

Abstract
Polyhydroxybutyrate synthase (PhaC) catalyzes the polymerization of 3-(R)-hydroxybutyryl-coenzyme A as a means of carbon storage in many bacteria. The resulting polymers can be used to make biodegradable materials with properties similar to those of thermoplastics and are an environmentally friendly alternative to traditional petroleum-based plastics. A full biochemical and mechanistic understanding of this process has been hindered in part by a lack of structural information on PhaC. Here we present the first structure of the catalytic domain (residues 201–589) of the class I PhaC from Cupriavidus necator (formerly Ralstonia eutropha) to 1.80 Å resolution. We observe a symmetrical dimeric architecture in which the active site of each monomer is separated from the other by ∼33 Å across an extensive dimer interface, suggesting a mechanism in which polyhydroxybutyrate biosynthesis occurs at a single active site. The structure additionally highlights key side chain interactions within the active site that play likely roles in facilitating catalysis, leading to the proposal of a modified mechanistic scheme involving two distinct roles for the active site histidine. We also identify putative substrate entrance and product egress routes within the enzyme, which are discussed in the context of previously reported biochemical observations. Our structure lays a foundation for further biochemical and structural characterization of PhaC, which could assist in engineering efforts for the production of eco-friendly materials.
Keywords: bacterial metabolism, biosynthesis, biotechnology, enzyme mechanism, X-ray crystallography
Introduction
Polyhydroxyalkanoic acids (PHAs)5 are polyoxoesters synthesized by many bacterial species as a means of carbon storage under nutrient-limited conditions in which carbon is abundant (1–3). These polymers have garnered considerable biotechnological interest, because they have properties ranging from thermoplastics to elastomers, depending on how the monomer units are substituted, with alkyl substituents ranging from CH3 to C9H19 (3, 4). Additionally, PHAs are biodegradable, making them an environmentally friendly and sustainable alternative to petroleum-based plastics (5, 6). Such biologically derived materials are used in specialty markets, such as medical devices; however, they are currently not economically competitive in large-scale production with traditional petroleum-based plastics. This disparity in economic viability highlights a need for a more in-depth mechanistic and structural understanding of PHA biosynthesis to engineer the production of cost-effective materials.
PHAs are generated through the polymerization of 3-(R)-hydroxyalkyl-CoA substrates by the enzyme polyhydroxyalkanoate synthase, PhaC. The synthases are divided into four classes depending on their substrate specificity and subunit composition, although all classes are thought to have a common catalytic mechanism and a conserved active site architecture reminiscent of that seen in lipases (7–10). The class I and class III synthases are the best studied and share a common substrate specificity for 3-(R)-hydroxybutyryl-CoA (HB-CoA) to form polyhydroxybutyrate (PHB) of high molecular mass (∼1–2 MDa) and low polydispersity (1, 2, 8, 11, 12). The class I synthases, as typified by the PhaC from Cupriavidus necator (formerly Ralstonia eutropha), are composed of a single polypeptide chain (65 kDa) and contain an N-terminal domain of unknown function (residues 1–200) and a C-terminal catalytic domain (residues 201–589) (13–15). The class III synthases are made up of a catalytic PhaC subunit (∼40 kDa) and a poorly understood, although functionally necessary, second subunit, PhaE (also ∼40 kDa), as characterized from Allochromatium vinosum (16, 17). The catalytic domain of the PhaC from C. necator (CnPhaC) shares 29% sequence identity with the PhaC subunit from A. vinosum, suggesting structural similarity. Although they are less well characterized, the catalytic domains of class II and IV PhaCs are likely to also be homologous based on sequence conservation, sharing ∼40 and 30% identity, respectively, with the CnPhaC catalytic domain. In contrast, no sequence similarity is observed between the N-terminal domain of the class I synthases and the PhaE subunit of the class III synthases, and it is unclear whether the two fulfill similar functional roles.
Two mechanisms for PHB synthesis have been proposed to date, each involving an active site composed of a cysteine, a histidine, and an aspartate residue (Fig. 1) (1, 2). The active site cysteine has been shown to become acylated with HB units after deprotonation of its thiol, thought to be performed by the histidine residue (8, 10, 13, 17). Esterification is then promoted by deprotonation of the HB hydroxyl group, with the aspartate residue serving as the suggested general base catalyst (18). The original PhaC mechanism (Fig. 1A) was proposed based on the mechanism of fatty acid synthases and required the participation of two sets of active site residues, with the PHB chain transferred between active site cysteine residues across a dimer interface (13, 19). The second, currently favored, mechanism (Fig. 1B) uses a single active site and requires both covalent and noncovalent intermediates during the catalytic cycle (9, 10, 20, 21).
FIGURE 1.

Proposed mechanisms for polyhydroxybutyrate formation catalyzed by PhaC. A, mechanism invoking the use of two PhaC active sites, suggesting that catalysis must take place at the dimer interface. B, mechanism invoking the use of a single PhaC active site with a CoA-bound thioester as an intermediate in catalysis. The inset shows the structure of CoA. We note that in both mechanisms, regeneration of the catalytic bases (i.e. histidine and aspartate) is ultimately achieved through proton transfer to the CoASH leaving group. Proton transfer steps have been omitted from the mechanisms as drawn for simplicity.
Over the last 25 years, biochemical work on these systems has established that all PhaCs require the same residues for catalysis but have distinct kinetics of PHB formation using CoA release to monitor activity (8, 11, 14, 15, 22). For CnPhaC, a variable lag phase in CoA release is observed, which can be overcome by priming the enzyme through acylation with synthetic (HB)n-CoA analogs, where n = 2–4. This acylation is accompanied by conversion of the predominant monomeric form of CnPhaC to the dimeric form, which is accompanied by an increase in its catalytic activity, suggesting that the dimer is the active form of the enzyme (14). Despite these significant insights, there is much that remains to be understood, including the origin of odd kinetic behaviors, which are distinct within and between different classes of PhaCs (14, 17, 22, 23); the process of chain termination; the molecular basis of substrate specificity; and the ability of the synthase to control polymer length and polydispersity. A full understanding of these aspects has been hindered in part by a lack of direct structural data on any PhaC. Here we present the X-ray crystal structure of the catalytic domain of the class I PhaC from C. necator, providing mechanistic insight and a structural context for understanding substrate binding and product egress.
Results
The Catalytic Domain of CnPhaC Has an α/β-Hydrolase Fold
To visualize the molecular architecture of a class I PhaC, we determined the structure of the C-terminal catalytic domain of CnPhaC to 1.80 Å resolution using tantalum multiwavelength anomalous dispersion (Table 1 and Fig. 2, A and B). Crystallization experiments were set up using full-length CnPhaC(C319A), a construct in which the active site cysteine (Cys319) was mutated to alanine to improve protein stability in the absence of detergent. Proteolysis of CnPhaC(C319A) occurred in the crystallization drop, as indicated by gel electrophoresis and a lack of electron density for residues 1–200. The net result is that the structure obtained is of the catalytic domain of CnPhaC, containing residues 201–368 and 378–589 (residues 369–377 are disordered). This domain has an α/β-hydrolase fold featuring a central mixed β-sheet flanked by α-helices on both sides (Fig. 2). This architecture is reminiscent of that seen in lipases, as had been suggested based on sequence similarity and threading models (9, 10, 24, 25) (Table 2). Structural comparison of the CnPhaC catalytic domain with these lipases reveals high structural similarity in the β-sheet core with variations in the lengths and relative placements of the surrounding α-helices (Fig. 2C). Structural similarity to an archaeal aminopeptidase and various bacterial haloperoxidases is also observed (Table 2).
TABLE 1.
Data collection and refinement statistics
| Native CnPhaC(C319A) | TaBr-derivatized CnPhaC(C319A) |
||
|---|---|---|---|
| Ta peaka | Ta inflection | ||
| Data collection | |||
| Space group | I222 | I222 | I222 |
| Cell dimensions (a, b, c (Å)) | 72.08, 87.82, 137.10 | 72.67, 87.16, 137.43 | 72.65, 87.00, 137.49 |
| Wavelength (Å) | 0.9792 | 1.0611 | 1.2553 |
| Resolution (Å)b | 100–1.80 (1.84–1.80) | 100–1.97 (2.01–1.97) | 100–2.10 (2.14–2.10) |
| No. unique reflectionsb | 40,649 (2966) | 56,214 (4181) | 46,607 (3423) |
| Rsym (%)b | 4.6 (85.2) | 3.6 (45.2) | 3.4 (41.1) |
| CC1/2b | 100 (76.2) | 99.9 (76.9) | 99.9 (82.3) |
| <I/σ(I)>b | 25.76 (2.24) | 18.93 (2.32) | 21.55 (2.93) |
| Completeness (%)b | 99.9 (100) | 94.3 (94.4) | 94.8 (94.6) |
| Multiplicityb | 6.6 (6.7) | 2.8 (2.7) | 3.2 (3.2) |
| Refinement | |||
| Resolution (Å) | 73.95–1.80 | ||
| Rwork/Rfreec | 0.172/0.200 | ||
| No. atoms | |||
| Protein | 2942 | ||
| Water | 283 | ||
| Sulfate | 20 | ||
| Average B-factors (Å2) | |||
| Protein | 31.6 | ||
| Water | 38.8 | ||
| Sulfate | 56.7 | ||
| Root mean square deviations | |||
| Bond lengths (Å) | 0.006 | ||
| Bond angles (°) | 0.82 | ||
| Rotamer outliers (%) | 0.0 | ||
| Ramachandran statistics (%) | |||
| Favored | 96.6 | ||
| Allowed | 2.9 | ||
| Disallowed | 0.5 | ||
a For this data set, Bijvoet pairs were not merged during data processing.
b The values in parentheses indicate highest resolution bin.
c A total of 5% of reflections were set aside during refinement for cross-validation.
FIGURE 2.

The catalytic domain of CnPhaC has an α/β-hydrolase fold and is structurally similar to lipases. A, overall structure of the catalytic domain of CnPhaC(C319A). The monomer is shown in ribbon representation in violet with active site residues shown as balls and sticks with black and purple carbons for the green and violet monomers, respectively. The active site cysteine has been modeled for illustrative purposes. The second protomer of the dimer is colored green. Disordered residues within the catalytic domain are indicated as black dashed lines. The N and C termini are shown as spheres. The distance between the active site cysteine residues of the two monomers is ∼33 Å and is indicated as a black solid line. B, topology diagram of the CnPhaC catalytic domain monomer, colored as in A with active site residues indicated as colored circles. Disordered residues are indicated as a dashed line. C, overlay of the CnPhaC catalytic domain colored as in A with the gastric lipase from Canis lupus (Protein Data Bank code 1K8Q) colored in gray (Cα root mean square deviation = 3.8 Å). D, stereo view of the overall Cα trace of the structure colored in rainbow mode with the N terminus in blue and the C terminus in red. Active site residues are shown as spheres and labeled with single-letter amino acid codes.
TABLE 2.
Structural homologs of the CnPhaC(C319A) catalytic domain
Alignments were performed using the DaliLite server (57).
| Protein | Protein Data Bank code (source) | Z-score | No. residues aligned | Cα root mean square deviation (Å) |
|---|---|---|---|---|
| Canis lupus gastric lipase | 1K8Q (Ref. 31) | 23.7 | 285 | 3.8 |
| Homo sapiens gastric lipase | 1HLG (Ref. 64) | 22.6 | 274 | 3.4 |
| Sulfolobus solfataricus lipase | 2RAUa | 20.1 | 350 | 3.1 |
| Saccharomyces cerevisiae lipase | 4ZWN (Ref. 32) | 20.0 | 263 | 3. 3 |
| H. sapiens monoglyceride lipase | 3HJU (Ref. 65) | 19.0 | 240 | 3.3 |
| Thermoplasma acidophilum aminopeptidase | 1XQX (Ref. 33) | 18.5 | 233 | 3.1 |
| Bacillus anthracis bromoperoxidase | 3FOBa | 18.4 | 227 | 2.7 |
| Pseudomonas fluorescens chloroperoxidase | 1A8S (Ref. 34) | 18.3 | 228 | 3.0 |
a Unpublished.
The CnPhaC catalytic domain crystallized as a dimer with one protomer in the asymmetric unit (Fig. 2). The symmetric dimer is formed by a small helical domain that extends away from the core of each monomer and by the extension of a partially disordered loop from one chain into the other. The interface is composed of 66 residues and buries a surface area of ∼2600 Å2 from solvent on each monomer. As noted above, CnPhaC is known to exist in an equilibrium between monomer and dimer in solution, with the dimer representing the more catalytically active form (13, 14, 22). Our structure likely represents the catalytically relevant dimeric form, favored by the high protein concentrations (490 μm) used for crystallography.
The Location of Cys319, His508, and Asp480 Defines the Active Site
Extensive biochemical studies have revealed that Cys319 and His508 form a catalytic dyad, with His508 deprotonating the thiol of the catalytically essential Cys319, allowing it to become covalently loaded with HB units (Fig. 1) (8, 10, 13, 17). Asp480 has been suggested to deprotonate the hydroxyl group of the second and subsequent HB-CoAs, allowing for ester formation and elongation of the PHB chain (8, 9, 18). Our structure of the catalytic domain of CnPhaC reveals these residues arranged together in a cavity that is ∼10 Å from the nearest surface of the protein, defining the location of the active site (Fig. 2, A and D). In the dimer, the active sites are ∼33 Å apart (Fig. 2A), a distance that excludes the mechanism presented in Fig. 1A, in which catalysis takes place at the dimer interface with the growing PHB chain shuttled back and forth between two active sites. Instead, the structure supports a mechanism similar to that in Fig. 1B, involving a single active site and invoking the use of a CoA-bound intermediate during the catalytic cycle.
The catalytic cysteine of CnPhaC (Cys319; mutated to alanine in our structure) is located at the junction of a β-strand and an α-helix on the so-called “nucleophile elbow” characteristic of the α/β-hydrolase fold (Fig. 3). His508 is positioned on a loop across from Cys319 such that, when cysteine is modeled into our structure, the Nϵ atom of the histidine side chain comes within 3.5 Å of the cysteine thiol, consistent with a role for His508 in deprotonation of the catalytic cysteine. Asp480 is located on a loop just behind His508 and is 7.5 Å from the modeled side chain thiol of Cys319 (Fig. 3). The side chain carboxylate forms a hydrogen bond with the Nδ atom of His508 (2.8 Å) and additional hydrogen bonds with the backbone amide groups of Ile 482 and Val 483 (2.9 and 3.0 Å, respectively) from the Asp480 turn (Fig. 3). The interactions of Asp480 are similar to those of the catalytic acid residue in other α/β-hydrolase enzymes, in which this residue appears to form part of a catalytic triad (26, 27).
FIGURE 3.
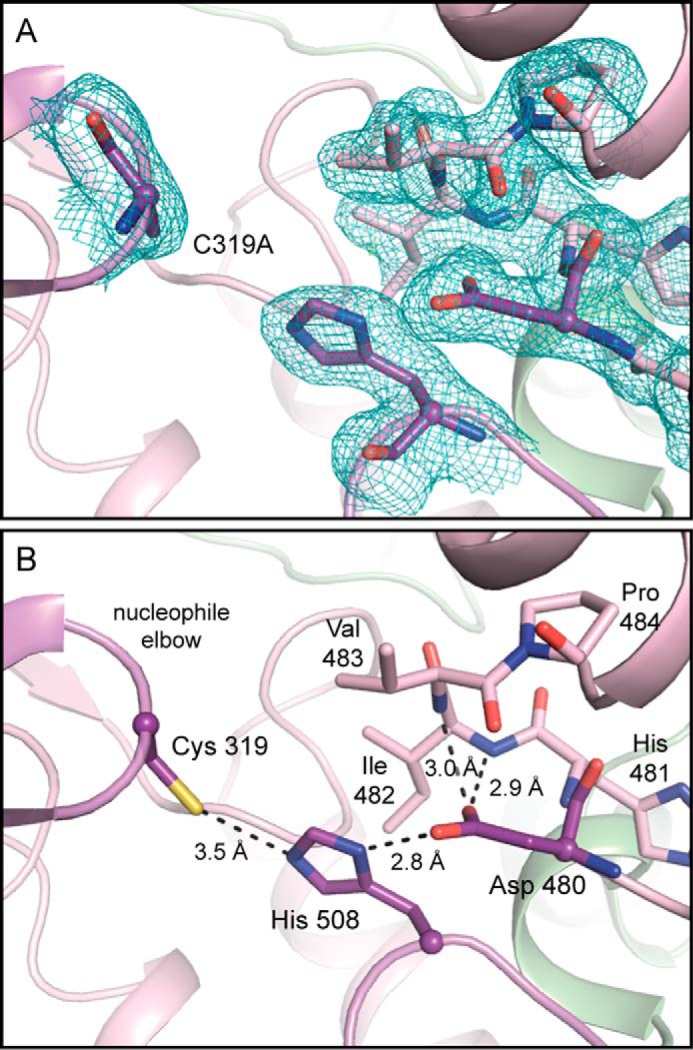
The active site of PhaC is composed of Cys319, Asp480, and His508. A, view of the CnPhaC(C319A) active site with electron density. 2Fo − Fc density (teal) contoured to 1σ. No Fo − Fc density is visible in this orientation when contoured to ±3σ. B, interactions within the active site with Cys319 modeled for illustrative purposes. Catalytic residues are shown in purple; residues on the Asp480 turn are shown in violet. Hydrogen bonds are indicated by black dashed lines.
A Proposed Substrate Access Channel Can Be Inferred from the Structure
To investigate how HB-CoA could access the active site, we searched for channels leading into C319A. A solvent channel running from the surface of the protein at the dimer interface to a water-filled cavity directly adjacent to the active site was immediately obvious by inspection and was additionally visualized using the software tool CAVER (28) (Fig. 4). This channel is ∼18 Å in length, which is sufficient to accommodate the pantetheine arm of CoA (Fig. 1), allowing the HB moiety to access the active site. The opening of the channel at the protein surface is in close proximity to two arginine residues, one from each chain of the dimer (Fig. 4). This arginine residue (Arg398) is strictly conserved in class I PhaCs, and the two together could function in binding the CoA nucleotide 5′-pyrophosphate (Fig. 4D). Similar binding of the 5′-pyrophosphate by arginine residues was observed in a CoA-bound structure of an archaeal esterase (another member of the α/β-hydrolase superfamily), although in this case binding was not at an interface, and the two arginine residues were contributed by a single chain (29). Binding of CoA by residues from each chain in PhaC could explain the increased activity of the dimeric form. A second conserved residue, His481, is also in the vicinity of the opening to the channel (Fig. 4, B and C). Mutagenesis studies have shown that the H481Q variant of CnPhaC retains 20% of the activity of the wild-type enzyme, suggesting that this residue, although not essential for activity, does play a role (8). We propose that His481 could serve to stabilize the 3′-phosphate of CoA, which has been previously implicated in substrate recognition (Fig. 4D) (30).
FIGURE 4.

The active site of CnPhaC is accessible via a water-filled channel. A, overall view of the CnPhaC catalytic domain with the substrate channel as visualized using CAVER (blue surface). The PhaC monomer is shown in violet; the second protomer of the dimer is in green. Active site residues are shown as balls and sticks in purple. Arg398 from each monomer are shown as sticks. Dashed lines represent the disordered region of the structure. B, view of the solvent channel from the dimer interface. Protein is shown in surface representation with underlying ribbons. Waters are shown as red spheres. C, view of the channel as in B with visualization from CAVER (blue surface). D, model of HB-CoA (carbon atoms in cyan) bound in the proposed substrate channel. Modeled interactions of Arg398 and His481 with HB-CoA are shown as gray dashes.
A Putative PHB Egress Route Is Stabilized by Conserved Structural Motifs
The PhaC reaction cycle requires both an entrance route for HB-CoA and an egress route for the nascent PHB chain, although unlike the putative entrance channel mentioned above, no single egress route was immediately obvious in the structure. However, through inspection of the catalytic domain of CnPhaC, we identified a putative product channel lined by a series of hydrophobic residues leading from the active site to the surface of the protein at a ∼95° angle to the proposed substrate entrance channel (Fig. 5A). The side chains form a narrow hydrophobic conduit ∼12.5 Å long that extends away from the β-sheet core of the CnPhaC catalytic domain and widens into a small solvent pocket near the surface of the protein in the vicinity of the N-terminal residue of the structure (Ser201) and a conserved aspartate residue (Asp421), which could be involved in PHB chain termination (see “Discussion”) (Fig. 5A). We suggest that this conduit serves as a possible egress route for the growing PHB chain.
FIGURE 5.

Conserved residues in CnPhaC form structural motifs that appear to stabilize a proposed PHB exit channel. A, stereo view of the CnPhaC catalytic domain, colored as in Fig. 4A, showing the proposed substrate channel (blue surface) and product egress channel (yellow surface). Conserved structural motifs are represented as sticks and colored as in B–E. Nonconserved residues that participate in structural motifs through backbone atoms are shown in violet. Asp421 is shown in pink. Cys319, Asp480, and His508 are shown in purple. B, sequence of the C-terminal domain of CnPhaC. Strictly conserved residues are in bold; active site residues are colored in purple. Residues forming structural motifs are colored as follows: Asp254–Ser260 motif in orange, WNXD motif in teal, and GSWW motif in green. Arg398, Asp421, and His481 are in pink. C–E, close-up views of structural motifs as colored in A and B.
Visualization of this putative egress pathway using CAVER (Fig. 5A) reveals that at its narrowest point, the conduit is ∼0.7 Å in diameter and would therefore need to expand to accommodate the growing polymer (the average radius of an HB polymer is ∼2 Å). From the structure, it appears that some degree of expansion is possible through simple rearrangement of the hydrophobic side chains lining the channel; however, larger conformational changes would also be necessary to allow for passage of the PHB chain. These dynamics could be mediated by a series of conserved structural motifs that we identified surrounding the putative egress channel (Fig. 5). Alignment of 300 class I PhaC sequences reveals 43 residues within the catalytic domain with >98% sequence conservation (Fig. 5B). Mapping of these conserved residues onto the catalytic domain of CnPhaC illustrates that many of them surround the hydrophobic core of the proposed exit route (Fig. 5A). Closer inspection indicates that these residues participate in hydrogen-bonding networks to form small structural motifs, which could serve to stabilize channel expansion and egress of the nascent PHB chain (Fig. 5, C–E). The high degree of sequence conservation, at the very least, suggests a likely functional role for these residues and underscores the importance of this region of the structure.
Substrate Can Be Modeled into CnPhaC Based on the Proposed Channels
Using Arg398 and His481 as anchoring points for the 5′- and 3′-phosphates, respectively, HB-CoA was modeled into the structure of the CnPhaC catalytic domain (Fig. 4D). Two binding modes for HB-CoA were modeled: one to represent the orientation of the substrate for initiation, and one to represent elongation; energy minimization was performed to minimize clashes for both binding modes (Fig. 6). In each case, the pantetheine arm of CoA runs through the proposed substrate channel in a partially extended conformation, and the adenine base has been tucked into a small solvent pocket on the surface of the protein. Additionally, for the elongation mode, an HB monomer was modeled onto Cys319 such that it is oriented in the direction of the proposed egress route (Fig. 6C). We can expect modest rearrangements in the active site upon substrate binding; however, the active site cavity, based on our modeling, in general appears large enough to facilitate catalysis for both polymer initiation and elongation.
FIGURE 6.

Stereo views of HB-CoA modeled into the CnPhaC catalytic domain. A, active site of CnPhaC(C319A) in the absence of substrate. Black dashed arrow indicates deprotonation of Cys319 by His508. The position of the Cys319 sulfur atom is modeled. Blue and yellow dashed arrows represent the directions of the proposed substrate entrance and product egress channels, respectively. B, active site of CnPhaC(C319A) with HB-CoA (carbon atoms in cyan) modeled for initiation. Black dashed arrow represents path of nucleophilic attack of Cys319 on the HB-CoA thioester. The position of the sulfur atom of Cys319 is modeled. Gray dashed line represents a modeled hydrogen bond between the carbonyl oxygen of HB-CoA and the amide group of Val320. Cys246 and Ile247 are located on a loop just behind the nucleophile elbow. The amide group of one of these residues, in combination with the Val320 amide, could form the oxyanion hole to stabilize the tetrahedral intermediate during catalysis, although a conformational change within the active site upon substrate binding would be necessary to bring the loop close enough to form the appropriate interaction with the substrate carbonyl. C, active site of CnPhaC(C319A) with HB monomer (carbon atoms in gray) modeled onto Cys319 and HB-CoA (carbon atoms in cyan) modeled for polymer elongation. Black dashed arrow represents path of nucleophilic attack of the HB-CoA hydroxyl group on the protein-bound thioester.
Initiation of polymer synthesis involves the deprotonation of Cys319 by His508 followed by nucleophilic attack of the deprotonated Cys319 on the CoA thioester of HB-CoA (8, 10, 13, 17). The position of the Cys319 side chain that is closest to His508 is not ideal for such a nucleophilic attack because is it positioned away from the substrate channel (Fig. 6A), suggesting that at least two Cys319 side chain conformations may be necessary to fulfill the catalytic cycle. When the Cys319 side chain is modeled to point toward the substrate channel, a close distance to the modeled HB-CoA thioester of 2.2 Å is obtained (Fig. 6B). Interaction of the HB-CoA carbonyl oxygen with the Val320 amide group could serve as a partial oxyanion hole to stabilize the tetrahedral intermediate (Fig. 6B). Stabilization of the tetrahedral intermediate by the amide group of the residue immediately following the catalytic nucleophile is a canonical feature of α/β-hydrolase enzymes (26, 27) and is consistent with these structural data. In addition to the use of the adjacent amide group to form the oxyanion hole, lipases also employ an amide group located on a loop in the vicinity of the nucleophile elbow corresponding to the approximate location of Cys246 or Ile247 in CnPhaC (Fig. 6B) (31–34). In the apo CnPhaC structure, the amide nitrogen atoms of these residues are 5.8 and 5.3 Å, respectively, from the modeled carbonyl oxygen atom, requiring only a modest loop movement to involve one of these amide groups in formation of an oxyanion hole. Following HB-Cys adduct formation, protonated CoA would exit the active site, and a second HB-CoA would enter. The arrival of this second HB-CoA into the active site would likely push the HB-Cys adduct to the rear of the active site pocket, positioning it at the start of the putative egress channel (Fig. 6C).
For elongation, the second HB-CoA needs to be oriented to allow for nucleophilic attack of the HB hydroxyl group on the protein-bound HB-thioester (Fig. 1B). In contrast to our proposed binding mode for initiation, functional groups that could contribute to oxyanion hole formation during polymer elongation are not apparent from the structure. Without obvious protein contacts to guide the positioning of the HB-Cys carbonyl oxygen atom, modeling of the adduct was guided by the required geometry of the thioester bond (i.e. the planarity of the bond). This modeling places the HB-Cys adduct at a distance of ∼2.8 Å from the hydroxyl group of HB-CoA such that nucleophilic attack on the protein-bound thioester is feasible (Fig. 6C). In this orientation, the hydroxyl group of the HB moiety is 6.7 Å from the Asp480 carboxylate, a distance that is too far to allow for proton transfer. This long distance and the observation that the Asp480 carboxylate is already involved in several hydrogen bonds to other protein atoms (discussed above and shown in Fig. 3) argue against the direct involvement of Asp480 as a general base for the deprotonation of substrate as depicted in Fig. 1. Asp480 may play an indirect role, however, because it is positioned to hydrogen bond with His508 in such a way that the histidine could serve as the general base catalyst in analogy to its role in the catalytic triads of other systems. In the model, His508 is 2.8 Å from the HB hydroxyl moiety. We note that for histidine to fulfill this role, the relationship between His508 and Asp480 must be such that the pKa of the histidine is perturbed and that this relationship could be induced by substrate binding, as has been proposed for serine proteases (35, 36).
Discussion
The polymerization of hydroxybutryate represents a fascinating biological phenomenon in the context of both microbial physiology and biotechnological application. Despite nearly three decades of research, our understanding of the enzyme responsible for this process remains incomplete, in part because of a lack of structural information. Herein, we present the first structure of the catalytic domain of a PHB synthase, PhaC from C. necator, providing insight into the catalytic mechanism and a framework for understanding substrate binding and product egress.
Our structure of the class I CnPhaC catalytic domain reveals a dimeric architecture that likely represents the physiologically relevant oligomeric state of the enzyme, given the substantial amount of buried surface area. The active sites of the dimer are found on the interior of each monomer and are separated by a distance of ∼33 Å, providing direct structural evidence that the mechanism of PHB synthesis very likely involves catalysis at a single active site rather than transfer of the PHB chain between active sites across an interface. This observation, combined with recent studies in which noncovalent, CoA-bound intermediates were detected during polymer formation (20, 21), supports a mechanism similar to that presented in Fig. 1B. The observed arrangement of residues within the active site, however, calls for a modification of this proposed catalytic scheme. In particular, the placement of the active site aspartate residue (Asp480) argues against its involvement as the general base catalyst for the direct deprotonation of substrate during polymer elongation. The side chain carboxylate of Asp480 is relatively removed from the active site and hydrogen bonds with the active site histidine (His508), as well as with the backbone amide groups of neighboring residues. His508, on the other hand, appears ideally positioned for the deprotonation of the substrate hydroxyl group. Similar Asp-His interactions are seen in the canonical catalytic triads of a number of other enzymes, including serine proteases, esterases, and lipases, in which the arrangement of residues is thought to facilitate nucleophilic activation of an active site serine residue. In addition to deprotonation of serine, catalytic triads have also been implicated in the deprotonation of substrate hydroxyl groups in lipase-catalyzed transesterification reactions, as well as in a recently described mechanism of peptidoglycan O-acetylation (37–39). Therefore, it is possible that it is the histidine residue, rather than the aspartate residue in PhaC, that deprotonates the hydroxyl group of HB-CoA.
A modified mechanistic scheme for catalysis by PhaC is presented in Fig. 7. As in the previously proposed mechanisms, the first step of our revised mechanism is deprotonation of Cys319 by His508 followed by acylation of the cysteine. Given the similarity in the standard pKa values of histidine and cysteine (6 and 8, respectively), modulation of the His508 basicity through the Asp-His interaction is, in principle, unnecessary for this step. However, mutation of Asp480 to Asn in CnPhaC was shown to result in a 50-fold decrease in the rate of enzyme acylation, suggesting that Asp480 could play a role in nucleophilic activation, although mutation of the equivalent residue in the class III synthase from A. vinosum had no effect on the acylation rate (8, 9). We note that it is also possible that the resting state of the enzyme is zwitterionic, containing a preformed cysteine thiolate and histidine imidazolium pair, as has been demonstrated in cysteine proteases (40, 41). In either case, the histidine base is (re)generated through donation of the imidazolium proton to the CoASH leaving group. Our revised mechanism continues with deprotonation of the hydroxyl group of a second HB-CoA substrate. In analogy to the well characterized mechanism of serine proteases (36, 42, 43), interaction with the negatively charged Asp480 effectively increases the pKa of His508, making it a better proton acceptor for deprotonation of the highly basic substrate hydroxyl group (pKa = ∼16). As noted above, substrate binding may be required to induce the appropriate hydrogen bonding distance between aspartate and histidine for pKa modulation to occur. The substrate-based alkoxide then performs nucleophilic attack on the acyl-enzyme adduct, resulting in transfer of the (HB)n chain to HB-CoA (forming (HB)n+1-CoA) followed by enzyme reacylation, thereby completing one cycle of chain elongation. This modified mechanistic scheme is consistent with previous biochemical studies in which mutation of Asp480 results in synthase that can still be acylated but that is severely impaired in its ability to catalyze chain elongation (8, 9, 18).
FIGURE 7.

Modified mechanistic scheme for PhaC. His508 deprotonates Cys319, allowing for nucleophilic attack on the HB-CoA thioester; the histidine base is regenerated by transfer of the proton to the CoASH leaving group. A second HB-CoA substrate binds, and the HB hydroxyl group is deprotonated by His508, facilitated through modulation of the histidine basicity by Asp480. The newly formed HB alkoxide attacks the Cys-HB thioester, generating a noncovalent, CoA-bound intermediate. The growing PHB chain is then transferred back to Cys319.
Another long-standing structural question is how PhaC is able to accommodate binding of a relatively large substrate (HB-CoA) while at the same time facilitating formation of a large, polymeric product. Our structure of the CnPhaC catalytic domain now reveals the presence of a likely substrate entrance channel and can be used to infer a putative product egress route. The relative orientations of our proposed channels suggest a mechanism in which the substrate HB-CoA enters the active site near the dimer interface and the budding polymer exits at an angle of ∼95° (Fig. 5A).
The proposed substrate channel is large enough to accommodate HB-CoA in multiple binding modes without large scale conformational changes. The solvent cavity at the base of the channel has a volume of ∼315 Å3, as calculated from the molecular surface area of the cavity, and the van der Waals volume of HB is ∼100 Å3 (44). This accommodation within the active site is essential because we expect the HB moiety of HB-CoA to adopt different binding modes for initiation and elongation to establish the appropriate geometry for catalysis in each case. Additionally, given the proposed mechanism, which involves transfer of the growing PHB chain from the active site cysteine to HB-CoA and back to cysteine, we expect that there will be some degree of motion in the pantetheine arm of CoA. Our proposed substrate channel is ∼4 Å wide, which should provide room for such movements to take place.
Interestingly, the opening to the proposed entrance channel sits at the intersection of the strictly conserved Arg398 residue from each chain of the dimer. We propose that these arginine residues bind the 5′-pyrophosphate of the CoA nucleotide. Binding of HB-CoA by residues across the dimer interface in PhaC could explain, at least in part, the requirement of dimerization for activity (13, 14, 22). Furthermore, incubation of PhaC with oligomers of (HB)n-CoA, where n = 2–4, or with (HB)3-CoA in which the terminal hydroxyl group is replaced with a hydrogen (saturated trimer, sTCoA), has been shown to induce formation of the dimer (14). Combined with our structural data, this biochemical result suggests a scenario in which binding of HB-CoA by one monomer stabilizes its interaction with a second monomer via electrostatic interactions between the CoA phosphates and Arg398. We expect that conformational changes in the region of the dimer interface could also occur upon substrate binding and/or enzyme acylation, additionally facilitating dimerization. In particular, ordering of the disordered region of our structure (residues 369–377) could result in additional contacts between chains.
The putative product egress channel in CnPhaC is made up of a hydrophobic core surrounded by a series of small structural motifs that are formed by residues that are largely conserved in class I PhaCs. Although this channel as it is observed in our structure is too narrow to accommodate PHB, simple side chain rearrangements could lead to a moderate expansion of the channel. Larger scale conformational changes will likely also be necessary to open the channel wide enough to allow passage of the PHB chain, and we predict that this expansion will be stabilized in part by the conserved structural motifs that circumscribe the passageway. These motifs display intricate hydrogen bonding schemes that appear to tie secondary structural elements together in a way that should secure the protein fold under stress. It is certainly intriguing that instead of lining the active site or domain interface, 11 of 43 highly conserved residues form this arc-like network extending away from the core of the PhaC α/β-hydrolase fold. The exact mechanism that would trigger channel opening is unclear, although we favor the possibility that the polymer itself could facilitate the opening of the channel and hold the channel open during product egress.
The idea that the budding polymer itself facilitates opening and stabilization of the exit channel is appealing when viewed in the context of the unusual, biphasic kinetics displayed by CnPhaC. The activity of CnPhaC exhibits a characteristic lag phase, as measured by both CoA release and polymer synthesis, followed by a rapid linear phase (13, 14). The basis of the lag phase is unknown but has been attributed to either the requirement for protein dimerization or a need for the synthase to be primed, with the likely possibility that both factors contribute. In addition to inducing dimerization, as discussed above, incubation of CnPhaC with synthetic (HB)n-CoA analogs, where n = 2–4, or with sTCoA also leads to a reduction in the lag phase and an increase in PhaC specific activity, with the HB dimers, trimers, or tetramers serving as primers for the synthase in vitro (14). Priming with the trimer or saturated trimer leads to the greatest reduction in the lag phase, highest extent of dimer formation, and the highest specific activity, which is interesting considering that the length of an extended HB trimer (∼11.5 Å) is close in length to the constricted hydrophobic core of our proposed exit channel (∼12.5 Å). This observation leads to the suggestion that priming of PhaC involves acylation of Cys319 with enough HB units to fill the product egress channel, providing the necessary expansion and stability for passage of the polymer. In this model, the HB dimer (∼8 Å) is not of sufficient length to entirely fill and stabilize the hydrophobic passageway and is therefore a less efficient primer. The HB tetramer (∼16 Å) should, in principle, be able to serve as an adequate primer based on our model; however, it is possible that (HB)4-CoA binds nonproductively to PhaC. Thus, (HB)3CoA may be the more efficient priming substrate in in vitro experiments because of its ability to productively bind PhaC and provide sufficient stabilization for exit channel opening. Structures of PhaC covalently loaded with synthetic (HB)n-CoA analogs will be needed to provide more definitive insight into the priming process.
Despite a number of studies investigating the process of chain termination in PhaCs, this event remains poorly understood. PhaC is thought to catalyze termination itself, although a phasin protein, PhaP, has been implicated in promoting the termination event (25, 45, 46). Proposals for the mechanism of termination include hydrolysis by a base-activated water molecule; transfer of the PHB chain to a nucleophilic residue on PhaC followed by hydrolysis; or chain transfer to an exogenous thiol- or hydroxyl-containing molecule (22, 47–49). Following chain termination and release of the polymer, it has been shown that the class III PhaC remains loaded with 3–10 HB units, suggesting that chain termination occurs at a site distant from the active site (47). Although similar studies have not been performed on the class I synthase, termination at a similarly positioned site may be generally applicable to all classes of PhaC. In our structure of the CnPhaC catalytic domain, there is a conserved aspartate residue (Asp421) on the surface of the protein directly at the end of the proposed exit channel (Fig. 5A). This aspartate is positioned 15.7 Å from the active site, a length corresponding to 4 fully extended HB units. It is possible that Asp421 is involved in chain termination by serving as either the general base catalyst for deprotonation of water for hydrolysis, or it could be the nucleophilic residue onto which the PHB chain is transferred. Mutagenesis studies on this residue could provide exciting insights into the chain termination event.
The N-terminal domain of CnPhaC is not visualized in our structure. This domain of class I PhaCs has poor sequence conservation and no predicted structural homologs, and its function is not well understood. The N-terminal residue of our structure (Ser201) lies near the exit of the proposed product egress channel (Fig. 5A), implying that the N-terminal domain would be in the general region of the polymer as it emerges from the catalytic domain. Thus, one possible role for the N-terminal domain could be in interacting with the nascent PHB chain, although the relevance of any such interaction remains elusive. A structure of a full-length class I PhaC will no doubt provide invaluable insight into the function of this enigmatic N-terminal domain.
Our structure of the catalytic domain of CnPhaC provides molecular insight into how a single enzyme can achieve the formation of a polymer over 15 times its size. In particular, we now have a foundation on which to base more guided mechanistic and mutagenesis studies to better understand catalysis and the interaction of the enzyme with its substrate and its product. We hope that a clearer understanding of these features will inform on engineering efforts toward the production of cost-effective and environmentally sustainable materials.
Experimental Procedures
Cloning of CnPhaC and CnPhaC(C319A)
The PhaC gene was amplified from C. necator H16 (formerly R. eutropha H16) genomic DNA by PCR using Phusion polymerase (NEB). The primers were purchased from IDT (forward, GCGGCCTGGTGCCGCGCGGCAGCCATATGGCGACCGGCAAAGGC; reverse, GGTGCTCGAGTGCGGCCGCAAGCTTCATGCCTTGGCTTTGACGTATCG). The gene was inserted into pET28a (Novagen) linearized with NdeI and HindIII, by Gibson isothermal assembly, yielding the expression plasmid pET28a-CnPhaC. The plasmid pET28a-CnPhaC(C319A) was constructed by site-directed mutagenesis from pET28a-CnPhaC (primer ACGTGCTCGGCTTCGCCGTGGGCGGCACCA, with the mutation underlined). All constructs were confirmed by DNA sequencing at the Massachusetts Institute of Technology Biopolymers Laboratory. The pET28a plasmid contains an N-terminal His6 affinity purification tag followed by a thrombin cleavage site (MGSSHHHHHHSSGLVPRGSH).
Expression and Purification of CnPhaC(C319A)
CnPhaC(C319A) was heterologously expressed in Escherichia coli. A single colony of E. coli BL21-CodonPlus(DE3)-RIL (Agilent Technologies) transformed with pET28a-CnPhaC(C319A) was inoculated into 5 ml of LB medium supplemented with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol and grown to saturation at 37 °C overnight. The overnight culture was diluted into 200 ml of LB medium with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol in a 500-ml Erlenmeyer flask and grown at 37 °C with shaking at 200 rpm. Once the culture reached an A600 of 0.8, the temperature was decreased to 20 °C, and isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.1 mm. After 16 h, the cells were harvested by centrifugation at 4000 × g and 4 °C for 10 min, yielding 2 g of cell paste, and stored at −80 °C until purification.
All of the purification steps were carried out at 4 °C. We note that, in contrast to the wild-type enzyme, which in our hands required the presence of the nonionic detergent Hecameg for purification (13), the CnPhaC(C319A) variant was well behaved in the absence of Hecameg, and no detergent was included in the purification buffers. The cells were resuspended in 25 ml of buffer A (20 mm sodium phosphate, pH 7.0, 5 mm TCEP) containing 0.5 mm PMSF and lysed by three passages through a French pressure cell at 14,000 p.s.i. A 5-ml solution of 6% streptomycin sulfate was added dropwise, followed by centrifugation at 20,000 × g and 4 °C for 10 min to remove cell debris. Proteins in the supernatant were precipitated by successive addition of solid ammonium sulfate to 40, 60, and 80% saturation. The fractions precipitating at 40 and 60% were redissolved in 40 ml of buffer A and incubated with 2 ml of TALON resin (Clontech) for 30 min with shaking. The resin was poured into a column and washed with 20 column volumes (CV) of buffer A supplemented with 0.2 m KCl and 10 CV containing 5 mm imidazole, followed by elution with 5 CV containing 150 mm imidazole. The eluate was diluted 3-fold and precipitated with 60% ammonium sulfate. The pellet was dissolved in a minimal volume of buffer A (∼0.5 ml) and desalted on a Sephadex G-25 column (15 ml, 1 × 20 cm; GE Healthcare) equilibrated with 50 mm Tris-HCl, pH 7.5, 10 mm β-mercaptoethanol. The protein-containing fractions were concentrated to 490 μm (∼30 mg/ml), as judged by absorbance at 280 nm using a calculated ϵ280 = 83,840 m−1 cm−1, yielding ∼6 mg of protein (∼3 mg/g cells).
Crystallization of CnPhaC(C319A)
The C-terminal domain of CnPhaC(C319A) was crystallized at 25 °C by the hanging drop vapor diffusion method. A 1-μl aliquot of His-tagged, full-length CnPhaC(C319A) (30 mg/ml in 50 mm Tris-HCl, pH 7.5) was combined with 1 μl of a precipitant solution (0.9 m (NH4)2SO4, 0.1 m HEPES, pH 7.0, 0–0.5% (w/v) PEG 8000) on a glass coverslip and sealed over a reservoir containing 500 μl of precipitant solution. Crystals grew out of heavy precipitate after ∼4 weeks. Under these conditions, the protein underwent proteolysis at the linker region between the N-terminal and C-terminal domains, as indicated by gel electrophoresis. The crystals consisted only of the C-terminal catalytic domain. An underivatized crystal was streaked through Paratone (Hampton Research) for cryoprotection and cryo-cooled in liquid nitrogen.
To generate crystals derivatized with tantalum bromide for phase determination, crystals that had been growing for 1 year were transferred to a drop containing 1.8 m (NH4)2SO4, 0.2 m HEPES, pH 7.0, 2 mm hexatantalum tetradecabromide (Ta6Br12; Jena Biosciences) on a glass coverslip and sealed for 16 h over a reservoir containing 500 μl of 1.8 m (NH4)2SO4, 0.2 m HEPES, pH 7.0. The crystals were then transferred in three steps of increasing glycerol concentration and decreasing tantalum bromide concentration to a cryogenic solution containing 1.8 m (NH4)2SO4, 0.2 m HEPES, pH 7.0, 20% (v/v) glycerol; incubated in that solution for 5 s; and then cryo-cooled in liquid nitrogen.
Data Collection
All data were collected at the Advanced Photon Source (Argonne, IL) at Beamline 24-ID-C using a Pilatus 6M pixel detector at a temperature of 100 K. All crystals belong to space group I222. A fluorescence scan collected on a lower quality tantalum bromide-derivatized crystal was used to determine the peak and inflection wavelengths for data collection. Another crystal was then used for collection of anomalous peak and inflection data. Ta peak data were collected at a wavelength of 1.0611 Å (11,685 eV) in four wedges of 20° in 0.25° increments. The crystal was rotated by 180° after completion of each wedge. Ta inflection data were collected at a wavelength of 1.2553 Å (9877 eV) in a single wedge of 140° in 0.25° increments. Native data were collected on an underivatized crystal at a wavelength of 0.9792 Å (12,662 eV) in a single wedge of 180° in 0.25° increments. All data were integrated in XDS and scaled in XSCALE (50). The data collection statistics are summarized in Table 1.
Model Building and Refinement
The structure of the CnPhaC(C319A) catalytic domain was solved by Ta multiple wavelength anomalous dispersion using SHELX (51) and contains one molecule per asymmetric unit. Positions of 21 individual Ta ions, corresponding to three and a half Ta6Br12 clusters, were located in SHELXD in the HKL2MAP suite (52) using the peak and inflection data trimmed to 2.50 Å resolution. Refinement of the heavy atom sites, density modification, and automatic model building of a polyalanine model were carried out in SHELXE. The model-adjusted mean figure of merit from SHELXE was 0.64 as calculated to 1.97 Å resolution. The resulting model contained 343 alanine residues, corresponding to the catalytic domain of one CnPhaC(C319A) protomer. The electron density maps were of sufficient quality to trace the majority of the remaining protein residues as well as a large number of side chains.
When a nearly complete model of CnPhaC(C319A) was obtained (containing 77% of residues and 23% of side chains), the coordinate file was used for refinement of atomic coordinates and atomic displacement parameters (B-factors) in Phenix (53) against the native CnPhaC(C319A) data set using data across the entire resolution range. The resulting R-factors were 38.8% and 40.7% for the working and free R-factor, respectively. The model was completed by iterative rounds of model building in Coot (54) and refinement in Phenix. In advanced stages of refinement, water molecules were added automatically in Phenix and modified in Coot with placement of additional water molecules until their number was stable. Final cycles of refinement included translation, libration, screw (TLS) parametrization with one TLS group for the asymmetric unit (55). Side chains without visible electron density were truncated to the last atom with electron density, and amino acids without visible electron density were not included in the model. The final model contains residues 201–368 and 378–589 (369–377 are disordered and 589 is the C-terminal residue of the protein), 283 water molecules, and 4 bound sulfate ions.
Refinement of the CnPhaC(C319A) structure yielded a model with low free R-factors, excellent stereochemistry, and small root mean square deviations from ideal values for bond lengths and angles. All refinement statistics are summarized in Table 1. The model was validated using simulated annealing composite omit maps calculated in Phenix. Analysis of model geometry using MolProbity (56) indicated that 96.6, 2.9, and 0.5% of residues are in the favored, allowed, and disallowed regions of the Ramachandran plot, respectively, and 98.3% of residues have favorable rotamers. Two residues are Ramachandran outliers: Leu255and Ser506. Leu255 is adjacent to Asp254, which forms a conserved structural motif surrounding the proposed exit channel; and Ser506 forms a crystal contact. Structural homology was assessed using the DaliLite server (57), and analysis of the dimer interface was performed using the protein interfaces, surfaces, and assemblies service at the European Bioinformatics Institute (58). Protein channels were visualized using CAVER (28). The volume of the proposed substrate binding cavity was determined using CASTp (59). HB-CoA conformations were modeled manually in Coot with energy minimization performed in CNS (60, 61). The figures were generated in PyMOL (62). Crystallography packages were compiled by SBGrid (63).
Author Contributions
E. C. W. and C. L. D. wrote the manuscript with critical contributions from M. J., Y. W., and J. S. All authors contributed to the experimental design. Y. W. cloned and purified CnPhaC(C319A). M. J. crystallized CnPhaC(C319A) and performed the heavy atom derivatization. E. C. W. collected and processed the crystallographic data and performed the structure determination. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are indebted to Prof. Ping Li for providing an alignment of class I PhaC sequences. We additionally thank Tsehai Grell and David Born for helpful conversations. This work is based on research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences through National Institutes of Health Grant P41 GM103403. The Pilatus 6M detector on Beamline 24-ID-C is funded by National Institutes of Health-Office of Research Infrastructure Programs High End Instrumentation Grant S10 RR029205. This research used resources of the Advanced Photon Source, a Department of Energy Office of Science User Facility operated for the Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grants T32 GM008334 (to E. C. W.), GM069857 (to C. L. D.), and GM49171 (to J. S.); by a Massachusetts Institute of Technology Poitras predoctoral fellowship (to M. J.); and by funding from ASTAR, Singapore (to Y. W.). C. L. D. is a Howard Hughes Medical Institute Investigator. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5T6O) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PHA
- polyhydroxyalkanoic acid
- PHB
- polyhydroxybutyrate
- HB
- hydroxybutyryl
- CV
- column volumes.
References
- 1. Stubbe J., and Tian J. (2003) Polyhydroxyalkanoate (PHA) homeostasis: the role of the PHA synthase. Nat. Prod. Rep. 20, 445–457 [DOI] [PubMed] [Google Scholar]
- 2. Stubbe J., Tian J., He A., Sinskey A. J., Lawrence A. G., and Liu P. (2005) Nontemplate-dependent polymerization processes: polyhydroxyalkanoate synthases as a paradigm. Annu. Rev. Biochem. 74, 433–480 [DOI] [PubMed] [Google Scholar]
- 3. Steinbuchel A., and Valentin H. E. (1995) Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol. Lett. 128, 219–228 [Google Scholar]
- 4. Sudesh K., Abe H., and Doi Y. (2000) Synthesis, structure and properties of polyhydroxyalkanoates: biological polyesters. Prog. Polym. Sci. 25, 1503–1555 [Google Scholar]
- 5. Steinbuchel A. (2001) Perspectives for biotechnological production and utilization of biopolymers: metabolic engineering of polyhydroxyalkanoate biosynthesis pathways as a successful example. Macromol. Biosci. 1, 1–24 [Google Scholar]
- 6. Keshavarz T., and Roy I. (2010) Polyhydroxyalkanoates: bioplastics with a green agenda. Curr. Opin. Microbiol. 13, 321–326 [DOI] [PubMed] [Google Scholar]
- 7. Rehm B. H. (2003) Polyester synthases: natural catalysts for plastics. Biochem. J. 376, 15–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jia Y., Yuan W., Wodzinska J., Park C., Sinskey A. J., and Stubbe J. (2001) Mechanistic studies on class I polyhydroxybutyrate (PHB) synthase from Ralstonia eutropha: class I and III synthases share a similar catalytic mechanism. Biochemistry 40, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 9. Jia Y., Kappock T. J., Frick T., Sinskey A. J., and Stubbe J. (2000) Lipases provide a new mechanistic model for polyhydroxybutyrate (PHB) synthases: characterization of the functional residues in Chromatium vinosum PHB synthase. Biochemistry 39, 3927–3936 [DOI] [PubMed] [Google Scholar]
- 10. Rehm B. H., Antonio R. V., Spiekermann P., Amara A. A., and Steinbüchel A. (2002) Molecular characterization of the poly(3-hydroxybutyrate) (PHB) synthase from Ralstonia eutropha: in vitro evolution, site-specific mutagenesis and development of a PHB synthase protein model. Biochim. Biophys. Acta 1594, 178–190 [DOI] [PubMed] [Google Scholar]
- 11. Yuan W., Jia Y., Tian J., Snell K. D., Müh U., Sinskey A. J., Lambalot R. H., Walsh C. T., and Stubbe J. (2001) Class I and III polyhydroxyalkanoate synthases from Ralstonia eutropha and Allochromatium vinosum: characterization and substrate specificity studies. Arch. Biochem. Biophys. 394, 87–98 [DOI] [PubMed] [Google Scholar]
- 12. Gerngross T. U., and Martin D. P. (1995) Enzyme-catalyzed synthesis of poly[(R)-(−)-3-hydroxybutyrate]: formation of macroscopic granules in vitro. Proc. Natl. Acad. Sci. U.S.A. 92, 6279–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerngross T. U., Snell K. D., Peoples O. P., Sinskey A. J., Csuhai E., Masamune S., and Stubbe J. (1994) Overexpression and purification of the soluble polyhydroxyalkanoate synthase from Alcaligenes eutrophus: evidence for a required posttranslational modification for catalytic activity. Biochemistry 33, 9311–9320 [DOI] [PubMed] [Google Scholar]
- 14. Wodzinska J., Snell K. D., Rhomberg A., Sinskey A. J., Biemann K., and Stubbe J. (1996) Polyhydroxybutyrate synthase: Evidence for covalent catalysis. J. Am. Chem. Soc. 118, 6319–6320 [Google Scholar]
- 15. Zhang S., Yasuo T., Lenz R. W., and Goodwin S. (2000) Kinetic and mechanistic characterization of the polyhydroxybutyrate synthase from Ralstonia eutropha. Biomacromolecules 1, 244–251 [DOI] [PubMed] [Google Scholar]
- 16. Liebergesell M., and Steinbüchel A. (1992) Cloning and nucleotide sequences of genes relevant for biosynthesis of poly(3-hydroxybutyric acid) in Chromatium vinosum strain D. Eur. J. Biochem. 209, 135–150 [DOI] [PubMed] [Google Scholar]
- 17. Müh U., Sinskey A. J., Kirby D. P., Lane W. S., and Stubbe J. (1999) PHA synthase from Chromatium vinosum: cysteine 149 is involved in covalent catalysis. Biochemistry 38, 826–837 [DOI] [PubMed] [Google Scholar]
- 18. Tian J., Sinskey A. J., and Stubbe J. (2005) Detection of intermediates from the polymerization reaction catalyzed by a D302A mutant of class III polyhydroxyalkanoate (PHA) synthase. Biochemistry 44, 1495–1503 [DOI] [PubMed] [Google Scholar]
- 19. Griebel R., Smith Z., and Merrick J. M. (1968) Metabolism of poly-β-hydroxybutyrate: I. purification, composition, and properties of native poly-β-hydroxybutyrate granules from Bacillus megaterium. Biochemistry 7, 3676–3681 [DOI] [PubMed] [Google Scholar]
- 20. Li P., Chakraborty S., and Stubbe J. (2009) Detection of covalent and noncovalent intermediates in the polymerization reaction catalyzed by a C149S class III polyhydroxybutyrate synthase. Biochemistry 48, 9202–9211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C., Cao R., Shrestha R., Ward C., Katz B. B., Fischer C. J., Tomich J. M., and Li P. (2015) Trapping of intermediates with substrate analog HBOCoA in the polymerizations catalyzed by class III polyhydroxybutyrate (PHB) synthase from Allochromatium vinosum. ACS Chem. Biol. 10, 1330–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buckley R. M., and Stubbe J. (2015) Chemistry with an artificial primer of polyhydroxybutyrate synthase suggests a mechanism for chain termination. Biochemistry 54, 2117–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liebergesell M., Sonomoto K., Madkour M., Mayer F., and Steinbüchel A. (1994) Purification and characterization of the poly(hydroxyalkanoic acid) synthase from Chromatium vinosum and localization of the enzyme at the surface of poly(hydroxyalkanoic acid) granules. Eur. J. Biochem. 226, 71–80 [DOI] [PubMed] [Google Scholar]
- 24. Chen Y. J., Tsai P. C., Hsu C. H., and Lee C. Y. (2014) Critical residues of class II PHA synthase for expanding the substrate specificity and enhancing the biosynthesis of polyhydroxyalkanoate. Enzyme Microb. Technol. 56, 60–66 [DOI] [PubMed] [Google Scholar]
- 25. Buckley R. M. (2013) The Role of the Phasin PhaP in Promoting Polyhydroxybutyrate (PHB) Granule Formation in Caulobacter crescentus. Ph.D. Thesis, Massachusetts Institute of Technology [Google Scholar]
- 26. Ollis D. L., Cheah E., Cygler M., Dijkstra B., Frolow F., Franken S. M., Harel M., Remington S. J., Silman I., and Schrag J. (1992) The α/β hydrolase fold. Protein Eng. 5, 197–211 [DOI] [PubMed] [Google Scholar]
- 27. Holmquist M. (2000) α/β-Hydrolase fold enzymes: structures, functions and mechanisms. Curr. Protein Pept. Sci. 1, 209–235 [DOI] [PubMed] [Google Scholar]
- 28. Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., and Damborsky J. (2012) CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sayer C., Finnigan W., Isupov M. N., Levisson M., Kengen S. W., van der Oost J., Harmer N. J., and Littlechild J. A. (2016) Structural and biochemical characterisation of Archaeoglobus fulgidus esterase reveals a bound CoA molecule in the vicinity of the active site. Sci. Rep. 6, 25542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ushimaru K., Sangiambut S., Thomson N., Sivaniah E., and Tsuge T. (2013) New insights into activation and substrate recognition of polyhydroxyalkanoate synthase from Ralstonia eutropha. Appl. Microbiol. Biotechnol. 97, 1175–1182 [DOI] [PubMed] [Google Scholar]
- 31. Roussel A., Miled N., Berti-Dupuis L., Rivière M., Spinelli S., Berna P., Gruber V., Verger R., and Cambillau C. (2002) Crystal structure of the open form of dog gastric lipase in complex with a phosphonate inhibitor. J. Biol. Chem. 277, 2266–2274 [DOI] [PubMed] [Google Scholar]
- 32. Aschauer P., Rengachari S., Lichtenegger J., Schittmayer M., Das K. M., Mayer N., Breinbauer R., Birner-Gruenberger R., Gruber C. C., Zimmermann R., Gruber K., and Oberer M. (2016) Crystal structure of the Saccharomyces cerevisiae monoglyceride lipase Yju3p. Biochim. Biophys. Acta 1861, 462–470 [DOI] [PubMed] [Google Scholar]
- 33. Goettig P., Brandstetter H., Groll M., Göhring W., Konarev P. V., Svergun D. I., Huber R., and Kim J. S. (2005) X-ray snapshots of peptide processing in mutants of tricorn-interacting factor F1 from Thermoplasma acidophilum. J. Biol. Chem. 280, 33387–33396 [DOI] [PubMed] [Google Scholar]
- 34. Hofmann B., Tölzer S., Pelletier I., Altenbuchner J., van Pée K. H., and Hecht H. J. (1998) Structural investigation of the cofactor-free chloroperoxidases. J. Mol. Biol. 279, 889–900 [DOI] [PubMed] [Google Scholar]
- 35. Cassidy C. S., Lin J., and Frey P. A. (1997) A new concept for the mechanism of action of chymotrypsin: the role of the low-barrier hydrogen bond. Biochemistry 36, 4576–4584 [DOI] [PubMed] [Google Scholar]
- 36. Lin J., Cassidy C. S., and Frey P. A. (1998) Correlations of the basicity of His 57 with transition state analogue binding, substrate reactivity, and the strength of the low-barrier hydrogen bond in chymotrypsin. Biochemistry 37, 11940–11948 [DOI] [PubMed] [Google Scholar]
- 37. Yahya A. R. M., Anderson W. A., and Moo-Young M. (1998) Ester synthesis in lipase-catalyzed reactions. Enzyme Microb. Technol. 23, 438–450 [Google Scholar]
- 38. Gog A., Roman M., Tosa M., Paizs C., and Irimie F. D. (2012) Biodiesel production using enzymatic transesterification: current state and perspectives. Renew. Energ. 39, 10–16 [Google Scholar]
- 39. Moynihan P. J., and Clarke A. J. (2014) Mechanism of action of peptidoglycan O-acetyltransferase B involves a Ser-His-Asp catalytic triad. Biochemistry 53, 6243–6251 [DOI] [PubMed] [Google Scholar]
- 40. Storer A. C., and Menard R. (1994) Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 244, 486–500 [DOI] [PubMed] [Google Scholar]
- 41. Buller A. R., and Townsend C. A. (2013) Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. U.S.A. 110, E653–E661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frey P. A., Whitt S. A., and Tobin J. B. (1994) A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 264, 1927–1930 [DOI] [PubMed] [Google Scholar]
- 43. Ishida T. (2006) Low-barrier hydrogen bond hypothesis in the catalytic triad residue of serine proteases: correlation between structural rearrangement and chemical shifts in the acylation process. Biochemistry 45, 5413–5420 [DOI] [PubMed] [Google Scholar]
- 44. Zhao Y. H., Abraham M. H., and Zissimos A. M. (2003) Fast calculation of van der Waals volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 68, 7368–7373 [DOI] [PubMed] [Google Scholar]
- 45. Tian J., He A., Lawrence A. G., Liu P., Watson N., Sinskey A. J., and Stubbe J. (2005) Analysis of transient polyhydroxybutyrate production in Wautersia eutropha H16 by quantitative Western analysis and transmission electron microscopy. J. Bacteriol. 187, 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cho M. (2012) Studies on the Mechanism of Polyhydroxybutyrate (PHB) Granule Formation in Ralstonia eutropha H16. Ph.D. Thesis, Massachusetts Institute of Technology [Google Scholar]
- 47. Tian J., Sinskey A. J., and Stubbe J. (2005) Class III polyhydroxybutyrate synthase: involvement in chain termination and reinitiation. Biochemistry 44, 8369–8377 [DOI] [PubMed] [Google Scholar]
- 48. Madden L. A., Anderson A. J., Shah D. T., and Asrar J. (1999) Chain termination in polyhydroxyalkanoate synthesis: involvement of exogenous hydroxy-compounds as chain transfer agents. Int. J. Biol. Macromol. 25, 43–53 [DOI] [PubMed] [Google Scholar]
- 49. Lawrence A. G., Choi J., Rha C., Stubbe J., and Sinskey A. J. (2005) In vitro analysis of the chain termination reaction in the synthesis of poly-(R)-β-hydroxybutyrate by the class III synthase from Allochromatium vinosum. Biomacromolecules 6, 2113–2119 [DOI] [PubMed] [Google Scholar]
- 50. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pape T., and Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 53. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 56. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holm L., Kääriäinen S., Rosenström P., and Schenkel A. (2008) Searching protein structure databases with DaliLite v.3. Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 59. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 61. Brunger A. T. (2007) Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 62. (2015) The PyMOL Molecular Graphics System, Version 1.8, Schrodinger [Google Scholar]
- 63. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roussel A., Canaan S., Egloff M. P., Rivière M., Dupuis L., Verger R., and Cambillau C. (1999) Crystal structure of human gastric lipase and model of lysosomal acid lipase, two lipolytic enzymes of medical interest. J. Biol. Chem. 274, 16995–17002 [DOI] [PubMed] [Google Scholar]
- 65. Labar G., Bauvois C., Borel F., Ferrer J. L., Wouters J., and Lambert D. M. (2010) Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. ChemBioChem 11, 218–227 [DOI] [PubMed] [Google Scholar]