

Abstract
We highlight an under-recognised cause of acquired esotropia with this prospective observational case series of adults with diplopia secondary to cerebellar dysfunction. We also show deterioration of cerebellar esotropia over time, which has not been previously described. Seven adults (four women) developed diplopia at a median age of 63 years (range: 31–75 years), as the initial manifestation of the underlying cerebellar disorder. Causes of cerebellar dysfunction were familial cerebellar ataxia of unknown mutation (two patients), idiopathic cerebellar ataxia (four patients), and spinocerebellar ataxia 3 (one patient). At onset, three patients had unilateral and four had bilateral lateral rectus under-action. These were initially diagnosed as lateral rectus paresis, but the diagnosis was revised, as our examination showed no slowing of abducting saccades assessed clinically and full abduction with gaze-evoked nystagmus. Esotropia was concomitant and worse for distance, although at onset one patient’s esotropia was equal for near and distance. There was a trend of worsening esotropia over time, following a median interval follow-up of 4 years (range: 1–18). All patients were first observed to have cerebellar eye signs after a median interval of 5 years (range: 1–30) from presentation, i.e., impaired pursuit (7/7 patients), gaze-evoked nystagmus (7/7), hypometric saccades (3/7), downbeat nystagmus (2/7), and skew deviation (4/7). Only two patients have not developed non-ocular cerebellar eye signs, after 5 and 8 years from diplopia onset, respectively; the other five patients had gait ataxia, which could be mild. The patients were successfully treated with prisms (7/7), botulinum toxin injections (1/7), and strabismus surgery (1/7).
Keywords: Cerebellar degeneration, diplopia, esotropia, lateral rectus paresis
INTRODUCTION
Acquired esotropia in cerebellar dysfunction is well described but under-recognised. One of the first to highlight this was Zweifach, who described a 9-year-old boy with a medulloblastoma involving the cerebellum, diagnosed after 28 months of intermittent diplopia and concomitant esotropia.1 Esotropia has also been described in other posterior fossa structural lesions, Arnold-Chiari malformations, and degenerative cerebellar disease.2–7 The pathogenesis of the esotropia is controversial and may be a result of disruption to central vestibular pathways.8 We report a prospective observational case series of seven adults with diplopia due to “cerebellar” esotropia. Our aim in this article is to demonstrate that diplopia due to cerebellar esotropia can be the first manifestation of the underlying disorder and as such the condition may be misdiagnosed. We also show deterioration in the esotropia over time, which has not been described previously and discuss aspects of management.
CLINICAL DESCRIPTION OF ALL CASES
From 1994 to 2011, seven adults referred with horizontal binocular diplopia were found to have cerebellar dysfunction (Table 1).
TABLE 1. Summary of patients with cerebellar dysfunction presenting with binocular diplopia from esotropia.
| Patient | Sex | Genetic test results | Family history | Age of diplopia onset | Age of ataxia onset | Age first examined by us | Age at last review | Lateral rectus underaction | Esodeviation worse to Near(N) or Distance(D) | Vertical deviation present | Cerebellar eye signs | Non-ocular cerebellar signs | Brain maging* | Diagnosis | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | NEG | Y | 36 | 39 | 37 | 46 | L, then B | N = D, then D > N | Y | Broken pursuit, GEN | Y | Normal | ADCA | Prisms & surgery |
| 2 | M | NEG | Y | 63 | NA | 70 | 71 | B | D > N | Y | Broken pursuit, GEN | N | Small vessel disease | ADCA | Prisms |
| 3 | M | NEG | N | 73 | 74 | 74 | 76 | B | D > N | N | Broken pursuit, GEN, DBN | Y | Generalised atrophy | ICD | Prisms |
| 4 | F | NEG | N | 75 | 78 | 75 | 93 | R, then B | D > N | Y | Broken bursuit, hypometric saccade, DBN | Y | Normal | ICD | Prisms |
| 5 | F | NEG | N | 45 | 75 | 75 | 78 | B | D > N | Y | Broken bursuit, hypometric saccade, GEN | Y | Cerebellar atrophy | ICD | Prisms & botox |
| 6 | M | ND | N | 66 | NA | 69 | 75 | B | D > N | N | Broken pursuit, GEN | N | Small vessel disease | ICD | Prisms |
| 7 | F | SCA expansion | Y | 31 | 31 | 38 | 42 | L, then B | D > N | Y | Broken pursuit, GEN, hypometric saccade | Y | Cerebellar atrophy | ADCA | Prisms |
NA = not applicable; ND = not done; GEN = gaze-evoked nystagmus; DBN = down-beat nystagmus; NEG = negative for SCA 1, 2, 3, 6, 7; L = left; R = right; B = both; ADCA = autosomal dominant cerebellar ataxia; ICD = idiopathic cerebellar degeneration.
Only Patient 4 was examined by us at onset, but developed ataxia 3 years and DBN 7 years later. All other patients first presented elsewhere and were first observed to have cerebellar eye signs when first examined by us.
*Magnetic resonance imaging was undertaken except for Patient 4 who had computed tomography due to claustrophobia.
Median age at diplopia onset was 63 years (range: 31–75 years). Causes of cerebellar dysfunction were familial cerebellar ataxia of unknown mutation (Cases 1 and 2), idiopathic cerebellar degeneration (Cases 3–6), and autosomal dominant cerebellar ataxia with spinocerebellar ataxia 3 (SCA3) (Case 7).
At first presentation elsewhere, two were described as having unilateral and four bilateral lateral rectus under-action but cerebellar features were not reported. Patient 4 presented to us at onset of diplopia and had a unilateral lateral rectus under-action. She developed ataxia 3 years and downbeat nystagmus (DBN) 7 years later. Patients other than Patient 4 were first assessed by us after a median interval of 5 years (range: 1–30) from presentation and were observed to have cerebellar eye signs. Cerebellar eye signs included broken pursuit (7/7 patients), gaze-evoked nystagmus (7/7), hypometric saccades (Patients 4, 5, 7), and DBN (Patients 3, 4) and vertical deviation (skew deviation: Patients 1, 2, 4, 5, 7). For the patients with vertical deviation, the height was comitant (i.e. an over-action of superior oblique muscle), with no intorsion on examination or by patient report, which indicated a neurological cause. No patients had alternating skew.
All patients had concomitant esotropia. All patients had worse esotropia for distance, although at onset one patient’s esotropia was initially equal for near and distance fixation. Although initial assessments had described under-action of the lateral rectus in the cases, there was no slowing of abducting saccades assessed clinically by us, and we found full abduction of either eye (with gaze-evoked nystagmus).
There were no findings in favour of these patients having a decompenstated congenital esotropia. In particular, all patients had good binocularity and neither dissociated vertical deviation nor latent nystagmus. Binocularity was assessed by prism fusion range and stereopsis. Stereoacuity was normal by Frisby stereotest.
There was a trend of worsening esotropia over time (see Figure 1 showing serial findings in Patient 1 and Figure 2, Patients 1, 2, 5, 6), following a median interval follow-up of 4 years (range: 1–18).
FIGURE 1.
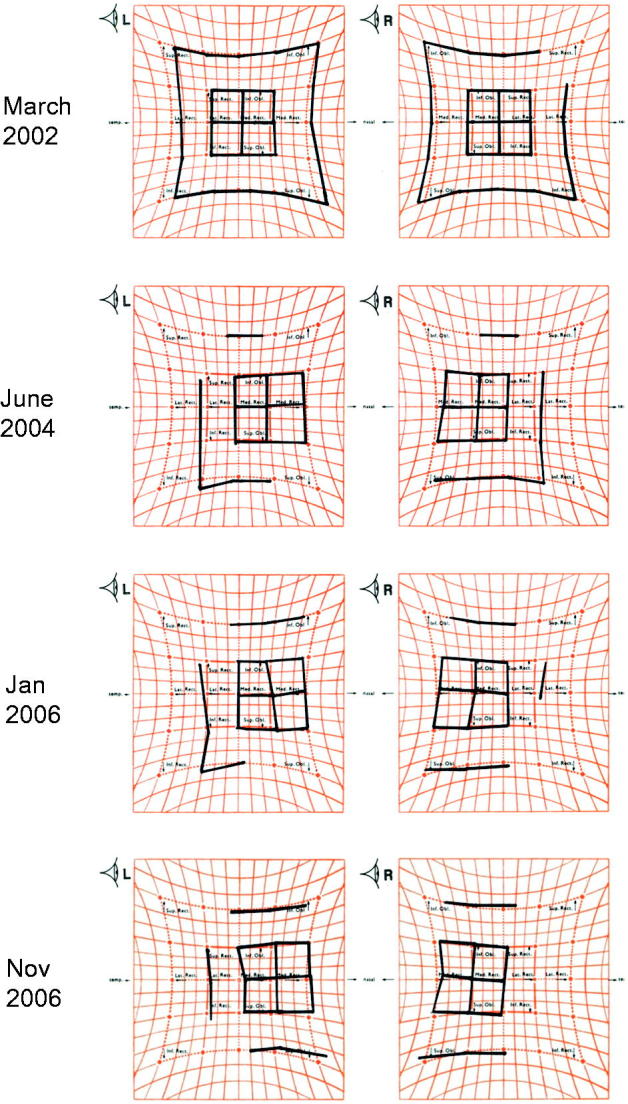
Serial Hess charts of Patient 1 demonstrating the progressively worsening esotropia. March 2002: At initial presentation, a small esodeviation was found in primary position with small bilateral under-action of lateral recti, without over-action of medial recti. Additionally, there was no over-action of superior obliques nor inferior obliques; therefore, no A or V pattern was seen. June 2004: There was an increase in the esodeviation and the under-action of the lateral recti. No A or V pattern was seen. January 2006 and November 2006: There was an increase in the esodeviation and lateral recti under-actions. There was also a small right over left deviation.
FIGURE 2.

Change in esodeviation over time in cerebellar esotropia. This graph shows a possible trend of worsening esotropia over time, particularly in Patients 1, 2, 5, and 6. Mann-Whitney U test comparing the measurements for Patients 1, 2, 4, 6, and 7 from onset with the measurements recorded at approximately 55 months (i.e. the last measurement before Patient 1 had surgery) showed a p value of 0.059. Patient 3 was excluded due to the short follow-up (2 years), and Patient 5 was excluded due to the treatment with botulinum toxin at 7 months from onset.
Only two (Patients 2, 6) have not developed non-ocular cerebellar signs, after 5 and 8 years from diplopia onset. The other five patients had gait ataxia, although this could be mild.
All patients had cranial imaging. Cerebellar atrophy (Patients 5 and 7), generalised atrophy (Patient 3), and small vessel disease (Patients 2 and 6) were the only notable abnormalities. All imaging was done by magnetic resonance apart from Patient 4 who had computed tomography due to claustrophobia.
All patients were successfully treated with prisms. The diplopia was fused with Fresnel prisms, and they achieved normal convergence and a good level of binocularity. As their condition progressed, the base in range on prism fusion range testing reduced, thereby indicating a stronger Fresnel prism was required. Patient 1 required additional strabismus surgery, and Patient 5 had recurrent botulinum toxin injection of her right medial rectus muscle (see Figure 2). Post surgery, Patient 1 still has a slightly reduced divergence range on prism fusion range test, although Fresnel prisms were no longer required.
DISCUSSION
Our series highlights that diplopia secondary to a comitant esotropia may be the presenting feature of cerebellar dysfunction. In all cases this was initially misdiagnosed as unilateral or bilateral lateral rectus paresis. Only Patient 4 was seen by us at symptom onset, ataxia developed 3 years and DBN 7 years later. In this case we can confirm that the esotropia preceded the development of other signs of cerebellar dysfunction. All other cases were first seen elsewhere, and we cannot be sure whether cerebellar signs were absent or overlooked: all these cases had cerebellar eye signs when first examined by us.
Patients 2 and 6 have not developed non-ocular cerebellar signs (ataxia) 8 and 9 years after onset of symptoms, respectively. In all patients in our series, the diplopia has been successfully managed with prism correction. However, the esotropia has in some been progressive over time requiring surgery (Patient 1) and botulinum toxin injection in another (Patient 5), in addition to ongoing correction with use of a small horizontal Fresnel prism (Figure 2).
Acquired esotropia due to posterior fossa tumours, tonsillar ectopia, and hydrocephalus, also termed “divergence insufficiency” or “divergence palsy”, is well recognised.3,6,7 The mechanism underlying this may be abducens palsy or raised intracranial pressure, but can occur in the absence of either.2
Cerebellar esotropia may also arise from excessive convergence tone,4 i.e., as a supranuclear phenomenon possibly resulting from disruption to the central vestibular system.8 Animal9,10 and human11 studies have shown that the cerebellar vermis is particularly important in vergence eye movements, whereby lesions could lead to excessive convergence tone. This would be in keeping with our patients whose esodeviation was greater for distance than near and had relative preservation of near fusion, i.e. findings compatible with divergence paresis. Our patients’ convergence was normal when diplopia was fused with prisms; we did not measure the amplitude of convergence in this preliminary study.
We have considered the possibility that the patients may have had age-related distance esotropia.12,13 Five of seven patients had onset of symptoms under 67 years of age. Furthermore, as all patients had cerebellar eye signs, this would indicate a collision of two unrelated ocular motility disorders. This question could only be answered in a population study.
One possible way to differentiate lateral rectus paresis from excessive convergence is the slowing of abducting saccades in lateral rectus paresis. It is difficult to be certain that our patients do not have lateral rectus paresis without further quantitative studies confirming our clinical observation of normal saccadic speeds. The comitance of the esotropia makes a bilateral lateral rectus paresis less likely. If there is a component of lateral rectus paresis in these patients, it is mild and the expected finding would be esotropia for distance and esophoria for near, as the vergence mechanism can be used to compensate for a mild lateral rectus paresis at near. Additionally, our patients did not show an over-action of the contralateral medial recti, which would be expected initially in lateral rectus paresis. The fact that all our patients, showing mild under-action of the lateral recti at onset, have an esotropia for near is also in favour of a supranuclear mechanism such as divergence paresis rather than lateral rectus paresis. We are planning further studies to elucidate the pathogenesis of esotropia in these patients.
This preliminary report highlights the importance of examining for cerebellar eye signs in patients who present with new-onset esotropia. All patients presenting with lateral rectus under-action without spontaneous recovery or demonstrating progression should raise the index of suspicion for possible cerebellar dysfunction, and a careful examination looking for cerebellar eye signs should be carried out.
Acknowledgements
This work was presented as a poster at the joint American Neurological Association and Association of British Neurologists (ABN) meeting in Boston, October 2012. Dr. Wong was awarded a bursary from the ABN for attendance at the meeting.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Zweifach PH. Childhood esotropia with delayed appearance of cerebellar tumour. Neuro-ophthalmology 1981;1:291–293 [Google Scholar]
- 2.Williams AS, Hoyt CS. Acute comitant esotropia in children with brain tumors. Arch Ophthalmol 1989;107:376–378 [DOI] [PubMed] [Google Scholar]
- 3.Hoyt CS, Good WV. Acute onset concomitant esotropia: when is it a sign of serious neurological disease? Br J Ophthalmol 1995;79:498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Versino M, Hurko O, Zee DS. Disorders of binocular control of eye movements in patients with cerebellar dysfunction. Brain 1996;119:1933–1950 [DOI] [PubMed] [Google Scholar]
- 5.Ohyagi Y, Yamada T, Okayama A, Sakae N, Yamasaki T, Ohshima T, Sakamoto T, Fujii N, Kira J. Vergence disorders in patients with spinocerebellar ataxia 3/Machado-Joseph disease: a synoptophore study. J Neurol Sci 2000;173:120–123 [DOI] [PubMed] [Google Scholar]
- 6.Akman A, Dayanir V, Sanaç AS, Kansu T. Acquired esotropia as presenting sign of cranio-cervical junction anomalies. Neuro-ophthalmology 1995;15:311–314 [Google Scholar]
- 7.Lewis AR, Kline LB, Sharpe JA. Acquired esotropia due to Arnold-Chiari I malformation. J Neuroophthalmol 1996;16:49–54 [PubMed] [Google Scholar]
- 8.Brodsky MC. Three dimensions of skew deviation. Br J Ophthalmol 2003;87:1440–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nitta T, Akao T, Kurkin S, Fukushima K. Vergence eye movement signals in the cerebellar dorsal vermis. Prog Brain Res 2008;171:173–176 [DOI] [PubMed] [Google Scholar]
- 10.Takagi M, Tamargo R, Zee DS. Effects of lesions of the cerebellar oculomotor vermis on eye movements in primate: binocular control. Prog Brain Res 2003;142:19–33 [DOI] [PubMed] [Google Scholar]
- 11.Sander T, Sprenger A, Neumann G, et al. Vergence deficits in patients with cerebellar lesions. Brain 2009;132:103–115 [DOI] [PubMed] [Google Scholar]
- 12.Mittelman D. Age-related distance esotropia. J AAPOS 2006;10:212–213 [DOI] [PubMed] [Google Scholar]
- 13.Godts D, Mathysen DGP. Distance esotropia in the elderly. Br J Ophthalmol 2013;97:1415–1419 [DOI] [PubMed] [Google Scholar]