

Abstract
Although exercise is linked with improved health, the specific molecular mechanisms underlying its various benefits require further clarification. Here we report that exercise increases the nuclear localization and activity of p53 by acutely down-regulating coiled-coil-helix-coiled-coil-helix domain 4 (CHCHD4), a carrier protein that mediates p53 import into the mitochondria. This response to exercise is lost in transgenic mice with constitutive expression of CHCHD4. Mechanistically, exercise-induced nuclear transcription factor FOXO3 binds to the CHCHD4 promoter and represses its expression, preventing the translocation of p53 to the mitochondria and thereby increasing p53 nuclear localization. The synergistic increase in nuclear p53 and FOXO3 by exercise can facilitate their known interaction in transactivating Sirtuin 1 (SIRT1), a NAD+-dependent histone deacetylase that mediates adaptation to various stresses. Thus, our results reveal one mechanism by which exercise could be involved in preventing cancer and potentially other diseases associated with aging.
Keywords: exercise, FOXO, mitochondria, mitochondrial transport, p53
Introduction
The nuclear transcriptional activities of p53, well established to play a critical role in tumor suppression, are dynamically regulated by multiple factors, including posttranslational modifications and protein-protein interactions (1–3). Accumulating evidence also shows that p53 localization in non-nuclear subcellular compartments, such as the cytosol and mitochondria, can regulate cellular processes like autophagy and metabolism (4–6). Although stress-mediated p53 translocation to the mitochondria and its role in cell death have been well delineated, the effect of p53 translocation into mitochondria under normal states is less clear (7, 8). Thus, understanding the regulation of p53 partitioning into the mitochondria under physiologic conditions may provide new insights into its tumor-suppressive functions.
We reported previously that CHCHD4,2 the mammalian homolog of the yeast mitochondrial disulfide relay system carrier Mia40, interacts with p53 and mediates its translocation into mitochondria (9). Unlike other mechanisms by which proteins can be imported into mitochondria, the disulfide relay system is coupled to respiration, perhaps reflecting a homeostatic mechanism by which the import of p53 can be regulated by mitochondrial activity (10, 11). Subsequently, p53 import into mitochondria by CHCHD4 facilitates the repair of oxidative mtDNA damage, complementing the nuclear role of p53 in regulating mitochondrial respiration (9, 12, 13). However, these observations also raise questions regarding the physiological significance of the partitioning of p53 between the nucleus and mitochondria and whether such a mechanism can be demonstrated in vivo under normal conditions.
Exercise elicits many complex physiological responses, including the induction of specific transcription factors such as PGC-1α and peroxisome proliferator-activated receptors, which transactivate mitochondrial biogenesis genes (14, 15). As a mitochondrial protein import carrier, CHCHD4 is necessary for mitochondrial biogenesis. Therefore, we hypothesized that exercise may regulate CHCHD4 with subsequent effects on the subcellular distribution of p53. To test this hypothesis, we generated a CHCHD4 transgenic mouse model to investigate how the interaction between CHCHD4 and p53 is regulated by exercise.
Results
CHCHD4 Prevents Nuclear Localization and Transcriptional Activities of p53 in Vivo
To investigate the physiological significance of our previous finding that CHCHD4 mediates the import of p53 into mitochondria (9), we created a CHCHD4 transgenic (Tg) mouse line that overexpresses the mouse CHCHD4 gene under control of the mouse ROSA26 promoter. Confocal immunofluorescence of CHCHD4 in cross-sections of gastrocnemius skeletal muscle revealed an intense green fluorescence signal in CHCHD4 Tg mice that co-localized with the red fluorescence signal of COX4 as a mitochondrial marker (Fig. 1A). The merged fluorescence signal pattern was consistent with the overexpression of CHCHD4 in a largely subsarcolemma mitochondrial pattern surrounding the myofibrils and the Hoechst 33324-stained blue nuclei. The specificity of the immunofluorescence signal was supported by the increase in CHCHD4 mRNA and protein levels in the gastrocnemius skeletal muscle of CHCHD4 Tg relative to wild-type control mice (Fig. 1B). The overexpression of CHCHD4 mRNA and protein was also confirmed in multiple skeletal muscle groups and tissues of CHCHD4 Tg mice (supplemental Fig. S1).
FIGURE 1.

CHCHD4 regulates p53 subcellular localization and nuclear activity in vivo. A, confocal immunofluorescence images of CHCHD4 (green) expression in cross-sections of skeletal muscle of WT and CHCHD4 Tg mice. Immunofluorescence labeling of cytochrome c oxidase subunit 4 (COX4) (red) and Hoechst 33342 (blue) staining served as mitochondrial and nuclear markers, respectively. B, mRNA and protein expression of CHCHD4 and p21 in skeletal muscle of WT and CHCHD4 Tg mice. n ≥ 4. C, mRNA expression of p53-regulated mitochondrial biogenesis genes in skeletal muscle of WT and CHCHD4 Tg mice. n ≥ 4. D, relative mRNA expression (Tg versus WT) of p53-regulated genes analyzed by screening RT-PCR array in skeletal muscle. n ≥ 4. E, comparison of p53 protein in the mitochondrial (mito) and nuclear fractions of skeletal muscle and liver of WT and CHCHD4 Tg mice treated with doxorubicin for 18 h. VDAC and histone H3 (Hist. H3) served as mitochondrial and nuclear protein loading controls, respectively. Shown is a representative immunoblot image of four independent experiments used for quantification. Values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar = 20 μm.
We next examined whether the increased expression of CHCHD4 in vivo would promote the translocation of p53 into mitochondria, thereby decreasing nuclear p53 localization and activity. The levels of p21 mRNA and protein, encoded by the prototypical p53 target gene CDKN1A, were decreased in skeletal muscle and other tissues of CHCHD4 Tg mice (Fig. 1B and supplemental Fig. S1). Interestingly, the expression of three p53-regulated mitochondrial biogenesis genes, p53R2, TFAM, and PGC1α, was also decreased in CHCHD4 skeletal muscle (Fig. 1C). To more broadly assess p53 nuclear transcriptional activity, a screening real-time PCR (RT-PCR) array of 84 p53-regulated genes showed a baseline pattern of diminished activity in the skeletal muscle of CHCHD4 Tg compared with wild-type mice (supplemental Table S1). Of the 34 genes with expression changes of at least 1.5-fold, 30 genes showed decreased expression in CHCHD4 Tg mice, consistent with our previous finding that CHCHD4 overexpression in cells decreases p53 nuclear activity (Fig. 1D) (9). Notably, among the four genes having increased expression in CHCHD4 Tg mice, IL6 transcription has been reported to be repressed by p53 (Fig. 1D) (16).
To demonstrate that the decrease in p53 nuclear activity of CHCHD4 Tg mice is due to its selective subcellular partitioning, we examined p53 protein levels in the mitochondrial and nuclear fractions. p53 protein was first induced by doxorubicin treatment because of low endogenous wild-type p53 levels. Under this condition, the levels of p53 were higher in both the skeletal muscle and liver mitochondria of CHCHD4 Tg mice compared with wild-type mice, whereas this pattern was reversed in the nuclear fraction (Fig. 1E). As a positive control, CHCHD4 overexpression increased the level of the mitochondrial protein translocase of inner mitochondrial membrane 9 (TIMM9), a protein known to be imported into the intermembrane space by CHCHD4 (Fig. 1E) (11). Taken together, these results demonstrated that the overexpression of CHCHD4 in vivo can increase the translocation of p53 into mitochondria, thereby decreasing the levels of p53 protein and its activity in the nucleus (Fig. 1, B–D).
Exercise Decreases CHCHD4 Expression and Increases p53 Nuclear Activity
Given the important role of CHCHD4 on mitochondrial biogenesis and p53 on exercise capacity, we next examined how exercise affects CHCHD4-p53 interaction in skeletal muscle (17–19). Mice were submaximally exercised on a treadmill for 60 min, and then hind limb skeletal muscles were harvested at the indicated time points. In contrast to PGC-1α mRNA, which served as an exercise-induced mitochondrial biogenesis marker, CHCHD4 mRNA levels declined in a time-dependent manner, plateauing at ∼12 h after exercise (Fig. 2A). Although unexpected, given the earlier observation that CHCHD4 can decrease p53-induced mitochondrial biogenesis genes (Fig. 1C), the decrease in CHCHD4 expression by exercise would be consistent with the effect of exercise in promoting mitochondrial biogenesis. The functional consequence of inhibiting CHCHD4 expression was also reflected by a decrease in the mitochondrial level of its substrate TIMM9 (Fig. 2A). Based on our previous in vitro observation, the acute decrease in CHCHD4 expression with exercise would be predicted to increase the nuclear levels of p53 and its transcriptional activity (9). Indeed, skeletal muscle p21 mRNA and protein levels were increased with exercise stimulation in a p53-dependent manner (Fig. 2B). The increase in p53 nuclear activity was also supported by an increase in the mRNA levels of another p53 target gene, GADD45A (supplemental Fig. S2).
FIGURE 2.
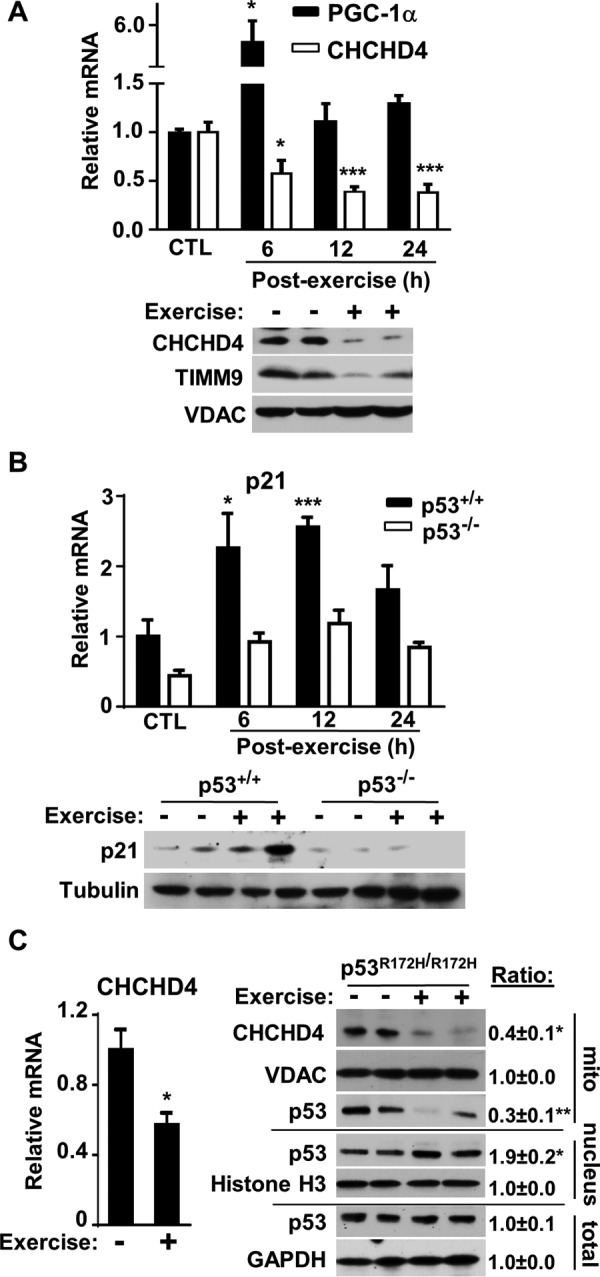
Exercise decreases CHCHD4 expression and increases p53 nuclear localization and activity. A, relative mRNA levels of CHCHD4 and PGC-1α in skeletal muscle of WT mice in the unexercised control state (CTL) and at the indicated time points after 60 min of submaximal treadmill exercise (Post-exercise). Bottom panel, immunoblot of CHCHD4 protein and its substrate TIMM9 in skeletal muscle mitochondria at 12 h post-exercise (+) compared with unexercised control (−). VDAC served as a mitochondrial protein loading control. n = 3. B, relative p21 mRNA level in skeletal muscle of unexercised control and post-exercise WT (p53+/+) and p53−/− mice. Bottom panel, immunoblot of p21 protein at 12 h post-exercise (+) muscle compared with unexercised control (−). Tubulin served as protein loading control. n = 3. C, relative CHCHD4 mRNA level in skeletal muscle of unexercised control (−) and 12 h post-exercise (+) mutant p53 R172H (p53R172H/R172H) knockin mice. Right panel, immunoblots of the indicated proteins in total skeletal muscle tissue and its nuclear and mitochondrial fractions. GAPDH, histone H3, and VDAC served as total, nuclear, and mitochondrial fraction protein loading controls, respectively. Shown is a representative immunoblot image of three independent experiments used for quantification. Values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To detect endogenous p53 protein in the absence of DNA damage, we investigated whether a mutant p53 protein, which is overexpressed because of loss of negative feedback activity but retains interaction with CHCHD4, could demonstrate the subcellular partitioning of p53 in response to exercise (9). Mice with homozygous knockin of the p53 R172H mutation (p53R172H/R172H), homologous to the human p53 R175H Li-Fraumeni syndrome mutation, were exercised for 60 min on a treadmill, and their skeletal muscles were harvested after 12 h. As observed in wild-type mice, p53R172H/R172H mice also displayed a significant reduction in the expression of CHCHD4 mRNA and protein with exercise stimulation (Fig. 2C). Although loss of wild-type activity by the p53 R172H mutation precluded assessing p21 mRNA levels, the nuclear and mitochondrial fractions of unexercised control and 12 h post-exercise mice were examined for p53 protein levels. Exercise stimulation did not change the total level of skeletal muscle p53 protein; however, there were reciprocal changes in the mitochondrial and nuclear levels of p53 consistent with its subcellular partitioning to the nucleus by CHCHD4 down-regulation (Fig. 2C, right panel).
Constitutive CHCHD4 Expression Blocks Exercise-induced p53 Activation
Given the in vivo partitioning of p53 by exercise-regulated CHCHD4, we speculated that the constitutive expression of CHCHD4 would block the nuclear localization and activation of p53 in response to exercise. Indeed, in contrast to wild-type mice, CHCHD4 Tg mice showed no p21 induction at both the mRNA and protein levels in skeletal muscle after 60 min of submaximal exercise (Fig. 3A). Consistent with blunted p21 induction by exercise in CHCHD4 Tg mice, the reciprocal changes in the nuclear and mitochondrial levels of p53 were no longer evident after exercise in transgenic mice (supplemental Fig. S3). Furthermore, the RT-PCR array of p53-regulated genes showed a pattern of increased expression in wild-type control mice after exercise, whereas the induction of these same genes by exercise in CHCHD4 Tg mice was globally diminished (Fig. 3B). Individual RT-PCR of representative p53 target genes, such as GADD45A, BNIP3, and SIRT1, confirmed the pattern observed in the screening assay (Fig. 3C).
FIGURE 3.

Overexpression of CHCHD4 blocks exercise-induced p53 nuclear activity. A, p21 mRNA and protein levels in skeletal muscle of WT and CHCHD4 Tg mice in unexercised control (−) and 12 h post-exercise (+) states. Tubulin and VDAC served as protein loading controls for whole tissue (total) and the mitochondrial fraction, respectively. n = 4. B, mRNA expression of p53-regulated genes analyzed by RT-PCR array in skeletal muscle of WT and CHCHD4 Tg mice 12 h after a 60-min period of submaximal treadmill exercise. Of the 84 p53 target genes, 26 were induced 2.5-fold or higher only in WT mice after exercise. Color-coded -fold change in gene expression after exercise compared with non-exercised controls is shown (red, induced; green, repressed). n = 3. C, confirmation of some representative exercise-induced p53-regulated genes identified by RT-PCR array. n = 3. Values are mean ± S.E. *, p < 0.05; **, p < 0.01.
FOXO3 Mediates the Regulation of CHCHD4 by Exercise
The relatively early time course of decreased CHCHD4 mRNA levels after exercise suggested a mechanism involving its transcriptional regulation. Examination of the promoter regions 5′ from the start codon of both mouse (−2080 and −1298 bp) and human (−3708, −2364, and −2278 bp) CHCHD4 genes revealed the conserved FOXO3 DNA binding elements (DBEs) (G/C)(T/C)AAA(C/T)A. This was notable, as FOXO3 is a known adaptive stress response gene that can be induced directly by exercise (20, 21). The time course of increase in the total cellular levels of FOXO3 in skeletal muscle after exercise was relatively modest (Fig. 4A). However, measuring the nuclear fraction of FOXO3 protein revealed a more marked increase at 12 h after exercise, suggesting a larger role for its regulation by subcellular redistribution as also observed with p53 (Figs. 4A and 2C, right panels). To demonstrate an interaction between the FOXO3 protein and CHCHD4 gene, FOXO3 was induced in HCT116 cells by treatment with the mitochondrial uncoupler CCCP, and its interaction with FOXO3 DBEs was examined by ChIP assay (Fig. 4B) (22). FOXO3 specifically bound to DBE2 (−2364 bp) of human CHCHD4 with a 12-fold enrichment in amplification after immunoprecipitation versus the nonspecific IgG antibody control (Fig. 4B).
FIGURE 4.
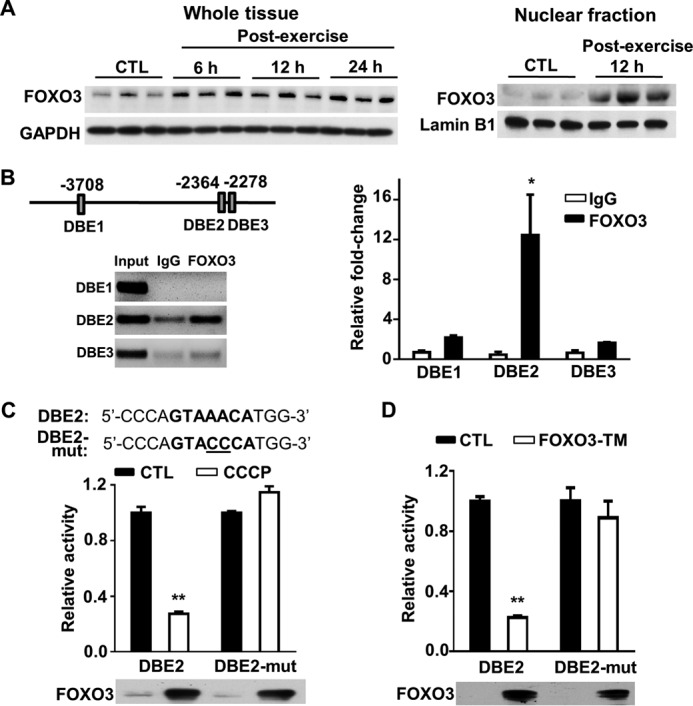
Exercise-induced FOXO3 protein interacts with the FOXO3 DBE of the CHCHD4 gene. A, FOXO3 immunoblot of wild-type mouse skeletal muscle in the unexercised state (control, CTL) and post-exercise state at the indicated time points. Whole tissue and nuclear fraction samples are shown. GAPDH and lamin B1 served as total and nuclear protein loading controls, respectively. B, schematic of the three putative FOXO3 DBEs on the human CHCHD4 promoter relative to the start codon. Shown is the ChIP analysis of the interaction between FOXO3 and the DBEs of the CHCHD4 gene in HCT116 cells treated with CCCP for 6 h to induce FOXO3. Input DNA samples were diluted 1:20 for the agarose gel image. n = 3. C, luciferase activity of CHCHD4 promoter constructs containing wild-type or mutated (mut) DBE2 sequence. HCT116 cells were treated with CCCP to induce FOXO3 protein after transfection with the constructs. Control is without CCCP treatment. n = 4. D, effect of the constitutively active triple phosphorylation mutant FOXO3 (FOXO3-TM) on DBE2 promoter construct activity. HCT116 cells were co-transfected with FOXO3-TM cDNA 36 h prior to the luciferase assay. Control is without FOXO3-TM co-transfection. n = 4. Values are mean ± S.E. *, p < 0.05; **, p < 0.001.
To test the transcriptional regulation of CHCHD4 by DBE2, a luciferase reporter construct was made using an ∼4-kb CHCHD4 gene fragment that contained either wild-type DBE2 or its mutated sequence (Fig. 4C). These constructs were transfected into HCT116 cells and then treated with the mitochondrial uncoupler CCCP to induce FOXO3 for 6 h. FOXO3 markedly suppressed the activity of the luciferase reporter driven by the promoter containing wild-type DBE2 but not that containing mutated DBE2 (Fig. 4C). Furthermore, the transient expression of a constitutively active triple mutant of FOXO3 (FOXO3-TM) inhibited the activity of the CHCHD4 promoter fragment containing wild-type but not mutated FOXO3 DBE2 sequence (Fig. 4D). These data suggested that mitochondrial stress-activated FOXO3 can suppress the expression of CHCHD4 at the transcriptional level.
To assess whether FOXO3 is sufficient to suppress CHCHD4 expression, we modulated FOXO3 expression and examined its effects on CHCHD4 and nuclear p53 levels. The transient transfection of constitutively active FOXO3-TM into HCT116 cells dose-dependently decreased CHCHD4 mRNA and protein levels while concomitantly increasing p21 expression (Fig. 5A). We further confirmed that FOXO3-TM can increase nuclear p53 localization by subcellular fractionation and immunofluorescence imaging studies (Fig. 5, B and C). We next examined whether suppressing FOXO3 by shRNA would prevent the inhibition of CHCHD4 expression. Stable transduction of HCT116 cells with FOXO3 shRNA markedly attenuated mitochondrial depolarization-induced FOXO3 levels and prevented both the down-regulation of CHCHD4 and induction of p21 mRNA and protein levels (Fig. 6A). As another demonstration of this mechanism, FOXO3 induced by serum starvation also decreased CHCHD4 and increased p21 expression, whereas there was marked blunting of these effects by FOXO3-specific shRNA (Fig. 6B). Nuclear fractionation and immunofluorescence imaging studies were performed to ensure corresponding changes in the subcellular localization of p53 (Fig. 6, C and D). Together, these observations indicated that FOXO3 is both necessary and sufficient for suppressing CHCHD4 expression and further validated the specific translocation mechanism by which exercise can regulate nuclear p53 activity, as summarized in Fig. 7.
FIGURE 5.
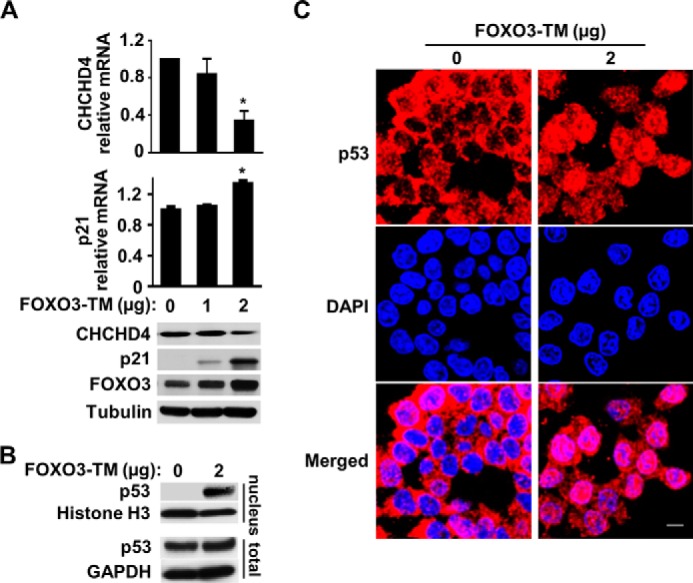
FOXO3 represses CHCHD4 expression. A, expression levels of CHCHD4 and p21 in HCT116 cells transfected with the FOXO3-TM plasmid. n = 3. B, p53 immunoblot of nuclear fraction and total cell extract of HCT116 cells transfected with the FOXO3-TM plasmid. C, representative confocal immunofluorescence images of p53 (red) in FOXO3-TM-transfected cells. DAPI stain (blue) shows nuclei. Values are mean ± S.E. *, p < 0.001. Scale bar = 10 μm.
FIGURE 6.

Knocking down FOXO3 increases CHCHD4 expression. A, CHCHD4 and p21 expression in HCT116 cells with knockdown of CCCP-induced FOXO3 with shRNA. NS, nonspecific shRNA. n = 3. B, CHCHD4 and p21 expression in HCT116 cells with knockdown of serum starvation-induced FOXO3 with shRNA. n = 3. C, p53 immunoblot of nuclear and total cell fractions of HCT116 cells after knockdown of FOXO3 induced by CCCP or serum starvation. D, representative confocal immunofluorescence image of p53 (red) in HCT116 cells after knockdown of FOXO3 induced by CCCP or serum starvation. DAPI stain (blue) shows nuclei. Values are mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar = 10 μm.
FIGURE 7.
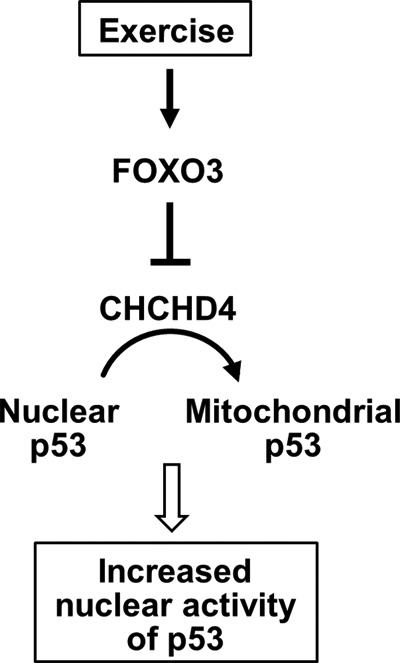
Summary of exercise regulation of p53 subcellular partitioning. The exercise-induced transcription factor FOXO3 represses CHCHD4 expression, thereby preventing the translocation of p53 into the mitochondria and increasing its nuclear level and activity.
Discussion
In this study, we have demonstrated the physiological significance of the subcellular partitioning of p53 by the mitochondrial disulfide relay carrier protein CHCHD4. Mechanistically, we have shown that exercise-induced FOXO3, a well known adaptive stress response transcription factor, can directly repress CHCHD4 expression and effectively promote the nuclear localization of p53 (Fig. 7). The in vivo specificity of this mechanism is highlighted in the CHCHD4 transgenic mouse, whereby the constitutive expression of CHCHD4 in skeletal muscle can abolish the increase in nuclear p53 activity induced by exercise. The subcellular partitioning of p53 is likely to have profound effects on cellular activities that mediate not only the effects of exercise but also of other stresses such as caloric restriction or even chemotherapy. For example, the down-regulation of CHCHD4 can increase the apoptotic response of cancer cells to 5-fluorouracil, a commonly used treatment for various human cancers, and decrease cell proliferation (supplemental Fig. S4).
Physical exercise has been associated with numerous health benefits, including cancer prevention, but the pleiotropic nature of its effects has made it difficult to delineate specific molecular mechanisms (23). Compromised exercise capacity in p53-null mice and the effect of mutated p53 on skeletal muscle oxidative metabolism in a human translational study have underscored the importance of this tumor suppressor in exercise physiology (13, 17, 18, 24). A number of studies have explored the effects of exercise on p53. In mouse models, increases in both nuclear and mitochondrial p53 have been reported within 0–3 h after exercise stimulation (19, 25, 26). In humans, nuclear p53 and phosphorylation of p53 Ser-15 have been reported to be increased in skeletal muscle 3 h after a bout of endurance exercise (27, 28). Although the time course of this study is different from these prior publications, it may provide insights into further delineating the specific underlying molecular mechanisms.
The negative regulation of CHCHD4 by FOXO3 in response to exercise is unexpected because exercise is generally thought to promote mitochondrial biogenesis. FOXO genes can act as regulators of cellular redox homeostasis under conditions of stress such as exercise (29, 30). In this regard, the partitioning of p53 to the nucleus may be important for initiating mitochondrial biogenesis as well as antioxidant responses, which can be tumor-suppressive (31). Interestingly, FOXO3, a putative tumor suppressor (32), requires nutrient-sensitive physical interaction with p53 to transactivate SIRT1 at its promoter (33). Therefore, exercise-induced FOXO3 and the subsequent translocation of p53 to the nucleus may have a synergistic effect on promoting the expression of SIRT1, an established mediator of mitochondrial biogenesis and the benefits of caloric restriction (34). Taken together, it is tempting to speculate that the regulation of CHCHD4-mediated p53 translocation may play profound roles not only in exercise biology but also in cancer and other age-associated diseases.
Experimental Procedures
Cell Culture
The HCT116 cell line (wild-type p53) was obtained from the American Type Culture Collection and cultured in McCoy's 5A medium with 10% FBS.
Mice
Wild-type and p53+/− mice (The Jackson Laboratory) and p53+/R172H mice (strain 01XL9, NCI, National Institutes of Health, Frederick Mouse Repository) were of the C57BL6 strain or backcrossed at least five generations into the C57BL6 background. All mice were maintained and handled in accordance with the NHLBI, National Institutes of Health Animal Care and Use Committee protocol.
CHCHD4 Transgenic Mice
A full-length untagged cDNA encoding mouse CHCHD4 was subcloned into the HindIII and EcoR I sites of the pBROAD3 vector (InvivoGen, San Diego, CA), which contains the mouse ROSA26 promoter and the rabbit β-globin polyadenylation sequence for overexpressing CHCHD4 ubiquitously. The cloning vector was removed from the transgenic construct (pBROAD3-CHCHD4) by PacI digestion, and the transgene alone was microinjected in the pronuclei of C57BL6 zygotes. After culturing overnight in M16 medium (EMD Millipore), embryos that reached the two-cell stage of development were implanted into the oviducts of pseudopregnant surrogate mothers. Offspring born to these foster mothers was genotyped by analyzing tail tip DNA using PCR.
Mouse Studies
Male mice (∼10 weeks old) were acclimated to a treadmill (10% incline) for 5 days as follows: days 1 and 2, 10 min at 10 m/min; days 3–5, 10 min at 10 m/min followed by another 10 min at 15 m/min. Submaximal exercise was performed on day 7 for 60 min, starting at 12 m/min and increasing the speed by 2 m/min every 20 min. To induce and detect endogenous wild-type p53 protein in tissues, mice were injected with doxorubicin (20 mg/kg, i.p.) 18 h prior to sample collection.
Antibodies and Immunoblot Analysis
The following antibodies were used: CHCHD4 (polyclonal antibody H107, sc-98628, Santa Cruz Biotechnology), FOXO3 (2497, Cell Signaling Technology), GADPH (Ambion), histone H3 (ab1791, Abcam), lamin B1 (ab16048, Abcam), human p21 (OP64, Calbiochem), mouse p21 (OP76, Calbiochem), p53 (monoclonal antibody DO-1, sc-126, Santa Cruz Biotechnology), TIMM9 (11479-1-AP, Proteintech), tubulin (A11126, Invitrogen), and VDAC (Rockland). Proteins were resolved and subjected to immunoblot analysis using standard SDS/PAGE and ECL (GE Healthcare).
Immunofluorescence Imaging
Gastrocnemius skeletal muscle was fixed overnight in 10% formalin and embedded in paraffin. After antigen retrieval with citrate buffer (pH 6), paraffin sections were blocked with 10% goat serum at room temperature for 30 min, followed by incubation with rabbit polyclonal anti-CHCHD4 or mouse monoclonal anti-COX4 primary antibody at 4 °C overnight. The sections were then incubated with goat anti-rabbit secondary antibody conjugated with Alexa Fluor 488 or goat anti-mouse secondary antibody conjugated with Alexa Fluor 555 at room temperature for 1 h and counterstained with Hoechst 33342. For cell imaging studies, HCT116 cells were transfected with the indicated constructs for 48 h, fixed in 4% paraformaldehyde/PBS (Electron Microscopy Sciences), and permeabilized in 0.1% Triton X-100/PBS. Immunofluorescence labeling was performed by blocking with 5% goat serum at room temperature and incubating in p53 primary antibody at 4 °C overnight, followed by Alexa Fluor 555-labeled secondary antibody (Invitrogen) for 37 °C for 1 h. Cell nuclei were stained with DAPI. Both tissue and cell images were captured using a confocal laser-scanning microscope (Zeiss LSM 780).
Tissue Dissection and Subcellular Fractionation
The gastrocnemius muscle was used as skeletal muscle in most experiments because of its relatively large volume. Mitochondria were isolated from mouse tissues after perfusion with ice-cold PBS as described previously (35). Nuclear fractions were prepared from freshly isolated skeletal muscle and liver after perfusion using a commercially available nuclear extraction kit (Pierce, NE-PERTM).
Real-time PCR
RT-PCR was performed using the HT7900 real-time PCR thermal cycler (ABI) as described previously (36). The average cycle threshold of the respective genes (see “Primer Sequences”) was normalized to the housekeeping gene translation initiation factor EIF3S5 (TIF). The mouse p53 signaling pathway screening RT2 PCR array was obtained from Qiagen.
Gene Knockdown and Overexpression
For knockdown experiments, plasmids containing sequences of nonspecific or human FOXO3-specific shRNA (see “Primer Sequences”) were obtained from Open Biosystems, and shRNA lentivirus was prepared according to the instructions of the manufacturer (Sigma-Aldrich). Cells were incubated with virus (multiplicity of infection, ∼1) for 24 h, followed by selection in 2 μg/ml puromycin. Constitutively active mutant FOXO3 (FOXO3-TM), containing three consensus AKT/PKB phosphorylation sites mutated to alanine and known to localize to the nucleus (37), was used for FOXO3 overexpression experiments.
Luciferase Assay
The ∼4-kb human CHCHD4 fragment containing the putative FOXO3 DBE sequences was cloned into the pGL4.71-luciferase vector (Clontech). After treatment with CCCP (6 h) to induce FOXO3 or co-transfection with FOXO3-TM, transactivation was measured in HCT116 cells 36 h after transfection with the pGL4.74 construct containing the TK promoter and Renilla luciferase as a transfection efficiency control. The QuikChange II kit (Stratagene) was used to introduce point mutations into the CHCHD4 promoter (see “Primer Sequences”).
ChIP Assay
The chromatin immunoprecipitation assay was carried out using ChIP-IT Express (Active Motif) according to the protocol of the manufacturer. Briefly, HCT116 cells were treated with CCCP for 6 h, fixed in 1% formaldehyde containing complete medium at room temperature for 10 min, and sonicated to obtain the nuclear lysates. Rabbit control IgG serum or polyclonal anti-FOXO3 antibody (10 μg/ml) was used to immunoprecipitate the fixed chromatin for PCR amplification.
Annexin V and Propidium Iodide Staining
HCT116 cells were treated with 5-fluorouracil (50 μg/ml) for 24 h and then stained with Annexin V (BD Pharmingen) and propidium iodide (Invitrogen) according to the protocols of the manufacturer. The cells were analyzed by flow cytometry (LSRII instrument, BD Biosciences).
Colony Formation Assay
HCT116 cells were transduced with nonspecific or CHCHD4-specific shRNA, plated at the indicated densities, cultured for 7 days, and stained with crystal violet solution for quantification.
Primer Sequences
CHCHD4 transgenic mice genotyping primers were as follows: 5′-GATGACCCCAATGATCCCTATG-3′ (forward) and 5′-CTGTAGTGGAAGCAGGAGAAG-3′ (reverse). Human CHCHD4 gene putative FOXO3 DNA binding elements were as follows: DBE1, 5′-CAAACAGTGGCTGAGCTTTC-3′ (forward) and 5′-CCTCTGTGTGACCTTCTAGTATTG-3′ (reverse); DBE2, 5′-TGGGCAGTGACATGACAAA-3′ (forward) and 5′-AGATCCTTTATGATCCTCCATGTT-3′ (reverse); DBE3, 5′-CACACATTATATAAACACACACACACAC-3′ (forward) and 5′-ACTCATTGCTCATTTCTTCTAAGTCC-3′ (reverse); and human FOXO3 shRNA, 5′-TGCGAAGCATCACGTTCCG-3′. Human mRNA quantification primers were as follows: CHCHD4, 5′-GCTTGGCTGTTCCTTGTTATTC-3′ (forward) and 5′-GTTTCCTCTCTTGCTGCTACTC-3′ (reverse); FOXO3, 5′-AGAGCTGAGACCAGGGTAAA-3′ (forward) and 5′-GACAGGCTTCACTACCAGATTC-3′ (reverse); and p21, 5′-CCCGTCTCAGTGTTGAGCCTT-3′ (forward) and 5′-GTTCCGCTGCTAATCAAAGTGC-3′ (reverse). Mouse mRNA quantification primers were as follows: BNIP3, 5′-CTGATGGGTATGGAGAAGGAGG-3′ (forward) and 5′-CCAACTTCAGCCATCTGCTCTTC-3′ (reverse); CHCHD4, 5′-CGCTGCCTCTGCGAGCTGCAG-3′ (forward) and 5′-TCTGTGCTGTAGTGGAAGCAG-3′ (reverse); FOXO3, 5′-AGTGGATGGTGCGCTGTGT-3′ (forward) and 5′-CTGTGCAGGGACAGGTTGT-3′ (reverse); GADD45A, 5′-CCACGCTGATGCAAGGATTA-3′ (forward) and 5′-TTCTTCAGGCTCACCTCTCT-3′ (reverse); p21, 5′-AGGGCAACTTCGTCTGGGAG-3′ (forward) and 5′-TTGGAGACTGGGAGAGGGCA-3′ (reverse); PGC-1α, 5′-ACGGTTTACATGAACACAGCTGC-3′ (forward) and 5′-CTTGTTCGTTCTGTTCAGGTGC-3′ (reverse); and SIRT1, 5′-GGAACCTTTGCCTCATCTACA-3′ (forward) and 5′-CACCTAGCCTATGACACAACTC-3′ (reverse).
Author Contributions
J. Z., P. Y. W., and P. H. conceived the study. J. Z., W. K., J. L., C. L., P. Y. W., and J. G. K. performed the research. J. Z., W. K., P. Y. W., and P. H. analyzed the data. J. Z., P. Y. W., and P. H. wrote the paper.
Supplementary Material
Acknowledgments
We thank Zu-Xi Yu of the NHLBI, National Institutes of Health Pathology Core for histology and immunofluorescence studies, John M. Sabandal for technical assistance, Toren Finkel for the FOXO3 constructs, and Ji-Hoon Park for helpful comments and suggestions.
This work was supported by the Division of Intramural Research of the NHLBI, National Institutes of Health Grant HL005101 (to P. M. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S4 and Table S1.
- CHCHD
- coiled-coil-helix-coiled-coil-helix domain
- Tg
- transgenic
- VDAC
- voltage-dependent anion channel
- RT-PCR
- real-time PCR
- DBE
- DNA binding element
- CCCP
- carbonyl cyanide p-chlorophenylhydrazone
- TM
- triple mutant.
References
- 1. Toledo F., and Wahl G. M. (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923 [DOI] [PubMed] [Google Scholar]
- 2. Kruse J. P., and Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Batchelor E., Loewer A., and Lahav G. (2009) The ups and downs of p53: understanding protein dynamics in single cells. Nat. Rev. Cancer 9, 371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Green D. R., and Kroemer G. (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park J. H., Zhuang J., Li J., and Hwang P. M. (2016) p53 as guardian of the mitochondrial genome. FEBS Lett. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kamp W. M., Wang P. Y., and Hwang P. M. (2016) TP53 mutation, mitochondria and cancer. Curr. Opin. Genet. Dev. 38, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chipuk J. E., Bouchier-Hayes L., Kuwana T., Newmeyer D. D., and Green D. R. (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309, 1732–1735 [DOI] [PubMed] [Google Scholar]
- 8. Vaseva A. V., Marchenko N. D., Ji K., Tsirka S. E., Holzmann S., and Moll U. M. (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhuang J., Wang P. Y., Huang X., Chen X., Kang J. G., and Hwang P. M. (2013) Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc. Natl. Acad. Sci. U.S.A. 110, 17356–17361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bihlmaier K., Mesecke N., Terziyska N., Bien M., Hell K., and Herrmann J. M. (2007) The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 179, 389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herrmann J. M., and Riemer J. (2012) Mitochondrial disulfide relay: redox-regulated protein import into the intermembrane space. J. Biol. Chem. 287, 4426–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vahsen N., Candé C., Brière J. J., Bénit P., Joza N., Larochette N., Mastroberardino P. G., Pequignot M. O., Casares N., Lazar V., Feraud O., Debili N., Wissing S., Engelhardt S., Madeo F., et al. (2004) AIF deficiency compromises oxidative phosphorylation. EMBO J. 23, 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matoba S., Kang J. G., Patino W. D., Wragg A., Boehm M., Gavrilova O., Hurley P. J., Bunz F., and Hwang P. M. (2006) p53 regulates mitochondrial respiration. Science 312, 1650–1653 [DOI] [PubMed] [Google Scholar]
- 14. Holloszy J. O., and Coyle E. F. (1984) Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. 56, 831–838 [DOI] [PubMed] [Google Scholar]
- 15. Hood D. A., Tryon L. D., Vainshtein A., Memme J., Chen C., Pauly M., Crilly M. J., and Carter H. (2015) Exercise and the regulation of mitochondrial turnover. Prog. Mol. Biol. Transl. Sci. 135, 99–127 [DOI] [PubMed] [Google Scholar]
- 16. Santhanam U., Ray A., and Sehgal P. B. (1991) Repression of the interleukin 6 gene promoter by p53 and the retinoblastoma susceptibility gene product. Proc. Natl. Acad. Sci. U.S.A. 88, 7605–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park J. Y., Wang P. Y., Matsumoto T., Sung H. J., Ma W., Choi J. W., Anderson S. A., Leary S. C., Balaban R. S., Kang J. G., and Hwang P. M. (2009) p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 105, 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saleem A., Adhihetty P. J., and Hood D. A. (2009) Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genomics 37, 58–66 [DOI] [PubMed] [Google Scholar]
- 19. Bartlett J. D., Close G. L., Drust B., and Morton J. P. (2014) The emerging role of p53 in exercise metabolism. Sports Med. 44, 303–309 [DOI] [PubMed] [Google Scholar]
- 20. Slopack D., Roudier E., Liu S. T., Nwadozi E., Birot O., and Haas T. L. (2014) Forkhead BoxO transcription factors restrain exercise-induced angiogenesis. J. Physiol. 592, 4069–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stefanetti R. J., Lamon S., Wallace M., Vendelbo M. H., Russell A. P., and Vissing K. (2015) Regulation of ubiquitin proteasome pathway molecular markers in response to endurance and resistance exercise and training. Pflugers Arch. 467, 1523–1537 [DOI] [PubMed] [Google Scholar]
- 22. Liu J., Peng Y., Wang X., Fan Y., Qin C., Shi L., Tang Y., Cao K., Li H., Long J., and Liu J. (2016) Mitochondrial dysfunction launches dexamethasone-induced skeletal muscle atrophy via AMPK/FOXO3 signaling. Mol. Pharm. 13, 73–84 [DOI] [PubMed] [Google Scholar]
- 23. Rogers C. J., Colbert L. H., Greiner J. W., Perkins S. N., and Hursting S. D. (2008) Physical activity and cancer prevention: pathways and targets for intervention. Sports Med. 38, 271–296 [DOI] [PubMed] [Google Scholar]
- 24. Wang P. Y., Ma W., Park J. Y., Celi F. S., Arena R., Choi J. W., Ali Q. A., Tripodi D. J., Zhuang J., Lago C. U., Strong L. C., Talagala S. L., Balaban R. S., Kang J. G., and Hwang P. M. (2013) Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med. 368, 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Philp A., Chen A., Lan D., Meyer G. A., Murphy A. N., Knapp A. E., Olfert I. M., McCurdy C. E., Marcotte G. R., Hogan M. C., Baar K., and Schenk S. (2011) Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation following endurance exercise. J. Biol. Chem. 286, 30561–30570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saleem A., and Hood D. A. (2013) Acute exercise induces p53 translocation to the mitochondria and promotes a p53-Tfam-mtDNA complex in skeletal muscle. J. Physiol. 591, 3625–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartlett J. D., Hwa Joo C., Jeong T. S., Louhelainen J., Cochran A. J., Gibala M. J., Gregson W., Close G. L., Drust B., and Morton J. P. (2012) Matched work high-intensity interval and continuous running induce similar increases in PGC-1α mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J. Appl. Physiol. 112, 1135–1143 [DOI] [PubMed] [Google Scholar]
- 28. Tachtsis B., Smiles W. J., Lane S. C., Hawley J. A., and Camera D. M. (2016) Acute endurance exercise induces nuclear p53 abundance in human skeletal muscle. Front. Physiol. 7, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eijkelenboom A., and Burgering B. M. (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 [DOI] [PubMed] [Google Scholar]
- 30. Hagenbuchner J., and Ausserlechner M. J. (2013) Mitochondria and FOXO3: breath or die. Front. Physiol. 4, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhuang J., Ma W., Lago C. U., and Hwang P. M. (2012) Metabolic regulation of oxygen and redox homeostasis by p53: lessons from evolutionary biology? Free Radic. Biol. Med. 53, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dansen T. B., and Burgering B. M. (2008) Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 18, 421–429 [DOI] [PubMed] [Google Scholar]
- 33. Nemoto S., Fergusson M. M., and Finkel T. (2004) Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306, 2105–2108 [DOI] [PubMed] [Google Scholar]
- 34. Menzies K. J., and Hood D. A. (2012) The role of SirT1 in muscle mitochondrial turnover. Mitochondrion 12, 5–13 [DOI] [PubMed] [Google Scholar]
- 35. Frezza C., Cipolat S., and Scorrano L. (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2, 287–295 [DOI] [PubMed] [Google Scholar]
- 36. Patino W. D., Kang J. G., Matoba S., Mian O. Y., Gochuico B. R., and Hwang P. M. (2006) Atherosclerotic plaque macrophage transcriptional regulators are expressed in blood and modulated by tristetraprolin. Circ. Res. 98, 1282–1289 [DOI] [PubMed] [Google Scholar]
- 37. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., and Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.