

Abstract
HIV, type 1 overcomes host restriction factor apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3) proteins by organizing an E3 ubiquitin ligase complex together with viral infectivity factor (Vif) and a host transcription cofactor core binding factor β (CBFβ). CBFβ is essential for Vif to counteract APOBEC3 by enabling the recruitment of cullin 5 to the complex and increasing the steady-state level of Vif protein; however, the mechanisms by which CBFβ up-regulates Vif protein remains unclear. Because we have reported previously that mouse double minute 2 homolog (MDM2) is an E3 ligase for Vif, we hypothesized that CBFβ might protect Vif from MDM2-mediated degradation. Co-immunoprecipitation analyses showed that Vif mutants that do not bind to CBFβ preferentially interact with MDM2 and that overexpression of CBFβ disrupts the interaction between MDM2 and Vif. Knockdown of CBFβ reduced the steady-state level of Vif in MDM2-proficient cells but not in MDM2-null cells. Cycloheximide chase analyses revealed that Vif E88A/W89A, which does not interact with CBFβ, degraded faster than wild-type Vif in MDM2-proficient cells but not in MDM2-null cells, suggesting that Vif stabilization by CBFβ is mainly caused by impairing MDM2-mediated degradation. We identified Vif R93E as a Vif variant that does not bind to MDM2, and the virus with this substitution mutation was more resistant to APOBEC3G than the parental virus. Combinatory substitution of Vif residues required for CBFβ binding and MDM2 binding showed full recovery of Vif steady-state levels, supporting our hypothesis. Our data provide new insights into the mechanism of Vif augmentation by CBFβ.
Keywords: host-pathogen interaction, HIV, mouse double minute 2 homolog (MDM2), proteasome, protein-protein interaction, ubiquitin, viral protein
Introduction
HIV-12 encodes the Vif accessory protein to counteract host restriction factor APOBEC3 proteins expressed in natural target cells such as CD4-positive T lymphocytes and macrophages (1–5). APOBEC3s are host DNA cytosine deaminases that convert cytosines in single-stranded DNA to uracils and can introduce mutations in the genome or DNA intermediates of several viruses, including HIV-1 (reviewed in Ref. 6). In the absence of Vif, at least four APOBEC3 proteins, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H, are encapsidated into budding virions and deaminate cytosine residues to uracils in the nascent minus-strand HIV-1 DNA, and uracils then template the insertion of adenosines during synthesis of the plus-strand DNA, consequently rising to G-to-A hypermutations (7). A high density of mutations results in the incorporation of premature stop codons and protein defects that do not support viral replication. Vif behaves as a substrate recognition subunit of the Cullin-RING ubiquitin ligase complex that consists of cullin 5 (CUL5), RING box protein 2 (RBX2), elongin C (ELOC), and elongin B (ELOB) and degrades APOBEC3 proteins by the ubiquitin-proteasome pathway (8–10). CBFβ, a non-DNA-binding subunit of the heterodimeric core-binding factor family of transcription factors, is also involved in the Vif ubiquitin ligase complex and is essential for APOBEC3 degradation and viral replication (11, 12). Mutational analyses suggested several Vif residues required for CBFβ binding, and some of those, Leu64, Ile66, Glu88, and Trp89, actually appeared to be on the interface between Vif and CBFβ in the crystal structure of the Vif-CBFβ-ELOB-ELOC-CUL5 complex (13–15). A couple of mechanisms by which CBFβ supports Vif functions have been reported. CBFβ enables the recruitment of CUL5 to Vif, which is required for ubiquitination and degradation of APOBEC3s (12). CBFβ also up-regulates the steady-state levels of Vif expression by reducing proteasomal degradation (11) and enhancing biosynthesis (16); however, it is still unclear how CBFβ up-regulates Vif levels.
Besides Vif-mediated APOBEC3 ubiquitination, Vif ubiquitination has also been manifested. The homologous to the E6AP carboxyl terminus domain family E3 ligases NEDD4 and itchy E3 ubiquitin protein ligase (also known as AIP4) posttranslationally modify Vif by monoubiquitination (17). The Cullin 5-RING ubiquitin ligase complex mediates polyubiquitination of Vif, likely by autoubiquitination (11, 18). Vif expression is controlled at low levels by the proteasome pathway (19), and MDM2 is responsible for the down-regulation of Vif (20). MDM2 is well known as a RING finger E3 ubiquitin ligase that specifically mediates p53 ubiquitination and degradation (21–23). MDM2 shuttles between cytoplasm and the nucleus constantly through the nuclear pore complex (24). Earlier report indicated that MDM2-mediated p53 ubiquitination takes place efficiently in the cytoplasm (22, 23). Hence, we hypothesized that Vif up-regulation by CBFβ might be due to protection from MDM2-mediated proteasomal degradation.
In present study, we employed Vif substitution mutant E88A/W89A, which does not bind CBFβ, to test our hypothesis. Co-immunoprecipitation experiments show that Vif binding to CBFβ impairs MDM2 binding to Vif. Kinetic studies using cycloheximide demonstrate that CBFβ binding increases the metabolic stability of Vif only in the presence of MDM2, not in the absence of MDM2. By mutational analyses, we identified that Vif arginine residue at position 93 is required for MDM2 binding, and that the substitution of this residue to glutamate conferred resistance to MDM2-mediated degradation. Additionally, the virus containing Vif R93E substitution mutation is more resistant to APOBEC3G than wild-type virus. Our data combined with the structural data of Vif-complex support the idea that CBFβ protein functions as a protector of Vif protein.
Results
Vif Mutants That Are Defective in CBFβ Binding Preferentially Interact with MDM2
To test our hypothesis that CBFβ might inhibit MDM2-mediated proteasomal degradation of Vif, we initially generated expression vectors for Vif amino acid substitution mutants that are reported to be defective in CBFβ binding: L64S, I66S (13), and E88A/W89A (25). To determine whether the substitution of these residues alters binding to MDM2, we then co-transfected HA-tagged MDM2 and myc-tagged Vif wild-type or one of the mutants, and cell lysates were immunoprecipitated by anti-myc serum. Endogenous CBFβ co-precipitated with wild-type Vif but not with L64S, I66S, or E88A/W89A, as expected (Fig. 1A). Despite lower levels of the Vif mutants, much larger amounts of MDM2 co-precipitated with L64S, I66S, and E88A/W89A compared with wild-type Vif (Fig. 1A). To confirm these findings, we performed reciprocal co-immunoprecipitation experiments by overexpression of myc-tagged MDM2 and untagged Vif wild-type or mutant. Large amounts of Vif mutants co-precipitated with MDM2 but very little of wild-type Vif (Fig. 1B). These results suggest that Vif mutants that are defective in CBFβ binding preferentially interact with MDM2.
FIGURE 1.

CBFβ interferes with the interaction between Vif and MDM2. A, co-immunoprecipitation of MDM2 with Vif wild-type and variants that do not interact with CBFβ. 293T cells transiently expressing myc-tagged Vif wild-type or the variant and HA-tagged MDM2 were lysed and immunoprecipitated by anti-myc serum, and samples were analyzed by immunoblotting (IB) with anti-HA, anti-myc, and anti-CBFβ sera. B, reciprocal co-immunoprecipitation of Vif wild-type and variants with MDM2. Lysates of 293T cells with overexpression of untagged Vif wild-type or the variant and myc-tagged MDM2 were immunoprecipitated by anti-myc serum, and samples were analyzed by immunoblotting with anti-Vif and anti-myc sera. C, co-immunoprecipitation of MDM2 with Vif wild-type in the presence of CBFβ overexpression. Lysates of 293T cells transiently expressing myc-tagged Vif, HA-tagged MDM2, and various amounts of CBFβ were immunoprecipitated by anti-myc serum and analyzed for co-precipitation of MDM2 by immunoblotting.
Overexpression of CBFβ Inhibits MDM2 Binding to Vif
To determine whether overexpression of CBFβ impairs MDM2 binding to Vif, we performed additional co-immunoprecipitation experiments in 293T cells with overexpression of myc-tagged Vif, HA-tagged MDM2, and untagged CBFβ. Overexpression of CBFβ reduced the amount of co-precipitated MDM2 in a dose-dependent manner (Fig. 1C), suggesting that CBFβ inhibits MDM2 binding to Vif.
CBFβ Is Not Required for Vif Augmentation in MDM2-null Cells
To determine whether the counteraction of MDM2-mediated degradation is a relevant function of CBFβ for Vif augmentation, we took advantage of MDM2-null cells. We employed p53−/−; Mdm2−/− double knockout mouse embryonic fibroblasts (MEFs) as Mdm2-null cells and p53−/− MEFs as an Mdm2-positive control because Mdm2 knockout mice are embryonic lethal (26, 27) and there are no Mdm2−/− MEFs available. These cells were transduced with the wild-type or E88A/W89A vif gene by murine leukemia virus-based retrovirus vectors and selected clones that express comparable levels of Vif (Fig. 2A). To assess whether CBFβ is supportive of Vif steady-state levels in these cells, we transfected siRNA against murine CBFβ and analyzed Vif levels by immunoblotting. Knockdown of CBFβ impaired the Vif wild-type level in Mdm2-proficient cells but not in Mdm2-null cells (Fig. 2B), suggesting that CBFβ is not supportive of Vif levels in the absence of MDM2. The levels of Vif E88A/W89A, which does not interact with CBFβ, were unaffected by CBFβ knockdown, as expected. To analyze Vif stability in these cells, cells were treated with cycloheximide for various durations, and cell lysates were subjected to immunoblotting (Fig. 2C). The intensity of Vif bands was quantified by densitometry, and each value was normalized to that without cycloheximide treatment (Fig. 2D). In Mdm2-proficient cells, E88A/W89A, which does not bind to CBFβ, decayed much faster than wild-type Vif (Fig. 2C, first and third panels, and Fig. 2D, dotted lines), as expected, whereas in Mdm2-null cells, both E88A/W89A and wild-type Vif showed similar kinetics (Fig. 2C, fifth and seventh panels, and Fig. 2D, solid lines). These results clearly demonstrate that Vif augmentation by CBFβ is mainly due to the counteraction of MDM2-mediated degradation.
FIGURE 2.

Protection of Vif from MDM2-mediated degradation is responsible for Vif augmentation by CBFβ. A, p53−/− and p53−/−; MDM2−/− MEF cells were stably transfected with Vif wild-type or E88A/W89A, and clones that expressed comparable amount of Vif protein were picked according to immunoblotting. B, siRNA against murine Cbfb or control siRNA was transfected into MDM2-proficient and MDM2-null cells stably expressing Vif, and Vif levels were analyzed by immunoblotting. C, cycloheximide (CHX) chase analyses of Vif degradation. After cycloheximide treatment for the indicated time, lysates of p53−/− and p53−/−; MDM2−/− MEF cells stably expressing Vif wild-type or E88A/W89A were analyzed by immunoblotting with anti-Vif serum. Tubulin was also analyzed as a loading control. D, the amounts of Vif in C were quantified by densitometry, and those without cycloheximide treatment were normalized to 100%.
HIV-1 Vif R93 Is Crucial for the Interaction with MDM2
To further assess the relevance of MDM2-mediated degradation of Vif for CBFβ function, we next explored Vif residues that interact with MDM2. Because one previous report proposed that the charged motif 90RKKR93 is related to steady-state levels of Vif protein (28), we generated Vif amino acid substitution mutants on these residues: R90A, K91A, K92A, R93E, Y94D, K91A/K92A, 91KKR93/3A, and 90RKKR93/4A. To determine whether these mutants are degraded by MDM2, 293T cells were co-transfected with expression vectors for MDM2 and Vif wild-type or variants, and protein levels of Vif were analyzed by immunoblotting. The electrophoretic mobility of some of the mutants appeared to be different from wild-type Vif, likely because of differences in charge and molecular weight (Fig. 3A, top panel). Co-transfection of MDM2 reduced the expression levels of wild-type Vif, R90A, K91A, K92A, Y94D, and K91A/K92A (Fig. 3A, lanes 1–8, 11, and 12, top panel) but not that of R93E, 91KKR93/3A, or 90RKKR94/4A (Fig. 3A, lanes 9, 10, and 13–16, top panel). These results suggest that Arg93 of Vif is required for MDM2-mediated degradation. To determine whether Arg93 is actually involved in MDM2 binding, we next performed co-immunoprecipitation experiments in 293T cells with overexpression of myc-tagged Vif and HA-tagged MDM2. MDM2 co-precipitated with wild-type Vif, R90A, K91A, K92A, and Y94D but not with R93E (Fig. 3B). Taken together, Vif R93 is involved in MDM2 binding and consequent degradation of Vif.
FIGURE 3.

Vif R93 is required for MDM2 binding. A, degradation of Vif variants by MDM2. 293T cells were co-transfected with expression vectors for MDM2 and Vif wild-type or variant, and protein levels of Vif were analyzed by immunoblotting with anti-Vif serum. B, co-immunoprecipitation of MDM2 with Vif variants. 293T cells transiently expressing HA-tagged MDM2 and myc-tagged Vif wild-type or variant were lysed and immunoprecipitated by anti-myc serum. Samples were analyzed by immunoblotting (IB) with anti-HA and anti-myc sera.
The Substitution of R93 to Glutamate Enhances the Ability to Counteract the Restriction by APOBEC3G
To further confirm that Vif R93 is involved in MDM2 binding, we performed single-cycle infection experiments using luciferase reporter viruses harboring the Vif R93E substitution mutation in a setting with various amounts of APOBEC3G. The substitution was introduced into pNL4–3 ΔEnv-Luc and transfected into 293T cells with co-transfection of the VSV-G expression plasmid in the presence or absence of co-transfection of the APOBEC3G expression plasmid. Virus-containing supernatant was harvested and added to fresh 293T cells, and the luciferase activities of the cell lysates were quantified. The R93E-harboring virus was more potent for counteracting the restriction by APOBEC3G than wild-type Vif-harboring virus (Fig. 4, top panel). The amount of APOBEC3G encapsidated into R93E-harboring virions was less than that of wild-type Vif-harboring virions (Fig. 4, bottom panel). These results are consistent with the idea that Vif is down-regulated by MDM2-mediated degradation and that Vif R93 is required for MDM2 binding.
FIGURE 4.
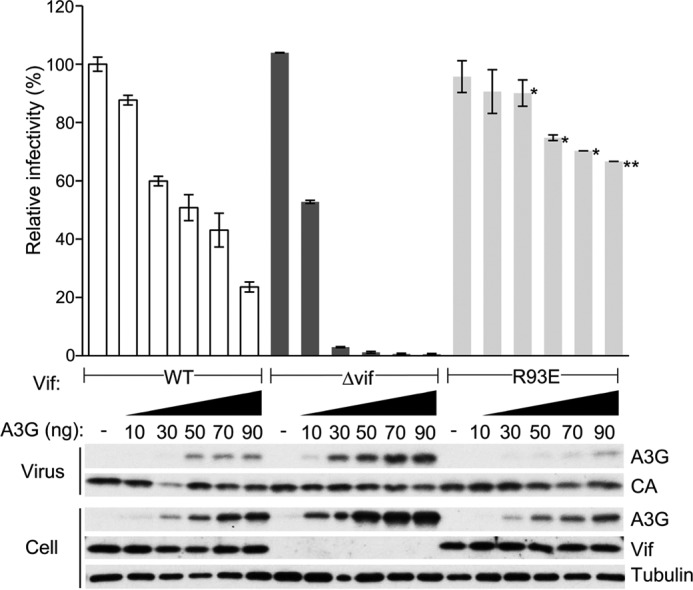
Enhanced counteraction of Vif R93E against the restriction by APOBEC3G. Single-cycle infection experiments were performed by using VSV-G pseudotyped luciferase reporter viruses produced in 293T cells by co-transfection of pNL43/ΔEnv-Luc with the indicated Vif phenotype and pVSV-G in the presence or absence of co-transfection of pcDNA3/HA-A3G. Virus-containing supernatant was added to fresh 293T cells, and the luciferase activity of the cell lysates was measured by adding substrate and using a luminometer. The value of the virus without vif mutation in the absence of APOBEC3G was normalized to 100%, and mean ± S.E. of three independent experiments is shown (top panel). *, p < 0.05; **, p < 0.01 (statistically significant differences of infectivity between wild-type and R93E-harboring viruses). The levels of APOBEC3G in cells and virions and Vif in cells were also analyzed by immunoblotting (bottom panel). Virus p24 capsid protein (CA) and cellular tubulin were used as control.
Additional R93E Substitution to E88A/W89A Restores Vif Steady-state Levels but not Counteraction of the Restriction by APOBEC3G
To further assess whether Vif augmentation by CBFβ is relevant to interference with MDM2-mediated degradation, we combinationally introduced amino acid substitutions that disrupt CBFβ binding and MDM2 binding into pNL4–3 ΔEnv-Luc and performed single-cycle infection experiments of a VSV-G pseudotyped virus produced in 293T cells with or without overexpression of APOBEC3G. The Vif levels of E88A/W89A were lower than those of wild-type Vif, as expected, and those of E88A/W89A/R93E markedly recovered (Fig. 5, bottom panel, fourth row, lanes 1, 2, and 5–8). These results further confirmed that Vif stabilization by CBFβ is due to the counteraction of MDM2. More interestingly, the infectivity of the E88A/W89A/R93E-harboring virus in the presence of APOBEC3G was less than that of the wild-type or R93E-harboring virus (Fig. 5, top panel). These results suggest that Vif stabilization is not the only function of CBFβ to support Vif counteraction to APOBEC3G. In other words, CBFβ plays multiple roles for Vif to enable sufficient ubiquitination and subsequent degradation of APOBEC3G.
FIGURE 5.
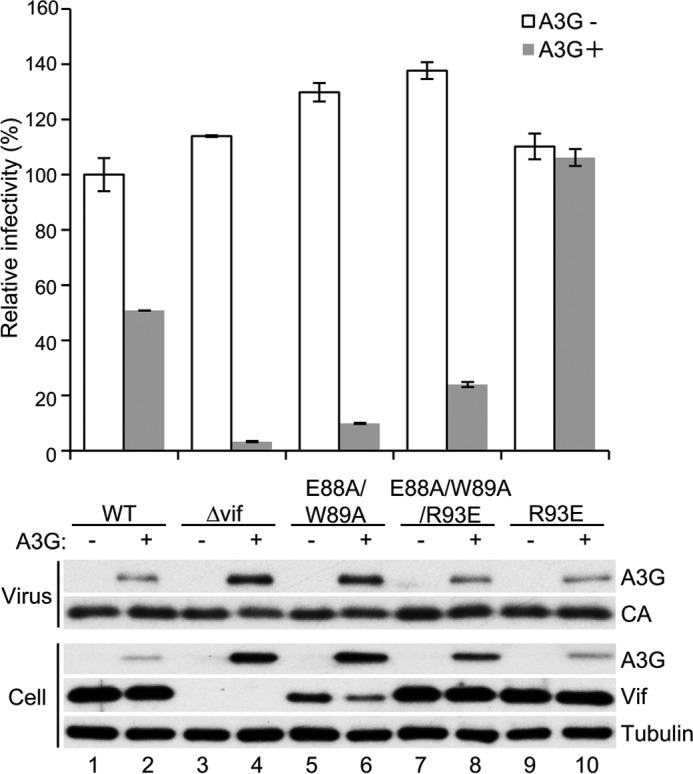
The disruption of MDM2 binding restores the protein levels of the Vif variant that does not bind to CBFβ, but it does not fully restore the function to counteract APOBEC3G. Single-cycle infection experiments were performed by using VSV-G pseudotyped luciferase reporter viruses produced in 293T cells by co-transfection of pNL43/ΔEnv-Luc with the indicated vif phenotype and pVSV-G in the presence or absence of co-transfection of pcDNA3/HA-A3G. Virus-containing supernatant was added to fresh 293T cells, and the luciferase activity of the cell lysates was measured by adding substrate and using a luminometer. The value of the virus without vif mutation in the absence of APOBEC3G was normalized to 100%, and mean ± S.E. of three independent experiments is shown (top panel). The levels of APOBEC3G in cells and virions and Vif in cells were also analyzed by immunoblotting (bottom panel). Capsid (CA) and tubulin were used as control.
Discussion
In this study, we propose the novel hypothesis that CBFβ protects HIV-1 Vif from MDM2-mediated degradation and use multiple ways to prove this hypothesis. First, co-immunoprecipitation analyses show that Vif mutants that are defective in CBFβ binding interact with a much larger amount of MDM2 compared with wild-type Vif. Additionally, overexpression of CBFβ reduces MDM2 binding to Vif. Second, kinetic studies reveal that the inhibition of MDM2-mediated Vif degradation is a critical role of CBFβ for the stabilization of Vif protein. Finally, we show that the Vif arginine residue at position 93 is crucial for MDM2 binding. Arg93 is located near the CBFβ binding site in the crystal structure of the Vif complex (PDB code 4N9F), which supports the idea that CBFβ protects, although not completely, Vif from MDM2-mediated ubiquitination and subsequent degradation via the proteasome pathway. Therefore, all of our data are consistent with the hypothesis and provide novel insights into the mechanism of how CBFβ stabilizes Vif protein.
Malbec et al. (29) reported that MDM2 affects the infectivity of a few retroviruses, including HIV-1, by modulating TRIM5α-mediated restriction. However, that phenomenon is not related to our findings because we used human cells that express non-restrictive human TIRM5α for HIV-1 infection studies.
Miyagi et al. (16) reported that the up-regulation of Vif protein in the presence of CBFβ is mainly caused at the translation step using CBFβ knockdown cells. However, knockdown of CBFβ in Mdm2-depleted cells did not affect Vif levels, suggesting that Vif up-regulation by CBFβ is mainly caused by protecting Vif from proteasomal degradation induced by MDM2. One explanation for this discrepancy is the difference in cell species. Miyagi et al. (16) used human cells, and we used murine cells that express murine CBFβ. Although CBFβ is conserved between these two species with more than 95% amino acid identity, we cannot exclude the possibility of functional deficiency of murine CBFβ.
We show that Arg93 is crucial for Vif to be recognized by MDM2. The substitution of this single residue with glutamate disrupts the interaction between Vif and MDM2 as well as MDM2-mediated down-regulation of Vif. We further demonstrate that the substitution makes the virus more resistant to APOBEC3G by single-cycle infection experiments of the reporter viruses. This residue is located very close to, but not fully covered by, CBFβ in the reported crystal structure of the Vif-CBFβ-ELOB-ELOC-CUL5 complex (PDB code 4N9F, Fig. 6A), supporting our hypothesis that Vif is protected from MDM2-mediated degradation by CBFβ. According to the HIV-1 sequence database by Los Alamos National Laboratory, the Vif residue at position 93 is conserved as arginine for a major part of the strains; there exist natural variants that harbor glutamate or other residues at this position (http://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html). Our data suggest that Glu93-harvoring viruses are more resistant to APOBEC3G, but considering that these viruses are not major strains, Vif Arg93 may be powerful enough to counteract APOBEC3G. We also show that a combination of the substitution of Vif residues required for binding to CBFβ and MDM2 restores Vif protein levels to that of wild-type Vif but not fully for the Vif function to counteract APOBEC3G. Our results are consistent with previous studies showing that CBFβ binding is required for CUL5 recruitment and/or organization of a functional ubiquitin ligase complex (12, 13, 30).
FIGURE 6.
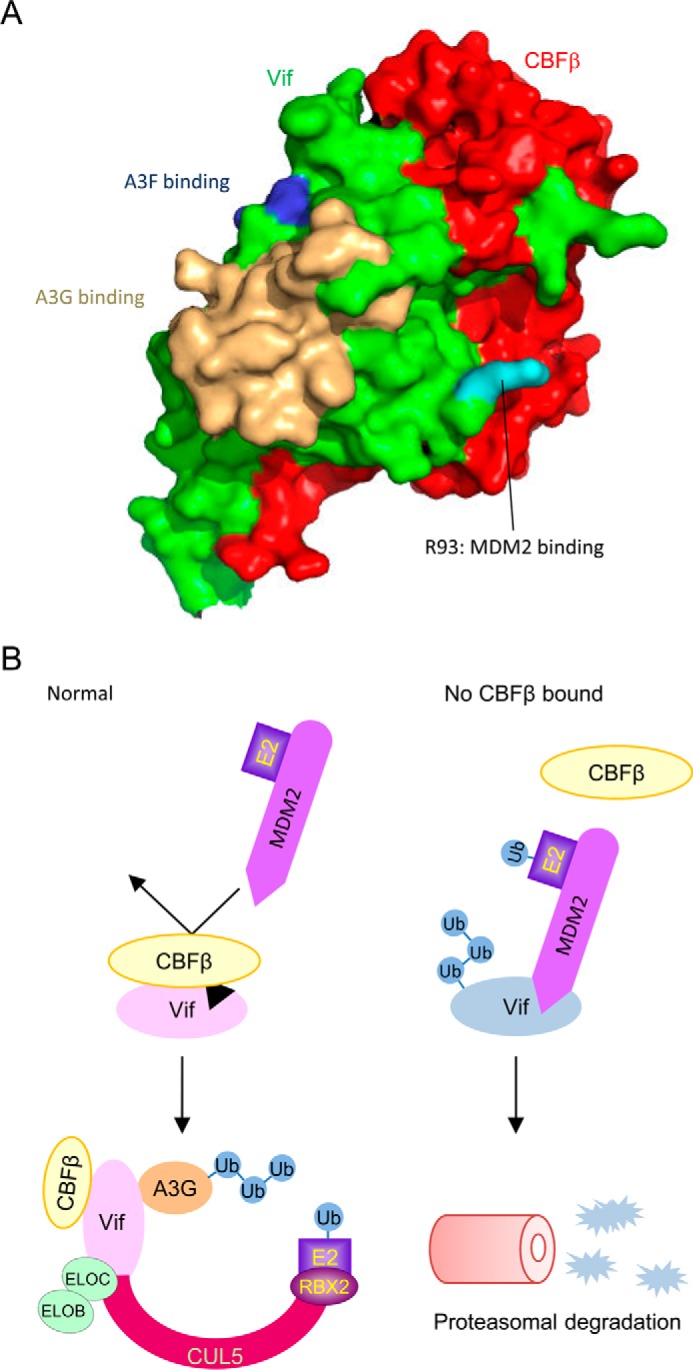
CBFβ protects Vif from MDM2. A, surface model of the Vif-CBFβ heterodimer based on the reported structure of the Vif-CBFβ-ELOB-ELOC-CUL5 complex (PDB code 4N9F). Vif and CBFβ are shown in green and red, respectively. Vif R93 is highlighted in cyan, and APOBEC3G and APOBEC3F binding sites are marked in wheat and blue, respectively. B, proposed model in which CBFβ stabilizes Vif protein. Under normal conditions, CBFβ interacts with Vif extensively just near the MDM2 binding site and sequesters MDM2 from Vif. The Vif variant that is defective in CBFβ binding is preferentially captured by MDM2 and rapidly cleared by the proteasome pathway. Ub, ubiquitin.
In our model, CBFβ shields the close site of MDM2 binding and sequesters Vif from MDM2. On the other hand, Vif mutants that are defective in CBFβ binding are preferentially captured by MDM2 and rapidly cleared by proteasomal degradation (Fig. 6B). Our kinetic studies imply that MDM2 is responsible for the short half-life of Vif protein. MDM2 physiologically interacts with the p53 tumor suppressor protein and negatively modulates its transcriptional activity and stability (12, 13, 30, 31). It is well known that overexpression of MDM2 is involved in many malignancies (12, 13, 30, 32). Hence, there are safety mechanisms to prevent MDM2 from running amok. MDM2 has intrinsic RING finger-dependent E3 activity toward itself (33). Furthermore, an MDM2-binding protein, p19ARF, binds upstream of its RING finger and exposes a cryptic nucleolar localization signal co-linear with the MDM2 RING finger (34). As a result, MDM2 is maintained at low levels in normal cells. We reported previously reported that Vif enhances p53 stability and releases MDM2-mediated inhibition of nuclear export of p53 to induce G2 cell cycle arrest (35). In HIV-1 infected cells, activated p53 might induce the transcription of several genes, including MDM2, and enhanced expression of MDM2 might lead to more Vif ubiquitination and degradation. We speculate that Vif requires CBFβ as a protector against the induced MDM2. Further studies of the relationship among these three molecules will be valuable in understanding the viral strategies to accomplish productive infection and also for developing pharmaceutical inhibition of HIV-1 replication.
Inhibition of the Vif function to promote proteasomal degradation of host restriction factors by small molecules has emerged as a promising target for therapeutics for patients with HIV-1 infection (36, 37). There are several target protein-protein interactions for this strategy because Vif directly interacts with three of the components of the ubiquitin ligase complex as well as substrate APOBEC3 proteins. Of these, the Vif-CBFβ interaction seems to be the most attractive target because of high effectiveness and few side effects. Disruption of this interaction would recover all APOBEC3 proteins that are targeted by Vif and would not affect host protein-protein interactions except the runt related transcription factor-CBFβ interaction, which is a physiological interaction but is interfered by Vif protein in HIV-1 infected cells (13, 15). Moreover, considering our data, even during interruption of Vif-CBFβ for a short duration, MDM2 would rapidly clear Vif protein through the proteasome pathway (Fig. 6B). Our data provide fundamental insights into the development of effective Vif inhibitors.
We propose the novel hypothesis that CBFβ protects HIV-1 Vif protein from MDM2-mediated degradation by inhibiting MDM2 binding to Vif and clearly demonstrate this using multiple experimental methods. Our data help understand Vif protein metabolism in infected cells and provide supportive evidence for promising therapeutics for HIV-1 infection that disrupt the Vif-CBFβ protein-protein interaction.
Experimental Procedures
Plasmid Construction
The C-terminally myc-tagged Vif expression plasmid pDON-Vif-myc and its E88A/W89A derivative were described previously (25). Other derivative amino acid substitution mutants were generated by a PCR-based method using properly designed primers. The untagged Vif expression plasmid and N-terminally HA-tagged MDM2 expression plasmid were described previously (20). The expression plasmid for CBFβ was generated by insertion of CBFβ complementary DNA into pcDNA3 at XhoI/KpnI sites. The luciferase reporter HIV-1 plasmids for single-cycle infection, pNL43/ΔEnv-Luc and pNL43/ΔEnvΔvif-Luc, were described previously (38). Mutations in the vif region of pNL43/ΔEnv-Luc and pNL4–3 were introduced by a PCR-based method using internal restriction sites, MscI at position 4553 and EcoRI at position 5743.
Cell Culture and Transfection
293T cells and murine embryonic fibroblasts were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin, streptomycin, and glutamine. Cells on 6-well plates were transfected with about 1 μg of plasmid DNA in total using X-tremegene HP DNA transfection reagent (Roche) according to the instructions of the manufacturer. For generating stable cell lines, cells were diluted and plated on 96-well plates 1 day after transfection, selected by adding G418 (Nacalai) at a final concentration to 1.6 mg/ml, and analyzed for expression of Vif protein by immunoblotting.
Immunoblotting
We used a standard chemiluminescence protocol for immunoblotting with a PVDF membrane (Millipore). Primary antibodies for immunoblotting against Vif, APOBEC3G, and p24Gag were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. Rabbit anti-CBFβ serum was purchased from Santa Cruz Biotechnology. Rabbit anti-myc serum and anti-HA serum were purchased from Sigma. Mouse anti-tubulin monoclonal antibody was purchased from Covance. HRP-conjugated secondary antibodies against mouse and rabbit were purchased from GE Healthcare.
Co-immunoprecipitation
293T cells were transfected with expression vectors as indicated in Fig. 1, treated with MG132 at a concentration of 2.5 μm for 16 h, and lysed with co-IP buffer (25 mm HEPES (pH 7.4), 150 mm NaCl, 0.1% Triton X-100, 1 mm EDTA, and 1 mm MgCl2) supplemented with protease inhibitor mixture (Roche) and 50 μm MG132. After centrifugation at 20,000 × g for 10 min, the supernatant was mixed with 2.5 μg of anti-myc polyclonal antibody (Sigma-Aldrich, C3956) or 500 ng of anti-HA monoclonal antibody (Roche, clone 3F10) for 1 h, and then mixed with 20 μl of protein A-Sepharose (GE Healthcare) for 1 h. Beads were washed with co-IP buffer three times, and bound protein was eluted with 1× SDS sample buffer. Samples were analyzed by immunoblotting as described above.
Knockdown of CBFβ in Mouse Embryonic Fibroblasts
Mouse embryonic fibroblasts stably expressing Vif wild-type or the E88A/W89A mutant at 50% confluency were transfected with SMARTpool siRNA against CBFβ (Dharmacon, M-062486–01) or Stealth RNAi negative control duplexes (Invitrogen) at a concentration of 50 nm using X-tremegene siRNA transfection reagent (Roche) in serum-free medium. The serum was replenished 1 day after transfection, and cells were harvested 2 days after transfection and analyzed for Vif levels by immunoblotting.
Cycloheximide Chase
p53−/− and p53−/−; Mdm2−/− mouse embryonic fibroblasts stably expressing Vif wild-type or the E88A/W89A mutant were treated with 140 μg/ml cycloheximide for 30, 60, 90, or 120 min, harvested by washing with PBS twice, lysed with co-IP buffer, and analyzed for Vif levels by immunoblotting. Tubulin levels were also analyzed as a loading control.
Single-cycle Infection of Luciferase Reporter HIV-1
Luciferase encoding HIV-1 particles was produced by transiently transfecting 293T cells at 50% confluency using 0.75 μg of pNL43/ΔEnv-Luc or derivative mutant, 0.3 μg of pVSV-G and 0.1 μg of pcDNA3/HA-A3G, or empty vector. After 48 h, virus-containing supernatants were harvested through a PVDF filter with 0.45-μm pores (Millipore) and added to fresh 293T cells. After 48 h, cells were lysed with passive lysis buffer (Promega), and luciferase activity was determined by luminometer (2030 Arvo X, PerkinElmer Life Sciences) using a luciferase assay system (Promega). Sample preparation of producer cells and virus for immunoblotting was performed as described previously (39).
Author Contributions
Y. M. performed most of the experiments, analyzed data, and contributed to writing of the manuscript. K. Shindo designed the experiments, analyzed data, and wrote the manuscript. K. N. and N. Y. performed experiments. K. Shirakawa, M. K., and A. T. K. participated in data analysis and writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank the laboratory members for helpful discussions. The following materials were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program: rabbit anti-Vif serum 2221 from Dr. Dana Gabuzda and anti-p24 Gag monoclonal antibody 6457 from Dr. Michael H. Malim.
This study was partly supported by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology (KAKENHI #JP24115004), Japan Society for the Promotion of Science (KAKENHI #JP16K08807), and the Ministry of Health, Labor, and Welfare in Japan. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HIV-1
- HIV, type 1
- Vif
- viral infectivity factor
- CBF
- core binding factor
- MEF
- mouse embryonic fibroblast
- IP
- immunoprecipitation
- VSV-G
- vesicular stomatitis virus G glycoprotein.
References
- 1. Gabuzda D. H., Lawrence K., Langhoff E., Terwilliger E., Dorfman T., Haseltine W. A., and Sodroski J. (1992) Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 66, 6489–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gabuzda D. H., Li H., Lawrence K., Vasir B. S., Crawford K., and Langhoff E. (1994) Essential role of vif in establishing productive HIV-1 infection in peripheral blood T lymphocytes and monocyte/macrophages. J. Acquir. Immune Defic. Syndr. 7, 908–915 [PubMed] [Google Scholar]
- 3. Sheehy A. M., Gaddis N. C., Choi J. D., and Malim M. H. (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650 [DOI] [PubMed] [Google Scholar]
- 4. Madani N., and Kabat D. (1998) An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J. Virol. 72, 10251–10255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simon J. H., Gaddis N. C., Fouchier R. A., and Malim M. H. (1998) Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 4, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 6. Harris R. S., and Dudley J. P. (2015) APOBECs and virus restriction. Virology 479–480, 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hultquist J. F., Lengyel J. A., Refsland E. W., LaRue R. S., Lackey L., Brown W. L., and Harris R. S. (2011) Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu X., Yu Y., Liu B., Luo K., Kong W., Mao P., and Yu X. F. (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060 [DOI] [PubMed] [Google Scholar]
- 9. Sheehy A. M., Gaddis N. C., and Malim M. H. (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407 [DOI] [PubMed] [Google Scholar]
- 10. Kobayashi M., Takaori-Kondo A., Shindo K., Abudu A., Fukunaga K., and Uchiyama T. (2004) APOBEC3G targets specific virus species. J. Virol. 78, 8238–8244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jäger S., Kim D. Y., Hultquist J. F., Shindo K., LaRue R. S., Kwon E., Li M., Anderson B. D., Yen L., Stanley D., Mahon C., Kane J., Franks-Skiba K., Cimermancic P., Burlingame A., et al. (2011) Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang W., Du J., Evans S. L., Yu Y., and Yu X. F. (2012) T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481, 376–379 [DOI] [PubMed] [Google Scholar]
- 13. Kim D. Y., Kwon E., Hartley P. D., Crosby D. C., Mann S., Krogan N. J., and Gross J. D. (2013) CBFβ stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 49, 632–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsui M., Shindo K., Izumi T., Io K., Shinohara M., Komano J., Kobayashi M., Kadowaki N., Harris R. S., and Takaori-Kondo A. (2014) Small molecules that inhibit Vif-induced degradation of APOBEC3G. Virol. J. 11, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo Y., Dong L., Qiu X., Wang Y., Zhang B., Liu H., Yu Y., Zang Y., Yang M., and Huang Z. (2014) Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505, 229–233 [DOI] [PubMed] [Google Scholar]
- 16. Miyagi E., Kao S., Yedavalli V., and Strebel K. (2014) CBFβ enhances de novo protein biosynthesis of its binding partners HIV-1 Vif and RUNX1 and potentiates the Vif-induced degradation of APOBEC3G. J. Virol. 88, 4839–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dussart S., Courcoul M., Bessou G., Douaisi M., Duverger Y., Vigne R., and Decroly E. (2004) The Vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin. Biochem. Biophys. Res. Commun. 315, 66–72 [DOI] [PubMed] [Google Scholar]
- 18. Mehle A., Goncalves J., Santa-Marta M., McPike M., and Gabuzda D. (2004) Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18, 2861–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujita M., Akari H., Sakurai A., Yoshida A., Chiba T., Tanaka K., Strebel K., and Adachi A. (2004) Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 6, 791–798 [DOI] [PubMed] [Google Scholar]
- 20. Izumi T., Takaori-Kondo A., Shirakawa K., Higashitsuji H., Itoh K., Io K., Matsui M., Iwai K., Kondoh H., Sato T., Tomonaga M., Ikeda S., Akari H., Koyanagi Y., Fujita J., and Uchiyama T. (2009) MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Honda R., Tanaka H., and Yasuda H. (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420, 25–27 [DOI] [PubMed] [Google Scholar]
- 22. Haupt Y., Maya R., Kazaz A., and Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 23. Kubbutat M. H., Jones S. N., and Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 24. Roth J., Dobbelstein M., Freedman D. A., Shenk T., and Levine A. J. (1998) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 17, 554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsui Y., Shindo K., Nagata K., Io K., Tada K., Iwai F., Kobayashi M., Kadowaki N., Harris R. S., and Takaori-Kondo A. (2014) Defining HIV-1 Vif residues that interact with CBFβ by site-directed mutagenesis. Virology 449, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones S. N., Roe A. E., Donehower L. A., and Bradley A. (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 [DOI] [PubMed] [Google Scholar]
- 27. Montes de Oca Luna R., Wagner D. S., and Lozano G. (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206 [DOI] [PubMed] [Google Scholar]
- 28. Fujita M., Sakurai A., Yoshida A., Miyaura M., Koyama A. H., Sakai K., and Adachi A. (2003) Amino acid residues 88 and 89 in the central hydrophilic region of human immunodeficiency virus type 1 Vif are critical for viral infectivity by enhancing the steady-state expression of Vif. J. Virol. 77, 1626–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malbec M., Pham Q. T., Plourde M. B., Létourneau-Hogan A., Nepveu-Traversy M. E., and Berthoux L. (2010) Murine double minute 2 as a modulator of retroviral restrictions mediated by TRIM5α. Virology 405, 414–423 [DOI] [PubMed] [Google Scholar]
- 30. Fribourgh J. L., Nguyen H. C., Wolfe L. S., Dewitt D. C., Zhang W., Yu X. F., Rhoades E., and Xiong Y. (2014) Core binding factor β plays a critical role by facilitating the assembly of the Vif-cullin 5 E3 ubiquitin ligase. J. Virol. 88, 3309–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu X., Bayle J. H., Olson D., and Levine A. J. (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7, 1126–1132 [DOI] [PubMed] [Google Scholar]
- 32. Momand J., Jung D., Wilczynski S., and Niland J. (1998) The MDM2 gene amplification database. Nucleic Acids Res. 26, 3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fang S., Jensen J. P., Ludwig R. L., Vousden K. H., and Weissman A. M. (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 275, 8945–8951 [DOI] [PubMed] [Google Scholar]
- 34. Lohrum M. A., Ashcroft M., Kubbutat M. H., and Vousden K. H. (2000) Identification of a cryptic nucleolar-localization signal in MDM2. Nat. Cell Biol. 2, 179–181 [DOI] [PubMed] [Google Scholar]
- 35. Izumi T., Io K., Matsui M., Shirakawa K., Shinohara M., Nagai Y., Kawahara M., Kobayashi M., Kondoh H., Misawa N., Koyanagi Y., Uchiyama T., and Takaori-Kondo A. (2010) HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U.S.A. 107, 20798–20803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salter J. D., Morales G. A., and Smith H. C. (2014) Structural insights for HIV-1 therapeutic strategies targeting Vif. Trends Biochem. Sci. 39, 373–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Albin J. S., and Harris R. S. (2010) Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shindo K., Takaori-Kondo A., Kobayashi M., Abudu A., Fukunaga K., and Uchiyama T. (2003) The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 278, 44412–44416 [DOI] [PubMed] [Google Scholar]
- 39. Haché G., Shindo K., Albin J. S., and Harris R. S. (2008) Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr. Biol. 18, 819–824 [DOI] [PMC free article] [PubMed] [Google Scholar]