

Abstract
The proper establishment of epithelial polarity allows cells to sense and respond to signals that arise from the microenvironment in a spatiotemporally controlled manner. Atypical PKCs (aPKCs) are implicated as key regulators of epithelial polarity. However, the molecular mechanism underlying the negative regulation of aPKCs remains largely unknown. In this study, we demonstrated that PH domain leucine-rich repeat protein phosphatase (PHLPP), a novel family of Ser/Thr protein phosphatases, plays an important role in regulating epithelial polarity by controlling the phosphorylation of both aPKC isoforms. Altered expression of PHLPP1 or PHLPP2 disrupted polarization of Caco2 cells grown in 3D cell cultures as indicated by the formation of aberrant multi-lumen structures. Overexpression of PHLPP resulted in a decrease in aPKC phosphorylation at both the activation loop and the turn motif sites; conversely, knockdown of PHLPP increased aPKC phosphorylation. Moreover, in vitro dephosphorylation experiments revealed that both aPKC isoforms were substrates of PHLPP. Interestingly, knockdown of PKCζ, but not PKCι, led to similar disruption of the polarized lumen structure, suggesting that PKCζ likely controls the polarization process of Caco2 cells. Furthermore, knockdown of PHLPP altered the apical membrane localization of aPKCs and reduced the formation of aPKC-Par3 complex. Taken together, our results identify a novel role of PHLPP in regulating aPKC and cell polarity.
Keywords: Akt PKB, cell polarity, phosphatase, protein kinase C (PKC), protein phosphorylation, PHLPP, atypical PKC, dephosphorylation
Introduction
Establishing the polarity of epithelial cells is crucial for the maintenance of tissue homeostasis. Increasing evidence has suggested that disruption of polarity promotes the malignant progression of cancer cells. It has been well documented that epithelial cells (including those in the gastrointestinal tract) become polarized during the differentiation process (1). The polarization process is characterized by the formation of specialized junctions between neighboring cells and subsequent separation of two plasma membrane domains: the apical surface facing the external medium and the basolateral surface connected to adjacent cells and extracellular matrix (2). The apical and basolateral membranes are segregated by two highly organized junctions, the tight and adherens junctions, assembled at the site of mammalian cell-cell contacts (3–5). As a major component of the Par complex, aPKCs2 including PKCζ and PKCι, are known to phosphorylate a number of polarity proteins, including Par3, LgL, Crumbs, and Lin5/NuMA, thereby exerting its regulatory roles in cellular polarization (6). Previous studies have suggested that a precise control of aPKC activity is required for the proper establishment of epithelial cell-cell junction and cell polarity (7, 8). Loss of polarity has been associated with increased cell migration and metastasis during tumor progression (1, 9).
Differing from conventional and novel PKCs, aPKCs are insensitive to diacylglycerol- and Ca2+-mediated activation due to the lack of functional C1 and C2 domains (10). As a result, the phosphorylation of aPKCs plays a major role in regulating the kinase activity (10, 11). Specifically, aPKCs are phosphorylated at two conserved phosphorylation sites, namely the activation loop (A-loop) and the turn motif (TM) (10, 11), and the phosphorylation of both sites is required to achieve full activation of aPKCs (12). In addition, the N-terminal pseudosubstrate domain of aPKCs plays an important role in controlling the kinase activity (10, 13). Interestingly, the hydrophobic motif site, which is basally phosphorylated in the conventional and novel PKCs, is replaced with a phospho-mimetic residue in aPKCs (10, 11). Although PDK-1 and mTOR have been identified as upstream kinases that are responsible for phosphorylating aPKCs at the A-loop and TM (12, 14), respectively, phosphatases that dephosphorylate aPKC remain largely unknown.
PHLPP belongs to a novel family of Ser/Thr protein phosphatases that consists of PHLPP1 and PHLPP2 isoforms. Two splice variants of PHLPP1, PHLPP1α and PHLPP1β, have been reported, of which PHLPP1β contains an in-frame extension at the N terminus of PHLPP1α (15, 16). Increasing evidence indicates that both PHLPP isoforms serve as tumor suppressors in breast, colorectal, lung, liver, and pancreatic cancers (17–20). It has been shown that PHLPP negatively regulates multiple oncogenic pathways by directly dephosphorylating and inactivating key signaling molecules, including Akt, S6K, and RAF1 (17, 21–25). Moreover, PHLPP promotes the degradation of conventional PKCs by dephosphorylating the hydrophobic motif (21). In this study, we report the identification of both aPKC isozymes, including PKCζ and PKCι, as novel substrates of PHLPP. The results from our study reveal that PHLPP plays an important role in regulating apical-basolateral polarity by dephosphorylating PKCζ in Caco2 cells.
Results
PHLPP Regulates Caco2 Morphogenesis and Apical-Basolateral Polarity in 3D Culture
In our effort to investigate the role of PHLPP in controlling cancer cell growth in three dimensions, we discovered that knockdown of PHLPP in Caco2 cells resulted in abnormal lumen formation (Fig. 1). As Caco2 cells grow in 3D matrix, cells localized in the outer layer keep proliferating, whereas the inner part of the cell clusters gradually undergoes apoptosis due to the lack of contact with the matrix. The morphogenesis process of these cells involves the formation of one lumen structure with defined apical-basolateral polarity (27). Consistent with previous studies (28), the majority of Caco2 sh-Con cells formed a cyst-like structure with a single hollow lumen after growing in a mixture of Matrigel and collagen I for 14 days (Fig. 1A). In contrast, the aberrant lumen structures, including irregular and multi-lumen formation and filled lumens, were observed in PHLPP knockdown cells (Fig. 1A). The size of cysts formed by PHLPP knockdown cells was significantly larger than that of the control cells (Fig. 1B). The decreased PHLPP expression in the knockdown cells was confirmed using Western blotting analysis (Fig. 1C). In addition, the cysts grown in 3D cell cultures were fixed and stained with the Ki67 antibody to label the proliferating cells. As shown in Fig. 1D, although Ki67-positive cells were found only in the cell layer that formed the lumen structure in sh-Con cells, they were detected at the center of the cysts formed by PHLPP knockdown cells, indicating that increased cell proliferation and disruption of the morphogenesis process (Fig. 1D).
FIGURE 1.
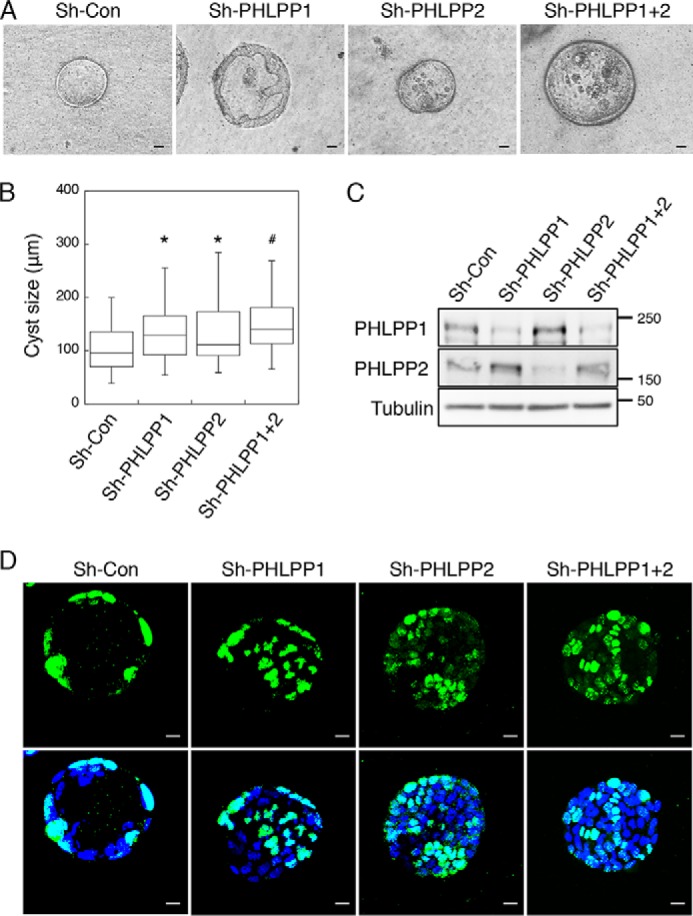
Knockdown of PHLPP alters morphogenesis of Caco2 cells grown in three dimensions. Caco2 cells infected with shRNA lentivirus targeting PHLPP1, PHLPP2, or both PHLPP isoforms were seeded in 3D matrix as a single cell suspension and allowed to grow for 14 days. A, the representative phase-contrast images of the cyst-like structure formed by control and PHLPP knockdown cells. The images were obtained using a Nikon TE2000 inverted microscope with 10× objective. Scale bar, 20 μm. B, the longest diameter of cysts formed by the control and PHLPP knockdown cells was measured using the Nikon Elements AR software. The size distributions of 50 randomly chosen cysts were analyzed for each group of cells and are shown in the box-whisker plot. The average cyst sizes for the following cells are (means ± S.E., in μm): sh-Con, 105.5 ± 5.7; sh-PHLPP1, 140.2 ± 8.5; sh-PHLPP2, 132.4 ± 7.8; and sh-PHLPP1+2, 146.8 ± 6.5 (n = 50, * indicates p < 0.01 and # indicates p < 0.001 by Student's t test when compared with sh-Con cells). C, the expression of PHLPP1 and PHLPP2 in the control and PHLPP knockdown cells as determined using Western blotting. D, the control and PHLPP knockdown cells grown in three dimensions were fixed and stained with the Ki67 antibody (green) and DAPI (blue). Confocal images of stained cells were obtained using an Olympus FluoView FV1000 confocal laser-scanning microscope. Scale bar, 20 μm. Note that the cysts formed by PHLPP knockdown cells show a lack of fully polarized structure and an increased number of Ki67-positive cells.
Furthermore, Caco2 cells grown in 3D cell cultures were stained with phalloidin and ZO-1 to evaluate whether the cells are polarized within the lumen structure. The Caco2 sh-Con cysts showed a spherical architecture, and the cells surrounding the lumen were polarized with F-actin and ZO-1 both concentrated at the apical side of the lumen, indicating a well defined apical-basolateral polarity (Fig. 2A). In marked contrast, PHLPP depletion altered the cyst-like structure by inducing the formation of multiple lumens (Fig. 2A). The polarity was lost in those lumen structures as F-actin was localized to both the apical and basolateral membranes and the cells inside the lumen showed non-polarized membrane distribution of ZO-1 (Fig. 2A). Quantitative analysis revealed that the percentage of cysts with one lumen was significantly reduced in all PHLPP knockdown Caco2 cells when compared with sh-Con cells (Fig. 2B).
FIGURE 2.

Knockdown of PHLPP alters the apical-basolateral polarity in Caco2 cells grown in three dimensions. Caco2 cells infected with shRNA lentivirus targeting PHLPP1, PHLPP2, or both PHLPP isoforms were seeded in 3D matrix as a single cell suspension and allowed to grow for 14 days. The cells were fixed and co-stained with the ZO-1 antibody (red), Alexa Fluor 488-conjugated phalloidin (green), and DAPI (blue). A, representative confocal images were taken from control and PHLPP knockdown cells showing the localization pattern of actin and ZO-1. Scale bar, 20 μm. B, the percentages of control and PHLPP knockdown cells with a single lumen were quantified based on the pattern of actin staining and expressed graphically. Fifty randomly chosen cysts were scored. The shaded bars represent cysts with one lumen, and the open bars represent cysts with multiple or filled lumens.
We next determined the effect of PHLPP overexpression in Caco2 cells grown in 3D cell cultures. Consistent with the role of PHLPP in negatively regulating cell proliferation (17), overexpression of either PHLPP isoform significantly reduced the size of cysts (Fig. 3, A and B). Overexpression of PHLPP was confirmed using Western blotting analysis (Fig. 3C). Intriguingly, we found that overexpression of either PHLPP isoform resulted in aberrant polarization in Caco2 cells similar to that observed in PHLPP knockdown cells (Fig. 3D). The percentage of cysts with one lumen was decreased in PHLPP-overexpressing cells (Fig. 3E). Collectively, these findings suggested that PHLPP plays an important role in regulating epithelial polarity and altered expression of either PHLPP isoform disrupts the morphogenesis process.
FIGURE 3.
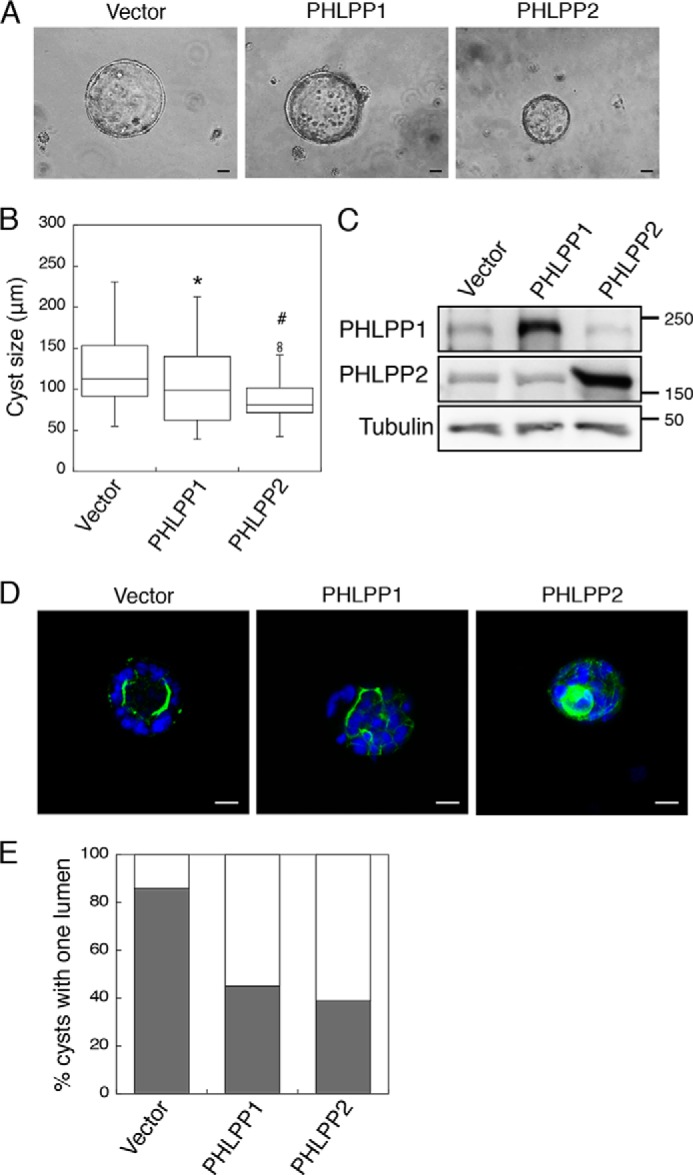
Overexpression of PHLPP alters the apical-basolateral polarity in Caco2 cells grown in three dimensions. Caco2 cells infected with retrovirus encoding vector, HA-PHLPP1, and HA-PHLPP2 were seeded in 3D matrix as a single cell suspension and allowed to grow for 14 days. A, the representative phase-contrast images of the cyst-like structure formed by the control and PHLPP knockdown cells. The images were obtained using a Nikon TE2000 inverted microscope with 10× objective. Scale bar, 20 μm. B, the sizes of 50 randomly chosen cysts were analyzed for each group of cells using the Nikon Elements AR software. The box-whisker plot shows the size distribution. The average cyst sizes for the following cells are (means ± S.E., in μm): vector, 127.2 ± 6.3; HA-PHLPP1, 103.9 ± 6.3; HA-PHLPP2, 89.9 ± 4.9 (* indicates p < 0.05 and # indicates p < 0.001 by Student's t test when compared with vector control cells). C, the expression of PHLPP1 and PHLPP2 in the control and PHLPP-overexpressing cells was determined using Western blotting. D, the control and PHLPP-overexpressing cells grown in three dimensions were fixed and stained with Alexa Fluor 488-conjugated phalloidin (green) and DAPI (blue). Representative confocal images were taken from control and PHLPP knockdown cells showing the localization pattern of actin. Scale bar, 20 μm. E, the percentages of control and PHLPP knockdown cells with a single lumen were quantified based on the pattern of actin staining and expressed graphically. Fifty randomly chosen cysts were scored. The shaded bars represent cysts with one lumen, and the open bars represent cysts with multiple or filled lumens.
PHLPP Negatively Regulates the Phosphorylation of Atypical PKCs
Because aPKCs are known regulators of cell polarity, we next investigated whether the PHLPP-induced polarity defect is mediated through aPKCs. To this end, we determined whether aPKCs are substrates of PHLPP. Silencing either PHLPP isoform resulted in a significant increase in the phosphorylation of both the A-loop and TM sites in PKCι and PKCζ in both SW480 and Caco2 colon cancer cells (Fig. 4). Because the phospho-specific antibodies against the A-loop and the TM site of aPKCs recognize both phosphorylated PKCι and phosphorylated PKCζ, each aPKC isoform was first immunoprecipitated from the cells using isoform-specific antibodies and then analyzed for changes in phosphorylation. Interestingly, knockdown of either PHLPP isoform had similar effects on promoting the phosphorylation at both phosphorylation sites in PKCι and PKCζ, and knockdown of both PHLPP1 and PHLPP2 isoforms did not induce further increase in phosphorylation, suggesting that loss of one PHLPP isoform is sufficient to enhance the phosphorylation of aPKCs (Fig. 4, B and D).
FIGURE 4.

Knockdown of PHLPP increases aPKC phosphorylation. A and B, cell lysates prepared from stable sh-Con, sh-PHLPP1, sh-PHLPP2, and sh-PHLPP1+2 SW480 (A) and Caco2 (B) cells were immunoprecipitated with PKCι- or PKCζ-specific antibody as indicated and subsequently analyzed for the phosphorylation (p) status at the A-loop and TM sites. C, and D, the relative phosphorylation levels of PKCι and PKCζ in SW480 and Caco2 cells, respectively. The relative phosphorylation of PKCι and PKCζ at the A-loop and TM sites was quantified by normalizing the ECL signals generated by the phospho-specific antibodies to that of total protein. Data shown in the graphs represent the means ± S.E. (n = 3, * indicates p < 0.05 by Student's t test when compared with sh-Con cells).
We have previously generated PHLPP1 and PHLPP2 knock-out mice (22, 26, 29). Here we examined whether phosphorylation of aPKCs is elevated in PHLPP knock-out MEF cells. As shown in our previous study, PKCζ is the predominant aPKC expressed in MEF cells, whereas PKCι/λ is not detected at the protein level (12). Knock-out of either PHLPP isoform resulted in an increase in phosphorylation at both the A-loop and TM sites in PKCζ (Fig. 5A). As a control, the phosphorylation of the hydrophobic motif of AKT, a known substrate of PHLPP, was elevated in PHLPP knock-out MEF cells as well (Fig. 5A). In addition, we examined the phosphorylation status of aPKCs in colon tissues of PHLPP knock-out mice. Consistently, the phosphorylation of PKCι and PKCζ at both the A-loop and TM sites was increased in the colon of PHLPP knock-out mice (Fig. 5, B and C).
FIGURE 5.
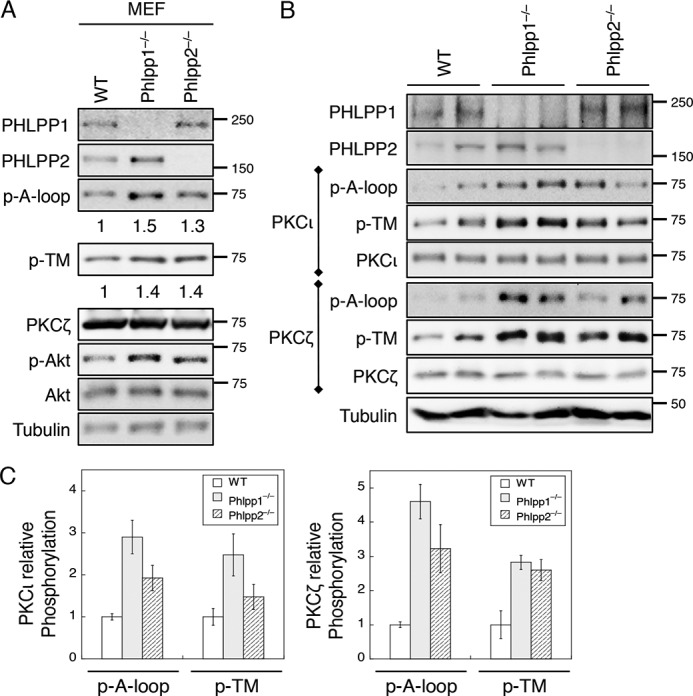
The phosphorylation of aPKCs is increased in PHLPP knock-out MEF cells and mouse tissues. A, cell lysates prepared from WT, PHLPP1−/−, and PHLPP2−/− MEF cells were analyzed for the phosphorylation (p) of PKCζ at the A-loop and TM sites using the phospho-specific antibodies. The relative phosphorylation was quantified by normalizing the amount of phosphorylation as detected by the phospho-specific antibody to that of total protein and shown below the phosphoblots. B, fresh colon tissues were isolated from WT, PHLPP1−/−, and PHLPP2−/− mice. Two mice from each genotype were analyzed. Protein extracts were prepared and first immunoprecipitated with the PKCι or PKCζ antibody. The phosphorylation status of PKCι and PKCζ was determined using the phospho-specific antibodies. C, Western blots as shown in B were quantified by normalizing the amount of phosphorylation as detected by the phospho-specific antibody to that of total protein. Graphs show the average results of two mice. Data shown in the graphs represent the means ± S.D.
Furthermore, we found that overexpression of either PHLPP isoform decreased the phosphorylation of endogenous PKCι and PKCζ at both sites in SW480 and Caco2 cells (Fig. 6A). To further determine whether PHLPP dephosphorylates aPKCs directly, we performed in vitro dephosphorylation experiments using purified PP2C domains of PHLPP1 and PHLPP2. PKCι and PKCζ overexpressed in 293T cells were immunoprecipitated and used as substrates in the dephosphorylation reactions. Our results showed that PHLPP was able to dephosphorylate both the A-loop and TM sites in PKCι and PKCζ in vitro (Fig. 6B). In addition, PHLPP-mediated dephosphorylation of aPKCs occurred rapidly, and no significant differences in the time course and extent of dephosphorylation at both sites of aPKCs were observed (Fig. 6, C and D). Because the phosphorylation of aPKCs is sensitive to PI3K activation (14), we next examined whether knockdown of PHLPP prevents dephosphorylation of aPKCs upon inhibition of PI3K. Indeed, treating cells with PI3K inhibitor LY294002 induced rapid dephosphorylation of both PKCι and PKCζ at the A-loop and TM sites in sh-Con Caco2 cells; however, this dephosphorylation of aPKCs was largely inhibited in PHLPP knockdown cells (Fig. 6E). To further examine the specificity of PHLPP-mediated dephosphorylation of aPKCs, we conducted in vitro dephosphorylation experiments to compare PHLPP-dependent dephosphorylation of PKCζ with Akt dephosphorylation. Interestingly, both Akt and PKCζ were readily dephosphorylated by the PP2C domains of PHLPP1 and PHLPP2 in a dose-dependent manner (Fig. 6F), suggesting that PHLPP functions similarly as a phosphatase toward both Akt and aPKC in vitro. Taken together, our results demonstrated that PHLPP is a key regulator mediating aPKC dephosphorylation both in vitro and in vivo.
FIGURE 6.

PKCζ and PKCι are substrates of PHLPP. A, cell lysates prepared from SW480 and Caco2 cells stably overexpressing PHLPP1 or PHLPP2 were immunoprecipitated using antibodies against PKCζ and PKCι and subsequently analyzed by Western blotting. The phosphorylation status of PKCι and PKCζ at the A-loop and the TM site was detected using the phospho-specific antibodies. The relative phosphorylation was quantified by normalizing ECL signals generated by the phospho-specific antibody to that of total protein and is shown below the phosphoblots. B, the time course of aPKC dephosphorylation in vitro. 293T cells transfected with PKCζ and PKCι expression plasmids were lysed and immunoprecipitated using antibodies against PKCι or PKCζ. Dephosphorylation reactions were carried out by incubating the immunoprecipitates with the purified PP2C domains of PHLPP1 or PHLPP2 at room temperature for 0–30 min. The phosphorylation of aPKCs was detected using the phospho-antibodies. C and D, the levels of PKCι and PKCζ phosphorylation at the A-loop and TM sites were quantified by normalizing ECL signals generated by the phospho-specific antibodies to that of total protein. Data shown in the graph represent the means ± S.E. (n = 3). E, knockdown of PHLPP prevents dephosphorylation of PKCι and PKCζ. Stable sh-Con, sh-PHLPP1, and sh-PHLPP2 knockdown Caco2 cells were treated with LY294002 (LY, 20 μm) for 30 min. The phosphorylation status of immunoprecipitated PKCι and PKCζ was analyzed using the phospho-specific antibodies. F, the dose-dependent dephosphorylation of Akt and PKCζ in vitro. 293T cells transfected with Akt and PKCζ expression plasmids were lysed and immunoprecipitated using antibodies against Akt or PKCζ. Dephosphorylation reactions were carried out by incubating the immunoprecipitates with increasing amounts of PP2C domains of PHLPP1 or PHLPP2 at room temperature for 10 min. The phosphorylation of Akt and PKCζ was detected using the phospho-antibodies.
PHLPP Regulates the Localization of aPKCs
Atypical PKCs are known to localize to the apical side of polarized epithelial cells, and this specialized localization is required to maintain the cell polarity (7, 30). As we found that silencing PHLPP resulted in the disruption of cell polarity and increased phosphorylation of aPKCs, we next tested whether loss of PHLPP alters aPKC localization in Caco2 cells. The control and PHLPP knockdown cells grown in 3D cell cultures were stained with the PKCζ antibody. The results showed that the localization of PKCζ was restricted to the apical side of the single lumen structure in the sh-Con cells (Fig. 7A). However, PKCζ was also found to localize to the internal membrane in the multi-lumen structure and the membrane of individual cells inside the lumen in PHLPP knockdown cells (Fig. 7A). Similar changes in membrane localization were observed for PKCι in PHLPP knockdown cells as well (data not shown). Furthermore, the localization of aPKCs was analyzed in confluent Caco2 cells grown on glass coverslips. The confluent monolayer Caco2 cells are known to polarize on 2D surfaces as well. Confocal images of cells stained with PKCζ or PKCι antibodies showed membrane localization of both aPKC isoforms (Fig. 7, B and C). Interestingly, both PKCζ and PKCι were predominately localized to the apical surface above the adherens junction as marked by E-cadherin staining in the control cells (Fig. 7D); however, both aPKC isoforms moved away from the apical membrane and became mislocalized to the cell-cell junction with significant co-localization with E-cadherin in PHLPP knockdown cells (Fig. 7D).
FIGURE 7.

PHLPP controls PKCζ localization in Caco2 cells. A, stable sh-Con, sh-PHLPP1, and sh-PHLPP2 Caco2 cells seeded in 3D matrix as a single cell suspension and allowed to grow for 14 days. The cells were fixed and stained with the PKCζ antibody (green) and DAPI. Representative confocal images show the localization of PKCζ in the cyst-like structure. Scale bar, 20 μm. B and C, stable sh-Con, sh-PHLPP1, and sh-PHLPP2 Caco2 cells were co-stained with antibodies against PKCζ or PKCι (green) and E-cadherin (red). Nuclei were stained with DAPI (blue). The merged images are shown. Confocal images were obtained using an Olympus FluoView FV1000 confocal laser scanning microscope with 60× objective. Scale bars, 20 μm. Note that the non-overlapping staining of aPKCs and E-cadherin indicates that aPKCs are localized to different membrane domains along the cell-cell junction when compared with E-cadherin. D, confocal xz images of PKCζ or PKCι (green), E-cadherin (red), and nuclei (blue) were generated from merged xy images of control and PHLPP knockdown Caco2 cells co-stained for these proteins.
To determine the mechanism by which PHLPP loss disrupts aPKC membrane localization, we examined the formation of aPKC-Par3 complex. It has been shown previously that Par3 is phosphorylated by aPKC and this phosphorylation reduces the interaction between aPKC and Par3 (31, 32). Consistent with increased aPKC phosphorylation in PHLPP knockdown cells, the amount of Par3 that co-immunoprecipitated with PKCι and PKCζ was largely reduced (Fig. 8A). The PHLPP loss-mediated increase in phosphorylation of aPKCs and Akt was confirmed in cell lysates of control and PHLPP knockdown cells (Fig. 8B). Taken together, these results suggested that PHLPP-mediated dephosphorylation of aPKC may alter the subcellular localization of aPKC by modulating the formation of aPKC-Par3 complex in polarized cells.
FIGURE 8.

Knockdown of PHLPP alters the formation of aPKC-Par3 complex. A, cell lysates prepared from sh-Con, sh-PHLPP1, and sh-PHLPP2 Caco2 cells were immunoprecipitated with protein A/G beads alone (Mock) or with antibodies against PKCι and PKCζ. The presence of Par3, as well as PKCι and PKCζ, in the cell lysate input and immunoprecipitates (IP) was detected using the Par3, PKCι, and PKCζ antibodies, respectively. B, the expression of PHLPP and phosphorylation status of Akt, PKCι, and PKCζ in the cell lysates was analyzed using Western blotting.
PKCζ but Not PKCι Regulates Caco2 Morphogenesis
Previous studies have shown that inhibition of aPKCs disrupts the proper establishment of polarity in Caco2 cells grown in 3D culture (7, 28). Because both PKCζ and PKCι have been implicated in apical-basolateral polarity regulation (7, 8) and both are substrates of PHLPP in cells, we next investigated the functional contribution of each aPKC isoform in the morphogenesis of Caco2 cells. Stable PKCζ and PKCι knockdown Caco2 cells were generated using two different lentiviral shRNAs (Fig. 9A). Control and PKC knockdown Caco2 cells were then cultured in 3D matrix and allowed to grow for 14 days. Consistent with the pro-proliferative function of aPKCs, knockdown of either PKCζ or PKCι reduced the size of cyst-like structures formed in three dimensions (Fig. 9B). Interestingly, although knocking down PKCζ significantly decreased the percentage of cysts with one lumen, silencing PKCι had little effect on the morphogenesis of Caco2 cells in three dimensions (Fig. 9, C and D). Knockdown of both aPKCs had a similar effect on disrupting the polarization process of Caco2 cells as silencing PKCζ alone (Fig. 9D). These data indicated that PKCζ is likely the downstream effector of PHLPP in maintaining apical-basal polarity in Caco2 cells.
FIGURE 9.

Knockdown of PKCζ but not PKCι disrupts apical-basolateral polarity in Caco2 cells grown in three dimensions. A, stable control (sh-Con) and aPKC knockdown Caco2 cells were analyzed for the expression of PKCι and PKCζ by Western blotting. Two different targeting shRNAs were used to silence each aPKC isoform. B, the sizes of 50 randomly chosen cysts formed by the control and aPKC knockdown cells were measured using the Nikon Elements AR software and are shown in the box-whisker plot. The average cyst sizes for the following cells are (means ± S.E., in μm): sh-Con, 134.5 ± 6.8; sh-PKCι#1, 100.0 ± 5.8; sh-PKCι#2, 110.4 ± 6.9; sh-PKCζ#1, 101.1 ± 6.6; sh-PKCζ#2, 114.6 ± 7.8; and sh-PKCι+ζ, 104.7 ± 7.7 (* indicates p < 0.05 and # indicates p < 0.001 by Student's t test when compared with sh-Con cells). C, the lumen structure of the cysts formed by the control and aPKC knockdown cells. The cells were stained with Alexa Fluor 488-conjugated phalloidin (green) and DAPI (blue). Scale bars, 10 μm. D, the percentages of control and aPKC knockdown cells with a single lumen were quantified based on the pattern of actin staining and expressed graphically. Fifty randomly chosen cysts were scored. The shaded bars represent cysts with one lumen, and the open bars represent cysts with multiple or filled lumens.
To further determine the specificity of PHLPP substrates on regulating cell polarity, control and PHLPP knockdown cells grown in three dimensions were treated with inhibitors of Akt or PKCζ (Fig. 10A). Treating cells with either Akt or PKCζ inhibitor (Akt-VIII and myristoylated pseudosubstrate of PKCζ, respectively) significantly decreased the size of cysts formed by both control and PHLPP knockdown cells (Fig. 10B). In addition, the PKCζ inhibitor resulted in a significant disruption of lumen structure similar to that observed in PKCζ knockdown cells, whereas the Akt inhibitor had little effect (Fig. 10C). However, inhibition of either Akt or PKCζ was unable to rescue the polarity defect caused by PHLPP loss (Fig. 10C). Collectively, our results indicated that elevated Akt and aPKC activity likely both contribute to increased cell proliferation seen in PHLPP knockdown cells grown in three dimensions. However, inhibition of Akt activity does not affect cell polarity.
FIGURE 10.

Inhibition of Akt does not affect cell polarity. A, stable sh-Con, sh-PHLPP1, and sh-PHLPP2 Caco2 cells seeded in 3D matrix as a single cell suspension. At day 7, the cells were left untreated or treated with Akt-VIII (0.5 μm) or PKCζ inhibitor (PKCζ-i, 20 μm) for an additional 7 days. The cells were subsequently fixed and stained with Alexa Fluor 488-conjugated phalloidin (green) and DAPI (blue). Scale bars, 10 μm. B, the sizes of 50 randomly chosen cysts as shown in A were measured using the Nikon Elements AR software. The size distribution is shown in the box-whisker plot. The average cyst sizes for the following cells are (means ± S.E., in μm): sh-Con, 84.2 ± 4.7; sh-Con+Akt-VIII, 59.6 ± 4.0; sh-Con+PKCζ-i, 61.7 ± 4.8; sh-PHLPP1, 122.1 ± 7.2; sh-PHLPP1+Akt-VIII, 55.7 ± 2.6; sh-PHLPP1+PKCζ-i, 59.1 ± 3.2; sh-PHLPP2, 108.2 ± 6.7; sh-PHLPP2+Akt-VIII, 59.3 ± 3.2; and sh-PHLPP1+PKCζ-i, 73.1 ± 6.5 (# indicates p < 0.001 and * indicates p < 0.01 when compared with untreated sh-Con cells, ¶ indicates p < 0.001 when compared with untreated sh-PHLPP1 cells, and § indicates p < 0.001 when compared with untreated sh-PHLPP2 cells by Student's t test). C, the percentages of cysts with a single lumen were quantified based on the pattern of actin staining and expressed graphically. Fifty randomly chosen cysts from each group were scored. The shaded bars represent cysts with one lumen, and the open bars represent cysts with multiple or filled lumens.
Discussion
Atypical PKCs are major regulators of the establishment and maintenance of cell polarity (6–8, 10, 33). Previous studies have demonstrated that loss of polarity is associated with late stage or metastatic tumors where cancer cells undergo epithelial–mesenchymal transition (1, 34, 35). The function of several oncogenic proteins and tumor suppressors (such as HER2/ERBB2, KRAS, and LKB1) has also been connected to their ability to regulate the integrity of epithelial polarity (9, 35–38). In addition, it has been suggested that loss of epithelial polarity may lead to aberrant activation of receptor tyrosine kinase (RTK) due to disruption of asymmetrical distribution of the receptors (39). Here we identified PHLPP as an important regulator of cell polarity by negatively controlling the phosphorylation of aPKCs. We found that both the A-loop and TM sites in aPKCs can be dephosphorylated by PHLPP in vitro and in cells. Functionally, silencing either PHLPP isoform is sufficient to disrupt the apical-basal polarity of Caco2 cells grown in three dimensions as indicated by multi-lumen formation. Interestingly, although both aPKC isoforms have been implicated in regulating cell polarity, our results showed that PKCζ, but not PKCι, is required for the proper morphogenesis of Caco2 cells.
Previous studies have indicated that the full activation of aPKCs requires phosphorylation at both the A-loop and TM sites (10, 12). However, the phosphatase-mediated dephosphorylation of these sites in aPKCs has not been studied extensively. It has been suggested that PKCλ may be regulated by PP2A as overexpression of SV40 small t-antigen, an inhibitor of PP2A, leads to increased phosphorylation at the A-loop site in 3T3-L1 adipocytes, but the functional effect of this PP2A-mediated dephosphorylation is unclear (40). Our results here provide strong evidence indicating that PHLPP negatively regulates the phosphorylation of aPKCs both in vitro and in cells. However, because PHLPP has been shown to regulate a number of important signaling pathways (such as PI3K/Akt and RAS/RAF the pathways), we cannot rule out the possibility that the negative correlation between PHLPP expression and aPKC phosphorylation observed in our study is an indirect effect of PHLPP-mediated regulation of other signaling molecules. Future studies are needed to define how the specificity of PHLPP-mediated regulation of different substrates is controlled in vivo. Although it has been shown that PHLPP preferentially dephosphorylates the hydrophobic motif of Akt, we found here that both the A-loop and TM sites of aPKCs are similarly controlled by PHLPP. This difference may due to the fact that both the A-loop and TM sites in aPKCs are readily accessible, as shown in the crystal structure of PKCι (41), whereas the A-loop of Akt may become less accessible when the hydrophobic motif is phosphorylated (42).
Both aPKC isoforms have been linked to the establishment and maintenance of apical-basal polarity in different cell systems. However, the specific contribution of each aPKC isoform is less understood as most of the previous studies employed reagents targeting both aPKCs (7, 43). Although we found that knockdown of PKCζ, but not PKCι, alters the apical-basal polarity in Caco2 cells, PKCι has been shown to regulate the polarization process in Madin-Darby canine kidney cells (8). PKCι is expressed at a lower level and with more restricted membrane localization when compared with PKCζ in Caco2 cells. Thus, the functional involvement of a specific aPKC isoform in maintaining cell polarity is likely cell type- and cell context-dependent.
Consistent with the notion that the activity of aPKC needs to be tightly controlled to maintain cell polarity, increased aPKC activity as a result of PHLPP knockdown or decreased aPKC activity a result of PHLPP overexpression, as well as silencing PKCζ or treating cells with PKCζ inhibitor, has the same effect on disrupting apical-basal polarity. In addition, we found that inhibition of Akt activity decreases cell proliferation but has no effect on polarity. Similar results have been reported in breast cancer cells (44), suggesting that polarity and proliferation are regulated by independent mechanisms. Although up-regulation of both aPKC and Akt activity may contribute to increased proliferation seen in PHLPP knockdown cells, aPKC rather than Akt downstream of PHLPP likely controls cell polarity. Intriguingly, inhibition of PKCζ in PHLPP knockdown cells was sufficient to block the pro-proliferation effect of silencing PHLPP but failed to rescue the polarity defect. These results are consistent with the notion that a threshold of aPKC activity is required for the correct establishment of epithelial cell polarity (8). Thus, both knockdown and overexpression of PHLPP may alter the balance of aPKC activity and result in similar disruption of epithelial morphogenesis.
Mechanistically, we found that knockdown of PHLPP results in redistribution of aPKCs from the apical to the basolateral membrane in Caco2 cells. Previous studies have shown that the localization of aPKC to the apical membrane depends on its ability to phosphorylate Par3 and phosphorylation of Par3 partially dissociates aPKC from Par3 (35, 45). This is consistent with our finding that the interaction between aPKC and Par3 is decreased in PHLPP knockdown cells as the activity of aPKC increases. Because aPKC is known to function in the PAR6-CDC42-aPKC complex exclusively on the apical side of the apical-basolateral membrane border in polarized epithelial cells (35), this PHLPP loss-induced mislocalization of aPKC as the result of aberrant aPKC-Par3 interaction likely disrupts the spatiotemporally regulated epithelial polarity.
In summary, our study identified a novel role of PHLPP in controlling epithelial polarity by maintaining the balance of aPKC activity. Previous studies have shown that PHLPP expression is frequently down-regulated in various cancer types and PHLPP loss promotes tumor progression by promoting cell growth and proliferation and inhibiting apoptosis (17, 24, 25, 46, 47). Because loss of epithelial polarity is an important hallmark of advanced malignant tumors, the ability of PHLPP to maintain proper polarization of epithelial cells likely contributes to its tumor suppressor function. Interestingly, several well characterized tumor suppressors, including LKB1 and phosphatase and tensin homolog (PTEN), are known to control cell polarity in addition to their ability to negatively regulate cell survival and proliferation (1). Thus, maintaining cell polarity is likely a common strategy utilized by different tumor suppressors in regulating cellular homeostasis.
Experimental Procedures
Antibodies and Reagents
Antibodies against PHLPP1 and PHLPP2 were purchased from Proteintech and Bethyl Laboratories, respectively. The antibodies against PKCι (including N-20 and H-12), PKCζ (including C-20 and H-1), Ki67, and Akt1 were from Santa Cruz Biotechnology. The phospho-PKCι/ζ (the A-loop site, Thr-412/Thr-410 in human), phospho-Akt (the Ser-473 site), and E-cadherin antibodies were obtained from Cell Signaling. The ZO-1, phospho-PKCι/ζ (the TM site, Thr-564/Thr-560 in human), and Par-3 (anti-PAR-3) antibodies were from Thermo Fisher, and the γ-tubulin antibody was from Sigma-Aldrich. The expression plasmids for GST-tagged PP2C domains of PHLPP1 and PHLPP2 have been described in previous studies (22). Akt inhibitor VIII (0.5 μm), myristoylated-PKCζ pseudosubstrate inhibitor (20 μm), and PI3K inhibitor LY294002 (20 μm) were purchased from Millipore. The FLAG-tagged PKCι, HA-tagged PKCζ, and HA-Akt1 expression plasmids were obtained from Addgene.
Cells
Human colon cancer cell lines SW480 and Caco2 and mouse embryonic fibroblast (MEF) cells were cultured in DMEM supplemented with 10% FBS (Sigma-Aldrich) and penicillin/streptomycin. Cells stably overexpressing HA-PHLPP1β and HA-PHLPP2 were generated by infecting with retrovirus encoding each PHLPP isoform and selecting against puromycin as described previously (48). The shRNA targeting sequences were constructed in pLKO.1-puro vector (Sigma-Aldrich), and the lentivirus-mediated delivery of shRNA and selection for stable knockdown cells were carried out as described previously (17, 22). The targeting sequences for human PKCι and PKCζ are the following: for PKCζ, 5′-GCCTCCAGTAGACGACAAGAA-3′ (#1) and 5′-CCCGACATGAACACAGAGGA-3′ (#2); and for PKCι, 5′-GCCTGGATACAATTAACCATT-3′(#1) and 5′-CCTGAAGAACATGCCAGATTT-3′ (#2). The shPHLPP1+2 double knockdown cells were generated by infecting stable single PHLPP knockdown cells with shRNA lentivirus targeting the other PHLPP isoform. The Phlpp1 (Phlpp1−/−) and Phlpp2 (Phlpp2−/−) knock-out mice were described previously (26, 29, 49). MEF cells were isolated from wild-type, Phlpp1−/−, and Phlpp2−/− mouse embryos at day 14 of gestation by following standard protocols (22). The primary MEF cells were immortalized using lentivirus-mediated knockdown of p53 using pBabe-puro-shp53 (Addgene).
Western Blotting Analysis
Cells were harvested and lysed in lysis buffer (50 mm Na2HPO4, 1 mm sodium pyrophosphate, 20 mm NaF, 2 mm EDTA, 2 mm EGTA, 1% Triton X-100, 1 mm DTT, 200 μm benzamidine, 40 μg ml−1 leupeptin, 200 μm PMSF), and the detergent-solubilized cell lysates were obtained after centrifugation for 5 min at 16,000 × g at 4 °C. Equal amounts of cell lysates as determined by Bradford assays were resolved by SDS-PAGE and subjected to Western blotting analysis. The density of ECL signals was obtained and quantified using a FluorChem digital imaging system (Alpha Innotech).
Immunoprecipitation and in Vitro Dephosphorylation Assay
The PP2C domains of PHLPP1 or PHLPP2 were produced as GST-tagged fusion proteins and purified from bacteria as described previously (24, 25). The GST fusion proteins were treated with PreScission Protease to release PP2C recombinant proteins from the GST tag. The in vitro dephosphorylation experiments were performed according to procedures described previously (22). Briefly, 293T cells were transfected with PKCζ or PKCι expression plasmids, and cell lysates were prepared in lysis buffer. The detergent-solubilized cell lysates were incubated with the PKCζ and PKCι antibodies to obtain PKCζ and PKCι proteins. The dephosphorylation reactions were carried out by incubating the immunoprecipitates with purified PHLPP1-PP2C or PHLPP2-PP2C (0.1 μg) at room temperature for 0–30 min in the phosphatase buffer (0.1 m sodium acetate, 0.05 m bis-Tris, 0.05 m Tris, 2 mm MnCl2, and 10 mm DTT, pH 7.5). For dose-dependent dephosphorylation of Akt and PKCζ, the amounts of purified PP2C proteins used are 0.01, 0.05, and 0.1 μg in 50-μl reaction.
Three-dimensional Morphogenesis Assay and Immunofluorescence Staining
To monitor the polarization process of epithelial cells, Caco2 cells were cultured in 3D matrix according to previously reported protocols with minor modifications (7, 27, 28). Briefly, single cell suspensions of Caco2 cells were prepared in serum-free DMEM medium and embedded into a collagen/Matrigel (1:1) mixture. The cells were allowed to grow into cyst-like structures in 3D matrix for 2 weeks. The size and morphology of the cysts were examined by phase-contrast microscopy. For immunofluorescence staining, the cysts were fixed in 4% paraformaldehyde and permeabilized using 1% Triton X-100 in PBS. Primary antibodies were diluted in labeling buffer (1% BSA/PBS) and incubated with cells overnight at 4 °C. The Alexa Fluor 594- or Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibodies (Thermo Fisher) were used subsequently. Actin was stained using Alexa Fluor 488-conjugated phalloidin, whereas the nuclei of the cells were stained with DAPI-containing mounting medium. The cellular distributions of endogenous proteins were visualized using an Olympus FluoView FV1000 confocal laser-scanning microscope. Confocal images of cysts grown in three dimensions and cells grown in two dimensions were obtained with 20× and 60× objective, respectively.
Author Contributions
X. X. conducted most of the experiments, analyzed the results, and wrote most of the paper. X. L. conceived the idea for the project and conducted initial experiments on determining the role of PHLPP in regulating cell polarity and PKCζ phosphorylation. Y. A. W. provided technical assistance and contributed to the preparation of the figures. T. G. designed the study and wrote the paper.
Acknowledgments
These studies were supported by the Biostatistics Shared Resources of the University of Kentucky Markey Cancer Center (Grant P30CA177558).
This work was supported by National Institutes of Health Grant R01 CA133429 (to T. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- aPKC
- atypical PKC
- PH
- pleckstrin homology
- PHLPP
- PH domain leucine-rich repeat protein phosphatase
- A-loop
- activation loop
- TM
- turn motif
- PP2A
- protein phosphatase 2
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MEF
- mouse embryonic fibroblast
- Con
- control.
References
- 1. Wodarz A., and Näthke I. (2007) Cell polarity in development and cancer. Nat. Cell Biol. 9, 1016–1024 [DOI] [PubMed] [Google Scholar]
- 2. Assémat E., Bazellières E., Pallesi-Pocachard E., Le Bivic A., and Massey-Harroche D. (2008) Polarity complex proteins. Biochim. Biophys. Acta 1778, 614–630 [DOI] [PubMed] [Google Scholar]
- 3. Kemler R. (1992) Classical cadherins. Semin. Cell Biol. 3, 149–155 [DOI] [PubMed] [Google Scholar]
- 4. Fleming T. P., Papenbrock T., Fesenko I., Hausen P., and Sheth B. (2000) Assembly of tight junctions during early vertebrate development. Semin. Cell Dev. Biol. 11, 291–299 [DOI] [PubMed] [Google Scholar]
- 5. Baas A. F., Smit L., and Clevers H. (2004) LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends Cell Biol. 14, 312–319 [DOI] [PubMed] [Google Scholar]
- 6. Chen J., and Zhang M. (2013) The Par3/Par6/aPKC complex and epithelial cell polarity. Exp. Cell Res. 319, 1357–1364 [DOI] [PubMed] [Google Scholar]
- 7. Durgan J., Kaji N., Jin D., and Hall A. (2011) Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J. Biol. Chem. 286, 12461–12474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Linch M., Sanz-Garcia M., Rosse C., Riou P., Peel N., Madsen C. D., Sahai E., Downward J., Khwaja A., Dillon C., Roffey J., Cameron A. J., and Parker P. J. (2014) Regulation of polarized morphogenesis by protein kinase C ι in oncogenic epithelial spheroids. Carcinogenesis 35, 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Royer C., and Lu X. (2011) Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 18, 1470–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki A., Akimoto K., and Ohno S. (2003) Protein kinase C λ/ι (PKCλ/ι): a PKC isotype essential for the development of multicellular organisms. J. Biochem. 133, 9–16 [DOI] [PubMed] [Google Scholar]
- 11. Newton A. C. (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 370, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X., and Gao T. (2014) mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 15, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ivey R. A., Sajan M. P., and Farese R. V. (2014) Requirements for pseudosubstrate arginine residues during autoinhibition and phosphatidylinositol 3,4,5-(PO4)3-dependent activation of atypical PKC. J. Biol. Chem. 289, 25021–25030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou M. M., Hou W., Johnson J., Graham L. K., Lee M. H., Chen C. S., Newton A. C., Schaffhausen B. S., and Toker A. (1998) Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol. 8, 1069–1077 [DOI] [PubMed] [Google Scholar]
- 15. Brognard J., and Newton A. C. (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Neill A. K., Niederst M. J., and Newton A. C. (2013) Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 280, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J., Weiss H. L., Rychahou P., Jackson L. N., Evers B. M., and Gao T. (2009) Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene 28, 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai J., Fang L., Huang Y., Li R., Yuan J., Yang Y., Zhu X., Chen B., Wu J., and Li M. (2013) miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 73, 5402–5415 [DOI] [PubMed] [Google Scholar]
- 19. Chang R. M., Yang H., Fang F., Xu J. F., and Yang L. Y. (2014) MicroRNA-331–3p promotes proliferation and metastasis of hepatocellular carcinoma by targeting PH domain and leucine-rich repeat protein phosphatase. Hepatology 60, 1251–1263 [DOI] [PubMed] [Google Scholar]
- 20. Nitsche C., Edderkaoui M., Moore R. M., Eibl G., Kasahara N., Treger J., Grippo P. J., Mayerle J., Lerch M. M., and Gukovskaya A. S. (2012) The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology 142, 377–387 e371–e375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao T., Brognard J., and Newton A. C. (2008) The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 283, 6300–6311 [DOI] [PubMed] [Google Scholar]
- 22. Liu J., Stevens P. D., Li X., Schmidt M. D., and Gao T. (2011) PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol. Cell. Biol. 31, 4917–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X., Stevens P. D., Liu J., Yang H., Wang W., Wang C., Zeng Z., Schmidt M. D., Yang M., Lee E. Y., and Gao T. (2014) PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology 146, 1301–1312 e1301–e1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao T., Furnari F., and Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 25. Brognard J., Sierecki E., Gao T., and Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 26. Chen M., Pratt C. P., Zeeman M. E., Schultz N., Taylor B. S., O'Neill A., Castillo-Martin M., Nowak D. G., Naguib A., Grace D. M., Murn J., Navin N., Atwal G. S., Sander C., Gerald W. L., et al. (2011) Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 20, 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Debnath J., Muthuswamy S. K., and Brugge J. S. (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- 28. Jaffe A. B., Kaji N., Durgan J., and Hall A. (2008) Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J. Cell Biol. 183, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wen Y. A., Li X., Goretsky T., Weiss H. L., Barrett T. A., and Gao T. (2015) Loss of PHLPP protects against colitis by inhibiting intestinal epithelial cell apoptosis. Biochim. Biophys. Acta 1852, 2013–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horikoshi Y., Suzuki A., Yamanaka T., Sasaki K., Mizuno K., Sawada H., Yonemura S., and Ohno S. (2009) Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J. Cell Sci. 122, 1595–1606 [DOI] [PubMed] [Google Scholar]
- 31. Nagai-Tamai Y., Mizuno K., Hirose T., Suzuki A., and Ohno S. (2002) Regulated protein-protein interaction between aPKC and PAR-3 plays an essential role in the polarization of epithelial cells. Genes Cells 7, 1161–1171 [DOI] [PubMed] [Google Scholar]
- 32. McCaffrey L. M., and Macara I. G. (2012) Signaling pathways in cell polarity. Cold Spring Harb. Perspect. Biol. 4, a009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray N. R., Kalari K. R., and Fields A. P. (2011) Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J. Cell. Physiol. 226, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huber M. A., Kraut N., and Beug H. (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 17, 548–558 [DOI] [PubMed] [Google Scholar]
- 35. Martin-Belmonte F., and Perez-Moreno M. (2011) Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 12, 23–38 [DOI] [PubMed] [Google Scholar]
- 36. Etienne-Manneville S. (2008) Polarity proteins in migration and invasion. Oncogene 27, 6970–6980 [DOI] [PubMed] [Google Scholar]
- 37. Aranda V., Haire T., Nolan M. E., Calarco J. P., Rosenberg A. Z., Fawcett J. P., Pawson T., and Muthuswamy S. K. (2006) Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 8, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 38. Magudia K., Lahoz A., and Hall A. (2012) K-Ras and B-Raf oncogenes inhibit colon epithelial polarity establishment through up-regulation of c-myc. J. Cell Biol. 198, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Casaletto J. B., and McClatchey A. I. (2012) Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ugi S., Imamura T., Maegawa H., Egawa K., Yoshizaki T., Shi K., Obata T., Ebina Y., Kashiwagi A., and Olefsky J. M. (2004) Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24, 8778–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takimura T., Kamata K., Fukasawa K., Ohsawa H., Komatani H., Yoshizumi T., Takahashi I., Kotani H., and Iwasawa Y. (2010) Structures of the PKC-ι kinase domain in its ATP-bound and apo forms reveal defined structures of residues 533–551 in the C-terminal tail and their roles in ATP binding. Acta Crystallogr. D Biol. Crystallogr. 66, 577–583 [DOI] [PubMed] [Google Scholar]
- 42. Kumar C. C., and Madison V. (2005) AKT crystal structure and AKT-specific inhibitors. Oncogene 24, 7493–7501 [DOI] [PubMed] [Google Scholar]
- 43. Eder A. M., Sui X., Rosen D. G., Nolden L. K., Cheng K. W., Lahad J. P., Kango-Singh M., Lu K. H., Warneke C. L., Atkinson E. N., Bedrosian I., Keyomarsi K., Kuo W. L., Gray J. W., Yin J. C., et al. (2005) Atypical PKCι contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 12519–12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu H., Radisky D. C., Wang F., and Bissell M. J. (2004) Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J. Cell Biol. 164, 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morais-de-Sá E., Mirouse V., and St Johnston D. (2010) aPKC phosphorylation of Bazooka defines the apical/lateral border in Drosophila epithelial cells. Cell 141, 509–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qiao M., Wang Y., Xu X., Lu J., Dong Y., Tao W., Stein J., Stein G. S., Iglehart J. D., Shi Q., and Pardee A. B. (2010) Mst1 is an interacting protein that mediates PHLPPs' induced apoptosis. Mol. Cell 38, 512–523 [DOI] [PubMed] [Google Scholar]
- 47. Wen Y. A., Stevens P. D., Gasser M. L., Andrei R., and Gao T. (2013) Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol. Cell. Biol. 33, 4594–4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X., Yang H., Liu J., Schmidt M. D., and Gao T. (2011) Scribble-mediated membrane targeting of PHLPP1 is required for its negative regulation of Akt. EMBO Rep. 12, 818–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Masubuchi S., Gao T., O'Neill A., Eckel-Mahan K., Newton A. C., and Sassone-Corsi P. (2010) Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc. Natl. Acad. Sci. U.S.A. 107, 1642–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]