

Abstract
Lamprey angiotensinogen (l-ANT) is a hormone carrier in the regulation of blood pressure, but it is also a heparin-dependent thrombin inhibitor in lamprey blood coagulation system. The detailed mechanisms on how angiotensin is carried by l-ANT and how heparin binds l-ANT and mediates thrombin inhibition are unclear. Here we have solved the crystal structure of cleaved l-ANT at 2.7 Å resolution and characterized its properties in heparin binding and protease inhibition. The structure reveals that l-ANT has a conserved serpin fold with a labile N-terminal angiotensin peptide and undergoes a typical stressed-to-relaxed conformational change when the reactive center loop is cleaved. Heparin binds l-ANT tightly with a dissociation constant of ∼10 nm involving ∼8 monosaccharides and ∼6 ionic interactions. The heparin binding site is located in an extensive positively charged surface area around helix D involving residues Lys-148, Lys-151, Arg-155, and Arg-380. Although l-ANT by itself is a poor thrombin inhibitor with a second order rate constant of 500 m−1 s−1, its interaction with thrombin is accelerated 90-fold by high molecular weight heparin following a bell-shaped dose-dependent curve. Short heparin chains of 6–20 monosaccharide units are insufficient to promote thrombin inhibition. Furthermore, an l-ANT mutant with the P1 Ile mutated to Arg inhibits thrombin nearly 1500-fold faster than the wild type, which is further accelerated by high molecular weight heparin. Taken together, these results suggest that heparin binds l-ANT at a conserved heparin binding site around helix D and promotes the interaction between l-ANT and thrombin through a template mechanism conserved in vertebrates.
Keywords: crystal structure, heparin, hypertension, serpin, thrombin, angiotensinogen
Introduction
Angiotensinogen genes exist throughout all vertebrates and have evolved over 500 million years from early cyclostome (lamprey) to human (1–3). Angiotensinogen functions as the carrier of active angiotensin hormones of the renin-angiotensin system that regulates body fluid homeostasis, blood pressure, and blood vessel formation (4, 5).
Early studies have shown that angiotensinogen with the angiotensin peptide located at its N terminus belongs to the serine protease inhibitor (serpin) superfamily (6, 7). All serpins are known to share similar tertiary structures with three β-sheets, 8–9 α-helices, and an exposed reactive center loop (RCL)4 that serves as a bait for proteolytic attack (8, 9). Once the target protease recognizes and cleaves RCL, the serpin undergoes a typical serpin stressed-to-relaxed (S-to-R) transition with the insertion of the cleaved RCL into the central β-sheet as a middle strand, which leads to translocation and inactivation of the covalently linked protease (10, 11).
Classically, angiotensinogen is regarded as a passive hormone carrier and a non-inhibitory serpin that lacks serpin S-to-R conformational rearrangements upon RCL cleavage (12, 13); however, recent studies have found that angiotensinogen derived from lamprey, a representative of an early lineage of vertebrate, to be an inhibitor of thrombin, which is unknown for other orthologues (14, 15). Thrombin is the central enzyme of blood coagulation that has various substrates and cofactors. Thrombin inhibition by serpins is crucial in regulation of blood coagulation, and its control requires several serpins in varying physiological settings in mammals (16). Antithrombin is the principal inhibitor of thrombin during thrombosis and the normal hemostatic response (17). Heparin cofactor II (HCII) is also an important inhibitor of thrombin in the arterial vasculature (18, 19). Lampreys possess a simpler coagulation system with lesser components and less than 10 serpins (20, 21). The gene of antithrombin has not been found in the lampreys (Lampetra fluviatilis, Petromyzon marinus, and Lethenteron japonicum) (14, 22), but genes for angiotensinogen and HCII have been cloned and shown to play key roles in thrombin inhibition in lamprey (15). Notably, lamprey angiotensinogen (l-ANT) has an Ile as the P1 residue instead of a thrombin-preferred Arg.
The interactions between serpins and proteases of blood coagulation are often regulated by cofactors such as heparin (23). Heparin binds residues of helix D or helix H of serpins, such as antithrombin (10, 11, 24–26), HCII (27–29), protease nexin 1 (PN1) (30, 31), plasminogen activator inhibitor 1 (PAI-1) (32), protein Z-dependent protease inhibitor (ZPI) (33, 34), and protein C inhibitor (PCI) (35, 36), and promotes the interaction between serpin and protease either by an allosteric mechanism where heparin induces conformational changes in serpin or by a template mechanism where heparin bridges serpin and protease together (8, 37). The reactions between l-ANT and thrombin are also accelerated by heparin (14) and heparan sulfate (15), but it is unclear how heparin binds and activates l-ANT. Here we have solved the crystal structure of RCL-cleaved l-ANT and characterized its properties in heparin binding and protease inhibition. This study provides information on how the angiotensin system and the blood coagulation system overlapped and diverged during evolution.
Experimental Procedures
Materials
The angiotensinogen cDNA of L. fluviatilis was amplified from a cDNA library as described previously (14, 22). Proteinases, including human α-thrombin (factor IIa), factor IXa, factor Xa, factor XIa, kallikrein-related peptidase 7 (KLK7), kallikrein-related peptidase 1 (KLK1), trypsin, and activated protein C were purchased from Hematologic Technologies. Low molecular weight (LMW) heparin (average molecular weight of 5000) and high molecular weight (HMW) heparin (average molecular weight of 17,000–19,000) were purchased from Sigma. Heparin oligosaccharides of 6, 8, 12, 16, and 20 monosaccharide units (DP6, DP8, DP12, DP16, and DP20) were from Iduron (Manchester, UK). High affinity pentasaccharide (H5*) containing an extra sulfate group was a gift from Dr. Maurice Petitou (Sanofi Recherche-Centre Choay, Gentilly, France) (38). All columns for protein purification were purchased from GE Healthcare. All reagents and kits for crystallization were purchased from Hampton Research.
Cloning, Mutagenesis, Expression, and Purification of Recombinant Proteins
The angiotensinogen cDNA was cloned in expression vector pE-SUMO3 as described previously (39). All mutants were constructed by PCR mutagenesis using the KOD Plus mutagenesis kit (TYOBO). The mutant AAR of l-ANT was used for crystallization, where the P1 Ile was substituted with Arg and 2 Ala residues were added between positions P2 and P3 (Fig. 1A). Plasmids were transformed into BL21 (DE3) cells and grown in 2× TY at 37 °C until optical density at 600 nm reached 0.8. Then isopropyl-β-d- thiogalactopyranoside was added to a final concentration of 0.2 mm, and the culture was transferred to a 25 °C shaker for a further 12 h. The cells were collected by centrifugation and resuspended in ice-cold buffer A (20 mm Tris-HCl, pH 7.4, 0.3 m NaCl, and 20 mm imidazole) and disrupted by a high pressure cell breaker. The supernatant of the cell lysate was loaded onto a 5-ml HiTrap HisTrap FF crude column, and eluted by a 0.02–0.2 m imidazole gradient. The peak fractions were collected. Then protein was digested by protease SENP2 to release the SUMO tag and dialyzed against buffer B (20 mm Tris-HCl, pH 7.4, 0.3 m NaCl). Subsequently, proteins were loaded onto a 5-ml Hitrap heparin column and eluted by a NaCl gradient (0.3–1.5 m) with l-ANT eluted at about 0.8 m NaCl. The peak fractions were collected and dialyzed against buffer C (20 mm Tris-HCl, pH 7.4, 0.15 m NaCl). Proteins for crystallization were further purified by a Superose 12 gel filtration column.
FIGURE 1.
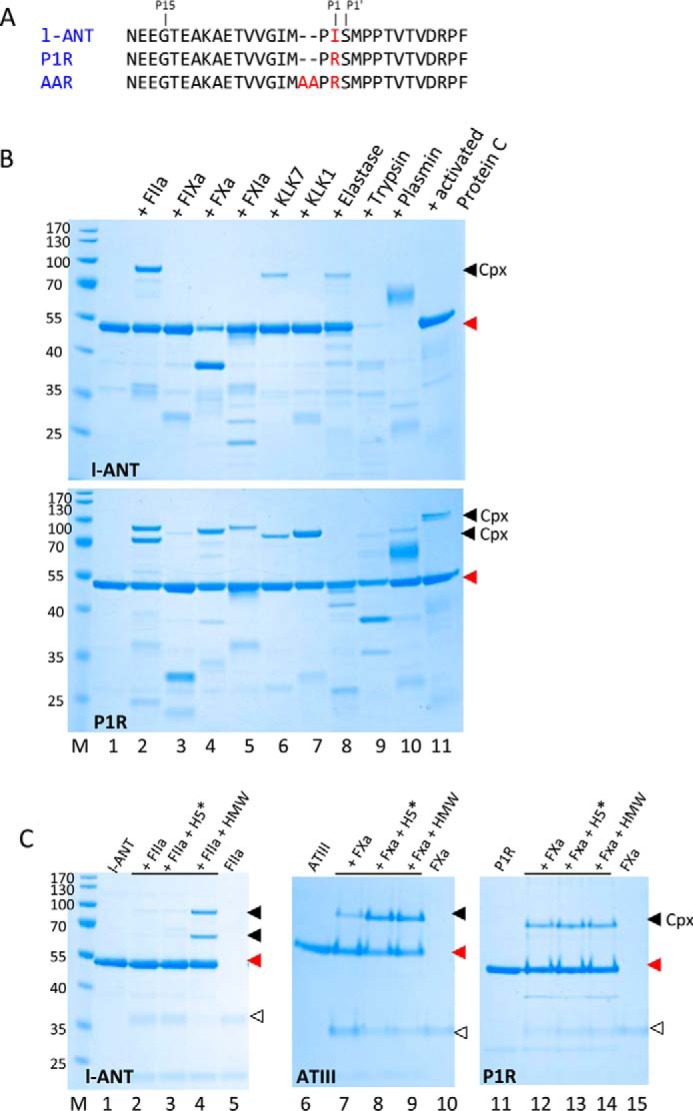
Inhibition of proteases by l-ANT variants. A, RCL sequence of l-ANT variants. B, protease inhibition by wild type l-ANT (top) or P1R (bottom). 2 μg of l-ANT or P1R were incubated with 0.5 μg of serine protease for 15 min at room temperature in 20 μl of PBS. Lane M, molecular weight marker; lane 1, l-ANT variants; lanes 2–11, l-ANT or P1R interacts with human α-thrombin (factor IIa), factor IXa, factor Xa, factor XIa, KLK7, KLK1, elastase, trypsin, plasmin, and activated protein C, respectively. C, effect of heparin on protease inhibition by l-ANT or antithrombin (ATIII). 0.5 μg of protease was incubated with 2 μg of l-ANT or P1R variant or antithrombin for 5 min in the absence or presence of HMW heparin or H5*. All samples were analyzed by reducing SDS-PAGE and stained by Coomassie Blue. The filled triangles show the positions of serpin·protease complexes (cpx), red triangles show the positions of native serpins, and open triangles show proteases.
Complex Formation Assay for Protease Inhibitor Function
Protease inhibitory function was checked by incubating 2 μg of l-ANT with 0.5 μg of serine protease for 15 min at room temperature in 20 μl of PBS. Ten proteases were selected, including human α-thrombin, factor IXa, factor Xa, factor XIa, KLK7, KLK1, elastase, trypsin, plasmin, and activated protein C. The same procedure was applied to variant l-ANT P1R variant, where the P1 residue Ile was mutated to Arg (Fig. 1A). The effect of heparin on protease inhibition by l-ANT was further checked by incubating 2 μg of l-ANT or P1R with 0.5 μg of proteases in the absence or presence of pentasaccharide/HMW heparin for 2 or 5 min at room temperature in 20 μl of PBS.
Crystallization, Data Collection, and Structure Determination
The l-ANT AAR variant was cleaved by human α-thrombin at a 2000:1 ratio (w/w) for 2 h at 4 °C and concentrated to 10 mg/ml. Crystallization was set up at 22 °C by mixing equal volumes of protein and precipitation buffer (25% PEG 4000, 0.1 m sodium acetate, pH 4.6, 0.2 m ammonium sulfate) by the sitting drop method. Crystals appeared after 20 days. For data collection, a single crystal was soaked in 27% PEG 3350, 0.1 m sodium acetate, pH 4.6, 0.2 m ammonium sulfate, and 20% glycerol and rapidly frozen in liquid nitrogen. The diffraction data were collected at SSRF BL17U and scaled using Imosfilm (40). The structure was solved by molecular replacement with Phaser (41) in the CCP4 suite using the structure of cleaved antitrypsin (PDB code 3NDD) as the initial search model. The structure was refined with Refmac (42), and model building was done with Wincoot (43). The surface electrostatic representation was analyzed by APBS (Adaptive Poisson-Boltzmann Solver) (44). The figures were created using the open source program PyMOL.
Heparin Affinity Chromatography of l-ANT Variants
The relative heparin affinities of recombinant l-ANT variants were determined by a 1-ml HiTrap heparin column. The proteins were loaded onto the column pre-equilibrated with buffer B and washed with 5 ml of buffer B followed by elution with a 20-column volume of 0.3-1.5 m NaCl gradient using the AKTA system (GE Healthcare).
Dissociation Constants of Heparin Binding to l-ANT
Fluorescence titration experiments were performed using an LS55 luminescence spectrometer (PerkinElmer Life Sciences). Because tryptophan fluorescence changes of l-ANT were not obvious when heparin was added, we followed the fluorescence change of 2-p-toluidinylnaphthalene-6-sulfonate (TNS) upon the addition of l-ANT and subsequently heparin as described previously for antithrombin, HCII, and PCI (18, 45, 46, 48). TNS binding to l-ANT resulted in a significant fluorescence increase, and heparin additions caused a 40–70% fluorescence quench with a 3–4-nm blue shift of the emission maximum. Cleaved l-ANT had a significantly smaller effect on fluorescence change of TNS and a smaller fluorescence decrease upon titration with heparin. TNS fluorescence was insensitive to the heparin additions in the absence of protein. Fluorescence of TNS was collected at emission wavelength of 430 nm with an excitation wavelength of 340 nm during l-ANT titration. Dissociation constants were calculated by Equation 1 (17, 18, 49).
| (Eq. 1) |
In this equation, ΔF represents the fluorescence change following each addition of heparin, and ΔFmax represents the maximal change of fluorescence intensity. [P]0 and [H]0 are total protein and heparin concentrations, and Kd is the dissociation constant. We confirmed that the dissociation constants were independent of the concentrations of TNS and proteins. All titrations were performed in solutions containing 5–10 μm TNS in 50 mm Tris-HCl, pH 7.4, 20% glycerol, 0.1% PEG 8000, and the ionic strengths were adjusted by adding NaCl. Variant proteins were used at 0.2–0.3 μm for experiments with LMW heparin (ionic strength 0.3 m NaCl) and 0.05–0.1 μm proteins for experiments with DP8 (ionic strength 0.15 m NaCl). To determine the effect of heparin length and ionic strength on binding affinity, 0.05–0.1 μm wild type proteins were used in combination with heparin oligosaccharides of 6, 8, 12, 16, and 20 monosaccharide units in length (DP6, DP8, DP12, DP16, and DP20) at different ionic strengths ranging from 0.1 to 0.35 m NaCl. The effect of heparin length was determined by a linear fit using Equation 2 (18, 50).
| (Eq. 2) |
In this equation, Kd is the dissociation constant obtained from Equation 1 for N-monosaccharides heparin. L is the minimal heparin size fully occupying the binding site of l-ANT, and Kd, int is its intrinsic dissociation constant. The influence of ionic strength was decided by a linear fit using Equation 3 (18, 50, 51).
| (Eq. 3) |
In this equation, Kd is the dissociation constant obtained from Equation 1. KNI is the dissociation constant for the nonionic interaction at 1 m Na+. Z is the number of ionic interactions. Ψ is the fraction of a counterion that is bound to the polyelectrolyte per ionic charge and that is released upon the binding of the protein. The ionic strength-independent constant of Ψ calculated for heparin from its axial charge density is ∼0.8 according to previous publications (51). All of the Kd measurements were repeated 2–4 times, and the means ± S.D. are shown in Tables 2 and 3.
TABLE 2.
Binding of l-ANT variants on heparin column and dissociation constants measured by TNS fluorescence titration
| Protein | Heparin column affinitya |
Dissociation constantb |
||||
|---|---|---|---|---|---|---|
| [NaCl] in peaks | [NaCl]WT − [NaCl]variant | DP8 |
LMW |
|||
| Kd | Kd/Kd,wt | Kd | Kd/Kd,wt | |||
| mm | mm | nm | μm | |||
| WT | 820 | 0 | 34 ± 1 | 1 | 0.17 ± 0.01 | 1 |
| Cleaved | 820 | 0 | 28 ± 1 | 0.80 | 0.18 ± 0.03 | 1.05 |
| K76A | 786 | 34 | 66 ± 2 | 1.93 | 0.21 ± 0.01 | 1.22 |
| R79A | 768 | 52 | 48 ± 04 | 1.38 | 0.20 ± 0.01 | 1.15 |
| K144A | 782 | 38 | 49 ± 3 | 1.44 | 0.24 ± 0.01 | 1.38 |
| K145A | 791 | 29 | 53 ± 3 | 1.54 | 0.24 ± 0.01 | 1.36 |
| K146A | 777 | 43 | 56 ± 6 | 1.63 | 0.28 ± 0.01 | 1.63 |
| K148A | 718 | 102 | 87 ± 1 | 2.54 | 0.44 ± 0.02 | 2.54 |
| K151A | 716 | 104 | 96 ± 5 | 2.80 | 0.52 ± 0.01 | 2.99 |
| R155A | 728 | 92 | 92 ± 1 | 2.67 | 0.31 ± 0.02 | 1.81 |
| K162A | 774 | 46 | 44 ± 1 | 1.28 | 0.17 ± 0.01 | 0.98 |
| R163A | 788 | 32 | 57 ± 1 | 1.62 | 0.18 ± 0.01 | 1.01 |
| R380A | 800 | 20 | 72 ± 2 | 2.10 | 0.33 ± 0.01 | 1.88 |
| K438A | 785 | 35 | 57 ± 1 | 1.66 | 0.20 ± 0.02 | 1.16 |
| R440A | 784 | 36 | 61 ± 7 | 1.79 | 0.16 ± 0.01 | 0.94 |
| K158A/K151A/R155A | 615 | 205 | 105 ± 3 | 3.04 | 1.62 ± 0.08 | 8.89 |
a l-ANT variants were loaded onto a 1-ml heparin column and eluted with a 0.3–1.5 m NaCl gradient. The NaCl concentrations ([NaCl]) of the elution peaks are shown on the left.
b The dissociation constant Kd values of l-ANT variants were determined at 0.15 m NaCl for DP8 and at 0.3 m NaCl for LMW heparin in the presence of 5–10 μm TNS in 50 mm Tris-HCl, pH 7.4, 20% glycerol, 0.1% polyethylene glycol 8000.
TABLE 3.
The effect of heparin length and ionic strength on heparin binding affinity of l-ANT
The effect of heparin length was determined by a linear fitting using the Equation 2, and the influence of ionic strength was decided by a linear fitting using Equation 3 (see Experimental Procedures). −, not determined.

Stoichiometry of Inhibition
The stoichiometry of inhibition (SI) was measured by incubating 0.25 μm thrombin with increasing concentrations of l-ANT (0–1 μm) without or with heparin. Reactions were incubated for 1–2 h with l-ANT P1R variant and for 12–24 h with wild type l-ANT in PBS with 0.1% PEG 8000 at room temperature. Residual protease activity was determined by diluting the reaction mixture into 0.1 mm chromogenic substrate S-2238 (Chromogenix) and measuring the rate of substrate hydrolysis, as described previously (17, 45, 46).
Effect of Heparin on the Rates of Thrombin Inhibition by l-ANT
The rates of thrombin inhibition by l-ANT were determined under pseudo-first-order conditions (17). For the heparin-catalyzed reaction, 10−5 to 10 μg/μl LMW or HMW heparin or 0.005–25 μm heparin oligosaccharides (8–20 monosaccharide units) were used. Thrombin (10 μl of 10 nm) was mixed with 10 μl of 2 μm l-ANT in PBS with 0.1 mg/ml BSA and 0.1% PEG 8000. For the P1R variant, 0.1 μm proteins were used. The residual protease activity at timed intervals was determined by diluting the reaction mixture into the assay buffer containing 0.1 mm S-2238. The observed rate constant, kobs, was obtained from the slope of a semi-log plot of the residual protease activity against time. The apparent second order rate constant, kapp, was calculated by linear fitting of kobs versus the initial inhibitor concentration (0.4–1.2 μm wild type, 0.03–0.06 μm P1R) without or with LMW or HMW heparin at an optimal concentration (17, 18, 45).
Results
Expression and Characterization of l-ANT
l-ANT was expressed in Escherichia coli using the SUMO fusion expression system and purified by nickel and heparin affinity columns. To test the effect of the P1 residue on the specificity and activity of l-ANT, we also prepared variant P1R, where the P1 Ile was substituted with Arg (Fig. 1A). Activities of l-ANT and P1R were analyzed by incubating these proteins with 10 serine proteases, including α-thrombin, factor IXa, factor Xa, factor XIa, KLK7, KLK1, elastase, trypsin, plasmin, and activated protein C. As shown in Fig. 1B, l-ANT could form a covalent SDS-stable complex with thrombin, KLK7, and elastase (Fig. 1B, top gel). However, P1R appeared to form complexes with almost all of the proteases tested here apart from elastase (Fig. 1B, bottom gel). Therefore, l-ANT was a relatively specific inhibitor toward thrombin but became less specific once its P1 residue was replaced by an Arg. The interaction between l-ANT and thrombin was enhanced by HMW heparin, with significantly more l-ANT·thrombin complexes formed (Fig. 2C, lane 4); however, the well characterized H5* that promotes factor Xa inhibition by antithrombin (lane 8) had no effect on the inhibition of thrombin by l-ANT (lane 3). l-ANT did not inhibit factor Xa, but the P1R variant did form a complex with factor Xa. Both HMW heparin and H5* had little effect on the activity of P1R variant toward factor Xa (lanes 13 and 14). Overall, these data confirm that l-ANT is a relatively specific thrombin inhibitor, and its activity is enhanced by HMW heparin but not heparin pentasaccharide.
FIGURE 2.

Overall structure of cleaved l-ANT. A, the structure of cleaved l-ANT exhibits a very typical serpin fold. The RCL (yellow) cleaved by human α-thrombin is completely inserted into central β-sheet A (red) as a middle strand. A seventh chain of β-sheet A (termed s1′A here) is seen above helix D (cyan), composed of residues 162–164. The N-terminal residues 1–72 and residues 138–147 between helix C and helix D (indicated with an orange dashed line) are not built into the structure because of poor electron density. Helix A is shown in green. B, overlaid structures of l-ANT and latent antithrombin (AT) (orange, PDB code 1AZX) indicate a large shift in helix F position and a different conformation of the connecting loop (blue) on top of helix D in antithrombin. C, overlaid structures of l-ANT (gray) and human angiotensinogen (h-ANT) (green, PDB code 2WXW). The N-terminal fragment of human angiotensinogen is shown in blue, and the hormone peptide is shown in red. The Cys-18–Cys-138 disulfide bond of human angiotensinogen is shown as spheres.
The Overall Structure of Cleaved l-ANT
To further characterize l-ANT, we tried to determine the crystal structure of native l-ANT; however, crystals of native l-ANT were small and very difficult to optimize. We then engineered an l-ANT mutant AAR, where the RCL was extended by 2 Ala residues and the P1 residue was mutated to Arg (Fig. 1A). This variant becomes a substrate of thrombin due to the extended RCL (52). The purified RCL variant was cleaved and prepared for crystallization. The crystal structure of cleaved l-ANT was determined at 2.7 Å resolution (Table 1). There are two copies of l-ANT in the asymmetric unit. The overall folds of the two molecules are almost the same, with a root mean square deviation (RMSD) of α carbon atoms of 0.18 Å. Molecule A was chosen for subsequent interpretation because it was more complete with clear electron density for 366 of the total 453 residues. Overall, l-ANT showed very typical cleaved serpin folds, with the cleaved RCL completely inserted into β-sheet A as a middle strand (Fig. 2A). When it is compared with other relaxed serpin structures, such as latent antithrombin (PDB code 1AZX) (Fig. 2B), cleaved α-antitrypsin (PDB code 3NDD), or cleaved PCI (PDB code 1LQ8), the RMSDs of atomic positions are between 1.4 and 1.7 Å although the sequence identity is about 33% with each of these three. Interestingly, in the l-ANT structure here, the connecting loop on top of helix D forms a short extra strand in β-sheet A (termed s1′A), and there is a large shift in position of helix F when compared with latent antithrombin (Fig. 2B). When the structure of l-ANT is overlaid with that of human angiotensinogen (PDB code 2WXW) (Fig. 2C), the RMSD of atomic positions is 2.7 Å. In crystal structures of human, mouse, and rat angiotensinogen, the angiotensin peptides are anchored by a disulfide bond and partially buried within the body of angiotensinogen (Fig. 2C); however, the N-terminal angiotensin sequence of l-ANT was not visible in the structure, indicating high flexibility of this part of the molecule.
TABLE 1.
Data collection and refinement statistics
| Data collection | |
| Beamline | SSRF 17U |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 42.40, 119.36, 141.59 |
| α, β, γ (degrees) | 90, 90, 90 |
| Wavelength (Å) | 0.97923 |
| Resolution (Å) | 91.26–2.70 (2.83–2.70)a |
| Total no. of observations | 127,579 (15,093) |
| Total no. of unique observations | 18,674 (2271) |
| Rmerge (%) | 0.095 (1.065) |
| I/σI | 12.3 (1.7) |
| Completeness (%) | 90.9 (84.6) |
| Multiplicity | 6.3 (6.6) |
| Refinement | |
| Resolution (Å) | 91.26–2.70 (2.77–2.70) |
| No. of reflections | 17,694 (1197) |
| No. of residues | 732 |
| No. of atoms | 5704 |
| Rwork/Rfree | 0.223/0.284 |
| B-factors (Å2) | 62.3 |
| RMSD | |
| Bond lengths (Å) | 0.0088 |
| Bond angles (degrees) | 1.3057 |
| Ramachandran plot | 97.77/0 |
a Values in parentheses are for the highest resolution shell.
Heparin Binding Sites
Surface electrostatic representation analysis of l-ANT shows an extensive positively charged area along helix D and on the edge of β-sheet B (Fig. 3A). Because numerous biochemical and structural studies have shown that helix D is often a key area in serpins for heparin binding (10, 11, 24–34), we systematically compared this surface area of all of the known heparin binding serpins, such as antithrombin (PDB code 1AZX), HCII (PDB code 1JMJ), ZPI (PDB code 3FIS), PN1 (PDB code 4DYO), PAI-1 (PDB code 1B3K), PCI (PDB code 2OL2), and C1 inhibitor (PDB code 2OAY) (supplemental Fig. 1). It appears that l-ANT has the largest positive charged surface around this region among all of the serpins. These positively charged residues in l-ANT include Lys-76 and Arg-79 of helix A; Lys-148, Lys-151, and Arg-155 in helix D; Lys-162 and Lys-163 on the s1′A chain; Arg-380 on the connecting loop between helix J and s5A; and Lys-438 and Arg-440 on the edge of β-sheet B (Fig. 3C). Residues Lys-144, Lys-145, and Lys-146 of the CD loop, invisible in the structure, are also located in this area. Antithrombin is the most well studied heparin binding serpin with several heparin·antithrombin complex structures solved (10, 11, 24). These residues of l-ANT spatially resemble those of antithrombin (Fig. 3D), with residues 155, 162, and 163 of l-ANT corresponding to 129, 132, and 133 of antithrombin. Also, sequence alignment of this region (from helix C to s2A) of various heparin binding serpins shows that l-ANT has 9 positively charged residues in this region with 8 for PN1 and 7 for antithrombin (Fig. 3E).
FIGURE 3.
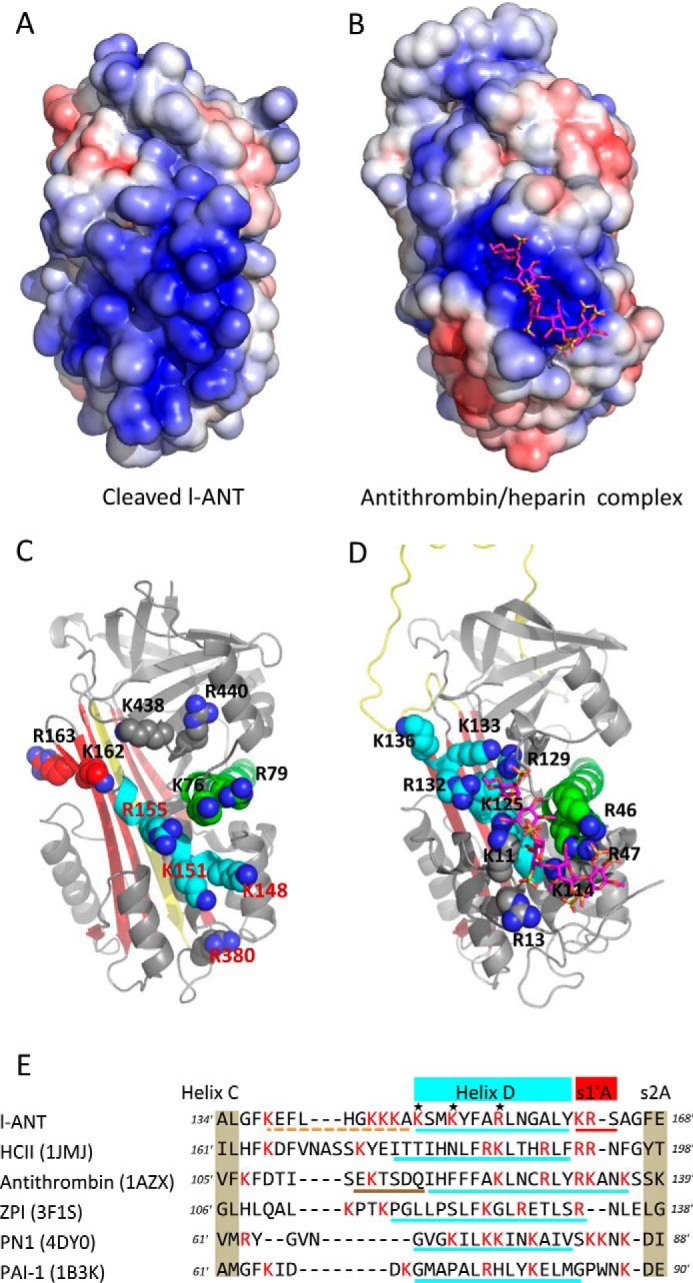
The heparin binding site near helix D. Surface electrostatic analysis shows that l-ANT (A) has a larger positively charged area than that seen in the native antithrombin·heparin complex structure (B) (PDB code 1AZX). All of the positively charged residues in this region are shown as spheres in C (l-ANT) and D (antithrombin), with these two structures shown as schematics with the same orientation. The RCL is colored in yellow, β-sheet A in red, helix A in green, and helix D in cyan. Heparin is shown as magenta sticks. E, sequence alignment of residues between helix C and strand 2A based on the structures of heparin-binding serpins (HCII, antithrombin, ZPI, PN1, and PAI-1). All of the Lys and Arg residues are colored in red. The residues of helix D are underlined in cyan with the CD loop of l-ANT indicated by an orange dashed line and helix P of antithrombin indicated by a brown dashed line.
To verify the role of these residues in heparin binding of l-ANT, we mutated these residues individually to Ala and then assessed the effect on heparin binding. Activity assays showed that all mutants formed SDS-stable complexes with thrombin, the same as the wild type (data not shown). The relative heparin binding affinities were determined by NaCl gradient elution from a heparin column (Fig. 4A). They displayed different elution profiles. First, the wild type l-ANT was eluted at about 820 mm NaCl, similar to that of α-antithrombin, indicating tight binding (53). Three variants (K148A, K151A, and R155A) were eluted at about 720 mm NaCl, significantly lower than the wild type. Other variants, including K76A, R79A, K144A, K145A, K146A, K162A, R163A, R380A, K438A, and R440A, were eluted at about 780 mm NaCl, slightly lower than the wild type (Table 2). When the 3 residues in helix D were mutated to Ala simultaneously, the variant K148A/K151A/R155A (termed AAA) was eluted with the lowest NaCl concentration of 615 mm.
FIGURE 4.

Identification of heparin binding site. A, heparin column affinity of l-ANT variants. l-ANT variants were loaded onto a 1-ml heparin column and eluted with a 20-column volume (x axis) with a gradient of 0.3–1.5 m NaCl (right y axis, brown dashed line). The left y axis shows the absorption value at 280-nm wavelength. Wild type l-ANT (black line) is eluted at 820 mm NaCl. Variants K144A (red line) and K151A (blue line) are eluted at about 780 mm NaCl and 716 mm NaCl, respectively. The variant K148A/K151A/R155A (termed AAA, green line) is eluted at about 615 mm NaCl. The exact values of NaCl concentrations in elution peaks are listed in Table 2. B, fluorescence spectrum of TNS. The fluorescence spectrum of 5 μm TNS alone is given as a black line. The TNS fluorescence intensity increases with the addition of 0.3 μm l-ANT (red line) and decreases in response to the addition of 0.1 μm LMW heparin (cyan line). C, dissociation constants are non-linearly fitted by Equation 1 as described under “Experimental Procedures.” All titrations were performed in solutions containing 5–10 μm TNS in 50 mm Tris-HCl, pH 7.4, 20% glycerol, 0.1% PEG 8000. The ionic strengths were adjusted by adding NaCl. Filled circles, wild type l-ANT; open circles, K151A. D and E, column diagram of the relative dissociation constant values (Kd/Kd,wt) of the variants binding to LMW heparin (D) or 8-monosaccharide heparin (DP8) (E). The exact values are listed in Table 2.
To quantify the binding affinity of l-ANT toward heparin, we established a method for dissociation constant measurement through fluorescence titration. Although l-ANT contains 5 tryptophan residues at positions 30, 182, 251, 266, and 333, the addition of heparin does not induce any fluorescence change of these tryptophan residues, so we used TNS as a fluorescence probe as described previously for antithrombin, HCII, and PCI (18, 45, 46, 48). l-ANT binding of TNS induces a TNS fluorescence increase at 430 nm, whereas the subsequent addition of heparin causes a 40–70% fluorescence quench with a 3–4-nm blue shift (Fig. 4B). The Kd values of these l-ANT variants were determined by non-linear fitting using Equation 1 described under “Experimental Procedures” (Fig. 4C). The measurements show that l-ANT binds LMW heparin very tightly, with a Kd of ∼10 nm at an ionic strength of 0.15 m NaCl. To allow accurate and easy measurements, we subsequently measured the affinity of l-ANT variants toward LMW heparin in buffer with 0.3 m NaCl. We also measured the affinity of l-ANT toward DP8 at an ionic strength of 0.15 m NaCl with detailed data shown in Table 2. Comparative values were plotted as a column diagram (Fig. 4, D and E). Cleaved l-ANT has a Kd of 28 nm for DP8 and 180 nm for LMW heparin, which are similar to those of wild type of 34 nm for DP8 and 170 nm for LMW heparin. This implies that the S-to-R transition does not influence l-ANT heparin binding affinity. The variants K148A and K151A have 2.5–3-fold reduction in binding affinity for both DP8 and LMW heparin. R155A has a 2.7-fold reduction for DP8 binding and 1.8-fold reduction for LMW heparin binding. K144A, K145A, and K146A also result in 1.4–1.6-fold reduction in affinity toward both heparins. Mutation of Arg-380 induces a 2-fold reduction in affinity toward both heparins. Moreover, the variant K148A/K151A/R155A exhibited remarkable reduced affinity to heparin, with Kd for LMW heparin binding decreased by 8.9-fold and Kd for DP8 binding decreased by 3-fold. Other variants only exhibited a slightly increased dissociation constant. Overall, these data are largely consistent with the results from heparin columns, indicating that residues from helix D (Lys-148, Lys-151, Arg-155) and residue Arg-380 contribute most to heparin binding. The lesser effect of this triple mutation on DP8 binding might reflect that the short oligosaccharide chain could bind l-ANT in an alternative orientation, as observed previously with PCI (36), whereas long oligosaccharide chain might have less freedom due to its bulkiness.
Heparin Binding Properties
To check the effect of heparin length and ionic strength on binding affinity, dissociation constants of l-ANT binding to DP6, DP8, DP12, DP16, or DP20 were measured at several NaCl concentrations (Table 3). The effect of heparin length using the linear fitting of the association constant (Ka = 1/Kd) on heparin length (N) according to Equation 2 was assessed at 0.2, 0.25, and 0.3 m NaCl, respectively (Fig. 5A), with the corresponding x intercepts of 5.9, 6.8, and 6.2. The minimal heparin binding sizes (L) are given by the x intercept + 1 (Table 3). Therefore, the minimal heparin length to fully occupy the heparin binding site of l-ANT is about 8 monosaccharide units. The calculated result is consistent with the experimental findings that Kd for DP8 is much lower than Kd for DP6 at the same ionic strength (>10 times lower at 0.15 m and 0.2 m NaCl) and is much closer to that of DP12, due to full occupancy. Furthermore, 8 monosaccharides appear to fit well with the height of the positively charged region near helix D of l-ANT (supplemental Fig. 1). The slope of the linear fitting (Fig. 5A) also gives the values of the intrinsic dissociation constant (Kd, int) for the minimal heparin chain at these three ionic strengths: 0.22, 0.77, and 1.6 μm for 0.2, 0.25, and 0.3 m NaCl, respectively (Table 3).
FIGURE 5.

Heparin-binding properties of l-ANT. A, effect of chain length on heparin affinity of l-ANT was determined by a linear fit using Equation 1/Kd = 1/Kd, int × (N - L + 1). The x intercept of the linear plot gives the minimal heparin length L (x intercept + 1) at NaCl concentrations of 0.2 m (filled triangles), 0.25 m (open circles), and 0.3 m (filled squares), respectively. B, the effect of ionic strength on heparin affinity of l-ANT was determined by a linear fitting using the equation, log Kd = log KNI + ZΨlog [Na+]. The ionic interaction (Z) was calculated from the slope for DP6 (filled circles), DP8 (open circles), DP12 (filled triangles), DP16 (open triangles), and DP20 (filled squares). The apparent strength of the nonionic interactions (KNI) was determined from the y intercept. The detailed values are listed in Table 3.
The dependence of heparin binding on ionic strength was derived from the plot of log Kd against log [Na+]. The slope of this plot when linearly fitted with Equation 3 gives the product of numbers of ionic interactions (Z) and Ψ, and the y intercept gives the nonionic dissociation constant (KNI). When the binding affinities of DP6, DP8, DP12, DP16, and DP20 at several NaCl concentrations were plotted (Fig. 5B), the slopes of these lines for DP6, DP8, DP12, DP16, and DP20 were 3.8, 4.5, 5.0, 4.8, and 4.5, respectively. The calculated Z values are 4.7, 5.7, 6.3, 6.0, and 5.6, correspondingly (Table 3), indicating that ∼5 ionic interactions contribute to DP6 binding to l-ANT and ∼6 ionic interactions contribute to 8–20-monosaccharide heparin. The fitting curves also determine the nonionic dissociation constants (KNI) of 0.89, 0.20, 0.13, 0.069, and 0.026 mm for DP6, DP8, DP12, DP16, and DP20, respectively (Table 3). These values are several thousand-fold greater than the observed dissociation constants of l-ANT (∼10 nm), illustrating that the heparin binding affinity mainly depends on ionic interactions.
The affinity measurements of the H5* toward l-ANT show that H5* binds l-ANT very tightly, with a Kd of 37 nm at 0.15 m NaCl, which is close to that of DP8 and about 14-fold smaller than that of DP6 under the same ionic strength. This increased binding affinity of H5* is probably attributable to its ability to form more ionic interactions with l-ANT than DP6 because H5* is highly sulfated.
Overall, affinity measurements show that l-ANT binds heparin tightly, with a Kd value of ∼10 nm involving ∼8 monosaccharides and ∼6 ionic interactions. In comparison, antithrombin binds heparin with a Kd of a few nanomolar and 4–5 ionic bonds (17). HC II binds heparin with Kd of 26 μm, involving 13 monosaccharides and 4–5 ionic interactions (18).
Effect of Heparin on the Rate of Thrombin Inhibition by l-ANT
Here we measured the second order association constants (k2) and the SI of the interactions between l-ANT and thrombin in the absence and presence of heparin by a discontinued method (Table 4). The SI measurement indicates that about 1.5 molecules of l-ANT are required to inhibit one thrombin molecule. In the presence of HMW heparin, about 2.5 molecules of l-ANT are required to inhibit one thrombin molecule. The apparent second rate constant (kapp) was determined from the slope of the plot of the apparent first order rate (kobs) versus inhibitor concentration (Fig. 6A). The k2 of the reaction between thrombin and l-ANT, given by the product of kapp × SI, is 500 m−1 s−1, which is comparable with the previous measurement by Wang and Ragg (14). To test the effect of heparin on the reaction between l-ANT and thrombin, kobs was measured at a fixed concentration of l-ANT in the presence of LMW or HMW heparin ranging from 10−5 to 10 μg/μl. HMW heparin accelerated the reaction with a bell-shaped dose-response curve (Fig. 6B) with a maximum k2 of 4.5 × 104 m−1 s−1 (Table 4), which is 90-fold faster. Rates of thrombin inhibition by l-ANT were also measured in the presence of short heparin chains of 8–20 monosaccharide units of heparin, but even 25 μm 20-monosaccharides had little effect on thrombin inhibition. Although LMW heparin (average molecular weight of 5000) also marginally accelerated thrombin inhibition by l-ANT in a similar bell-shaped curve (Fig. 6B), we suspect that this is due to the presence of a small amount of long chain heparin. Wang and Ragg (14) reported that heparin could accelerate the reaction between l-ANT and thrombin by nearly 810-fold instead of the 90-fold measured here. The difference may reflect the difference in heparin used and/or the different production systems of the recombinant serpin. Nevertheless, our results are consistent with their findings that heparin mediates thrombin inhibition following a bell-shaped dose-dependent curve.
TABLE 4.
Second order rate constants for the interactions between l-ANT and thrombin in the absence and presence of optimal concentration of heparin
The apparent second order rate constants (kapp) were determined from the slope of the linear plot of kobs versus inhibitor concentration. The product of kapp × SI represents the second order rate constant (k2) corrected for SI values.
| l-ANT variants | Rate of thrombin inhibition |
Rate of thrombin inhibition + LMW |
Rate of thrombin inhibition + HMW |
||||||
|---|---|---|---|---|---|---|---|---|---|
| kapp | SI | k2 (kapp × SI) | kapp | SI | k2 (kapp × SI) | kapp | SI | k2 (kapp × SI) | |
| m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | m−1 s−1 | ||||
| WT | 3.3 ± 0.1 × 102 | 1.5 ± 0.1 | 5.0 × 102 | 1.3 ± 0.1 × 103 | 2.2 ± 0.1 | 2.9 × 103 | 1.8 ± 0.1 × 104 | 2.5 ± 0.1 | 4.5 × 104 |
| P1R | 4.7 ± 0.1 × 105 | 1.6 ± 0.1 | 7.5 × 105 | 1.9 ± 0.2 × 106 | 2.8 ± 0.2 | 5.3 × 106 | ∼4.7 ± 0.5 × 106 | 2.9 ± 0.2 | 1.4 × 107 |
FIGURE 6.

Effect of heparin on thrombin inhibition by l-ANT. Shown are the discontinuous measurements for the rates of thrombin inhibition by wild type l-ANT (A) or P1R (C) without or with 0.1 μg/μl LMW or HMW heparin. The apparent second rate constants (kapp) were determined from the slope of the plot of kobs versus inhibitor concentrations, which are listed in Table 4. Apparent first order rate constants (kobs) for wild type (B) or P1R (D) were measured with LMW or HMW heparin ranging from 10−5 to 10 μg/μl under pseudo-first-order conditions containing 5 nm thrombin and 1.0 μm l-ANT or 0.05 μm P1R in PBS with 0.1 mg/ml BSA and 0.1% PEG 8000. The kobs values were obtained from the slope of semilog plots of residual enzyme activity measured from initial velocities of S-2238 hydrolysis (A405/min) versus time. Filled inverted triangles, l-ANT; open circles, l-ANT + LMW; filled squares, l-ANT + HMW; filled triangles, P1R; open squares, P1R + LMW; filled circles, P1R + HMW.
We also measured the k2 of the interaction between l-ANT variant P1R and thrombin and assessed the effect of heparin (Fig. 6, C and D). This substitution of P1 residues with Arg does not affect the SI of l-ANT/thrombin interaction but increases the association rate to 7.5 × 105 m−1 s−1 (Table 4), nearly 1500-fold faster than wild type l-ANT. This reaction is further enhanced by 20-fold in the presence of HMW heparin with a k2 of 1.4 × 107 m−1 s−1 (Table 4) in a similar bell-shaped dose-dependent curve (Fig. 6D).
Antithrombin is a principal inhibitor of thrombin in human. Its reaction with thrombin is not affected by short chain heparin but rapidly increases by longer heparins (54). Its heparin-mediated interaction also follows a bell-shaped curve, and the crystal structure of the ternary complex antithrombin·thrombin·heparin confirms that heparin promotes their interaction by a bridging mechanism (10, 11, 55). The bell-shaped dose-dependent curve indicates that at an optimal concentration of long chain heparin, the inhibitor and protease bind the same heparin chain, and at very high concentration, heparin attenuates the acceleration rate of protease inhibition due to binding of the inhibitor and protease to different heparin chains. The findings here that a short oligosaccharide chain is insufficient to enhance thrombin inhibition by l-ANT and that HMW heparin accelerates the reaction following a bell-shaped dose-dependent curve suggest that heparin mediates thrombin inhibition by l-ANT through a similar bridging mechanism.
Discussion
Serpins are known to participate in many physiological processes, such as blood coagulations, fibrinolysis, complement activation, tumor suppression, hormone delivery, etc. (8, 56). In this study, we have characterized lamprey angiotensinogen, which has a dual function both in blood pressure regulation and in blood coagulation in an early lineage of vertebrates. Apparently, this ancient serpin kept its hormone carrier function over 500 million years, as seen in mammalian angiotensinogen, whereas its function in the coagulation system has been assumed by other serpins. Understanding the structural roles of lamprey angiotensinogen in these two systems will shed light on how the angiotensin system evolved to regulate blood pressure and how thrombin activity was put under control in the primitive coagulation system.
Lamprey Angiotensinogen as a Hormone Carrier
Lamprey angiotensinogen shares about 30% sequence identity with angiotensinogen from other vertebrates, with the angiotensin peptide located at the N terminus (supplemental Fig. 2). Classically, angiotensinogen is described as a passive hormone carrier; however, recent findings indicate that human angiotensinogen could actively regulate angiotensin release through an adjustable disulfide bond between residues Cys-18, near the N terminus of angiotensinogen, and Cys-138 in the connecting loop linking helices C and D (57). Both oxidized and reduced forms of angiotensinogen are present in circulation, and angiotensin release is affected by the redox state of this disulfide bond. Interestingly, these 2 cysteines are largely conserved in all angiotensinogens of vertebrates but not in early diverged lampreys (supplemental Fig. 2). Also, it appears that only l-ANT possess a functional reactive center loop for protease inhibition. The crystal structure of lamprey angiotensinogen solved here (Fig. 2A) shows no electron density for the whole N-terminal tail (∼70 residues), which indicates that this part of the molecule is highly flexible in lamprey angiotensinogen and is not anchored closely to the body of the molecule, as seen with mammalian angiotensinogens (57). Clearly, the addition of this disulfide linkage between the N terminus and the helix C/D connecting loop in higher vertebrates during evolution provides fine tuning of hormone release from angiotensinogen, and the trade-off is the loss of serpin typical S-to-R transition in angiotensinogen of mammals.
Lamprey Angiotensinogen as a Thrombin Inhibitor
Thrombin, a key enzyme of blood coagulation, is present in all vertebrates and shares important structural features, including the 60-loop, the anion binding exosites I and II, and the sodium site (58). Its activity is tightly controlled by serpins in mammals, and the mechanisms by which serpins inhibit thrombin has been well elucidated by several structures of serpin-thrombin Michaelis complexes and many detailed biochemical analyses (10, 11, 27, 30, 35). Thrombin inhibition often involves cofactors, such as heparin or thrombomodulin, to allow localized control of thrombin activity. Heparin can promote thrombin inhibition either through an allosteric mechanism or a template bridging mechanism. Although genome DNA sequence analysis has shown that lampreys have a simple blood coagulation system with fewer components, very little is known about how lamprey thrombin is activated and how its activity is controlled in vivo. Nevertheless, the amino acid sequence alignment of the protease domain of lamprey and human thrombin shows that ∼80% of thrombin residues are conserved during evolution (supplemental Fig. 3A). More importantly, ∼90% of the key residues of thrombin that form critical interactions with heparin and serpins, as identified from crystal structures of human thrombin complexed with various serpins, such as antithrombin, HCII, PCI, and PN1 (10, 27, 30, 35), are well preserved (supplemental Fig. 3). Therefore, we have used human thrombin as a surrogate for lamprey thrombin and characterized the heparin-mediated thrombin inhibition by lamprey angiotensinogen.
Our study here shows that lamprey angiotensinogen has a typical serpin fold with the ability to undergo S-to-R transition and adopts the same suicidal inhibition mechanism in forming covalently linked serpin·protease complexes as seen with other inhibitory serpins. Furthermore, it has a much larger positively charged surface area around helices D and A than other heparin-binding serpins (Fig. 3 and supplemental Fig. 1). Our mutagenesis experiments indicate that the residues involved in heparin binding in lamprey angiotensinogen are spatially conserved when compared with antithrombin and HCII. Residues Lys-148, Lys-151, and Arg-155 in helix D are crucial for heparin binding (Fig. 4D). Interestingly, it appears that Arg-380 located in the connecting loop between helix J and s5A also plays a role in heparin binding. This indicates that lamprey angiotensinogen probably has an extended heparin binding site with heparin binding lamprey angiotensinogen in an orientation slightly different from that of antithrombin (10, 11) and PN1 (30). A short heparin with 8 monosaccharide units could be docked along helix D with 3 sulfate groups forming interactions with these 4 key residues (supplemental Fig. 1).
More importantly, we have found that heparin binding does not induce any significant conformational changes in lamprey angiotensinogen. There are no changes in fluorescence intensity and no changes in its inhibitory activity in the presence of short heparin oligosaccharides. This indicates that lamprey angiotensinogen does not possess a delicate allosteric mechanism adopted by antithrombin in inhibiting activated factor X (24), but it utilizes a template mechanism where heparin could bridge serpin and protease together by forming a ternary complex (16). The heparin-accelerated thrombin inhibition by lamprey angiotensinogen requires an optimal concentration of HMW heparin, where lamprey angiotensinogen and thrombin could bind to the same heparin chain. High concentrations of heparin attenuate thrombin inhibition due to binding of the inhibitor and protease to different heparin chains. Therefore, l-ANT inhibits thrombin through a heparin-mediated bridging mechanism, and this heparin-mediated mechanism is well preserved during evolution from ancient vertebrates, such as lampreys, to high vertebrates over millions of years.
Another strategy that was adopted by this ancient serpin and preserved in mammals is to maximize the inhibitor's specificity toward target protease by choosing a protease less favorable P1 residue and to increase activity through cofactor modulation. In general, the single most important residue in determining the inhibitor's activity and specificity is its P1 residue, which is directly recognized by the active site of protease (8, 59). Most serpins select a target protease via the use of preferred residues at the P1 position for efficient recognition and inhibition. For example, α1-antitrypsin has a Met at P1 that enables efficient inhibition of neutrophil elastase, and antithrombin, PAI-1, and antiplasmin have Arg at P1 for their corresponding target proteases. Mutations at P1 can result in serpins targeting wrong proteases, leading to devastating consequences in humans as observed with the α1-antitrypsin Pittsburgh variant (60–62), where the original neutrophil elastase inhibitor is converted to a potent inhibitor of thrombin and other trypsin-like proteases. However, some serpins efficiently inhibit target protease through non-conventional residues at P1 to avoid nonspecific recognition by other proteases. For example, HCII possess a Leu at P1 for thrombin inhibition, and ZPI holds a Tyr at P1 for inhibiting factor Xa, although both proteases favor Arg at P1 (28, 63). The activity of these serpins toward their target proteases could be enhanced and localized at sites where these cofactors are present (16, 47). As seen here, lamprey angiotensinogen chooses Ile as the P1 residue for thrombin inhibition, and its reaction with thrombin is accelerated by heparin or heparan sulfate (14). Once its P1 residue is mutated to an Arg, the mutant reacts with thrombin more >1500-fold faster but has far less selectivity toward other Arg favored proteases (Fig. 1A).
Overall, this study shows that lamprey angiotensinogen, an ancient serpin with dual functions in regulating blood pressure and blood coagulation, has a typical serpin fold with a labile angiotensin segment at its N terminus and inhibits its target protease with the unique S-to-R conformational change. Furthermore, it has an extended but spatially conserved heparin binding site and inhibits thrombin through a bridging mechanism, as seen with other heparin-binding serpins. Although during evolution angiotensinogen has been relieved from its duty in the blood coagulation system of higher vertebrates, the unique features of lamprey angiotensinogen, such as S-to-R transition, maximization of serpin specificity through selecting a non-canonical P1 residue, and the cofactor-mediated template mechanism in inhibiting protease, are persisting with other serpins.
Author Contributions
A. Z., Y. Z. and H. R. designed all experiments. H. W., H. C., J. W., Z. W., F. Z., X. H., L. M., L. F., R. Z., Y. W., H. R., Y. Z., and A. Z. performed the experiments and contributed to data analysis. H. W. and A. Z. wrote the paper.
Supplementary Material
Acknowledgments
We thank the staff from BL17U/BL19U beamline of National Center for Protein Sciences Shanghai (NCPSS) at Shanghai Synchrotron Radiation Facility, for assistance during data collection.
This work was supported in part by National Basic Research Program of China (973 Program) Grant 2014CB910304; National Natural Science Foundation of China Grants 31370727, 31170724, and 81572090; the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning; the Shanghai PuJiang Program; Innovation Program of Shanghai Municipal Education Commission Grant 12ZZ113. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5INW) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Figs. 1–3.
- RCL
- reactive center loop
- l-ANT
- lamprey angiotensinogen
- HCII
- heparin cofactor II
- PN1
- protease nexin 1
- PAI-1
- plasminogen activator inhibitor 1
- ZPI
- protein Z-dependent protease inhibitor
- PCI
- protein C inhibitor
- KLK7
- kallikrein-related peptidase 7
- KLK1
- kallikrein-related peptidase 1
- LMW
- low molecular weight
- HMW
- high molecular weight
- DP6 (DP8, DP12, DP16, DP20)
- heparin oligosaccharides of 6 (8, 12, 16, 20) monosaccharide units
- TNS
- 2-p-toluidinylnaphthalene-6-sulfonate
- S-to-R
- stressed-to-relaxed
- SI
- stoichiometry of inhibition
- RMSD
- root mean square deviation
- H5*
- high affinity pentasaccharide
- PDB
- Protein Data Bank.
References
- 1. Kumar A. (2015) Bayesian phylogeny analysis of vertebrate serpins illustrates evolutionary conservation of the intron and indels based six groups classification system from lampreys for approximately 500 MY. PeerJ 3, e1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong M. K., and Takei Y. (2011) Characterization of a native angiotensin from an anciently diverged serine protease inhibitor in lamprey. J. Endocrinol. 209, 127–137 [DOI] [PubMed] [Google Scholar]
- 3. Fournier D., Luft F. C., Bader M., Ganten D., and Andrade-Navarro M. A. (2012) Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. 90, 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atlas S. A. (2007) The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 13, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khakoo A. Y., Sidman R. L., Pasqualini R., and Arap W. (2008) Does the renin-angiotensin system participate in regulation of human vasculogenesis and angiogenesis? Cancer Res. 68, 9112–9115 [DOI] [PubMed] [Google Scholar]
- 6. Doolittle R. F. (1983) Angiotensinogen is related to the antitrypsin-antithrombin-ovalbumin family. Science 222, 417–419 [DOI] [PubMed] [Google Scholar]
- 7. Irving J. A., Pike R. N., Lesk A. M., and Whisstock J. C. (2000) Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 10, 1845–1864 [DOI] [PubMed] [Google Scholar]
- 8. Gettins P. G. (2002) Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 9. Silverman G. A., Bird P. I., Carrell R. W., Church F. C., Coughlin P. B., Gettins P. G., Irving J. A., Lomas D. A., Luke C. J., Moyer R. W., Pemberton P. A., Remold-O'Donnell E., Salvesen G. S., Travis J., and Whisstock J. C. (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins: evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 276, 33293–33296 [DOI] [PubMed] [Google Scholar]
- 10. Li W., Johnson D. J., Esmon C. T., and Huntington J. A. (2004) Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol. 11, 857–862 [DOI] [PubMed] [Google Scholar]
- 11. Dementiev A., Petitou M., Herbert J. M., and Gettins P. G. (2004) The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol. 11, 863–867 [DOI] [PubMed] [Google Scholar]
- 12. Stein P. E., Tewkesbury D. A., and Carrell R. W. (1989) Ovalbumin and angiotensinogen lack serpin S-R conformational change. Biochem. J. 262, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carrell R., Qi X., and Zhou A. (2011) Serpins as hormone carriers: modulation of release. Methods Enzymol. 501, 89–103 [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., and Ragg H. (2011) An unexpected link between angiotensinogen and thrombin. FEBS Lett. 585, 2395–2399 [DOI] [PubMed] [Google Scholar]
- 15. Wang Y., Köster K., Lummer M., and Ragg H. (2014) Origin of serpin-mediated regulation of coagulation and blood pressure. PLoS One 9, e97879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huntington J. A. (2013) Thrombin inhibition by the serpins. J. Thromb. Haemost. 11, 254–264 [DOI] [PubMed] [Google Scholar]
- 17. Olson S. T., Björk I., and Shore J. D. (1993) Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 222, 525–559 [DOI] [PubMed] [Google Scholar]
- 18. O'Keeffe D., Olson S. T., Gasiunas N., Gallagher J., Baglin T. P., and Huntington J. A. (2004) The heparin binding properties of heparin cofactor II suggest an antithrombin-like activation mechanism. J. Biol. Chem. 279, 50267–50273 [DOI] [PubMed] [Google Scholar]
- 19. Rau J. C., Mitchell J. W., Fortenberry Y. M., and Church F. C. (2011) Heparin cofactor II: discovery, properties, and role in controlling vascular homeostasis. Semin. Thromb. Hemost. 37, 339–348 [DOI] [PubMed] [Google Scholar]
- 20. Doolittle R. F., Jiang Y., and Nand J. (2008) Genomic evidence for a simpler clotting scheme in jawless vertebrates. J. Mol. Evol. 66, 185–196 [DOI] [PubMed] [Google Scholar]
- 21. Doolittle R. F. (2009) Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb. Symp. Quant. Biol. 74, 35–40 [DOI] [PubMed] [Google Scholar]
- 22. Ragg H., Kumar A., Köster K., Bentele C., Wang Y., Frese M. A., Prib N., and Krüger O. (2009) Multiple gains of spliceosomal introns in a superfamily of vertebrate protease inhibitor genes. BMC Evol. Biol. 9, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huntington J. A. (2011) Serpin structure, function and dysfunction. J. Thromb. Haemost. 9, 26–34 [DOI] [PubMed] [Google Scholar]
- 24. Johnson D. J., Li W., Adams T. E., and Huntington J. A. (2006) Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 25, 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ersdal-Badju E., Lu A., Zuo Y., Picard V., and Bock S. C. (1997) Identification of the antithrombin III heparin binding site. J. Biol. Chem. 272, 19393–19400 [DOI] [PubMed] [Google Scholar]
- 26. Jin L., Abrahams J. P., Skinner R., Petitou M., Pike R. N., and Carrell R. W. (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U.S.A. 94, 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baglin T. P., Carrell R. W., Church F. C., Esmon C. T., and Huntington J. A. (2002) Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc. Natl. Acad. Sci. U.S.A. 99, 11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tollefsen D. M. (1997) Heparin cofactor II. Adv. Exp. Med. Biol. 425, 35–44 [DOI] [PubMed] [Google Scholar]
- 29. Ragg H., Ulshöfer T., and Gerewitz J. (1990) Glycosaminoglycan-mediated leuserpin-2/thrombin interaction: structure-function relationships. J. Biol. Chem. 265, 22386–22391 [PubMed] [Google Scholar]
- 30. Li W., and Huntington J. A. (2012) Crystal structures of protease nexin-1 in complex with heparin and thrombin suggest a 2-step recognition mechanism. Blood 120, 459–467 [DOI] [PubMed] [Google Scholar]
- 31. Stone S. R., Brown-Luedi M. L., Rovelli G., Guidolin A., McGlynn E., and Monard D. (1994) Localization of the heparin-binding site of glia-derived nexin/protease nexin-1 by site-directed mutagenesis. Biochemistry 33, 7731–7735 [DOI] [PubMed] [Google Scholar]
- 32. Keijer J., Linders M., Wegman J. J., Ehrlich H. J., Mertens K., and Pannekoek H. (1991) On the target specificity of plasminogen activator inhibitor 1: the role of heparin, vitronectin, and the reactive site. Blood 78, 1254–1261 [PubMed] [Google Scholar]
- 33. Yang L., Ding Q., Huang X., Olson S. T., and Rezaie A. R. (2012) Characterization of the heparin-binding site of the protein z-dependent protease inhibitor. Biochemistry 51, 4078–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang X., Rezaie A. R., Broze G. J. Jr., and Olson S. T. (2011) Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J. Biol. Chem. 286, 8740–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W., Adams T. E., Nangalia J., Esmon C. T., and Huntington J. A. (2008) Molecular basis of thrombin recognition by protein C inhibitor revealed by the 1.6-Å structure of the heparin-bridged complex. Proc. Natl. Acad. Sci. U.S.A. 105, 4661–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li W., and Huntington J. A. (2008) The heparin binding site of protein C inhibitor is protease-dependent. J. Biol. Chem. 283, 36039–36045 [DOI] [PubMed] [Google Scholar]
- 37. Rein C. M., Desai U. R., and Church F. C. (2011) Serpin-glycosaminoglycan interactions. Methods Enzymol. 501, 105–137 [DOI] [PubMed] [Google Scholar]
- 38. Belzar K. J., Zhou A., Carrell R. W., Gettins P. G., and Huntington J. A. (2002) Helix D elongation and allosteric activation of antithrombin. J. Biol. Chem. 277, 8551–8558 [DOI] [PubMed] [Google Scholar]
- 39. Wei Z., Yan Y., Carrell R. W., and Zhou A. (2009) Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood 114, 3662–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 41. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., and Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 42. Winn M. D., Isupov M. N., and Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 43. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 44. Ritchie A. W., and Webb L. J. (2013) Optimizing electrostatic field calculations with the adaptive Poisson-Boltzmann Solver to predict electric fields at protein-protein interfaces. I. Sampling and focusing. J. Phys. Chem. B 117, 11473–11489 [DOI] [PubMed] [Google Scholar]
- 45. Meagher J. L., Olson S. T., and Gettins P. G. (2000) Critical role of the linker region between helix D and strand 2A in heparin activation of antithrombin. J. Biol. Chem. 275, 2698–2704 [DOI] [PubMed] [Google Scholar]
- 46. Schedin-Weiss S., Richard B., Hjelm R., and Olson S. T. (2008) Antiangiogenic forms of antithrombin specifically bind to the anticoagulant heparin sequence. Biochemistry 47, 13610–13619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gettins P. G., and Olson S. T. (2009) Exosite determinants of serpin specificity. J. Biol. Chem. 284, 20441–20445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li W., Adams T. E., Kjellberg M., Stenflo J., and Huntington J. A. (2007) Structure of native protein C inhibitor provides insight into its multiple functions. J. Biol. Chem. 282, 13759–13768 [DOI] [PubMed] [Google Scholar]
- 49. Qi X., Loiseau F., Chan W. L., Yan Y., Wei Z., Milroy L. G., Myers R. M., Ley S. V., Read R. J., Carrell R. W., and Zhou A. (2011) Allosteric modulation of hormone release from thyroxine and corticosteroid-binding globulins. J. Biol. Chem. 286, 16163–16173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olson S. T., Halvorson H. R., and Björk I. (1991) Quantitative characterization of the thrombin-heparin interaction: discrimination between specific and nonspecific binding models. J. Biol. Chem. 266, 6342–6352 [PubMed] [Google Scholar]
- 51. Olson S. T., and Björk I. (1991) Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin-thrombin reaction: elucidation from salt concentration effects. J. Biol. Chem. 266, 6353–6364 [PubMed] [Google Scholar]
- 52. Zhou A., Carrell R. W., and Huntington J. A. (2001) The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J. Biol. Chem. 276, 27541–27547 [DOI] [PubMed] [Google Scholar]
- 53. Beauchamp N. J., Pike R. N., Daly M., Butler L., Makris M., Dafforn T. R., Zhou A., Fitton H. L., Preston F. E., Peake I. R., and Carrell R. W. (1998) Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood 92, 2696–2706 [PubMed] [Google Scholar]
- 54. Bray B., Lane D. A., Freyssinet J. M., Pejler G., and Lindahl U. (1989) Anti-thrombin activities of heparin: effect of saccharide chain length on thrombin inhibition by heparin cofactor II and by antithrombin. Biochem. J. 262, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Griffith M. J. (1982) Kinetics of the heparin-enhanced antithrombin III/thrombin reaction: evidence for a template model for the mechanism of action of heparin. J. Biol. Chem. 257, 7360–7365 [PubMed] [Google Scholar]
- 56. Rau J. C., Beaulieu L. M., Huntington J. A., and Church F. C. (2007) Serpins in thrombosis, hemostasis and fibrinolysis. J. Thromb. Haemost. 5, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou A., Carrell R. W., Murphy M. P., Wei Z., Yan Y., Stanley P. L., Stein P. E., Broughton Pipkin F., and Read R. J. (2010) A redox switch in angiotensinogen modulates angiotensin release. Nature 468, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Di Cera E. (2007) Thrombin as procoagulant and anticoagulant. J. Thromb. Haemost. 5, 196–202 [DOI] [PubMed] [Google Scholar]
- 59. Ciaccia A. V., Willemze A. J., and Church F. C. (1997) Heparin promotes proteolytic inactivation by thrombin of a reactive site mutant (L444R) of recombinant heparin cofactor II. J. Biol. Chem. 272, 888–893 [DOI] [PubMed] [Google Scholar]
- 60. Owen M. C., Brennan S. O., Lewis J. H., and Carrell R. W. (1983) Mutation of antitrypsin to antithrombin: α1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N. Engl. J. Med. 309, 694–698 [DOI] [PubMed] [Google Scholar]
- 61. Hua B., Fan L., Liang Y., Zhao Y., and Tuddenham E. G. (2009) α1-Antitrypsin Pittsburgh in a family with bleeding tendency. Haematologica 94, 881–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vidaud D., Emmerich J., Alhenc-Gelas M., Yvart J., Fiessinger J. N., and Aiach M. (1992) Met 358 to Arg mutation of α1-antitrypsin associated with protein C deficiency in a patient with mild bleeding tendency. J. Clin. Invest. 89, 1537–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Han X., Fiehler R., and Broze G. J. Jr. (1998) Isolation of a protein Z-dependent plasma protease inhibitor. Proc. Natl. Acad. Sci. U.S.A. 95, 9250–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.