

Abstract
Apoptosis is thought to play a critical role in several pathological processes, such as neurodegenerative diseases (i.e. Parkinson's and Alzheimer's diseases) and various cardiovascular diseases. Despite the fact that apoptotic mechanisms are well defined, there is still no substantial therapeutic strategy to stop or even slow this process. Thus, there is an unmet need for therapeutic agents that are able to block or slow apoptosis in neurodegenerative and cardiovascular diseases. The outer mitochondrial membrane protein voltage-dependent anion channel 1 (VDAC1) is a convergence point for a variety of cell survival and death signals, including apoptosis. Recently, we demonstrated that VDAC1 oligomerization is involved in mitochondrion-mediated apoptosis. Thus, VDAC1 oligomerization represents a prime target for agents designed to modulate apoptosis. Here, high-throughput compound screening and medicinal chemistry were employed to develop compounds that directly interact with VDAC1 and prevent VDAC1 oligomerization, concomitant with an inhibition of apoptosis as induced by various means and in various cell lines. The compounds protected against apoptosis-associated mitochondrial dysfunction, restoring dissipated mitochondrial membrane potential, and thus cell energy and metabolism, decreasing reactive oxidative species production, and preventing detachment of hexokinase bound to mitochondria and disruption of intracellular Ca2+ levels. Thus, this study describes novel drug candidates with a defined mechanism of action that involves inhibition of VDAC1 oligomerization, apoptosis, and mitochondrial dysfunction. The compounds VBIT-3 and VBIT-4 offer a therapeutic strategy for treating different diseases associated with enhanced apoptosis and point to VDAC1 as a promising target for therapeutic intervention.
Keywords: apoptosis, bioluminescence resonance energy transfer (BRET), drug development, membrane protein, mitochondria, mitochondrial apoptosis, oligomerization, small molecule, voltage-dependent anion channel (VDAC), apoptosis
Introduction
Mitochondria play crucial roles in cellular energy generation and metabolism, maintenance of the cell redox potential, calcium homeostasis, pH control and fatty acid oxidation, cell signaling, proliferation, differentiation, aging, and death (1). It is therefore not surprising that mitochondrial dysfunction is associated with various human diseases (1, 2).
Located at the outer mitochondrial membrane (OMM),2 the voltage-dependent anion channel (VDAC) serves as a mitochondrial gatekeeper. Three VDAC isoforms have been discovered (3), but only for VDAC1 is there a complete set of structural and functional information available. VDAC1 controls the metabolic and energy cross-talk between mitochondria and the rest of the cell, mediating the fluxes of ions, nucleotides, and other metabolites across the OMM (4–7). VDAC1 is composed of 19 transmembrane β-strands connected by flexible loops to form a β-barrel, along with a 26-residue-long N-terminal region that lies inside the pore (8–10). Mitochondria also function in apoptosis, mediating the intrinsic pathway (11). This pathway is initiated in response to various stimuli, such as high cytoplasmic Ca2+, oxygen radicals, activation of pro-apoptotic Bcl-2 family proteins, UV damage, and various drugs (11, 12). These stimuli provoke permeabilization of the OMM, allowing the release of pro-apoptotic proteins, such as cytochrome c (Cyto c) and apoptosis-inducing factor, from the intermembrane space to the cytosol (13). This leads to the activation of caspases, which cleave targeted proteins, and subsequently to apoptosis. Defects in the regulation of apoptosis are associated with cancer and neurodegenerative diseases (14), with evasion of apoptosis being a hallmark of cancer (15) and enhancement of apoptosis being seen in neurodegenerative diseases (16, 17).
VDAC1 has also been recognized as a key protein in mitochondrion-mediated apoptosis, regulating the release of apoptogenic proteins, as well as interacting with anti-apoptotic proteins (5, 18–20). Release of Cyto c from mitochondria is considered a key initial step in the apoptotic process, although the precise mechanisms regulating Cyto c release remain unknown. To date, all of the mitochondrial components known to translocate to the cytoplasm following an apoptotic stimulus reside in the intermembrane space. Therefore, only the permeability of the OMM needs to be modified.
Among the many models that have been put forward for the release of apoptotic proteins, some suggest that release involves the formation of a channel large enough to allow the passage of apoptogenic proteins (1, 5, 6, 21–23), whereas others suggest disruption of OMM integrity (24–26).
Recently, we demonstrated that apoptosis induction leads to the oligomerization of VDAC1 into dimers, trimers, tetramers, and higher order oligomers (23, 27–35). We have also demonstrated that VDAC1 oligomerization is a general mechanism common to numerous apoptogens acting via different initiating cascades (30, 36, 37). Furthermore, apoptosis inhibitors (30, 38) and recently identified VDAC1-interacting molecules (e.g. diphenylamine-2-carboxylate (39)) inhibited VDAC oligomerization. These results led us to propose a novel model in which VDAC1 exists in a dynamic equilibrium between monomeric and oligomeric states, with apoptosis inducers shifting the equilibrium toward oligomers, forming a large channel that enables Cyto c release, leading to cell death.
Our results thus suggest not only that VDAC1 oligomerization is a molecular focal point in cellular life or death decision processes but that VDAC1 may also offer a prime target for therapeutic agents designed to modulate apoptosis. As such, targeting the oligomeric status of VDAC1, and hence apoptosis, offers a strategy for combating cancers and neurodegenerative diseases. Currently, there are no known specific inhibitors of VDAC1-mediated apoptosis. We have, however, developed new molecules that interact with VDAC1, inhibiting VDAC1 oligomerization and preventing apoptosis at pharmacologically relevant concentrations. These novel drug candidates, with defined modes of action, can serve to treat diseases associated with enhanced apoptosis and point to VDAC1 as a promising target for therapeutic intervention. Indeed, several studies have identified pharmacological agents that target VDAC1 so as to induce cancer cell death or protect against apoptosis (6). However, none of these molecules are specific to VDAC1 and could instead affect the cell via different biochemical pathways.
To identify a molecule with higher specificity toward VDAC, we screened a library of 1,468 drug-like compounds in living mammalian cells using a bioluminescence resonance energy transfer (BRET2)-based VDAC1 oligomerization assay. Several hit compounds were identified as inhibitors of VDAC1 oligomerization. Using medicinal chemistry approaches, these active compounds were structurally optimized to develop two novel active molecules, VBIT-3 and VBIT-4. These novel VDAC1 inhibitors represent drug candidates targeting apoptosis in a variety of diseases.
Results
This study addressed the need for inhibitors of apoptosis that act at an early stage of the apoptotic pathway, inhibiting the release of Cyto c from mitochondria. Our approach considered VDAC1-based compounds able to inhibit VDAC1 oligomerization, thereby protecting cells against apoptotic cell death.
Validation of the BRET-2 Assay Used for High-throughput Screening (HTS)
To directly monitor the oligomeric state of VDAC1 molecules in the native membrane, we used BRET2 technology (30). In BRET2 technology, VDAC1 is tagged with either Renilla luciferase (RLuc) as donor or a variant of GFP (GFP2) as acceptor and expressed in T-REx cells stably expressing shRNA-hVDAC1 and containing low levels of endogenous hVDAC1. Energy transfer between the two only occurs when the donor and acceptor are in spatial proximity to each other (<10 nm apart), making this an ideal technique for monitoring protein-protein interactions in biological systems (40). The detection of a BRET2 signal in this case corresponds to VDAC1 oligomerization, although attenuation of an apoptosis-enhanced BRET2 signal indicates an inhibition of VDAC1 oligomerization (Fig. 1A).
FIGURE 1.

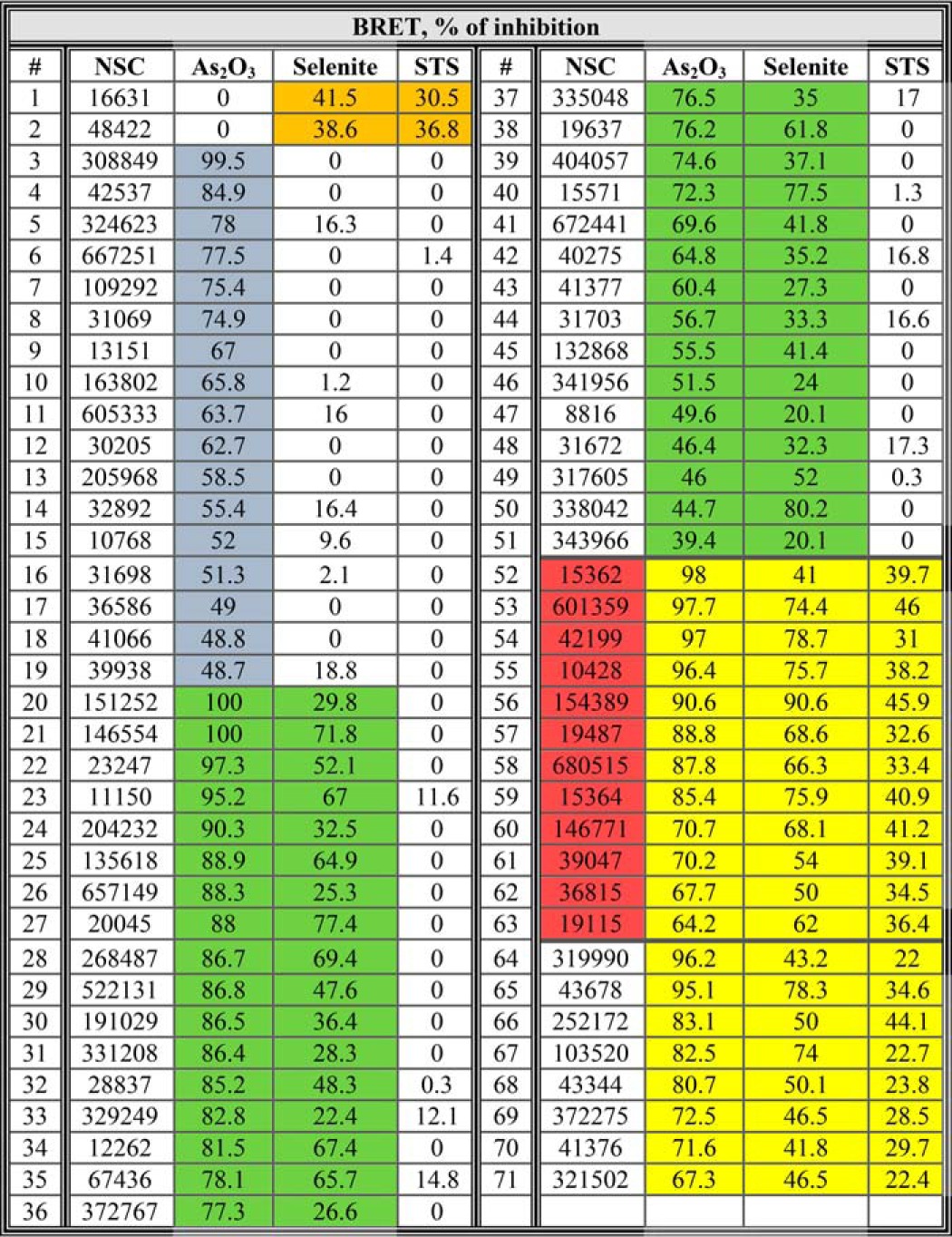
BRET assay for monitoring VDAC1 oligomerization and inhibition. A, schematic representation showing energy transfer between VDAC1-luciferase (RLuc, a light-producing enzyme) as donor and VDAC1-GFP2 (fluorophore) as acceptor. Energy transfer only occurs when the donor and the acceptor are in spatial proximity or physically interact. Compounds enhancing apoptosis lead to VDAC1 oligomerization and thus enhance the BRET2 signal, although apoptosis inhibitors inhibit VDAC1 oligomerization and therefore decrease the BRET2 signal. The luciferase substrate DBC emits light upon cleavage and thus causes excitation of the proximal GFP2 protein, thereby generating a BRET2 signal. B, DNDS inhibits the BRET2 signal induced by selenite. T-REx cells expressing hVDAC1 shRNA were co-transfected with plasmids encoding rVDAC1-Rluc (0.1 μg) and rVDAC1-GFP2 (0.8 μg). The BRET2 signals obtained in cells, treated with or without pre-treatment with DNDS (200 μm, 1 h) and selenite (30 μm, 3 h), were measured. A representative of three independent repeats is presented. C, T-REx cells were treated as described in B, harvested, cross-linked with EGS, and then analyzed by immunoblotting using anti-VDAC1 antibodies. The positions of VDAC1 monomers, dimers, and multimers (multi) are indicated. The white asterisk indicates monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular crossed-linked monomeric VDAC1. D, inhibition of BRET2 enhancement by the 12 most active compounds from the NCI library. BRET2 signals were obtained in cells treated with As2O3 (60 μm, 3 h; purple bars), selenite (30 μm, 3 h; green bars), or STS (1 μm, 3 h; red bars) with or without pre-treatment with the indicated compounds (10 μm, 1 h). Results are presented as percent inhibition of the BRET signal and correspond to mean ± S.E. (n = 2), p < 0.05(*). E, summary of BRET assay. Out of an NCI library of 1,468 compounds, 71 were able to inhibit the BRET signal (30–100%) induced by treatment with As2O3, selenite, or STS (the list is presented in Table 1). These inhibited BRET induced by either As2O3 or selenite or by selenite or STS (overlapping areas). Twelve of the 71 compounds (see Fig. 2A) were able to inhibit the BRET2 signal elicited by all three inducers (yellow).
To validate the assay, selenite, a known inducer of apoptosis and VDAC1 oligomerization (30), was used to enhance VDAC1 oligomerization. To inhibit the selenite-induced BRET2 signal, 4,4-diisothiocyanostilbene-trans-2,2-disulfonic acid (DNDS), an inhibitor of VDAC1 channel conductance and apoptosis (38, 39), was used (Fig. 1B). Chemical cross-linking using the cell-permeable cross-linker ethylene glycol bis(succinimidylsuccinate) (EGS) and Western blotting analysis also served to demonstrate any enhancement or inhibition of VDAC1 oligomerization (Fig. 1C). The results show that selenite provoked oligomerization, as reflected in the enhanced BRET2 signal (Fig. 1B) and in the appearance of protein bands corresponding to VDAC1 dimers (72 kDa) and multimers (Fig. 1C). The obtained results suggest that VDAC1 oligomerization is a dynamic process and can be activated or inhibited and thus can be used to search for compounds modulating this process. Here, we focused on inhibition of VDAC1 oligomerization, apoptosis, and protection against dysfunction associated with apoptosis induction.
HTS for Inhibitors of VDAC1 Oligomerization
To search for active molecules that inhibit apoptosis via blocking VDAC1 oligomerization, we screened a drug-like compound library (1,468 molecules) provided by NCI, National Institutes of Health (see under “Experimental Procedures”), using BRET2 technology (19, 30). The compound library was robotically screened for inhibitors of BRET2 signaling as induced by STS, selenite, or As2O3 in VDAC1-Luc- and VDAC1-GFP2-expressing cells (Table 1). Seventy one compounds caused a 40% decrease in BRET2 signaling induced by the apoptotic agents. The STS-enhanced BRET2 signal was reduced by 22 molecules, although the BRET2 signal generated upon As2O3- and selenite-induced apoptosis was decreased by 69 and 54 molecules, respectively. Twelve compounds inhibited the BRET2 signal induced by all three inducers (Fig. 1, D and E). Thus, the hit rate from the primary screen was 0.8% (i.e. 12 of 1,468 compounds).
TABLE 1.
Summary of BRET2-based screen results of the anti-VDAC1 oligomerization activity of compounds from the National Institutes of Health NCI library
T-Rex cells expressing hVDAC1 shRNA were co-transfected with plasmids encoding rVDAC1-Rluc (0.1 μg) and rVDAC1-GFP2 (0.8 μg) and treated with As2O3 (60 μM, 3 h), selenite (30 μm, 3 h), or STS (1 μm, 3 h), with or without pre-treatment with the indicated compounds (10 μm, 1 h), followed by BRET2 signal measurement. Results are presented as percent inhibition of the BRET2 signal induced by the indicated pro-apoptotic agent. The highlighted numbers indicate compounds which inhibited BRET2 signals induced by selenite and STS (orange), by As2O3 (blue), by As2O3 and selenite (green), and by As2O3, selenite, and STS (yellow). The 12 most active compounds are highlighted in red.
Structure-based Analysis of Hits from the First Round of Screening for Oligomerization Inhibitors
The 12 active compounds identified were divided in three structurally related groups (Fig. 2A), and based on these structures, a 3D pharmacophore model was generated from the lowest energy conformation of all 12 active compounds (Fig. 2B). The resulting structure consisted of aromatic ring areas, a hydrophobic region, and hydrogen bond donor and acceptor points. Next, based on this proposed pharmacophore model, we identified 34 commercially available compounds as potential inhibitors of VDAC1 oligomerization that were subsequently tested for such activity (Fig. 3).
FIGURE 2.

Structure-activity analysis from the first round of screening for inhibitors of VDAC1 oligomerization. A, chemical structure of the 12 most active inhibitors of VDAC1 oligomerization from the NCI library. B, pharmacophore search was applied to the compounds shown in A. Pharmacophore and the four anchoring points are presented (orange, aromatic points; purple, hydrogen bond donor points; blue, hydrogen bonds acceptor point; green, hydrophobic points). The six aligned active compounds that led to the pharmacophore are also presented.
FIGURE 3.

Second round of screening for inhibitors of VDAC1 oligomerization. A, HeLa cells were incubated with the indicated compounds obtained from Diverchim (10 μm, 1 h). The cells were then exposed to selenite (30 μm, 3 h; gray bars) or cisplatin (15 μm, 20 h; blue bars), harvested, analyzed for VDAC1 oligomerization by chemical cross-linking as described in the legend to Fig. 1C, and VDAC1 dimer levels were quantified. The results are presented as percent inhibition. The red squares indicate the most active compounds selected for analysis in round 3 (medicinal chemistry-based compound design). The results shown correspond to means ± S.E. (n = 3), p < 0.05 (*). B, chemical structures of the four most active inhibitors of VDAC1 oligomerization are shown. The yellow background highlights similar structural moieties in all four molecules. C and D, HeLa cells were incubated with the indicated compounds (20 μm, 1 h) and then with or without selenite (30 μm, 3 h) (C) or cisplatin (15 μm, 20 h) (D). The cells were harvested, cross-linked with EGS (300 μm, 15 min), and analyzed by immunoblotting using anti-VDAC1 antibodies. The positions of VDAC1 monomers, dimers, and multimers (multi) are indicated. The white asterisk corresponds to monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular crossed-linked monomeric VDAC1. The positions of molecular size protein standards are provided. The source of the molecules (AKOS or Diverchim) is indicated. E, quantitative analysis of the amount of the VDAC1 dimers obtained in C and D is shown. Gray and blue bars represent results obtained with selenite and cisplatin, respectively. The data are presented as percent inhibition of dimer formation. The results shown correspond to means ± S.E. (n = 3), p < 0.05(*).
Second Round of Compound Screening for Inhibitors of VDAC1 Oligomerization
The ability of the 34 compounds, selected on the basis of the pharmacophore model, to inhibit VDAC1 oligomerization as induced by selenite or cisplatin in HeLa cells, was analyzed by following chemical cross-linking with EGS and Western blotting analysis using anti-VDAC1 antibodies. The inhibition of dimeric VDAC1 formation by the compounds is presented (Fig. 3A).
Of the compounds tested, four showed high inhibition of VDAC1 oligomerization as induced by selenite or cisplatin (Fig. 3A, boxed in red) and shared several structural aspects. For example, in all four compounds, substituted or non-substituted benzyl rings were bound to a nitrogen-containing ring either directly (DIV03322, DIV02963, and DIV00996) or via a thiourea linker (DIV00284) (Fig. 3B). These structural similarities served as the basis for designing compounds for the third round of screening.
Third Round of Screening for Inhibitors of VDAC1 Oligomerization
A third round of screening was carried out with 13 compounds that were designed based on the results obtained in the second round screening. These compounds were commercially available. The active molecules in this screen were identified in terms of their ability to inhibit VDAC1 oligomerization as induced by selenite or cisplatin. The level of oligomerization was estimated by the chemical cross-linking method (Fig. 3, C–E). One compound, AKOS-022075291 (AKOS-022), was able to almost completely prevent selenite- and cisplatin-induced VDAC1 oligomerization at low micromolar concentrations (Fig. 3, C–E). Thus, this molecule was further studied with respect to its ability to interact with VDAC1 and inhibit apoptosis.
AKOS-022 Prevents VDAC1 Oligomerization and Apoptosis
AKOS-022 was found to inhibit VDAC1 oligomerization and apoptosis, as analyzed by annexin-V/propidium iodide staining and flow cytometry, regardless if induced by selenite (Fig. 4, A–C) or cisplatin (Fig. 4, B, D, and E). AKOS-22 inhibited both VDAC1 oligomerization and apoptosis in a concentration-dependent manner, with 50% inhibition of both apoptosis and VDAC1 oligomerization (Fig. 4, C and E) obtained at a similar concentration (7.5 μm). Moreover, quantitative analysis showed a linear relationship between the decrease in the amounts of VDAC1 dimers and the extent of apoptotic cell death (Fig. 4F), as analyzed at the identical AKOS-022 concentration, revealing the tight relationship between these processes.
FIGURE 4.

Correlation between the extent of AKOS-022 inhibition of apoptosis and VDAC1 oligomerization. HeLa cells were incubated with the indicated concentrations of AKOS-022 for 2 h and then with or without selenite (30 μm, 3 h) (A–C) or cisplatin (15 μm, 20 h) (B, D, and E). Cells were harvested and either cross-linked with EGS (300 μm, 15 min) and analyzed for VDAC1 oligomerization by immunoblot using anti-VDAC1 antibodies (A and D) or assayed for apoptotic cell death using annexin V-FITC/PI staining and FACS analysis (B, C, and E). The white asterisks in A and D indicate monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular cross-linked monomer VDAC1. Representative FACS results are presented (B). Quantitative analysis of the inhibition of VDAC1 dimer levels and cells undergoing apoptosis (annexin-positive cells) as induced by selenite (C) or cisplatin (E) are presented as a function of AKOS-022 concentration. The results reflect means ± S.D. (n = 3). F, quantitative analysis of the extent of apoptosis inhibition as a function of the inhibition of VDAC1 dimer formation as induced by selenite (■) or cisplatin (□), and as analyzed at the identical AKOS-022 concentration.
Development of New VDAC1-interacting Molecules Inhibiting Oligomerization, HK Detachment, Cytochrome c Release, and Apoptosis
AKOS-022 provided the structural basis for the design of several novel compounds. Two such molecules, VBIT-3 and VBIT-4, were successfully synthesized by ChemPartner. Two main changes were made to the core of AKOS-022 to obtain VBIT-3 and VBIT-4. First, the piperazine ring was conjugated with an aniline moiety in both molecules instead of a piperidine in AKOS-022, and second, the central part of the AKOS-022 core corresponding to a pyrrolidine-2,5-dione ring was linearized to obtain the butanamide linear moiety found in VBIT-4.
The parent molecule (AKOS-022) and its two derivatives (Fig. 5A) were tested for their ability to directly interact with VDAC1 and inhibit VDAC1 oligomerization and apoptosis as induced by a known pro-apoptotic agent (Figs. 5 and 6). The direct interaction between purified VDAC1 and the tested compounds was measured by assessing VDAC1 channel conductance, following its reconstitution into a planar lipid bilayer (PLB). AKOS-022, VBIT-3, and VBIT-4 interacted with purified VDAC1 and reduced its channel conductance, especially at voltages between −40 and +40 mV (Fig. 5, B and C). To obtain a quantitative analysis of the interaction of the compounds with VDAC1 and to derive dissociation constants, a microscale thermophoresis (MST) interaction assay was performed. The fraction of VDAC1 bound to the compound was analyzed as a function of their concentration (Fig. 5D). The dissociation values were derived from the curves showing that AKOS-022 and VBIT-4 interacted with VDAC1 with similar affinity but were 2-fold higher than that of VBIT-3 (Fig. 5, D and E).
FIGURE 5.

Compounds directly interact with purified VDAC1 and reduce channel activity. A, chemical structure of the parent molecule (AKOS-022) and two newly synthesized molecules (VBIT-3 and VBIT-4). B, purified VDAC1 was reconstituted into a PLB, and currents through VDAC1, in response to a voltage step from 0 to 10 mV, were recorded before and 30 min after the addition of 40 μm AKOS-022, VBIT-3, or VBIT-4. C, multichannel recordings of VDAC1 conductance as a function of voltage, and the average steady-state conductance of VDAC1 before (■) and 30 min after the addition of AKOS-022 (○),VBIT-3 (□), or VBIT-4 (●). Relative conductance (G/Go, conductance/maximal conductance) was determined at a given voltage. The data were normalized according to the conductance at −10 mV (maximal conductance). D, purified VDAC1 (133 nm), labeled using the NanoTemper fluorescent protein-labeling Kit BLUE, was incubated with increasing concentrations of AKOS-022 (○),VBIT-3 (□), or VBIT-4 (●) (0.3–100 μm). After 20 min of incubation, 3–5 μl of the samples were loaded into MST-grade glass capillaries, and the thermophoresis process was measured using the Monolith-NT115 apparatus. The results are presented as % of the bound fraction calculated as follows: fraction bound = 100 × (F − Fmin)/(Fmax − Fmin). E, VDAC1 binding affinities of the tested compounds derived from the MST measurements presented in D. The results shown correspond to means ± S.D. (n = 3). F, VBIT-4 binding to recombinant purified VDAC isoforms as revealed using the MST method. ○, ●, and □ indicate binding by VDAC1, VDAC2, and VDAC3, respectively. Inset shows Commassie Blue staining of the purified proteins used.
FIGURE 6.

Inhibition of selenite-induced VDAC1 oligomerization by the tested compounds. A, HEK-293 cells were incubated with the indicated concentration of AKOS-022, VBIT-3, or VBIT-4 for 2 h and then with or without selenite (15 μm, 4 h), harvested, cross-linked with EGS (300 μm, 15 min), and analyzed by immunoblot using anti-VDAC1 antibodies. The positions of VDAC1 monomers and multimers are indicated. The white asterisk indicates monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular crossed-linked monomeric VDAC1. The positions of molecular size protein standards are provided. B, quantitative analysis of selenite-induced VDAC1 dimer formation by the various compounds, presented as percent of inhibition. The results show means ± S.D. (n = 3). C, inhibition of selenite-induced apoptosis by the compounds as analyzed using annexin V-FITC/PI staining and FACS. ●, ○, and □ indicate VBIT-4, AKOS-022, and VBIT-3, respectively. D, inhibition of Cyto c release from the mitochondria as induced by selenite. To assess Cyto c release, cells were incubated on ice for 10 min with 0.025% digitonin and centrifuged, and the pellet (mitochondria) and supernatants (cytosol) were subjected to SDS-PAGE and immunoblotting, using anti-Cyto c, antibodies. Anti-VDAC1 and anti-GAPDH antibodies were used to verify that the cytosolic extracts are free of mitochondria. E, quantitative analysis of selenite-induced Cyto c release to the cytosol by the tested compounds. The data are presented as percent inhibition. The results shown correspond to means ± S.D. (n = 3). ●, ○, and □ indicate VBIT-4, AKOS-022, and VBIT-3, respectively. F, IC50 values (μm) of the compounds as derived from B, C, and E. The results shown correspond to means ± S.D. (n = 3). G, VBIT-4 and AKOS-022 protection against STS-induced cell death. HEK cells were incubated with VBIT-4 or AKOS-022 (15 μm, 2 h), and then apoptosis was induced by STS (0.2 μm, 3 h). Apoptotic cell death was analyzed by acridine orange and ethidium bromide staining. It should be noted that with STS the apoptotic cells undergo fragmentation, and FACS analysis was problematic. H, VBIT-4 effect on As2O3-induced cell death. HEK cells were incubated with VBIT-4 (15 μm, 2 h) and then apoptosis was induced by incubation with of As2O3 (16 h, 30 μm). Cell death was analyzed PI staining and FACS analysis. I, inhibition of HK-I detachment from the mitochondria as induced by selenite. To assess HK-I detachment, cells were treated as in D and subjected to SDS-PAGE and immunoblotting, using anti-HK-I, antibodies. Anti-VDAC1 and anti-GAPDH antibodies were used to verify that the cytosolic extracts are free of mitochondria. The levels of HK in the supernatants are presented as percentages of selenite-induced HK detachment (after accounting for the amount of cytosolic HK present in untreated cells).
The interaction of VBIT-4 with recombinant purified VDAC1, VDAC2, and VDAC3 was analyzed using the MST method (Fig. 5F). VBIT-4 bound to the three recombinant isoforms with a similar binding affinity, although 3-fold lower than that of VDAC1 purified from rat liver mitochondria.
The effects of AKOS-022 and its two synthetic derivatives, VBIT-3 and VBIT-4, on VDAC1 oligomerization, Cyto c release from mitochondria and apoptosis in HEK-293 cells, as induced by selenite, were analyzed (Fig. 6, A–E). The results indicated that VBIT-4 was more potent and more effective than VBIT-3 and AKOS-022 in inhibiting all three apoptosis-related activities. Importantly, the IC50 values of VBIT-4 in all three assays were in the same range of concentrations (about 1.8–2.9 μm) (Fig. 6F).
VBIT-4 and AKOS-022 also inhibited apoptosis as induced by STS (Fig. 6G). However, when apoptosis was induced by As2O3, addition of VBIT-4 or AKOS-022 resulted in slightly enhanced apoptosis rather than inhibition (Fig. 6H). The reasons for this effect is not clear (see under “Discussion”).
Finally, we also analyzed the effect of VBIT-4 on mitochondrion-bound HK (Fig. 6I). Apoptosis induction by selenite resulted in the detachment of mitochondrion-bound HK that was subsequently detected in the supernatant of digitonin-treated cells. This selenite-induced HK detachment was strongly inhibited in the presence of VBIT-4. The results also show that in the absence of apoptosis induction, 80% of the HK pool is bound to the mitochondria.
Separation and Activity Evaluation of Two VBIT-4 Enantiomers
VBIT-4 contains a chiral carbon, allowing for two enantiomers (Fig. 7A). VBIT-4 was synthesized as a racemate, and racemic VBIT-4 was used in all of the experiments described above. Given the possible lack of activity of one of the enantiomers, the two VBIT-4 enantiomers were separated by chiral column chromatography, and their effects on VDAC1 oligomerization and apoptosis were investigated in HEK-293 and HeLa cells (Fig. 7, B and C). Both enantiomers inhibited VDAC1 oligomerization and apoptosis as induced by selenite in an identical manner.
FIGURE 7.

VBIT-4 and its enantiomers are equally effective in inhibiting selenite-induced VDAC1 oligomerization and cell death. A, chemical structure of the VBIT-4 two enantiomers, VBIT-4-1 and VBIT-4-2. B, HEK-293 cells were incubated with VBIT-4, VBIT-4-1, or VBIT-4-2 (10 μm) for 2 h and then with or without selenite (15 μm, 4 h). The cells were harvested, cross-linked with EGS (300 μm, 15 min), and analyzed by immunoblot using anti-VDAC1 antibodies. The positions of VDAC1 monomers and multimers are indicated. The white asterisk indicates monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular crossed-linked monomeric VDAC1. Cell death was measured using PI staining and FACS analysis and is indicated at the bottom of the blot. C, HeLa cells were incubated with the indicated concentrations of the tested compounds for 1 h and then with or without selenite (25 μm, 3 h). Cells were harvested and assayed for cell death, using PI staining and FACS analysis. The results shown correspond to means ± S.D. (n = 3).
To demonstrate that VBIT-4 and AKOS-022 inhibited VDAC1 oligomerization and apoptosis regardless of the cell type used, we also tested the compound-mediated inhibition of cisplatin-induced apoptosis in the neuroblastoma cell line SH-SY5Y, in addition to HEK-293 and HeLa cells (Fig. 8, A and B). Both compounds inhibited cisplatin-induced VDAC1 oligomerization and apoptosis in a similar manner.
FIGURE 8.

Compounds inhibition of VDAC1 oligomerization and apoptosis in neuronal cells and Bax/Bak-lacking MEFs cells. A and B, SH-SY5Y cells were incubated with AKOS-022 or VBIT-4 (30 μm, 2 h) and then with or without cisplatin (20 μm, 20 h). The cells were harvested, cross-linked with EGS (200 μm, 15 min), and analyzed by immunoblot using anti-VDAC1 antibodies (A) or apoptosis (B) as analyzed using annexin V-FITC/PI staining and FACS. The white asterisk indicates monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular cross-linked monomer VDAC1. B, quantitative analysis of cisplatin-induced VDAC1 dimer formation (gray columns) and apoptosis (black columns). The results shown correspond to mean ± S.D. (n = 3), p < 0.001(***). C, Bax−/−/Bak−/− MEFs cells were incubated with AKOS-022 or VBIT-4 (20 μm, 2 h) and then with or without cisplatin (20 μm, 20 h). The cells were harvested, cross-linked with EGS (200 μm, 15 min). The white asterisk indicates monomeric VDAC1 with modified electrophoretic mobility, representing intramolecular cross-linked monomer VDAC1. D, Bax−/−/Bak−/− MEFs cells were treated as in C and assessed for Cyto c release. Cells were incubated on ice for 10 min with 0.025% digitonin and centrifuged, and the pellet (Mito, mitochondria) and supernatants (cytosol) were subjected to SDS-PAGE and immunoblotting, using anti-Cyto c antibodies. Anti-VDAC1 and anti-GAPDH antibodies were used to verify that the cytosolic extracts are free of mitochondria. E, quantitative analysis of cisplatin-induced VDAC1 dimer formation (gray bars) and Cyto c release (black bars). The results shown correspond to mean ± S.D. (n = 3), p < 0.01 (**) or <0.001 (***). The positions of molecular size protein standards are provided.
VBIT-4-mediated Inhibition of VDAC1 Oligomerization in Bax/Bak-lacking Cells
As the pro-apoptotic Bax and Bak proteins are considered central to apoptosis, we asked whether the protection against apoptosis conferred by the molecules tested was VDAC1-dependent but not Bax/Bak-dependent (Fig. 8, C–E). The results showed that AKOS-022 and VBIT-4 were effective in inhibiting cisplatin-induced VDAC1 oligomerization and Cyto c release in Bax−/−/Bak−/− mouse embryonic fibroblasts (MEFs), with VBIT-4 being the most effective inhibitor.
Compounds Providing Protection against Mitochondrial Depolarization, ROS Production, and [Ca2+]i Increase, as Induced by Apoptosis Induction
Apoptosis induction was shown to disrupt cellular Ca2+ homeostasis and energy production (41). Indeed, many anti-cancer drugs and other cytotoxic agents, such as thapsigargin, staurosporine, As2O3, and selenite, induce apoptotic cell death, as well as disrupt cell Ca2+ homeostasis (36, 37). Thus, we investigated a possible protective effect of VBIT-4 and AKOS-022 on the increased intracellular [Ca2+]i elicited by selenite in HEK-293 cells. As expected, selenite increased [Ca2+]i, as monitored using Fluo-4 and FACS analysis (Fig. 9A). [Ca2+]i was, however, maintained at the basal level when the cells were pre-incubated with AKOS-022 or VBIT-4.
FIGURE 9.

Compound inhibition of the selenite-induced increases in intracellular calcium and ROS levels and decreases in mitochondrial membrane potential. HEK-293 cells were incubated with AKOS-022 or VBIT-4 (15 μm, 2 h) and then with or without selenite (15 μm, 4 h). A, cells were harvested, and intracellular calcium ([Ca2+]i) levels were measured using Fluo-4 and FACS analysis, and representative FACS results are presented. Quantitative analysis of the results are presented as percent maximal [Ca2+]i at the bottom. B, mitochondrial membrane potential (ΔΨm) was analyzed with TMRM and FACS analysis. Representative FACS results are presented. CCCP (25 μm, 30 min) served as a positive control for ΔΨm dissipation, and the CCCP-sensitive TMRM fluorescence was considered as 100%, and the results are presented at the bottom. C, cellular ROS levels were analyzed with carboxy-H2-DCFDA and FACS analysis, with representative FACS results, and quantitative analyses (bottom) are shown. D, mitochondrial superoxide was detected with MitoSOX Red and flow cytometry with representative FACS results, and quantitative analyses (bottom) are shown. The results shown in A–D correspond to mean ± S.D. (n = 3), p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
Apoptosis induction may affect the mitochondria membrane potential (ΔΨm) directly or via increased [Ca2+]i, which led to an increase in mitochondrial Ca2+, a process expected to lead to dissipation of ΔΨm (42). We thus tested the effect of VBIT-4 and AKOS-022 on ΔΨm. Both compounds prevented the decrease in ΔΨm as measured using tetramethylrhodamine methyl ester (TMRM) (Fig. 9B). The compounds were further tested for their ability to inhibit overall cellular reactive oxidative species (ROS) production, as analyzed by 2′,7′-dichlorofluorescein (DCF) fluorescence, and in mitochondria as measured by MitoSOX RedTM, a mitochondrial superoxide indicator (Fig. 9, C and D). The increases in both cellular and mitochondrial ROS levels as induced by selenite were completely prevented by VBIT-4 and AKOS-022.
These results showed that the compounds inhibited not only VDAC1 oligomerization, Cyto c release, and apoptosis but also protected against the mitochondrial dysfunction associated with apoptosis induction and compromised cell energy production.
Discussion
Accumulating evidence points to VDAC1 as functioning in mitochondrion-mediated apoptosis involving the release of apoptogenic proteins, such as Cyto c, leading to caspase activation (4, 5, 43, 44). Based on our previous studies (19, 23, 30, 36, 37, 45), we proposed that Cyto c is transported across the OMM via a large flexible pore formed within oligomeric VDAC1 upon apoptosis induction. Thus, shifting the equilibrium of oligomeric toward monomeric VDAC1, thus preventing Cyto c release, is an effective approach for blocking apoptosis at an early stage.
Despite the fact that apoptotic mechanisms are well defined, there is still no substantial therapeutic strategy to stop or even slow this process. Thus, there is an unmet need for therapeutic agents able to block or slow apoptosis in neurodegenerative and cardiovascular diseases. Currently, the majority of known apoptosis inhibitors directly target end steps in the apoptosis pathway, such as blocking caspase activity (16, 46, 47). At the same time, known VDAC1 inhibitors are nonspecific and were effective in inhibiting apoptosis at comparatively high concentrations (19, 30, 38). Thus, we searched for a more effective VDAC-specific apoptosis inhibitor. Specifically, we sought anti-apoptotic drugs targeting VDAC1 to prevent its oligomerization, an early and critical step in the progression of apoptosis. These molecules would be potential candidates for treating neurodegenerative disorders (16, 17) and various cardiovascular diseases, where enhanced apoptosis also occurs (48–50).
A BRET-based method was used to follow VDAC1 oligomerization in living cells as part of a screen of a library of small compounds, with the aim of identifying possible potent and effective inhibitors of VDAC1 oligomerization and thus apoptosis. Following three cycles of structure-activity relationship studies, a commercially available molecule, AKOS-022, was identified as a compound interacting with VDAC1 and inhibiting both VDAC1 oligomerization and apoptosis (Fig. 10). AKOS-022 inhibited VDAC1 oligomerization and apoptosis at a pharmacological range of concentrations (Fig. 4). Furthermore, AKOS-022 interacts directly with purified VDAC1, as revealed by MST and its effect on bilayer-reconstituted channel conductance (Fig. 5).
FIGURE 10.
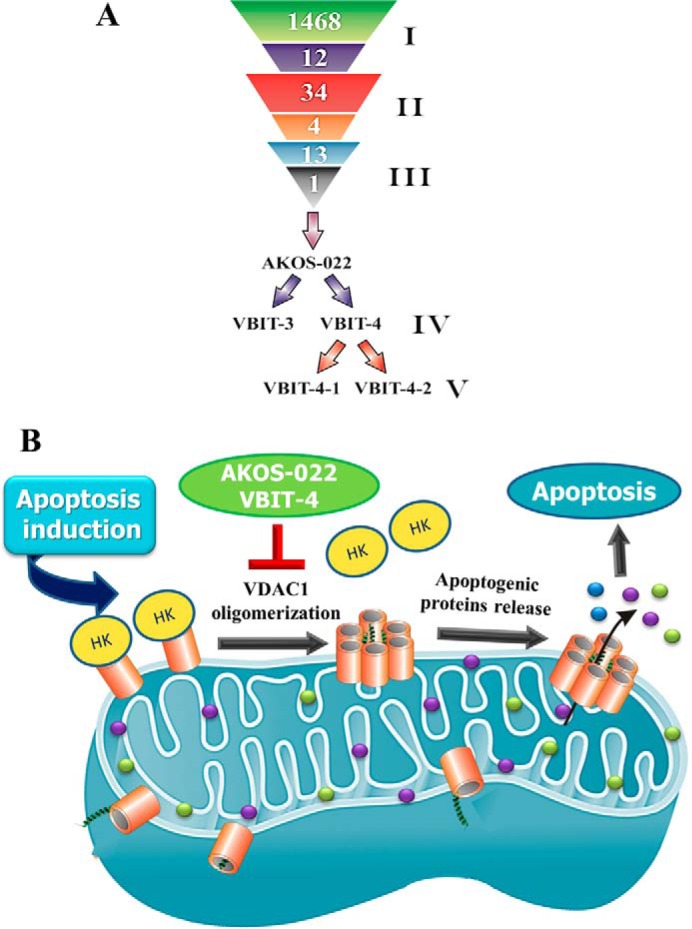
Summary of the three rounds of compound screening and the mode of VBIT-4- and AKOS-022-mediated protection against apoptosis. A, lead compound development: I, first screen, HTS of 1,468 small molecules by BRET2 resulted in the identification of 12 compounds that inhibited VDAC1 oligomerization. II, second round of screening of 34 molecules, selected based on the 12 compounds identified in the first screen. The screen identified four compounds displaying strong inhibition of VDAC1 oligomerization. III, third round of screening addressed 13 molecules designed based on the four compounds identified in the second screen. The most potent compound, AKOS-022, was identified. IV, AKOS-022 was used for the design and synthesis of the novel derivatives, VBIT-3 and VBIT-4. V, VBIT-4 enantiomers. B, proposed model for VDAC1-based apoptosis and its inhibition by VBIT-4 and AKOS-022. Apoptotic stimuli or pathological conditions lead to VDAC1 oligomerization, forming a pathway for the release of apoptogenic proteins (e.g. Cyto c and AIF). VBIT-4 and AKOS-022 directly interact with VDAC1 and inhibit its oligomerization, the subsequent HK detachment, and release of apoptogenic proteins from the mitochondrial inter-membrane space, such that apoptosis is inhibited.
Based on the structure of AKOS-022, we designed several new compounds of which two, VBIT-3 and VBIT-4, directly interacted with VDAC1 and strongly inhibited VDAC1 oligomerization and apoptosis as induced by various means in several cell types (Figs. 5–8). VBIT-4 was more potent and more effective than VBIT-3 and AKOS-022 in inhibiting VDAC1 oligomerization, Cyto c release, and apoptosis as induced by cisplatin, STS, and selenite. Interestingly, VBIT-4 enhanced As2O3-induced rather than inhibited apoptosis. This could be associated with one or more defined molecular targets of As2O3 and molecular mechanisms of action that that are not affected or may be enhanced by VBIT-4 inhibition of VDAC1 oligomerization. These include the following: induction of ROS formation; down-regulation of Bcl-2 expression (51); direct binding to promyelocytic leukemia, a tumor suppressor protein, and induction of its oligomerization (52); interaction with sulfhydryl groups (53); induction of VDAC homo-dimerization, which can be prevented by Bcl-xL (54), and VDAC1 oligomers (30); and the formation of Bax and VDAC hetero- and homo-oligomers (55). The linear relationship between inhibition of VDAC1 oligomerization and inhibition of apoptosis by AKOS-022 (Figs. 4 and 6) and VBIT-4 (Fig. 6) offers further support for the involvement of VDAC1 oligomerization in the induction of apoptosis.
VBIT-4 interacted with the three recombinant VDAC isoforms with a similar affinity of 53 ± 3 μm. This is about 3-fold lower than that of VDAC1 purified from rat liver mitochondria. The lower affinity of the recombinant proteins may result from the unfolding and refolding process resulting in modified conformations. The three VDAC isoforms share high homology and structural similarities (3). This can explain their capacity to bind VBIT-4 with similar affinity. In addition, the three recombinant isoforms share a short tail of histidines that was shown to be of no importance for VDAC activity in reconstituted membranes (56) but can influence the binding of VBIT-4 to the proteins.
VBIT-4 had no effect on cell growth and viability. Moreover, mice that were exposed to the compound for 4 months showed no toxic signs, as reflected by weight, behavior, organ histo-chemistry, and more (results not shown). It thus seems that VBIT-4 interaction with any of the VDAC isoforms has no effect on cell functional homeostasis. As VBIT-4 inhibits VDAC1 oligomerization, VDAC1 is the major isoform in most cell types, and no VDAC2 or VDAC3 oligomerization has been reported, it is reasonable to assume that the anti-apoptotic effect of VBIT-4 is mainly mediated via its interaction with VDAC1.
In addition to inhibition of apoptosis, VBIT-4, as well as AKOS-022, prevented the elevation of [Ca2+]i associated with apoptosis induction, and thus Ca2+ accumulation by the mitochondria. This prevents the collapse of the associated ΔΨm and the increase in ROS production (Fig. 9) (57) and apoptosis-associated mitochondrial dysfunction.
Interestingly, VBIT-4, the more active and potent compound, has only one structural difference that distinguishes it from VBIT-3, namely a linear 4-hydroxybutanamide moiety in place of a pyrrolidine-2,5-dione rigid ring. This change makes VBIT-4 more flexible (because of the presence of a linear moiety versus a cycle group) and hydrophilic (alcohol versus amide). The increased flexibility of VBIT-4 is important when considering its interference with protein-protein interactions associated with VDAC1 oligomerization. Moreover, VBIT-4 is a chiral molecule, yet the two enantiomers showed identical activity in inhibiting VDAC1 oligomerization (Fig. 7). This suggests that flexible VBIT-4 interferes with the interactions between VDAC1 monomers in areas that are widely accessible for both the VBIT-4 R and S conformations.
The VDAC1-interacting molecule VBIT-4 can be used to further explore the function of VDAC1 in controlling metabolism, energy production, transport of cholesterol, and apoptosis. Moreover, VBIT-4, as an apoptosis inhibitor, can be used for therapeutic purposes in apoptosis-associated disorders, such as neurodegenerative and cardiovascular diseases. Indeed, many dying neurons in brains of patients with neurodegenerative diseases appear to display morphological features of apoptosis, such as chromatin condensation, DNA fragmentation, and activation of caspases (58). There is also evidence suggesting that caspases assume a role in Alzheimer's and Parkinson's diseases (59). Thus, the activated cell death that occurs in neurological diseases makes inhibition of apoptosis with reagents such as VBIT-4 an attractive therapeutic approach.
In other types of human disease, such as heart failure, myocardial infarction, and cardiac ischemia/reperfusion injury, apoptosis, necrosis, and autophagy of cardiac myocytes were detected (48–50). Activation of the mitochondrial apoptotic pathway has also been implicated in ischemia/reperfusion injury, involving the release of Cyto c from mitochondria, followed by activation of caspase-9 in the myocardium (60). In addition, atrial fibrillation, the most common cardiac arrhythmia and associated with high morbidity and mortality rate in adults, was also linked to apoptotic processes in cardiomyocytes (61). Apoptosis is a contributing factor in the initiation and progression of fibrosis, which is a main factor in the occurrence, development, and poor prognosis of atrial fibrillation patients (62, 63). Thus, VBIT-4 can be used to inhibit apoptosis in cardiac myocytes as induced under pathological conditions.
The inhibition of mitochondrion-bound HK detachment by VBIT-4 (Fig. 6H) is extremely important. In addition to the metabolic function assigned to mitochondrion-bound HK (HK-I and HK-II), namely the coupling of cytosolic glycolysis to mitochondrial oxidative phosphorylation, it is now clear that VDAC1-bound HK also prevents the release of pro-apoptotic factors and subsequent apoptosis accompanied with detachment of HK (31, 32, 64–71). Indeed, several pro-apoptotic agents have been shown to induce VDAC1-HK complex dissociation (7, 72–74). The results presented here show that VBIT-4 inhibited HK detachment, as induced by apoptosis induction. This finding supports the concept of HK detachment being a prerequisite for apoptosis induction. VBIT-4 inhibition of HK detachment suggests that such detachment is associated with VDAC1 oligomerization, with VBIT-4 inhibiting oligomerization and preventing HK detachment.
In this respect, several pathological conditions, such as Parkinson's disease (75), mood and psychotic disorders (76), Alzheimer's disease (77), and schizophrenia (78) were related to HK detachment from the mitochondria. Thus, VBIT-4 may be valuable in treating these pathological conditions.
Finally, in this respect, VDAC1 overexpression was shown to lead to apoptotic cell death by shifting monomeric to oligomeric VDAC1, enabling Cyto c release and hence apoptosis in the absence of any apoptosis stimuli (37, 79–82). VDAC was shown to be overexpressed in brains of Alzheimer's disease patients (83–85), in pancreatic β-cells exposed to high glucose (86), and in cardiovascular diseases (87). This VDAC overexpression may be responsible for the apoptosis observed in these diseases. Thus, VBIT-4 interacting with VDAC and inhibiting apoptosis, HK detachment from the mitochondria, and the associated increase in ROS and [Ca2+]i may also protect against cell death in these diseases.
Experimental Procedures
Materials
Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), carboxymethylcellulose (CMC), cisplatin, cytochalasin B, dimethyl sulfoxide (DMSO), dl-dithiothreitol (DTT), EDTA, HEPES, leupeptin, phenylmethylsulfonyl fluoride (PMSF), N-decane, sodium selenite, soybean asolectin, STS, TMRM, and Tris were purchased from Sigma. N,N-Lauryl-(dimethyl)-amine oxide (LDAO) was obtained from Fluka (Buchs, Switzerland). Coelenterazine (DeepBlueC (DBC)) was obtained from Bioline (Taunton, MA). Hydroxyapatite (Bio-Gel HTP) was procured from Bio-Rad. Digitonin came from Calbiochem-Novabiochem (Nottingham, UK). Celite was purchased from the British Drug Houses (London, UK). Rabbit monoclonal antibodies against VDAC1 (ab154856) and against HK-I (ab150423) and mouse monoclonal antibodies against GAPDH (ab9484) were obtained from Abcam (Cambridge, UK). Monoclonal antibodies against actin were obtained from Millipore (Billerica, MA), and anti-Cyto c antibodies (556433) were obtained from BD Biosciences. Fluo-4 AM, carboxy-H2-2,7-dichlorofluorescein diacetate, and MitoSOX Red were acquired from Invitrogen. Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit antibodies were obtained from Promega (Madison, WI). EGS was obtained from Pierce. Annexin V-fluorescein isothiocyanate (FITC) was from Enzo Life Sciences (Lausen, Switzerland). Dulbecco's modified Eagle's medium (DMEM) and the supplements fetal bovine serum (FBS), l-glutamine, and penicillin/streptomycin were purchased from Biological Industries (Beit-Haemek, Israel).
The diversity set compound library was provided by NCI (National Institutes of Health) in frozen bar-coded 96-well plates (465030–466730), each well containing 20 μl of 10 mm stock solution in DMSO with plate map accession numbers. Compounds used for the second and third round of screenings were purchased from Diverchim (France) and AKos Consulting & Solutions (Germany). VBIT-3 and VBIT-4 were synthesized by ChemPartner (Chengdu, China).3
Plasmids
Plasmids encoding the fusion proteins rat (r)VDAC1-GFP2 and rVDAC1-luc were constructed using the BRET2 plasmids (PerkinElmer Life Sciences). The vdac1 gene was cloned into BamHI and HindIII sites of the BRET2 plasmids (N2 variants) and amplified using the forward primer CGAAGCTTATGGCTGTGCCACCCACGTATGCC and the reverse primer GGATCCGCCGCCGCCGGACCGCCGCCGCCTGCTTGAAATTC. The reverse primer was designed to contain a double linker sequence ((GGGS)2) connecting vdac1 and the RLuc or GFP2 genes that introduced flexibility to the region (88).
Plasmids encoding shRNA against human (h) VDAC1 (hVDAC1) for specific silencing of endogenous hVDAC1 were introduced into an shRNA-expressing vector. The hVDAC1-shRNA-encoding sequence was created using the two complementary oligonucleotides indicated below, each containing the 19-nucleotide target sequence of hVDAC1(337–355), followed by a short spacer and an antisense sequence of the target:oligonucleotide 1, AGCTTAAAAACACTAGGCACCGAGATTATCTCTTGAATAATCTCGGTGCCTAGTGTG, and oligonucleotide 2, GATCCACACTAGGCACCGAGATTATTCAAGAGATAATCTCGGTGCCTAGTGTTTTTTA, with the VDAC1-derived sequence underlined. The hVDAC1-shRNA-encoding sequence was cloned into the BglII and HindIII sites of the pSUPERretro plasmid (OligoEngine, Seattle, WA), containing a puromycin-resistance gene. Transcription of this sequence under the control of the H1 RNA promoter of RNA polymerase III produces a hairpin (hVDAC1-shRNA).
Tissue Culture
HEK, HeLa, SH-SY5Y, and K-Ras-transformed Bax−/−/Bak−/− MEF cell lines were grown at 37 °C under an atmosphere of 95% air and 5% CO2 in DMEM supplemented with 10% FBS, 2 mm l-glutamine, 1,000 units/ml penicillin, and 1 mg/ml streptomycin. T-REx-293 cells (HEK cells stably containing the pcDNA6/TR regulatory vector and thus expressing the tetracycline repressor; Invitrogen) stably expressing hVDAC1-shRNA and showing low (10–20%) endogenous VDAC1 expression (referred to as T-REx-pS10) were grown under the same conditions as HEK cells, except with an addition of 5 μg/ml blasticidin.
Cell Transfection
In the BRET experiments, T-REx-pS10 cells were transfected using calcium phosphate. Transfections were carried out with 0.1 μg of a plasmid coding for rVDAC1-Rluc and with 0.8 μg of a plasmid coding for rVDAC1-GFP2, respectively. Cells were analyzed for induction of apoptosis and BRET2 48–72 h post-transfection. For negative controls, cells were either transfected with plasmids encoding for rVDAC1-Rluc (0.1 μg of DNA) and GFP2 (0.8 μg) or with a plasmid encoding for rVDAC1-Rluc (0.1 μg) and plasmid pcDNA4/TO (0.8 μg).
Compound Library Testing
The drug-like compound library used in this study was provided by the NCI (National Institutes of Health) in frozen bar-coded 96-well plates, with each well (with a plate map accession number) containing 20 μl of a 10 mm stock solution in 100% DMSO diluted with DMSO to yield 2 mm of the tested compound and stored frozen. The screen was conducted using the cells in a 96-well format for enhancement of BRET2 signals to identify inhibitors of VDAC1 oligomerization. Compounds (1 μl of 2 mm stock solutions) were added to a final concentration of 10 μm in 100 μl (1% final DMSO concentration). The compounds used for the second and third rounds of screening were analyzed for their effects on VDAC1 oligomerization by chemical cross-linking, as described below.
BRET Assay
DNA encoding genetically engineered fusion proteins rVDAC1-Rluc (in which RLuc was connected to rVDAC1 at the C-terminal position through a linker (GGGS)) and rVDAC1-GFP2 (in which the GFP2 was fused to the rVDAC1 C terminus) was cloned into BRET2 vectors. rVDAC1-GFP2 and rVDAC1-Rluc were expressed in T-REx cells stably expressing shRNA-hVDAC1 and a low level of endogenous hVDAC1 (79). shRNA-VDAC1, being specific to human VDAC1, allowed the expression of rVDAC1 and decreased the participation of endogenous hVDAC in oligomerization, thereby enhancing the BRET2 signal. rVDAC1-GFP2 and rVDAC1-Rluc expression levels were correlated with the amount of plasmids used. Specifically, 0.8 μg of rVDAC1-GFP2 and 0.1 μg of rVDAC1-Rluc were found to give the best signal.
Following incubation, cells were harvested, washed twice with PBS, resuspended in 200 μl of PBS, and divided between two wells of a 96-well clear-bottom plate (Grenier). Luciferase activity was assayed using the membrane-permeable substrate DeepBlueC coelentrazine in PBS supplemented with MgCl2 (1 g/liter) and glucose (1 g/liter), with DBC being added to a final concentration of 5 μm just before luminescence was measured. Appropriate conditions for resolving VDAC1 oligomerization in mammalian living cells using BRET2 technology were developed. These conditions considered the number of cells to be plated, amounts and ratios of rVDAC1-Rluc and rVDAC1-GFP2 encoding plasmids, apoptosis inducer concentration, the time of incubation, and concentration of the luciferase substrate coelentrazine to be used. Cells transiently expressing rVDAC1-Rluc and rVDAC1-GFP2, as well as control cells, were incubated with the apoptosis inducer.
The BRET2 signal represents the ratio of the GFP2 fluorescence, measured at its emission wavelength (510 nm), over the light intensity (luminescence) emitted at 395 nm. All measurements were performed using the Infinite 200 ELISA reader (Tecan). BRET2 signals were defined as GFP2/Rluc intensity ratio and calculated as follows. (a) The BRET2 signals obtained in VDAC1-RLuc/pcDNA4/TO cells (control cells) were subtracted from the signals obtained in cells expressing VDAC1-Rluc and VDAC1-GFP2. (b) The net ratios of Renilla luciferase and GFP2 activities (GFP2/luciferase ratio after the subtraction of the BRET2 signals from control cells) were calculated. (c) The ratios of BRET2 signals between different cells exposed and not exposed to apoptosis inducers were compared.
High-throughput Screening to Identify Inhibitors of VDAC1 Oligomerization
The screen was conducted using the cells in a 96-well format for enhancement of BRET2 signals to identify inhibitors of VDAC1 oligomerization. T-REx cells with low VDAC1 levels were transfected to express rVDAC1-GFP2 and rVDAC1-Rluc and seeded at a density of 9,000 cells/well in a 96-well plate. Compounds (1 μl of 2 mm stock solutions) were added to a final concentration of 10 μm in 100 μl (1% final DMSO concentration). The cells were pre-incubated for 1 h with the NCI compounds and then incubated with the apoptosis inducers for an additional 3 h (STS, 1 μm; selenite, 30 μm; As2O3, 60 μm). The tested NCI compounds were dispensed by a robotic system into the 96-well plates. After treatment, the medium was removed and assayed for BRET2 signals as described above. Liquid handling was done with the Tecan (Männedorf, Switzerland) Freedom 150 Robotic & MCA Liquid Handling System, although luciferase luminescence and fluorescence readings were obtained a robot-integrated Tecan Infinite M1000 reader.
Cross-linking Experiments
Cells (2.5–3 mg/ml) in PBS were harvested after the appropriate treatment and incubated with the cross-linking reagent EGS, pH 8.3, for 15 min. Samples (60–80 μg of protein) were subjected to SDS-PAGE and immunoblotting using anti-VDAC1 antibodies. Quantitative analysis of immunoreactive VDAC1 dimer, trimer, and multimer bands was performed using FUSION-FX (Vilber Lourmat, France).
Gel Electrophoresis and Immunoblot Analyses
Samples (10–40 μg of protein) were subjected to SDS-PAGE and immunoblotting using monoclonal anti-VDAC1, anti-Cyto c, or anti-actin antibodies, followed by HRP-conjugated anti-mouse or anti-rabbit IgG, serving as secondary antibodies and detected by chemiluminescence. To detect VDAC1 oligomers, membranes were treated with 0.1 m glycine, pH 2.0, prior to immunoblotting and washed several times with 0.1% Tween 20 in Tris-buffered saline. Band intensities were imaged and quantified using FUSION-FX (Vilber Lourmat, France).
Flow Cytometry Using Propidium Iodide (PI) and Annexin V-FITC Staining
Cells (2 × 105), untreated or treated with apoptosis-inducing reagents, were analyzed for apoptotic cell death using PI, annexin V-FITC, and flow cytometer analysis. Cells were collected (1500 × g for 10 min), washed, and resuspended in 200 μl binding buffer (10 mm HEPES/NaOH, pH 7.4, 140 mm NaCl, and 2.5 mm CaCl2). Annexin V-FITC was added according to the recommended protocol (Enzo Life Sciences, Switzerland), and the cells were incubated in the dark for 15 min. Cells were then washed with binding buffer and resuspended in 200 μl of binding buffer, to which PI was added immediately before flow cytometry analysis. At least 10,000 events were collected, recorded on a dot plot, and analyzed by the FACSCalibur flow cytometer software (BD Biosciences).
Cytochrome c Release and HK Detachment from Mitochondria
Cells treated with apoptosis inducers in the absence or presence of the indicated reagent were harvested, washed twice with PBS, pH 7.4, gently resuspended at 6 mg/ml in ice-cold buffer (100 mm KCl, 2.5 mm MgCl2, 250 mm sucrose, 20 mm HEPES/KOH, pH 7.5, 0.2 mm EDTA, 1 mm dithiothreitol, 1 μg/ml leupeptin, 5 mg/ml cytochalasin B, and 0.1 mm PMSF) containing 0.025% digitonin, and incubated for 10 min on ice. Samples were centrifuged at 10,000 × g at 4 °C for 10 min to obtain supernatants (cytosolic extracts) and pellets (contains mitochondria). HK and Cyto c released to the cytosol was analyzed by immunoblotting using HK and Cyto c-specific antibodies. Anti-VDAC1 and anti-GAPDH antibodies were used to verify that the cytosolic extracts are free of mitochondria.
VDAC1 Purification
VDAC1 protein was purified from rat liver mitochondria as described previously (89). Briefly, rat liver mitochondria (5 mg/ml) in 10 mm Tris-HCl, pH 7.2, were incubated with 2% LDAO at 0 °C for 20 min, followed by centrifugation (30 min, 14,000 × g), and the obtained supernatant was loaded onto a dry celite/hydroxyapatite (2:1) column. VDAC1 was eluted with a solution containing 2% LDAO, 10 mm Tris-HCl, pH 7.2, 50 mm NaCl, and 22 mm NaH2PO4. The VDAC1-containing fractions were dialyzed against 10 mm Tris-HCl, pH 7.2, and subjected to a second chromatography step on a carboxymethylcellulose (CMC) column from which VDAC1 was eluted with a solution containing 10 mm Tris-HCl, pH 7.2, 0.1% LDAO, and 500 mm NaCl. The VDAC1-containing fractions were collected and used for VDAC1 channel conductance and MST assays.
Expression, Purification and Refolding of His6-tagged VDAC Isoforms
The preparation of refolded VDAC isoforms was performed essentially as described (90). In brief, Escherichia coli BL21(DE3) cells were transformed with plasmid pET21a containing the VDAC isoform-coding sequences. Protein expression was induced by addition of 1 mm isopropyl β-d-thiogalactopyranoside (Sigma) at an optical density (λ = 595 nm) of ∼0.6 at 37 °C for 3 h, as reported previously (91). The cells were resuspended in 8 m urea, phosphate buffer, pH 8.0, and shaken overnight at 4 °C. After pelleting cell debris by centrifugation, the clear lysate was loaded onto a nickel-nitrilotriacetic acid-agarose-packed column (Qiagen), pre-equilibrated with 10 column volumes of the same buffer. The column was then washed twice with 5 volumes of the same solution at pH 6.2, and the purified proteins were eluted with 5 volumes of the same solution at pH 3.5. The denatured protein mixture was added dropwise to a refolding buffer (25 mm Tris-HCl, pH 7.0, 100 mm NaCl, 1 mm EDTA, 1% (v/v) LDAO, Sigma), to obtain a 10-fold dilution of the urea concentration, and gently stirred overnight at 4 °C. The protein solution was dialyzed against 100 volumes of a dialysis buffer (25 mm Tris-HCl, pH 7.0, 1 mm EDTA, 0.1% LDAO) in Thermo Scientific Slide-A-Lyzer dialysis cassettes (3.5 kDa molecular mass cutoff) changing the dialysis buffer two times after 2 h of stirring and once more 24 h later, at 4 °C. Samples were then subjected to additional purification and concentration using a CMC column (89). Protein purity was verified by SDS-PAGE and Coomassie staining. Purified samples were stored at −20 °C. VDAC protein concentration was determined using SDS-PAGE and Commassie staining with ovalbumin as a standard.
VDAC1 Channel Conductance
The reconstitution of purified rat VDAC1 into a PLB and subsequent single and multiple channel current recordings and data analysis were carried out as described previously (92). Briefly, the PLB was prepared from soybean asolectin dissolved in n-decane (30 mg/ml). Purified VDAC1 (1 ng) was added to the chamber defined as the cis side containing 1 m NaCl, 10 mm HEPES, pH 7.4. Currents were recorded under voltage clamp using a Bilayer Clamp BC-535B amplifier (Warner Instrument, Hamden, CT). The currents, measured with respect to the trans side of the membrane (ground), were low-pass filtered at 1 kHz and digitized on line using a DigiData1440-interface board and pClampex 10.2 software (Axon Instruments, Union City, CA).
Measurement of Superoxide Generation
ROS production was monitored using the oxidant-sensitive dye DCFDA fluorescent probe, a cell-permeable indicator of ROS, which is converted by H2O2 and peroxidases to the 2′,7′-dichlorofluorescein fluorescent derivative and briefly incubated with DCFDA (4 μm) for 30 min. For mitochondrial accumulated ROS, MitoSOX Red (4 μm), mitochondrial superoxide indicator for live-cell imaging was used according to the manufacturer's protocol (Invitrogen). Fluorescence was measured using a FACSCalibur flow cytometer software (BD Biosciences).
MST Analysis
MST analysis was performed using a NanoTemper Monolith NT.115 apparatus as described previously (93). Briefly, purified VDAC1 was fluorescently labeled using NanoTemper Protein labeling kit BLUE (L001, NanoTemper Technologies). A constant concentration of the protein was incubated with different concentrations of the tested inhibitor in PBS. Afterward, 3–5 μl of the samples were loaded into a glass capillary (Monolith NT Capillaries), and thermophoresis analysis was performed (LED 20%, IR laser 20%).
Mitochondrial Membrane Potential Determination
Mitochondrial membrane potential (ΔΨ) was determined using TMRM, a potentially sensitive dye, and a plate reader. HEK-293 cells were treated with the compounds considered here and an apoptotic inducer and subsequently incubated with TMRM (0.5 μm, 20 min). The cells were then washed twice with PBS and examined with FACSCalibur flow cytometer software (BD Biosciences). CCCP-mediated ΔΨ dissipation served as control.
Cellular Ca2+ Analysis
Fluo-4-AM was used to monitor changes in cytosolic Ca2+ levels. HeLa cells (1 × 106 cells/ml) were harvested after the appropriate treatment, collected (1,500 × g for 10 min), washed with HBSS buffer (5.33 mm KCl, 0.44 mm KH2PO4, 138 mm NaCl, 4 mm NaHCO3, 0.3 mm Na2HPO4, 5.6 mm glucose, 0.03 mm phenol red) supplemented with 1.8 mm CaCl2, and incubated with 2.5 μm Fluo-4 in 200 μl of HBSS(+) buffer in the dark for 30 min at 37 °C. After washing the remaining dye, the cells were incubated with 200 μl of HBSS(+) buffer, and changes in cellular free Ca2+concentration were measured immediately via FACS analysis. At least 10,000 events were recorded on the FL1 detector, represented as a histogram, and analyzed with FACSCalibur flow cytometer software. Positive cells showed a shift to an enhanced level of green fluorescence (FL1).
Statistical Analysis
Data are expressed as means ± S.D. Statistical evaluation was carried out using Student's t test (two-tailed) to test for differences between control and experimental results. p values less than 0.05 were considered as significant.
Author Contributions
D. B.-H., M. S., A. S.-K., and R. B.-S. performed the research and analyzed the data. V. S.-B. and D. B.-H. designed the research and wrote the paper. A. G. designed the compounds, followed their synthesis, and wrote the paper. V. D.-P. and S. R. provided purified VDAC isoforms.
Acknowledgment
We thank Dr. Dalia Tyomkin for the help with the BRET assay.
This work was supported Israel Science Foundation Grant 307/13 and by Sima and Philip Needleman research funds. The authors declare that they have no conflicts of interest with the contents of this article.
The synthetic pathway and the analytical data for each compound (including two enantiomers of VBIT-4) will be made available upon request to the corresponding author.
- OMM
- outer mitochondrial membrane
- BRET2
- bioluminescence resonance energy transfer
- CCCP
- carbonyl cyanide m-chlorophenyl hydrazone
- Cyto c
- cytochrome c
- DBC
- DeepBlueC coelentrazine
- DNDS
- 4,4-diisothiocyanostilbene-trans-2,2-disulfonic acid
- EGS
- ethylene glycol bis(succinimidylsuccinate)
- HK
- hexokinase
- MST
- micro-scale thermophoresis
- PLB
- planar lipid bilayer
- STS
- staurosporine
- VDAC
- voltage-dependent anion channel
- ROS
- reactive oxidative species
- TMRM
- tetramethylrhodamine methyl ester
- MEF
- mouse embryonic fibroblast
- LDAO
- N,N-lauryl-(dimethyl)-amine oxide
- CMC
- carboxymethylcellulose
- r
- rat
- h
- human
- PI
- propidium iodide
- HTS
- high-throughput screening
- DCFDA
- 2′,7′-dichlorofluorescein diacetate.
References
- 1. Doran E., and Halestrap A. P. (2000) Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem. J. 348, 343–350 [PMC free article] [PubMed] [Google Scholar]
- 2. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Pinto V., Reina S., Gupta A., Messina A., and Mahalakshmi R. (2016) Role of cysteines in mammalian VDAC isoforms' function. Biochim. Biophys. Acta 1857, 1219–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shoshan-Barmatz V., Arbel N., and Arzoine l. (2008) VDAC, the voltage-dependent anion channel: function, regulation and mitochondrial signaling in cell life and death. Cell Sci. 4, 74–118 [Google Scholar]
- 5. Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., and Arbel N. (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 31, 227–285 [DOI] [PubMed] [Google Scholar]
- 6. Shoshan-Barmatz V., and Ben-Hail D. (2012) VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34 [DOI] [PubMed] [Google Scholar]
- 7. Shoshan-Barmatz V., Ben-Hail D., Admoni L., Krelin Y., and Tripathi S. S. (2015) The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 1848, 2547–2575 [DOI] [PubMed] [Google Scholar]
- 8. Bayrhuber M., Meins T., Habeck M., Becker S., Giller K., Villinger S., Vonrhein C., Griesinger C., Zweckstetter M., and Zeth K. (2008) Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U.S.A. 105, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hiller S., Garces R. G., Malia T. J., Orekhov V. Y., Colombini M., and Wagner G. (2008) Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ujwal R., Cascio D., Colletier J. P., Faham S., Zhang J., Toro L., Ping P., and Abramson J. (2008) The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U.S.A. 105, 17742–17747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kroemer G., Galluzzi L., and Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163 [DOI] [PubMed] [Google Scholar]
- 12. Keeble J. A., and Gilmore A. P. (2007) Apoptosis commitment-translating survival signals into decisions on mitochondria. Cell Res. 17, 976–984 [DOI] [PubMed] [Google Scholar]
- 13. van Loo G., Saelens X., van Gurp M., MacFarlane M., Martin S. J., and Vandenabeele P. (2002) The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 9, 1031–1042 [DOI] [PubMed] [Google Scholar]
- 14. Johnstone R. W., Ruefli A. A., and Lowe S. W. (2002) Apoptosis: a link between cancer genetics and chemotherapy. Cell 108, 153–164 [DOI] [PubMed] [Google Scholar]
- 15. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 16. Sureda F. X., Junyent F., Verdaguer E., Auladell C., Pelegri C., Vilaplana J., Folch J., Canudas A. M., Zarate C. B., Pallès M., and Camins A. (2011) Antiapoptotic drugs: a therapeutic strategy for the prevention of neurodegenerative diseases. Curr. Pharm. Des. 17, 230–245 [DOI] [PubMed] [Google Scholar]
- 17. Obulesu M., and Lakshmi M. J. (2014) Apoptosis in Alzheimer's disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 39, 2301–2312 [DOI] [PubMed] [Google Scholar]
- 18. Scharstuhl A., Mutsaers H. A., Pennings S. W., Russel F. G., and Wagener F. A. (2009) Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PloS One 4, e6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shoshan-Barmatz V., Keinan N., Abu-Hamad S., Tyomkin D., and Aram L. (2010) Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim. Biophys. Acta 1797, 1281–1291 [DOI] [PubMed] [Google Scholar]
- 20. Tajeddine N., Galluzzi L., Kepp O., Hangen E., Morselli E., Senovilla L., Araujo N., Pinna G., Larochette N., Zamzami N., Modjtahedi N., Harel-Bellan A., and Kroemer G. (2008) Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27, 4221–4232 [DOI] [PubMed] [Google Scholar]
- 21. Martinou J. C., Desagher S., and Antonsson B. (2000) Cytochrome c release from mitochondria: all or nothing. Nat. Cell Biol. 2, E41–E43 [DOI] [PubMed] [Google Scholar]
- 22. Tsujimoto Y., and Shimizu S. (2002) The voltage-dependent anion channel: an essential player in apoptosis. Biochimie 84, 187–193 [DOI] [PubMed] [Google Scholar]
- 23. Zalk R., Israelson A., Garty E. S., Azoulay-Zohar H., and Shoshan-Barmatz V. (2005) Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 386, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bernardi P. (1996) The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim. Biophys. Acta 1275, 5–9 [DOI] [PubMed] [Google Scholar]
- 25. Crompton M. (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 [PMC free article] [PubMed] [Google Scholar]
- 26. Halestrap A. P., McStay G. P., and Clarke S. J. (2002) The permeability transition pore complex: another view. Biochimie 84, 153–166 [DOI] [PubMed] [Google Scholar]
- 27. Betaneli V., Petrov E. P., and Schwille P. (2012) The role of lipids in VDAC oligomerization. Biophys. J. 102, 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonçalves R. P., Buzhynskyy N., Prima V., Sturgis J. N., and Scheuring S. (2007) Supramolecular assembly of VDAC in native mitochondrial outer membranes. J. Mol. Biol. 369, 413–418 [DOI] [PubMed] [Google Scholar]
- 29. Hoogenboom B. W., Suda K., Engel A., and Fotiadis D. (2007) The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J. Mol. Biol. 370, 246–255 [DOI] [PubMed] [Google Scholar]
- 30. Keinan N., Tyomkin D., and Shoshan-Barmatz V. (2010) Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 30, 5698–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abu-Hamad S., Zaid H., Israelson A., Nahon E., and Shoshan-Barmatz V. (2008) Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490 [DOI] [PubMed] [Google Scholar]
- 32. Shoshan-Barmatz V., Keinan N., and Zaid H. (2008) Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191 [DOI] [PubMed] [Google Scholar]
- 33. Zeth K., Meins T., and Vonrhein C. (2008) Approaching the structure of human VDAC1, a key molecule in mitochondrial cross-talk. J. Bioenerg. Biomembr. 40, 127–132 [DOI] [PubMed] [Google Scholar]
- 34. Ujwal R., Cascio D., Chaptal V., Ping P., and Abramson J. (2009) Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels 3, 167–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shoshan-Barmatz V., Mizrachi D., and Keinan N. (2013) Oligomerization of the mitochondrial protein VDAC1: from structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 117, 303–334 [DOI] [PubMed] [Google Scholar]
- 36. Keinan N., Pahima H., Ben-Hail D., and Shoshan-Barmatz V. (2013) The role of calcium in VDAC1 oligomerization and mitochondrion-mediated apoptosis. Biochim. Biophys. Acta 1833, 1745–1754 [DOI] [PubMed] [Google Scholar]
- 37. Weisthal S., Keinan N., Ben-Hail D., Arif T., and Shoshan-Barmatz V. (2014) Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 1843, 2270–2281 [DOI] [PubMed] [Google Scholar]
- 38. Huang L., Han J., Ben-Hail D., He L., Li B., Chen Z., Wang Y., Yang Y., Liu L., Zhu Y., Shoshan-Barmatz V., Liu H., and Chen Q. (2015) A new fungal diterpene induces VDAC1-dependent apoptosis in Bax/Bak-deficient cells. J. Biol. Chem. 290, 23563–23578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ben-Hail D., and Shoshan-Barmatz V. (2016) VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta 1863, 1612–1623 [DOI] [PubMed] [Google Scholar]
- 40. Bertrand L., Parent S., Caron M., Legault M., Joly E., Angers S., Bouvier M., Brown M., Houle B., and Ménard L. (2002) The BRET2/arrestin assay in stable recombinant cells: a platform to screen for compounds that interact with G protein-coupled receptors (GPCRS). J. Recept. Signal Transduct. Res. 22, 533–541 [DOI] [PubMed] [Google Scholar]
- 41. Anis Y. (2006) Involvement of Ca2+ in the apoptotic process–friends or foes. Pathways 2, 2–7 [Google Scholar]
- 42. Baumgartner H. K., Gerasimenko J. V., Thorne C., Ferdek P., Pozzan T., Tepikin A. V., Petersen O. H., Sutton R., Watson A. J., and Gerasimenko O. V. (2009) Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 284, 20796–20803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lemasters J. J., and Holmuhamedov E. (2006) Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim. Biophys. Acta 1762, 181–190 [DOI] [PubMed] [Google Scholar]
- 44. Shimizu S., Matsuoka Y., Shinohara Y., Yoneda Y., and Tsujimoto Y. (2001) Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 152, 237–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abu-Hamad S., Arbel N., Calo D., Arzoine L., Israelson A., Keinan N., Ben-Romano R., Friedman O., and Shoshan-Barmatz V. (2009) The VDAC1 N terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 122, 1906–1916 [DOI] [PubMed] [Google Scholar]
- 46. Cavallucci V., and D'Amelio M. (2011) Matter of life and death: the pharmacological approaches targeting apoptosis in brain diseases. Curr. Pharm. Des. 17, 215–229 [DOI] [PubMed] [Google Scholar]
- 47. Rohn T. T., Kokoulina P., Eaton C. R., and Poon W. W. (2009) Caspase activation in transgenic mice with Alzheimer-like pathology: results from a pilot study utilizing the caspase inhibitor, Q-VD-OPh. Int. J. Clin. Exp. Med. 2, 300–308 [PMC free article] [PubMed] [Google Scholar]
- 48. Kostin S., Pool L., Elsässer A., Hein S., Drexler H. C., Arnon E., Hayakawa Y., Zimmermann R., Bauer E., Klövekorn W. P., and Schaper J. (2003) Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 92, 715–724 [DOI] [PubMed] [Google Scholar]
- 49. Kroemer G., El-Deiry W. S., Golstein P., Peter M. E., Vaux D., Vandenabeele P., Zhivotovsky B., Blagosklonny M. V., Malorni W., Knight R. A., Piacentini M., Nagata S., Melino G., and Nomenclature Committee on Cell Death. (2005) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 12, 1463–1467 [DOI] [PubMed] [Google Scholar]
- 50. Marunouchi T., and Tanonaka K. (2015) Cell death in the cardiac myocyte. Biol. Pharm. Bull. 38, 1094–1097 [DOI] [PubMed] [Google Scholar]
- 51. Chen G. Q., Zhu J., Shi X. G., Ni J. H., Zhong H. J., Si G. Y., Jin X. L., Tang W., Li X. S., Xong S. M., Shen Z. X., Sun G. L., Ma J., Zhang P., Zhang T. D., et al. (1996) In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR α/PML proteins. Blood 88, 1052–1061 [PubMed] [Google Scholar]
- 52. Zhang X. W., Yan X. J., Zhou Z. R., Yang F. F., Wu Z. Y., Sun H. B., Liang W. X., Song A. X., Lallemand-Breitenbach V., Jeanne M., Zhang Q. Y., Yang H. Y., Huang Q. H., Zhou G. B., Tong J. H., et al. (2010) Arsenic trioxide controls the fate of the PML-RARα oncoprotein by directly binding PML. Science 328, 240–243 [DOI] [PubMed] [Google Scholar]
- 53. Carney D. A. (2008) Arsenic trioxide mechanisms of action–looking beyond acute promyelocytic leukemia. Leuk. Lymphoma 49, 1846–1851 [DOI] [PubMed] [Google Scholar]
- 54. Zheng Y., Shi Y., Tian C., Jiang C., Jin H., Chen J., Almasan A., Tang H., and Chen Q. (2004) Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 23, 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu J., Qian H., Li Y., Wang Y., Zhang X., Liang X., Fu M., and Lin C. (2007) Therapeutic effect of arsenic trioxide (As2O3) on cervical cancer in vitro and in vivo through apoptosis induction. Cancer Biol. Ther. 6, 580–586 [DOI] [PubMed] [Google Scholar]
- 56. Aiello R., Messina A., Schiffler B., Benz R., Tasco G., Casadio R., and De Pinto V. (2004) Functional characterization of a second porin isoform in Drosophila melanogaster. DmPorin2 forms voltage-independent cation-selective pores. J. Biol. Chem. 279, 25364–25373 [DOI] [PubMed] [Google Scholar]
- 57. Ly J. D., Grubb D. R., and Lawen A. (2003) The mitochondrial membrane potential (ΔΨ(m)) in apoptosis; an update. Apoptosis 8, 115–128 [DOI] [PubMed] [Google Scholar]
- 58. Radi E., Formichi P., Battisti C., and Federico A. (2014) Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 42, S125–S152 [DOI] [PubMed] [Google Scholar]
- 59. Friedlander R. M. (2003) Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 348, 1365–1375 [DOI] [PubMed] [Google Scholar]
- 60. Jiang X., and Wang X. (2000) Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 275, 31199–31203 [DOI] [PubMed] [Google Scholar]
- 61. Deedwania P. C., and Lardizabal J. A. (2010) Atrial fibrillation in heart failure: a comprehensive review. Am. J. Med. 123, 198–204 [DOI] [PubMed] [Google Scholar]
- 62. De Jong A. M., Maass A. H., Oberdorf-Maass S. U., Van Veldhuisen D. J., Van Gilst W. H., and Van Gelder I. C. (2011) Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc. Res. 89, 754–765 [DOI] [PubMed] [Google Scholar]
- 63. Müller P., Deneke T., Schiedat F., Bösche L., Strauch J., Dietrich J. W., Vogt M., Tannapfel A., Stiegler H., Mügge A., and Ewers A. (2013) Increased preoperative serum apoptosis marker fas ligand correlates with histopathology and new-onset of atrial fibrillation in patients after cardiac surgery. J. Cardiovasc. Electrophysiol. 24, 1110–1115 [DOI] [PubMed] [Google Scholar]
- 64. Azoulay-Zohar H., Israelson A., Abu-Hamad S., and Shoshan-Barmatz V. (2004) In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 377, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pastorino J. G., and Hoek J. B. (2008) Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pastorino J. G., Shulga N., and Hoek J. B. (2002) Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618 [DOI] [PubMed] [Google Scholar]
- 67. Shoshan-Barmatz V., Zakar M., Rosenthal K., and Abu-Hamad S. (2009) Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 1787, 421–430 [DOI] [PubMed] [Google Scholar]
- 68. Arzoine L., Zilberberg N., Ben-Romano R., and Shoshan-Barmatz V. (2009) Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 284, 3946–3955 [DOI] [PubMed] [Google Scholar]
- 69. Goldin N., Arzoine L., Heyfets A., Israelson A., Zaslavsky Z., Bravman T., Bronner V., Notcovich A., Shoshan-Barmatz V., and Flescher E. (2008) Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 27, 4636–4643 [DOI] [PubMed] [Google Scholar]
- 70. Pastorino J. G., Hoek J. B., and Shulga N. (2005) Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554 [DOI] [PubMed] [Google Scholar]
- 71. Shoshan-Barmatz V., Israelson A., Brdiczka D., and Sheu S. S. (2006) The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 12, 2249–2270 [DOI] [PubMed] [Google Scholar]
- 72. Krasnov G. S., Dmitriev A. A., Lakunina V. A., Kirpiy A. A., and Kudryavtseva A. V. (2013) Targeting VDAC-bound hexokinase II: a promising approach for concomitant anti-cancer therapy. Expert Opin. Ther. Targets 17, 1221–1233 [DOI] [PubMed] [Google Scholar]
- 73. Aram L., Geula S., Arbel N., and Shoshan-Barmatz V. (2010) VDAC1 cysteine residues: topology and function in channel activity and apoptosis. Biochem. J. 427, 445–454 [DOI] [PubMed] [Google Scholar]
- 74. Shoshan-Barmatz V., and Golan M. (2012) Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 19, 714–735 [DOI] [PubMed] [Google Scholar]
- 75. Okatsu K., Iemura S., Koyano F., Go E., Kimura M., Natsume T., Tanaka K., and Matsuda N. (2012) Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem. Biophys. Res. Commun. 428, 197–202 [DOI] [PubMed] [Google Scholar]
- 76. Regenold W. T., Pratt M., Nekkalapu S., Shapiro P. S., Kristian T., and Fiskum G. (2012) Mitochondrial detachment of hexokinase 1 in mood and psychotic disorders: implications for brain energy metabolism and neurotrophic signaling. J. Psychiatr. Res. 46, 95–104 [DOI] [PubMed] [Google Scholar]
- 77. Saraiva L. M., Seixas da Silva G. S., Galina A., da-Silva W. S., Klein W. L., Ferreira S. T., and De Felice F. G. (2010) Amyloid-β triggers the release of neuronal hexokinase 1 from mitochondria. PLoS ONE 5, e15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shan D., Mount D., Moore S., Haroutunian V., Meador-Woodruff J. H., and McCullumsmith R. E. (2014) Abnormal partitioning of hexokinase 1 suggests disruption of a glutamate transport protein complex in schizophrenia. Schizophr. Res. 154, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abu-Hamad S., Sivan S., and Shoshan-Barmatz V. (2006) The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. U.S.A. 103, 5787–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Godbole A., Varghese J., Sarin A., and Mathew M. K. (2003) VDAC is a conserved element of death pathways in plant and animal systems. Biochim. Biophys. Acta 1642, 87–96 [DOI] [PubMed] [Google Scholar]
- 81. Zaid H., Abu-Hamad S., Israelson A., Nathan I., and Shoshan-Barmatz V. (2005) The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 12, 751–760 [DOI] [PubMed] [Google Scholar]
- 82. Ghosh T., Pandey N., Maitra A., Brahmachari S. K., and Pillai B. (2007) A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS ONE 2, e1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Manczak M., and Reddy P. H. (2012) Abnormal interaction of VDAC1 with amyloid β and phosphorylated τ causes mitochondrial dysfunction in Alzheimer's disease. Hum. Mol. Genet. 21, 5131–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cuadrado-Tejedor M., Vilariño M., Cabodevilla F., Del Río J., Frechilla D., and Pérez-Mediavilla A. (2011) Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer's disease transgenic mice: an insight into the pathogenic effects of amyloid-β. J. Alzheimers Dis. 23, 195–206 [DOI] [PubMed] [Google Scholar]
- 85. Pérez-Gracia E., Torrejón-Escribano B., and Ferrer I. (2008) Dystrophic neurites of senile plaques in Alzheimer's disease are deficient in cytochrome c oxidase. Acta Neuropathol. 116, 261–268 [DOI] [PubMed] [Google Scholar]
- 86. Ahmed M., Muhammed S. J., Kessler B., and Salehi A. (2010) Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets 2, 283–292 [DOI] [PubMed] [Google Scholar]
- 87. Branco A. F., Pereira S. L., Moreira A. C., Holy J., Sardão V. A., and Oliveira P. J. (2011) Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc. Toxicol. 11, 191–203 [DOI] [PubMed] [Google Scholar]
- 88. Bacart J., Corbel C., Jockers R., Bach S., and Couturier C. (2008) The BRET technology and its application to screening assays. Biotechnol. J. 3, 311–324 [DOI] [PubMed] [Google Scholar]
- 89. Ben-Hail D., and Shoshan-Barmatz V. (2014a) Purification of VDAC1 from rat liver mitochondria. Cold Spring Harb. Protoc. 2014, 94–99 [DOI] [PubMed] [Google Scholar]
- 90. Reina S., Checchetto V., Saletti R., Gupta A., Chaturvedi D., Guardiani C., Guarino F., Scorciapino M. A., Magrì A., Foti S., Ceccarelli M., Messina A. A., Mahalakshmi R., Szabo I., and De Pinto V. (2016) VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: the putative role of cysteine residue modifications. Oncotarget 7, 2249–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Reina S., Magrì A., Lolicato M., Guarino F., Impellizzeri A., Maier E., Benz R., Ceccarelli M., De Pinto V., and Messina A. (2013) Deletion of β-strands 9 and 10 converts VDAC1 voltage-dependence in an asymmetrical process. Biochim. Biophys. Acta 1827, 793–805 [DOI] [PubMed] [Google Scholar]
- 92. Ben-Hail D., and Shoshan-Barmatz V. (2014b) Reconstitution of purified VDAC1 into a lipid bilayer and recording of channel conductance. Cold Spring Harb. Protoc. 2014, 100–105 [DOI] [PubMed] [Google Scholar]
- 93. Wienken C. J., Baaske P., Rothbauer U., Braun D., and Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]