

Abstract
Phosphatidylglycerol (PG) makes up 5–20% of the phospholipids of Escherichia coli and is essential for growth in wild-type cells. PG is synthesized from the dephosphorylation of its immediate precursor, phosphatidylglycerol phosphate (PGP) whose synthase in E. coli is PgsA. Using genetic, biochemical, and highly sensitive mass spectrometric approaches, we identified an alternative mechanism for PG synthesis in E. coli that is PgsA independent. The reaction of synthesis involves the conversion of phosphatidylethanolamine and glycerol into PG and is catalyzed by ClsB, a phospholipase D-type cardiolipin synthase. This enzymatic reaction is demonstrated herein both in vivo and in vitro as well as by using the purified ClsB protein. When the growth medium was supplemented with glycerol, the expression of E. coli ClsB significantly increased PG and cardiolipin levels, with the growth deficiency of pgsA null strain also being complemented under such conditions. Identification of this alternative mechanism for PG synthesis not only expands our knowledge of bacterial anionic phospholipid biosynthesis, but also sheds light on the biochemical functions of the cls gene redundancy in E. coli and other bacteria. Finally, the PGP-independent PG synthesis in E. coli may also have important implications for the understanding of PG biosynthesis in eukaryotes that remains incomplete.
Keywords: cardiolipin, Escherichia coli (E. coli), mass spectrometry (MS), phosphatidylethanolamine, phosphatidylglycerol, Phosphatidylglycerol
Introduction
In Escherichia coli, depending on growth phase and conditions, phosphatidylglycerol (PG)2 can account for 5–20% of the total phospholipid content, with the remainder mainly comprising phosphatidylethanolamine (PE) and cardiolipin (CL) (1, 2). Besides serving as a basic membrane component (2, 3), PG also plays critical roles in SecA-dependent protein translocation, is involved in the initiation of DNA replication at oriC, and is required for the proper location of the division septum at mid-cell (4–7). In E. coli, PG is synthesized by a system of enzymes first described by Eugene Kennedy and colleagues (1) (Fig. 1). In the initial steps of the pathway, the intermediate cytidine diphosphate-diacylglycerol (CDP-DAG) is formed from a condensation reaction between phosphatidic acid (PA) and cytidine triphosphate. The enzyme phosphatidylglycerol phosphate (PGP) synthase (PgsA) then catalyzes a reaction between CDP-DAG and glycerol-3-phosphate to yield PGP, which in turn is dephosphorylated to PG by three PGP phosphatases (PgpA, PgpB, and PgpC) (8). PG is a direct precursor of CL, whose synthesis is mediated by three CL synthase genes in E. coli (2).
FIGURE 1.
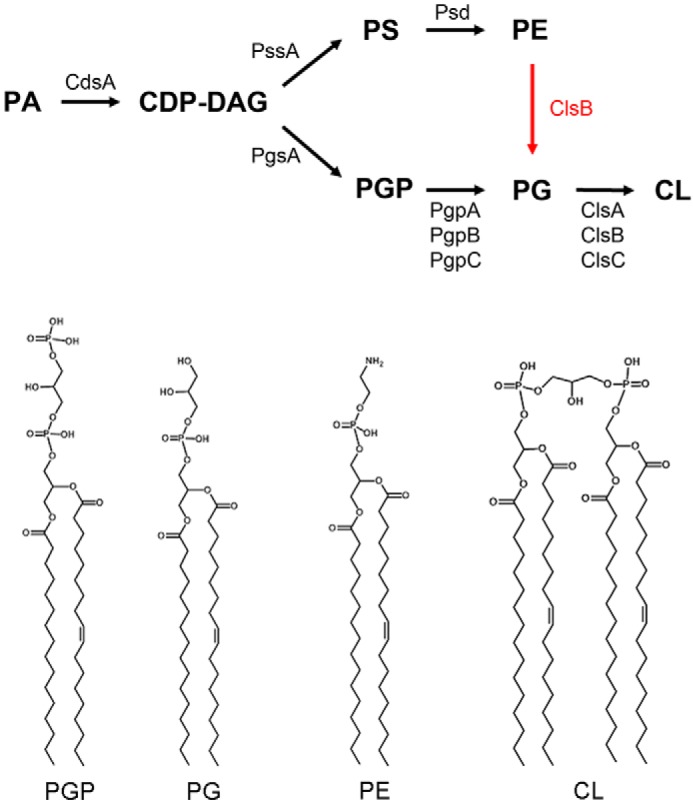
Biosynthetic pathways and representative structures of E. coli glycerophospholipids. A previously unrecognized mode of PG synthesis involves the conversion of PE into PG by ClsB is the subject of this study. The acyl compositions as drawn in the chemical structures only represent one of the major species.
Deletion of the pgsA gene in E. coli blocks the synthesis of PG and CL (5, 9, 10). The ΔpgsA mutant strain is not viable unless it also harbors mutations in the lpp and rcsF genes (10). The lpp mutation prevents accumulation of a nascent major outer membrane lipoprotein that requires PG for its modification (5, 9, 11), whereas the rcsF mutation prevents the lysis of ΔpgsA cells at the temperature of 37–42 °C (12). Thin-layer chromatography (TLC) and direct injection MS analysis have shown that the ΔpgsA strain (UE54) lacks detectable PG and CL, but accumulates phosphatidic acid, CDP-diacylglycerol, and N-acyl-phosphatidylethanolamine (N-acyl-PE) (13).
In this study, using highly sensitive normal phase liquid chromatography/mass spectrometry (LC/MS) (2, 14), we unexpectedly detected small amounts of PG species in ΔpgsA cells. We found that the formation of PG species in these mutant cells is catalyzed by cardiolipin synthases (Cls), with ClsB playing a dominant role. Furthermore, we purified the ClsB protein and demonstrated its PG synthesis activity using PE and glycerol as substrates. Elucidation of this PGP-independent PG synthesis not only expands our knowledge of anionic phospholipid biosynthesis and metabolism but also sheds light on the biochemical functions of the multiple cls genes found in E. coli and many other bacteria.
Results
LC/MS Reveals pgsA-independent and cls-dependent PG Formation in E. coli
Under the normal phase LC conditions used in this study, the major E. coli glycerophospholipids elute in the following order: PG (∼11–12 min), CL (∼12–14 min), PE (∼16–17 min), and PA (∼20–21 min). As identified by the exact mass measurement and MS/MS analysis, wild-type E. coli cells contain mainly PG (32:1), PG (33:1), PG (34:2), and PG (34:1), where the numbers of acyl chain carbon atoms and double bonds or double bond equivalents (such as cyclopropane) appear in parentheses (Fig. 2A). Surprisingly, when the total lipid extract of ΔpgsA (BKT25) cells was subjected to the same normal phase LC/MS analysis, PG species were still detectable, albeit at trace levels (<0.1% of the wild-type levels) (Fig. 2B). We confirmed that these PG species were, in fact, synthesized by the ΔpgsA strain by performing control experiments that ruled out the possibility that these lipids represented contaminants or were carried over from previous analyses. According to the Kennedy pathway (Fig. 1), pgsA deletion should completely block PG biosynthesis. This was indeed concluded based on TLC and direct injection MS analysis of pgsA null mutant (UE54) cells (13). Our detection of trace levels of PG species in the ΔpgsA (BKT25) mutant was only possible when the highly sensitive normal phase LC/MS was performed, thus providing an important clue of the existence of other mechanism(s) for PG synthesis. Moreover, it is worth noting that the acyl chain compositions of the residual PG species in the ΔpgsA (BKT25) strain (Fig. 2B) differed from those found in wild-type cells (Fig. 2A), implying that the PG species in these two strains might not be formed via the same biosynthetic route.
FIGURE 2.

PG species are observed in the E. coli ΔpgsA (BKT25) mutant but is completely absent in the ΔpgsA ΔclsABC (BKT29) mutant. Total lipids extracted from stationary phase cells were analyzed by normal phase LC-ESI/MS/MS in the negative ion mode. A, mass spectrum showing the [M-H]− ions of PG species from wild-type E. coli cells. B, residual PG ion species are detectable in the ΔpgsA (BKT25) mutant. C, PG is not detectable in the ΔpgsA ΔclsABC (BKT29) mutant.
While profiling the glycerophospholipids of other E. coli mutants, we found, unexpectedly, that PG was not detectable when all three paralogous cardiolipin synthase (cls) genes (i.e. clsA, clsB, and clsC) were deleted from the ΔpgsA (BKT25) strain (Fig. 2C). The combined pgsA and triple cls gene deletion mutant (ΔpgsA ΔclsABC (BKT29)) was previously generated in studies that led to our discovery of a third cardiolipin synthase (ClsC) in E. coli (2). The ΔpgsA ΔclsABC (BKT29) mutant lacks detectable PG and CL, regardless of growth phase or conditions (Fig. 2C). Intriguingly, PG was still detectable if any one of the three cls genes was not deleted, indicating that each of the cls genes might contribute to in vivo PG synthesis in the ΔpgsA mutant. However, the relative contributions of the three cls genes to PG synthesis remain to be delineated (for reasons that the relative PG levels in E. coli vary drastically with growth conditions).
To further assess whether Cls proteins are involved in the synthesis of PG in vivo, we cloned each of the cls genes into the arabinose-inducible pBAD30 expression vector (15) and introduced the plasmids into the ΔpgsA ΔclsABC (BKT29) mutant strain. The transformants were grown to stationary phase (A600 of 1∼2) in medium supplemented with carbenicillin and 0.02% arabinose. LC/MS/MS analysis of the total lipids extracted from these cells indicated that PG levels were increased upon ClsB expression but not when ClsA or ClsC were expressed (data not shown). In all cases, neither PG nor CL production was discernible by TLC analysis (Fig. 3A).
FIGURE 3.

Synthesis of PG and CL by ClsB in the ΔpgsA ΔclsABC mutant cells requires glycerol. A, TLC analysis of total lipid extracts of ΔpgsA ΔclsABC (BKT29) mutant cells expressing ClsA, ClsB, or ClsC. The trace spot detected near CL in the vector control sample was previously identified by LC/MS/MS to be undecaprenyl-phosphate (C55-P) (2). B, the addition of glycerol to the growth medium greatly increased the production of PG and CL by ClsB (to near wild-type levels). C and D, MS analysis of PG and CL produced as a result of ClsB expression and glycerol supplementation in BKT29. PG (C) and CL (D) were detected by LC/ESI/MS in the negative ion mode primarily as singly charged [M-H]− and doubly charged [M-2H]2− ions, respectively. The acyl compositions of PG species are almost identical to those of PE (E), hinting the phosphatidyl groups of PG were transferred from PE species.
Synthesis of PG by ClsB Requires Glycerol
All three E. coli Cls belong to the phospholipase D (PLD) superfamily, with each containing two HKD motifs (Fig. 4) (2, 16). PLD catalyzes the hydrolysis of phospholipids into PA and the corresponding headgroup (17). It is also known that in the presence of a primary alcohol (e.g. butanol), PLD can catalyze a trans-phosphatidylation reaction to yield a phosphatidyl alcohol (17, 18). We speculated that any PG formed by Cls would result from such a trans-phosphatidylation reaction, and thus supplemented the growth medium with glycerol to a final concentration of 0.4% (v/v). As shown by TLC analysis of the total lipids extracted from stationary phase cells (Fig. 3B), ClsB expression in conjunction with glycerol supplementation restored the production of PG and CL in the ΔpgsA ΔclsABC (BKT29) mutant to wild-type levels. The identities of ClsB-produced PG and CL species were further confirmed by normal phase LC/MS/MS analysis of total lipid extracts from cells expressing this protein (Fig. 3, C and D). In contrast, the levels of PG and CL upon expression of ClsA or ClsC remain very low (Fig. 3B). As such, we chose to focus on the role of ClsB in the cls-mediated PG formation. The glycerol-dependent PG synthesis by clsB was also demonstrated by an in vitro assay using clsB-expressing BKT29 membranes (supplemental Fig. S1).
FIGURE 4.

The three E. coli cardiolipin synthases (ClsA, ClsB, and ClsC) contain the two HKD motifs (boxed sequences) found in phospholipase D. The conserved His residues (indicated by asterisks) in the HKD motifs were subjected to mutagenesis.
ClsB Synthesizes PG Using PE as a Substrate
To identify the substrate (or phosphatidyl donor) used for PG synthesis by ClsB, we paid special attention to PE as it was noted that the composition of PG acyl chains (Fig. 3C) very closely matched those of PE (Fig. 3E), suggesting that the phosphatidyl groups of the PG species might be directly transferred from PE species in the ΔpgsA ΔclsABC (BKT29) mutant. To test whether PG could indeed be formed from PE by ClsB, 32P-labeled PE was prepared from BKT29 E. coli cells by supplementing the growth medium with [32P]PO4. Following fractionation on a DEAE column, the partially purified 32P-labeled PE substrate was incubated with cell membranes derived from ΔpgsA ΔclsABC (BKT29) cells expressing a vector control (pBAD30), wild-type ClsB, or a ClsB-H113A mutant. As shown by TLC and phosphorimaging analysis (Fig. 5), a significant amount of 32P-labeled PG was generated by membranes expressing wild-type ClsB, indicating that PE was indeed converted into PG by ClsB. In contrast, no PG was formed when membranes expressing the vector control or the ClsB-H113A mutant were used (Fig. 5).
FIGURE 5.
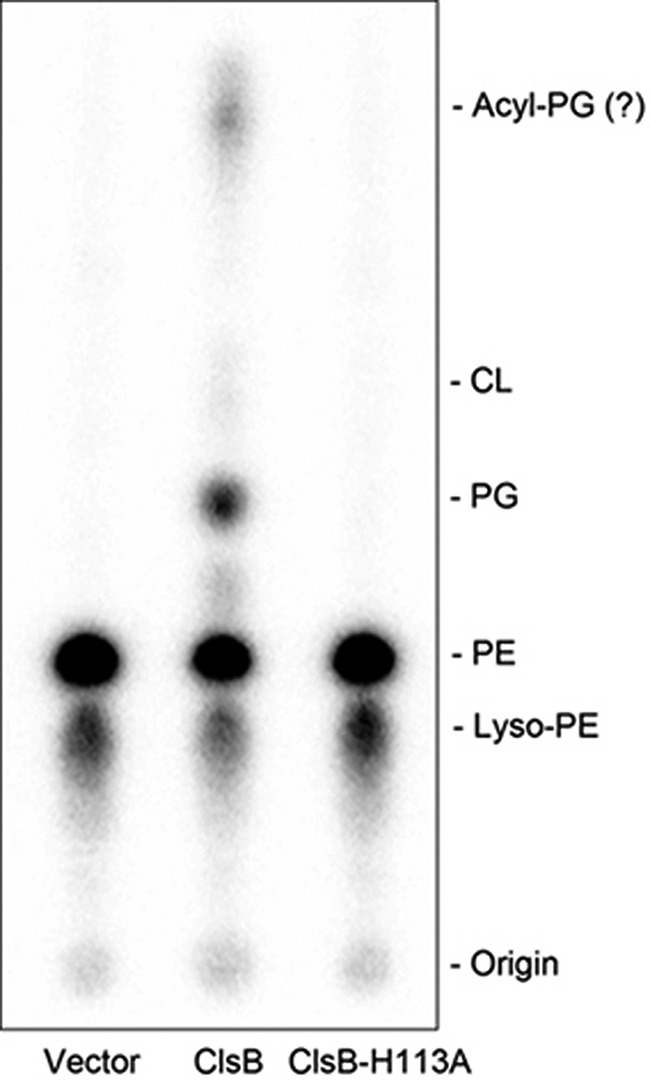
In vitro conversion of PE to PG by ClsB. [32P]PG was formed by mixing membranes derived from BKT29 cells expressing wild-type ClsB and partially purified 32P-labeled PE in the presence of 100 mm glycerol. In contrast, no PG was formed using membranes from cells transformed with an empty vector or expressing the ClsBH113A mutant. The presence of 32P-labeled lyso-PE likely resulted from partial hydrolysis of PE. The spot at the top of the second lane concurrent with PG formation is likely acyl-PG, a known PG derivative in E. coli.
To definitively demonstrate that ClsB catalyzes the conversion of PE to PG in vitro, we carried out this purification of ClsB protein. For this, pET vector constructs encoding C terminally FLAG-tagged wild-type ClsB or the protein containing mutations in either of the two HKD motifs (H113A and H291A) were transformed into E. coli strain C41 (DE3). The FLAG-tagged proteins were purified to homogeneity using anti-FLAG M2 affinity gel resin (Fig. 6A). To perform in vitro enzyme activity assays, synthetic PE (17:0/14:1) and penta-deuterated glycerol (d5-glycerol) were mixed with each of three recombinant ClsB proteins (Fig. 6B). After incubation, the reaction solutions were subjected to lipid extraction followed by normal phase LC/MS/MS analysis. As shown in Fig. 6C, the wild-type ClsB-FLAG protein produced the expected product, d5-PG (whose [M-H]− ion is observed at m/z 710.5). In contrast, the two catalytically inactive HKD mutant proteins, ClsBH113A-FLAG and ClsBH291A-FLAG, were unable to convert PE into PG (Fig. 6, D and E).
FIGURE 6.

Purified ClsB protein catalyzes the conversion of PE into PG in the presence of glycerol. A, SDS-PAGE analysis of purified wild-type ClsB-FLAG, ClsBH113A-FLAG, and ClsBH291A-FLAG proteins. B, proposed reaction scheme of the conversion of synthetic substrate PE (17:0/14:1) to product d5-PG (17:0/14:1) by ClsB in the presence of d5-glycerol. C-E, the expected d5-PG (17:0/14:1) ([M-H]− ion at m/z 710.5) is produced by wild-type ClsB-FLAG protein, but not by ClsBH113A-FLAG or ClsBH291A-FLAG mutant protein. F, MS/MS spectrum of d5-PG (17:0/14:1) ([M-H]− ion at m/z 710.5) and the fragmentation scheme for the major product ions. G, MS/MS spectrum of substrate PE (17:0/14:1) ([M-H]− ion at m/z 674.5) and the fragmentation scheme for the major product ions.
MS/MS analysis further confirmed the m/z 710.5 species produced by ClsB (Fig. 6C) as the [M-H]− ion of d5-PG derived from PE (17:0/14:1) and d5-glycerol. As shown in Fig. 6F, the m/z 158.03 product ion is derived from the cyclic phosphodiester of penta-deuterated glycerol; the corresponding m/z 153.0 ion is a signature product ion in the MS/MS spectra of non-deuteriated glycerophospholipids (19). The m/z 225.190 and 269.252 ions correspond to the carboxylic anions of the C17:0 and C14:1 fatty acids, respectively, with both being derived from the synthetic PE (17:0/14:1) substrate. The MS/MS spectrum of the [M-H]− ion of PE (17:0/14:1) and its fragmentation scheme are shown in Fig. 6G.
Other phospholipids were tested as potential substrates of ClsB, including synthetic standards of CDP-DAG, PA, and PS. None of these was utilized by ClsB to synthesize PG.
ClsB Uses Two PG Molecules to Synthesize CL
Although the role of ClsB as a cardiolipin synthase has been well established (2, 16), characterization of its substrate(s) remains incomplete. Previously, PG was identified as a substrate for ClsB, however, this finding was based on an in vitro study using membrane preparations (16), and as such could not rule out the possible use of other membrane-derived species as substrate(s). The availability of purified ClsB protein allowed us to address this question. Using synthetic PG (17:0/14:1) as substrate, ClsB-FLAG produced a single CL species whose molecular weight is consistent with the condensation of two synthetic PG (17:0/14:1) molecules (Fig. 7). This result provides the first experimental evidence that ClsB employs the classical “prokaryotic” mechanism to synthesize CL (3).
FIGURE 7.

Purified ClsB-FLAG protein uses two PG molecules to synthesize CL (the classical prokaryotic mode). After incubating purified ClsB-FLAG protein with synthetic PG (17:0/14:1) in a reaction buffer, the solution was subjected to lipid extraction and analysis by normal phase-LC/MS/MS. A, a single molecular species of CL was produced, and is detected as the [M-2H]2− ion at m/z 659.449. This corresponds to a molecular weight of 1320.914, consistent with CL formed from the condensation of two synthetic PG (17:0/14:1) molecules. B, MS/MS analysis further supports that the CL product was synthesized from the condensation of two synthetic PG (17:0/14:1) molecules (C). The fragmentation scheme of major product ions observed in the MS/MS spectrum is illustrated (C).
ClsB Complements Deficiencies of a ΔpgsA Mutant
An E. coli pgsA null mutant requires mutations in the lpp and rcsF genes to be viable (9, 10). Given that over-expression of ClsB leads to the production of near wild-type levels of PG and CL in the BKT29 mutant (ΔpgsA ΔclsABC, derived from UE54) when the growth medium was supplemented with glycerol, we tested whether ClsB expression could functionally complement the growth deficiency of a pgsA::KanR deletion in a wild-type background (i.e. without requiring additional suppressor mutations). Accordingly, two pBAD30 constructs containing either pgsA or clsB were individually transformed into the wild-type E. coli strain (W3110), followed by deletion of the pgsA gene from the chromosome by P1vir transduction, as described previously (2).
As demonstrated by a spot assay (Fig. 8A), the pBAD-clsB pgsA::KanR strain (CL15) required the addition of both arabinose and glycerol for growth. For comparison, the growth of a pBAD-pgsA pgsA::KanR strain (CL14) required neither additional arabinose nor glycerol, suggesting that a minimal level of pgsA expression is sufficient to maintain cell viability. In fact, over-expression of pgsA seems detrimental to cells, as reflected by the smaller colonies that appeared upon induction with arabinose (Fig. 8A).
FIGURE 8.

Expression of ClsB complements the defects of a ΔpgsA::KanR mutant in cell growth and in PG and CL synthesis. A, a spot assay was conducted with a 10-fold series dilution of E. coli ΔpgsA::KanR strains covered by plasmids expressing either pgsA or clsB. The strains were grown on LB plates in the presence or absence of 0.4% glycerol and/or 0.02% arabinose. The growth of the pBAD-clsB ΔpgsA::KanR strain required both arabinose induction and glycerol supplementation. In contrast, the pBAD-pgsA ΔpgsA::KanR strain required neither. B, TLC analysis of lipid extracts from wild-type, and pBAD-pgsA and pBAD-clsB ΔpgsA::KanR E. coli strains.
Total lipid analysis by TLC shows that expression of ClsB produced significant levels of PG and CL in the pgsA::KanR strain (CL15), comparable with those in the wild-type or the pgsA-covered ΔpgsA strains (Fig. 8B). These results demonstrated that with glycerol supplementation, ClsB expression could sufficiently complement the deficiencies of the ΔpgsA mutant in cell growth (without additional suppressor mutations), as well as in PG and CL synthesis.
Discussion
Pathways for the biosynthesis of the major E. coli glycerophospholipids (including PE, PG, and CL) were first described by Hirschberg and Kennedy (3) over 40 years ago. The key technique employed for their investigations was radioisotopic labeling (3), primarily involving the use of 32P-labeled phosphate and 14C-labeled glycerol or fatty acids as tracing substances. Radiolabeling techniques also played critical roles in the discovery and characterization of the biosynthetic enzymes of these glycerophospholipids (1). Although the power of radiolabeling techniques is unquestionable, such approaches are limited in terms of molecular specificity and in detecting minor species in a complex mixture.
The past decade has seen a dramatic increase in the application of MS to lipid research (4, 20–22). Lipid MS, like all other biological applications of MS, was revolutionized by the introduction of soft ionization techniques, most notably electrospray ionization (ESI) (23) and matrix-assisted laser desorption ionization (MALDI) (24). Furthermore, the combination of on-line chromatographic separation with ESI/MS detection has greatly improved the applicability of MS to the analysis of complex mixtures. In particular, LC/MS has greatly improved the detection of a minor species by separating them from major species, thus reducing or eliminating ion signal suppressions from the latter. Signal suppression is a common issue in direct injection MS, which is often responsible for the failure to detect the minor species in a mixture. As demonstrated in this study, normal phase LC/MS is particularly well suited for the detection and characterization of glycerophospholipids as these lipid species can be separated based on their headgroups (or charges). Our detection of residual PG species in E. coli ΔpgsA mutant cells benefited from the application of normal phase LC/MS. Indeed, the exceptional sensitivity and specificity afforded by modern high-resolution LC/MS/MS instruments have facilitated the discovery and characterization of numerous minor, novel lipid species (25–27), as well as functional elucidation of novel genes involved in lipid biosynthesis and metabolism (8, 28, 29), including the recent discovery of the first mammalian PGP phosphatase (29). In this study, the LC/MS-based approach was critical in revealing the PE to PG conversion previously unknown in E. coli, extending the s+tudies on the inter-conversion of phospholipids observed in other bacteria (30–32). For example, Lombardi and Fulco (31) reported that PE could be directly made from PG in Bacillus megaterium. Walton and Goldfine (32) reported in vitro trans-phosphatidylation activities that allowed remodeling of the headgroups of Clostridium butyricum membrane phospholipids, including the formation of PE from PG without the formation of PS intermediates. These observations were, however, all based exclusively on radioisotope-labeling techniques and lacked genetic and detailed biochemical characterization.
The physiological significance of the alternative mechanism of PG synthesis mediated by a CL synthase reported here is unknown. The ability to remodel pre-existing phospholipids may allow bacteria to adapt to changing environmental conditions without resorting to de novo phospholipid synthesis (32). The recent revelation that multiple paralogous cls genes exist in E. coli and many other bacteria calls for the study of their biochemical and physiological functions (1, 2, 33, 34). Given the dual functions of ClsB in E. coli phospholipid synthesis as described above, it is possible that some bacterial cls genes may be involved in inter-converting zwitterionic lipids (e.g. PE) to anionic phospholipids (e.g. PG) under certain environmental stress conditions.
Finally, the identification of an PGP-independent PG synthesis in E. coli may have important implications for fully understanding PG synthesis in eukaryotes (35). For example, the first yeast PGPase (Gep4) and mammalian PGPase (PTPMT1) were identified only during the last few years (29, 36). However, as reported in both studies, considerable amounts of PG species were still present in the PGPase-null yeast (ΔGep4) (36) and mammalian cell (ΔPTPMT1) mutants (29), indicating the existence of additional PGPase(s) or alternative mechanism(s) for PG synthesis in these eukaryotic cells. It is particularly intriguing that the acyl chain compositions of the PG species in ptpmt1-KO mouse embryonic fibroblasts differed from those of the accumulated PGP species, strongly implying that not all PG species were derived from PGP in such cells (29). Given that no PTPMT1 homologous genes have been identified in the mouse genome, it is tempting to speculate that some of these residual PG species in the ptpmt1-KO mouse embryonic fibroblast cells might be formed via a PLD-like trans-phosphatidylation activity. In a preliminary study, we demonstrated the conversion of PE into PG in yeast by overexpressing clsB in the ΔGep4 mutant cells (supplemental Fig. S2).
Experimental Procedures
Materials
Silica Gel 60 TLC plates, l-(+)-arabinose, and glycerol were obtained from EMD Chemicals (Gibbstown, NJ). Agar, tryptone, and yeast extract were purchased from Difco. Sodium chloride and HEPES were from VWR International (West Chester, PA). Tween 20 and the bicinchoninic acid protein concentration determination kit were from Thermo Fisher Scientific (Waltham, MA). DEAE-cellulose (type DE52) was from Whatman (Florham Park, NJ). Isopropyl 1-thio-β-d-galactopyranoside was from Invitrogen. Reagent grade chloroform, methanol, hydrochloric acid, sulfuric acid, ethanol, and d5-glycerol were from Sigma. Synthetic PG, PA, PE, and CDP-DAG were from Avanti Polar Lipids (Alabaster, AL). [32P]PO4 was from PerkinElmer Life & Analytical Sciences (Waltham, MA).
Bacterial Strains and Growth Conditions
All E. coli strains used in this study are listed in Table 1. The E. coli MG1655 and W3110 strains are designated as the wild-type strains with respect to their glycerophospholipid composition. Liquid LB medium (10 g/liter of tryptone, 5 g/liter of yeast extract, and 10 g/liter of NaCl) was used to culture E. coli. Solid medium consisted of LB medium with the addition of 15 g/liter of agar. For strain selection purposes, cell cultures were supplemented with kanamycin (50 mg/liter) or carbenicillin (100 mg/liter). All strains were grown at 30 or 37 °C as noted, and cell density was measured as absorption at 600 nm (A600) using a DU spectrophotometer (Beckman Coulter, Brea, CA).
TABLE 1.
E. coli strains used in this study
| Strain | Genotype | Source or Ref. |
|---|---|---|
| W3110 | Wild-type | 2 |
| MG1655 | Wild-type | 2 |
| JW1241 | BW25113 ΔclsA::KanR | 1 |
| JW0772 | BW25113 ΔclsB::KanR | 1 |
| JW5150 | BW25113 ΔclsC::KanR | 1 |
| UE54 | MG1655 lpp2 Δara714 rcsF::miniTn10cam ΔpgsA::FRT-Kan-FRT | 2 |
| BKT25 | MG1655 lpp2 Δara714 rcsF::miniTn10cam ΔpgsA | 2 |
| BKT29 | BKT25 ΔclsA, ΔclsB, ΔclsC, ΔymdB::KanR | 2 |
| BKT30 | BKT25 ΔclsB::KanR | 2 |
| CL01 | BKT25 ΔclsA::KanR | This work |
| CL02 | BKT25 ΔclsA (derived from CL01) | This work |
| CL03 | BKT25 ΔclsA, ΔclsB::KanR | This work |
| CL04 | BKT25 ΔclsA, ΔclsB (derived from CL03) | This work |
| CL06 | BKT25 ΔclsA, ΔclsC::KanR | This work |
| CL07 | BKT25 ΔclsA, ΔclsC (derived from CL06) | This work |
| CL08 | BKT25 ΔclsB (derived from BKT30) | This work |
| CL09 | BKT25 ΔclsB, ΔclsC::KanR | This work |
| CL10 | BKT25 ΔclsB, ΔclsC (derived from CL09) | This work |
| CL11 | BKT25 ΔclsC::KanR | This work |
| CL12 | BKT25 ΔclsC (derived from CL11) | This work |
| CL14 | W3110 pBAD30-pgsA, ΔpgsA::KanR | This work |
| CL15 | W3110 pBAD30-B, ΔpgsA::KanR | This work |
Construction of Chromosomal Mutants Lacking cls Genes
Chromosomal deletions of cls genes were constructed using the Keio Collection (the E. coli K12 single-gene deletion library), which served as the mutation donor (37). A gene of interest was replaced by a kanamycin cassette with flanking FLP sequences in each Keio mutant. P1vir transduction and subsequent kanamycin incision were performed according to a previous description (2). All strains with cls deletions were further verified by PCR.
DNA Manipulations and Plasmid Constructions
All plasmids used in this study are listed in Table 2. Plasmids were isolated using the Qiagen Spin Miniprep kit, and DNA fragments were isolated with the Qiaquick Spin Kits (Qiagen, Valencia, CA). Phusion High Fidelity DNA polymerase (New England Biolabs, Ipswich, MA), T4 DNA ligase (Invitrogen), and restriction endonucleases (New England Biolabs) were used according to the manufacturer's instructions. Expression of clsB from the low-copy pBAD30 plasmid was carried out as described previously, using pBAD-B (2). Site-directed mutagenesis was used to mutate His residues in each HKD motif of ClsB to Ala (Fig. 1). For protein expression and purification purposes, the clsB gene and DNA encoding the corresponding His-Ala mutants were also cloned into pET vectors. DNA encoding a C-terminal FLAG epitope was fused to each gene, which were cloned into the XbaI and HindIII sites of vector pET21b. The resulting FLAG-tagged ClsB constructs were designated as pET21b-B, pET21b-B-H113A, and pET21b-B-H291A, respectively. To generate the pBAD-pgsA vector, the pgsA gene from W3110 cells was amplified and cloned into the XbaI and HindIII sites of plasmid pBAD30. All constructs were verified by DNA sequencing at the Duke DNA Sequencing Facility.
TABLE 2.
E. coli plasmids used in this study
| Plasmid | Genotype | Source or Ref. |
|---|---|---|
| pCP20 | FLP recombinase expression; AmpR CamR; temperature-sensitive replicon | 2 |
| pBAD30 | Low copy number expression plasmid | 2 |
| pBAD-A | Expression of clsA on pBAD30 | 2 |
| pBAD-B | Expression of clsB on pBAD30 | 2 |
| pBAD-B-H113A | Expression of clsB with H113A substitution on pBAD30 | 2 |
| pBAD-B-H291A | Expression of clsB with H291A substitution on pBAD30 | 2 |
| pBAD-C | Expression of clsC on pBAD30 | 2 |
| pBAD-pgsA | Expression of pgsA on pBAD30 | This work |
| pET21b | Expression plasmid | Novagen |
| pET21b-B | Expression of clsB-FLAG on pET21b | This work |
| pET21b-B-H113A | Expression of clsB-FLAG with H113A substitution on pET21b | This work |
| pET21b-B-H291A | Expression of clsB-FLAG with H291A substitution on pET21b | This work |
Lipid Extraction and TLC Analysis
Lipid extraction was performed using a neutral Bligh-Dyer method (38). Cell pellets were washed twice with phosphate-buffered saline (PBS) before extraction. The washed pellets were then suspended in 1.9 ml of PBS, followed by the addition of 4.8 ml of methanol and 2.4 ml of chloroform to create a single-phase solution. The solution was incubated for 30 min at room temperature with intermittent mixing. After centrifugation at 3500 × g for 10 min, the supernatant was converted into a two-phase solution by adding 2.4 ml of PBS and 2.4 ml of chloroform. After centrifugation at 3500 × g for 10 min, the lower phase was recovered and dried under a stream of nitrogen gas. For TLC analysis, the dried lipid extracts were each dissolved in a 100 μl of chloroform/methanol (2:1, v/v). Approximately 1–5 μl of the solution was spotted onto a TLC plate. The TLC plate was developed in tanks equilibrated with chloroform/methanol/acetic acid (65:25:10, v/v). After drying the plate, lipids were visualized by spraying 10% sulfuric acid in ethanol (v/v), followed by charring on a hot plate (2).
Isolation of [32P]PE
To prepare radiolabeled [32P]PE, a 3-ml culture of E. coli BKT29 was grown overnight. Cells were harvested and washed three times by distilled water. The cells were diluted 100-fold with 10 ml of G56 medium (39) containing 100 μCi of [32P]orthophosphate and the cells were grown at 30 °C for 5 h. Lipid extraction and purification of PE using a DE52-cellulose column was carried out as previously described (40). To identify the PE-containing fractions, 10 μl of each fraction was loaded onto a silica TLC plate, which was developed using a solvent mixture consisting chloroform/methanol/acetic acid (65:25:10, v/v). After drying under hot air, the TLC plate was analyzed with a PhosphorImager system (Molecular Dynamics, Sunnyvale, CA). Fractions that contain [32P]PE were subjected to lipid extraction using a Bligh-Dyer method (38) as described above.
Liquid Chromatography Mass Spectrometry of Lipids
Normal phase LC was performed on an Agilent 1200 Quaternary LC system equipped with an Ascentis Silica HPLC column (5 μm, 25 cm × 2.1 mm) from Sigma. Mobile phase A consisted of chloroform/methanol/aqueous ammonium hydroxide (800:195:5, v/v); mobile phase B consisted of chloroform/methanol/water/aqueous ammonium hydroxide (600:340:50:5, v/v); mobile phase C consisted of chloroform/methanol/water/aqueous ammonium hydroxide (450:450:95:5, v/v). The elution program consisted of the following: 100% mobile phase A was held isocratically for 2 min and then linearly increased to 100% mobile phase B over 14 min and held at 100% B for 11 min. The LC gradient was then changed to 100% mobile phase C over 3 min and held at 100% C for 3 min, and finally returned to 100% A over 0.5 min and held at 100% A for 5 min. With a total flow rate of 300 μl/min, the LC eluent was injected into the ion spray source of a TripleTOF® 5600 quadrupole time-of-flight tandem mass spectrometer (AB SCIEX, Framingham, MA). Instrumental settings for negative ion ESI and MS/MS analysis of lipid species were as follows: electrospray ionization voltage (IS) = −4500 V; current gas (CUR) = 20 psi (pressure); gas-1 (GS1) = 20 p.s.i.; de-clustering potential (DP) = −55 V; and focusing potential (FP) = −150 V. The MS/MS analysis used nitrogen as the collision gas. Data analysis was performed using Analyst TF1.5 software from AB SCEIX.
Complementation of the ΔpgsA::kanR Mutant
Plasmids pBAD-B and pBAD-pgsA were individually transformed into wild-type E. coli W3110 cells. The P1 phage was prepared from UE54, a strain containing a chromosomal ΔpgsA::FRT-kanR-FRT cassette (Table 2). P1vir transductions of the W3110 strain containing either plasmid pBAD-B or pBAD-pgsA were performed as described above. After transduction, the recipient cells were incubated at 30 °C on LB agar plates containing 50 mg/liter of kanamycin, 100 mg/liter of carbenicillin, 5 mm sodium citrate, arabinose (0, 0.002, 0.02, or 0.2%), and glycerol (0 or 0.4%). The surviving colonies were purified twice with the same growth condition and the chromosomal ΔpgsA::kanR cassette in each colony was verified by PCR. The ΔpgsA::kanR strains in the W3110 background complemented by plasmid pBAD-pgsA or pBAD-B were designated as CL14 and CL15, respectively.
To assess the cell viability of the CL14 and CL15 strains, different growth conditions were employed. The CL14 strain was grown in LB medium containing 100 mg/liter of carbenicillin, whereas the CL15 strain was grown in LB medium containing 100 mg/liter of carbenicillin, 0.2% arabinose, and 0.4% glycerol. For each strain, 100 μl of cell culture was harvested at an A600 of 1.0, washed twice with 1 ml of LB medium, and re-suspended in 100 μl of LB medium. A series of 10-fold dilutions were generated for each strain and 3 μl of each diluted solution were spotted onto LB agar plates containing 100 mg/liter of carbenicillin, arabinose (0 and 0.02%), and glycerol (0 and 0.4%). Cells were grown at 30 °C overnight.
Expression and Purification of ClsB Proteins
For ClsB protein expression, the pET21b constructs were transformed into strain C41 (DE3) cells. 3-ml cultures were grown overnight at 37 °C and diluted 100-fold into 100 ml of LB medium containing 100 mg/liter of carbencillin. When the cells had grown to A600 of ∼0.5, expression of the FLAG-tagged ClsB protein was induced by adding isopropyl β-d-thiogalactoside to a final concentration of 1 mm. The cultures were grown at 17 °C for an additional 6 h with shaking. For ClsB purification, the following procedures were conducted at 4 °C, unless otherwise noted. Cell pellets were washed twice with ice-cold PBS and suspended in 3 ml of lysis buffer containing 20 mm HEPES (pH 8.0) and 150 mm NaCl. The cells were lysed using a French press twice at 16,000 p.s.i., and spun at 10,000 × g for 30 min to remove cell debris. The supernatant was centrifuged at 200,000 × g for 1 h to collect membranes, which were homogenized and suspended in a buffer containing 20 mm HEPES (pH 8.0), 150 mm NaCl, 10% glycerol, and 0.5% Tween 20. The protein concentration from the resulting suspension was maintained at 1 mg/ml. To solubilize the membranes, the suspension was incubated with gentle rotation overnight, followed by another centrifugation at 200,000 × g for 1 h to remove insoluble materials. The resulting soluble fraction was loaded onto a 0.5-ml column of anti-FLAG M2 affinity gel (Sigma) and incubated at 4 °C with gentle rotation for 2 h. The column was washed twice by 10 ml of high salt buffer containing 20 mm HEPES (pH 8.0), 500 mm NaCl, and 0.008% Tween 20. ClsB-FLAG protein was eluted four times using 0.5 ml of elution buffer containing 20 mm HEPES (pH 8.0), 150 mm NaCl, 0.008% Tween 20, and 150 mg/liter of 3× FLAG peptide (Sigma). The eluted protein was dialyzed against low salt buffer (20 mm HEPES (pH 8.0), 150 mm NaCl, and 0.008% Tween 20). The purification of ClsB was assessed by SDS-PAGE analysis. Purified ClsB was stored at −80 °C until enzyme assay.
Enzyme Assay of ClsB
To analyze membrane PLD activity in vitro, crude membranes were prepared from BKT29 cells expressing the pBAD30 control vector, the pBAD-B vector, or the pBAD-B-H113A vector. Membranes were prepared as described previously (2). To perform the in vitro assay, 20-μl reaction mixtures consisting of PBS (pH 7.4), 10 μm β-mercaptoethanol, 2 mm MgCl2, 100 mm glycerol (Sigma), 0.008% Tween 20, 1,000 cpm/μl of [32P]PE, and 1 mg/ml of cell membranes were incubated at 30 °C for 30 min. After incubation, a 3-μl aliquot of each reaction was spotted onto a TLC plate, which was developed in a solvent mixture consisting chloroform/methanol/acetic acid (65:25:10, v/v). The TLC plate was visualized using a PhosphorImager (Molecular Dynamics). To assess the PG synthesis activity of purified ClsB, the reaction solutions consisted of PBS (pH 7.4), 10 μm β-mercaptoethanol, 2 mm MgCl2, 100 mm d5-glycerol (Sigma), 0.008% Tween 20, 5 μm synthetic PE (17:0/14:1) (Avanti Polar Lipids), and 20 ng/ml of purified proteins. After incubation at 30 °C for 30 min, 88 μl of PBS, 120 μl of chloroform, and 120 μl of methanol were added to generate a two-phase Bligh-Dyer solution. The mixture was vortexed for 2 min and spun at 10,000 × g for 1 min. 10 μl of the lower phase was analyzed by normal phase LC/MS/MS. For assaying in vitro enzymatic activity of purified ClsB, a reaction mixture containing 320 mm potassium phosphate (pH 7.0), 10 mm l-mercaptoethanol, 0.008% Tween 20, 5 μm synthetic PG (17:0/14:1), and 20 ng/ml of protein was prepared. The reaction mixture was incubated at 30 °C for 30 min, followed by lipid extraction and analysis by normal phase LC/MS/MS, as described above.
Yeast
Wild-type BY4743 and gep4 KO yeast strains were ordered from ATCC. For protein expression in yeast, the E. coli ClsB gene was fused with N terminus su91–69 (36) by PCR and cloned onto p415GPD vector as described previously (29).
Author Contributions
C. L., B. K., and Z. G. designed and performed all experiments. J. Z. provide technical advice. C. L. and Z. G. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Edward Dennis, H. Alex Brown, Mikhail Bogdanov, and William Dowhan for advice and discussion, and Dr. Jerry Eichler for critically reading the manuscript.
This work was supported by the Lipid Maps Collaborative Grant GM069338 and EY023666 from the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1 and S2.
- PG
- phosphatidylglycerol
- PGP
- phosphatidylglycerophosphate
- CDP-DAG
- CDP-diacylglycerol
- CL
- cardiolipin
- ClsA
- ClsB, or ClsC, CL synthases encoded by the clsA, clsB, or clsC genes, respectively
- ESI
- electrospray ionization
- PA
- phosphatidic acid
- PE
- phosphatidylethanolamine
- PLD
- phospholipase D.
References
- 1. Dowhan W. (2013) A retrospective: use of Escherichia coli as a vehicle to study phospholipid synthesis and function. Biochim. Biophys. Acta 1831, 471–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tan B. K., Bogdanov M., Zhao J., Dowhan W., Raetz C. R., and Guan Z. (2012) Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl. Acad. Sci. U.S.A. 109, 16504–16509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hirschberg C. B., and Kennedy E. P. (1972) Mechanism of the enzymatic synthesis of cardiolipin in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 69, 648–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dowhan W. (2009) Molecular genetic approaches to defining lipid function. J. Lipid Res. 50, S305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsumoto K. (2001) Dispensable nature of phosphatidylglycerol in Escherichia coli: dual roles of anionic phospholipids. Mol. Microbiol. 39, 1427–1433 [DOI] [PubMed] [Google Scholar]
- 6. Mileykovskaya E., and Dowhan W. (2005) Role of membrane lipids in bacterial division-site selection. Curr. Opin. Microbiol. 8, 135–142 [DOI] [PubMed] [Google Scholar]
- 7. Xia W., and Dowhan W. (1995) In vivo evidence for the involvement of anionic phospholipids in initiation of DNA replication in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 783–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu Y. H., Guan Z., Zhao J., and Raetz C. R. (2011) Three phosphatidylglycerol-phosphate phosphatases in the inner membrane of Escherichia coli. J. Biol. Chem. 286, 5506–5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kikuchi S., Shibuya I., and Matsumoto K. (2000) Viability of an Escherichia coli pgsA null mutant lacking detectable phosphatidylglycerol and cardiolipin. J. Bacteriol. 182, 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shiba Y., Yokoyama Y., Aono Y., Kiuchi T., Kusaka J., Matsumoto K., and Hara H. (2004) Activation of the Rcs signal transduction system is responsible for the thermosensitive growth defect of an Escherichia coli mutant lacking phosphatidylglycerol and cardiolipin. J. Bacteriol. 186, 6526–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki M., Hara H., and Matsumoto K. (2002) Envelope disorder of Escherichia coli cells lacking phosphatidylglycerol. J. Bacteriol. 184, 5418–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagahama H., Sakamoto Y., Matsumoto K., and Hara H. (2006) RcsA-dependent and -independent growth defects caused by the activated Rcs phosphorelay system in the Escherichia coli pgsA null mutant. J. Gen. Appl. Microbiol. 52, 91–98 [DOI] [PubMed] [Google Scholar]
- 13. Mileykovskaya E., Ryan A. C., Mo X., Lin C. C., Khalaf K. I., Dowhan W., and Garrett T. A. (2009) Phosphatidic acid and N-acylphosphatidylethanolamine form membrane domains in Escherichia coli mutant lacking cardiolipin and phosphatidylglycerol. J. Biol. Chem. 284, 2990–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guan Z., and Eichler J. (2011) Liquid chromatography/tandem mass spectrometry of dolichols and polyprenols, lipid sugar carriers across evolution. Biochim. Biophys. Acta 1811, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guzman L. M., Belin D., Carson M. J., and Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo D., and Tropp B. E. (2000) A second Escherichia coli protein with CL synthase activity. Biochim. Biophys. Acta 1483, 263–274 [DOI] [PubMed] [Google Scholar]
- 17. Selvy P. E., Lavieri R. R., Lindsley C. W., and Brown H. A. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown H. A., Henage L. G., Preininger A. M., Xiang Y., and Exton J. H. (2007) Biochemical analysis of phospholipase D. Methods Enzymol. 434, 49–87 [DOI] [PubMed] [Google Scholar]
- 19. Pulfer M., and Murphy R. C. (2003) Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 22, 332–364 [DOI] [PubMed] [Google Scholar]
- 20. Han X., and Gross R. W. (2003) Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: a bridge to lipidomics. J. Lipid Res. 44, 1071–1079 [DOI] [PubMed] [Google Scholar]
- 21. Murphy R. C., Fiedler J., and Hevko J. (2001) Analysis of nonvolatile lipids by mass spectrometry. Chem. Rev. 101, 479–526 [DOI] [PubMed] [Google Scholar]
- 22. Quehenberger O., Armando A. M., Brown A. H., Milne S. B., Myers D. S., Merrill A. H., Bandyopadhyay S., Jones K. N., Kelly S., Shaner R. L., Sullards C. M., Wang E., Murphy R. C., Barkley R. M., Leiker T. J., et al. (2010) Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fenn J. B., Mann M., Meng C. K., Wong S. F., and Whitehouse C. M. (1989) Electrospray ionization for mass spectrometry of large biomolecules. Science 246, 64–71 [DOI] [PubMed] [Google Scholar]
- 24. Karas M., and Hillenkamp F. (1988) Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 60, 2299–2301 [DOI] [PubMed] [Google Scholar]
- 25. Guan Z., Breazeale S. D., and Raetz C. R. (2005) Extraction and identification by mass spectrometry of undecaprenyl diphosphate-MurNAc-pentapeptide-GlcNAc from Escherichia coli. Anal. Biochem. 345, 336–339 [DOI] [PubMed] [Google Scholar]
- 26. Guan Z., Li S., Smith D. C., Shaw W. A., and Raetz C. R. (2007) Identification of N-acylphosphatidylserine molecules in eukaryotic cells. Biochemistry 46, 14500–14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ward W. C., Guan Z., Zucca F. A., Fariello R. G., Kordestani R., Zecca L., Raetz C. R., and Simon J. D. (2007) Identification and quantification of dolichol and dolichoic acid in neuromelanin from substantia nigra of the human brain. J. Lipid Res. 48, 1457–1462 [DOI] [PubMed] [Google Scholar]
- 28. Cantagrel V., Lefeber D. J., Ng B. G., Guan Z., Silhavy J. L., Bielas S. L., Lehle L., Hombauer H., Adamowicz M., Swiezewska E., De Brouwer A. P., Blümel P., Sykut-Cegielska J., Houliston S., Swistun D., et al. (2010) SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142, 203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J., Guan Z., Murphy A. N., Wiley S. E., Perkins G. A., Worby C. A., Engel J. L., Heacock P., Nguyen O. K., Wang J. H., Raetz C. R., Dowhan W., and Dixon J. E. (2011) Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lombardi F. J., Chen S. L., and Fulco A. J. (1980) A rapidly metabolizing pool of phosphatidylglycerol as a precursor of phosphatidylethanolamine and diglyceride in Bacillus megaterium. J. Bacteriol. 141, 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lombardi F. J., and Fulco A. J. (1980) Two distinct pools of membrane phosphatidylglycerol in Bacillus megaterium. J. Bacteriol. 141, 618–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walton P. A., and Goldfine H. (1987) Transphosphatidylation activity in Clostridium butyricum: evidence for a secondary pathway by which membrane phospholipids may be synthesized and modified. J. Biol. Chem. 262, 10355–10361 [PubMed] [Google Scholar]
- 33. Kawai F., Shoda M., Harashima R., Sadaie Y., Hara H., and Matsumoto K. (2004) Cardiolipin domains in Bacillus subtilis Marburg membranes. J. Bacteriol. 186, 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koprivnjak T., Zhang D., Ernst C. M., Peschel A., Nauseef W. M., and Weiss J. P. (2011) Characterization of Staphylococcus aureus cardiolipin synthases 1 and 2 and their contribution to accumulation of cardiolipin in stationary phase and within phagocytes. J. Bacteriol. 193, 4134–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Henry S. A., Kohlwein S. D., and Carman G. M. (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Osman C., Haag M., Wieland F. T., Brügger B., and Langer T. (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., and Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 39. Galloway S. M., and Raetz C. R. (1990) A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J. Biol. Chem. 265, 6394–6402 [PubMed] [Google Scholar]
- 40. Kanipes M. I., Lin S., Cotter R. J., and Raetz C. R. (2001) Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-d-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide: a novel membrane enzyme dependent upon phosphatidylethanolamine. J. Biol. Chem. 276, 1156–1163 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.