative distribution of normalized LTP magnitude at 35–40 min after LTP induction for F.
(H) Summary graph of the mean LTP magnitude at 35–40 min after LTP induction for F.
(I–L) Same as E–H, but comparing control neurons to NL1 cKO neurons produced by stereotactic lentiviral injection at P21.
Data in summary graphs are means ± SEM; statistical comparisons were performed with the Kolmogorov-Smirnov test (cumulative distributions) or student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001; non-significant comparisons are not labeled). Numbers indicate number of cells/mice examined.
To more definitively explore the importance of the decrease in NMDAR EPSCs in contributing to the block of LTP caused by neuroligin KO, we used a method that induces LTP in an NMDAR-independent manner. In this protocol, NMDARs are blocked pharmacologically by high concentrations of AP5 (50 μM). Potentiation of AMPAR-EPSCs is then elicited by repetitive activation of voltage-gated Ca2+-channels (VGCCs), primarily L-type Ca2+-channels22,32,334 (Fig. S6). VGCC LTP was prevented by loading cells with tetanus toxin (TeTx) (Fig. 5E–H), which cleaves synaptobrevin-2 in a manner similar to botulinum toxin that blocks NMDAR-dependent LTP when loaded in CA1 pyramidal cells34. These results suggest that similar to NMDAR-dependent LTP, VGCC LTP also requires SNARE-mediated exocytosis, which is presumably used for the delivery of AMPARs to the plasma membrane35. Importantly, the NL1 cKO also completely blocked VGCC LTP (Fig. 5I–L). Together, these results strongly suggest that NL1 plays a critical role in LTP in CA1 pyramidal cells independent of its influence on NMDARs.
DISCUSSION
Neuroligins are postsynaptic cell-adhesion molecules that are ubiquitously present at excitatory and inhibitory synapses, are genetically associated with several neuropsychiatric disorders, and appear to play complex roles in synapse function1–6. Despite decades of work, the specific roles of neuroligins in synapse formation and synaptic function remain uncertain, even controversial (see ref. 7 for a summary). Both methodological differences and biologically relevant factors likely contribute to the variability in results obtained with molecular manipulations of neuroligins. Knockdown of neuroligins in culture and in vivo using shRNAs or microRNAs has proved useful but suffers from possible off-target effects and incomplete loss of targeted proteins. Constitutive knockout of neuroligins eliminates the targeted proteins but allows compensatory developmental adaptations. Thus, using a conditional KO approach currently provides the optimal manipulation by allowing complete genetic deletion of the targeted proteins in individual cells in a temporally controlled fashion. For proteins such as neuroligins, which may play distinct roles at different developmental stages, the ability to control the time point at which they are genetically eliminated is particularly important. However, even with conditional genetic deletion approaches, because it takes some time for the gene to be deleted and endogenous proteins to degrade, compensatory adaptations may still occur.
Using single and triple cKOs of NL1, NL2, and NL3, we examined their requisite roles in excitatory and inhibitory synapse formation and function in hippocampal CA1 pyramidal neurons. We chose to study these specific synapses because they are, arguably, the most extensively studied synapses in the mammalian brain. By expressing cre-recombinase in vivo at two different ages (P0 and P21), we were able to address the role of neuroligins in both synapse formation and mature synaptic function. Examining sparsely infected neurons enabled us to assess neuroligin functions under conditions where mutant neurons are surrounded by, and competing with, adjacent wild-type neurons. In this approach, our goal was to define, in the most rigorous fashion possible, the requisite synaptic roles of neuroligins.
Consistent with previous analyses of constitutive triple NL123 KO mice36, cKO of NL123 did not affect excitatory synapse formation or maintenance as assessed by measurements of dendritic spine density and mEPSCs. Also in line with previous studies8,12,37–40, we found that the triple NL123 cKO at P0 caused a robust decrease in mIPSC frequency and VGAT puncta density, likely due to the loss of NL2, the deletion of which alone largely phenocopied the triple NL123 cKO. It was surprising that NL3 cKO at P0 caused a small increase in mIPSC frequency. This may occur because NL3 modestly impairs inhibitory synapse formation or function when present alone at these inhibitory synapses. Alternatively, in the absence of NL3, NL2 may be able to stimulate inhibitory synapse formation and function more robustly. It is also conceivable that despite the use of cKO lines, the cre-recombinase mediated deletion allows sufficient time for compensatory adaptations to occur. Furthermore, since the genetic backgrounds of the NL3 cKO line and NL2 cKO line are different than the other cKO lines, it is formally possible that the synaptic effects of neuroligin deletion are influenced by the background genetic strain in which the deletion is performed. Despite these caveats, the in vivo conditional KO approach we have taken here clearly allows a much more rigorous test of the requisite function of neuroligins than previous attempts using KD or constitutive KO approaches.
In contrast to previous studies in which neuroligins were deleted in other brain regions, NL123 cKO at P21 in the hippocampus had only modest effects on inhibitory synaptic transmission. However, consistent with previous work using neuroligin KD and constitutive KO approaches23–25, cKO of NL123 at both P0 and P21 caused a large decrease in the ratio of NMDAR- to AMPAR EPSCs combined with a lack of effect on AMPAR-mediated mEPSCs. This decrease in synaptic NMDAR number and/or function was entirely due to loss of NL1 since cKO of NL1, but not cKO of NL3, phenocopied these synaptic changes.
Arguably, our most interesting finding is that the triple NL123 cKO, due to deletion of NL1, profoundly impairs LTP in CA1 pyramidal cells at both P0 and, importantly, at P21 after synapses are fully mature. Although impairments of LTP following neuroligin KD or KO have been reported23,25,27,30,31, previous work had produced conflicting conclusions, possibly because of the approaches used. Our data establish that NL1 is essential for LTP independent of development. The simplest explanation for the apparent difference in our results from previous ones using a knockdown of NL127 is that the NL1 knockdown in mature CA1 pyramidal cells did not reduce NL1 levels sufficiently to detect its critical role in LTP.
The fact that NL1 is essential for maintaining NMDAR responses24 raises the obvious possibility that the loss of LTP in NL1-deficient pyramidal neurons is a secondary effect of smaller NMDAR-mediated synaptic currents. Surprisingly, our data show that this is not the case. First, pharmacologically reducing NMDAR-mediated transmission to a degree greater than the reduction caused by NL1 deletion reduced the magnitude of LTP to a degree much less than the complete block caused by NL1 cKO (Fig. 5A–D). Second, activation of voltage-dependent Ca2+-channels to bypass NMDARs elicited a robust LTP in control neurons that was eliminated by cKO of NL1 (Fig. 5I–L). This form of LTP is incompletely understood22,32,33, but our results show that it is completely blocked by tetanus-toxin mediated cleavage of the SNARE-protein synaptobrevin-2 similar to NMDAR-dependent LTP34, suggesting a similar cellular mechanism induced by a different calcium source. The apparently independent effects of the NL1 cKO on NMDAR-mediated synaptic transmission and LTP demonstrate that NL1 is a critical component of the molecular machinery that underlies synaptic plasticity in CA1 pyramidal cells. It seems likely that NL1 enables LTP by recruiting crucial cytoplasmic proteins to synaptic junctions such as PSD95, which directly binds to NL141. In this manner, NL1 constitutes a central component of the postsynaptic machinery.
In summary, using a rigorous conditional KO approach, our data provide evidence for the following conclusions (summarized in Fig. S7). First, neuroligins are not absolutely necessary for the formation or maintenance of excitatory synapses on CA1 pyramidal cell dendritic spines, even if the mutant neuron is surrounded by wild-type neurons. Second, NL1 is essential for normal NMDAR-mediated synaptic transmission, presumably by influencing the numbers of synaptic NMDARs. The same decrease in NMDAR-mediated EPSCs was seen in slices of the basolateral amygdala following NL1 KD23, suggesting that this function of NL1 applies to many different cell types. Third, neuroligins make only a modest contribution to excitatory synaptic strength, but a much more significant contribution to inhibitory synaptic strength8,9. Fourth, at least in CA1 pyramidal neurons, NL1 plays a mandatory role in LTP, and this function of NL1 is independent of its influence on NMDARs. Although much work remains to be done to elucidate the molecular mechanisms underlying the ubiquitous and cell-type specific roles of neuroligins in excitatory and inhibitory synaptic function, these results provide a framework that can form the basis for specialized future studies.
Supplementary Material
Acknowledgments
We thank members of the Malenka and Südhof labs for many helpful conversations. This study was supported by a grant from NIH (P50MH086403) and the Simons Foundation Autism Research Initiative.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu X, Luo J-h, Xu J. The interplay between synaptic activity and neuroligin function in the CNS. BioMed Research International. 2015;2015:13. doi: 10.1155/2015/498957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang M, Owczarek S. A matter of balance: Role of neurexin and neuroligin at the synapse. Neurochemical Research. 2013;38(6):1174–1189. doi: 10.1007/s11064-013-1029-9. [DOI] [PubMed] [Google Scholar]
- 4.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 5.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, et al. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409(1):10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 7.Soler-Llavina GJ, Fuccillo MV, Ko J, Sudhof TC, Malenka RC. The neurexin ligands, neuroligins and leucine-rich repeat transmembrane proteins, perform convergent and divergent synaptic functions in vivo. Proc Natl Acad Sci USA. 2011;108(40):16502–16509. doi: 10.1073/pnas.1114028108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang J, Xu W, Hsu YT, Yee AX, Chen L, Sudhof TC. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol Psychiatry. 2015;20(7):850–859. doi: 10.1038/mp.2015.31. [DOI] [PubMed] [Google Scholar]
- 9.Zhang B, Chen L, Liu X, Maxeiner S, Lee SJ, Gokce O, et al. Neuroligins sculpt cerebellar purkinje-cell circuits by differential control of distinct classes of synapses. Neuron. 2015;87(4):781–796. doi: 10.1016/j.neuron.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158(1):198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song J-Y, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA. 1999;96(3):1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63(5):628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 13.Takacs VT, Freund TF, Nyiri G. Neuroligin 2 is expressed in synapses established by cholinergic cells in the mouse brain. PLoS One. 2013;8(9):e72450. doi: 10.1371/journal.pone.0072450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budreck EC, Scheiffele P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur J Neurosci. 2007;26(7):1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x. [DOI] [PubMed] [Google Scholar]
- 15.Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt K-F, et al. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci USA. 2011;108(7):3053–3058. doi: 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen AS, Lin DD, Coulter DA. Protracted postnatal development of inhibitory synaptic transmission in rat hippocampal area CA1 neurons. J Neurophysiol. 2000;84(5):2465–2476. doi: 10.1152/jn.2000.84.5.2465. [DOI] [PubMed] [Google Scholar]
- 17.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 18.Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Sudhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012;73(5):990–1001. doi: 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, et al. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144(2):282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Sudhof TC, et al. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73(2):260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skrede KK, Westgaard RH. The transverse hippocampal slice: a well-defined cortical structure maintained in vitro. Brain Res. 1971;35(2):589–593. doi: 10.1016/0006-8993(71)90508-7. [DOI] [PubMed] [Google Scholar]
- 22.Kato HK, Watabe AM, Manabe T. Non-Hebbian synaptic plasticity induced by repetitive postsynaptic action potentials. J Neurosci. 2009;29(36):11153–11160. doi: 10.1523/JNEUROSCI.5881-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ, et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc Natl Acad Sci USA. 2008;105(26):9087–9092. doi: 10.1073/pnas.0803448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, et al. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54(6):919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung SY, Kim J, Kwon OB, Jung JH, An K, Jeong AY, et al. Input-specific synaptic plasticity in the amygdala is regulated by neuroligin-1 via postsynaptic NMDA receptors. Proc Natl Acad Sci USA. 2010;107(10):4710–4715. doi: 10.1073/pnas.1001084107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, Nicoll RA. Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat Neurosci. 2011;14(6):718–726. doi: 10.1038/nn.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shipman SL, Nicoll RA. A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron. 2012;76(2):309–316. doi: 10.1016/j.neuron.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etherton M, Foldy C, Sharma M, Tabuchi K, Liu X, Shamloo M, et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011;108(33):13764–13769. doi: 10.1073/pnas.1111093108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 30.Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010;30(6):2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budreck EC, Kwon O-H, Jung JH, Baudouin S, Thommen A, Kim H-S, et al. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc Natl Acad Sci U S A. 2011;110(2):725–730. doi: 10.1073/pnas.1214718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyllie DJ, Nicoll RA. A role for protein kinases and phosphatases in the Ca2+-induced enhancement of hippocampal AMPA receptor-mediated synaptic responses. Neuron. 1994;13(3):635–643. doi: 10.1016/0896-6273(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 33.Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron. 1992;9(6):1175–1183. doi: 10.1016/0896-6273(92)90075-o. [DOI] [PubMed] [Google Scholar]
- 34.Lledo P-M, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 35.Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC, Malenka RC. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 2013;77(3):542–558. doi: 10.1016/j.neuron.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, et al. Neuroligins determine synapse maturation and function. Neuron. 2006;51(6):741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Fu Z, Vicini S. Neuroligin-2 accelerates GABAergic synapse maturation in cerebellar granule cells. Mol Cell Neurosci. 2009;42(1):45–55. doi: 10.1016/j.mcn.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hines RM, Wu L, Hines DJ, Steenland H, Mansour S, Dahlhaus R, et al. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J Neurosci. 2008;28(24):6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jedlicka P, Hoon M, Papadopoulos T, Vlachos A, Winkels R, Poulopoulos A, et al. Increased dentate gyrus excitability in neuroligin-2-deficient mice in vivo. Cereb Cortex. 2011;21(2):357–367. doi: 10.1093/cercor/bhq100. [DOI] [PubMed] [Google Scholar]
- 40.Gibson JR, Huber KM, Sudhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29(44):13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hiaro K, et al. Binding of neuroligins to PSD-95. Science. 1997;277(5331):1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.