

Abstract
Background
Hyperactivity of the classical axis of the renin-angiotensin system (RAS), mediated by angiotensin II (Ang II) activation of the angiotensin II type 1 receptor (AT1R), is implicated in the pathogenesis of Alzheimer’s disease (AD). Angiotensin-converting enzyme-2 (ACE-2) degrades Ang II to angiotensin 1–7 (Ang (1-7)) and counter-regulates the classical axis of RAS. We have investigated the expression and distribution of ACE-2 in post-mortem human brain tissue in relation to AD pathology and classical RAS axis activity.
Methods
We measured ACE-2 activity by fluorogenic peptide substrate assay in mid-frontal cortex (Brodmann area 9) in a cohort of AD (n = 90) and age-matched non-demented controls (n = 59) for which we have previous data on ACE-1 activity, amyloid β (Aβ) level and tau pathology, as well as known ACE1 (rs1799752) indel polymorphism, apolipoprotein E (APOE) genotype, and cerebral amyloid angiopathy severity scores.
Results
ACE-2 activity was significantly reduced in AD compared with age-matched controls (P < 0.0001) and correlated inversely with levels of Aβ (r = −0.267, P < 0.001) and phosphorylated tau (p-tau) pathology (r = −0.327, P < 0.01). ACE-2 was reduced in individuals possessing an APOE ε4 allele (P < 0.05) and was associated with ACE1 indel polymorphism (P < 0.05), with lower ACE-2 activity in individuals homozygous for the ACE1 insertion AD risk allele. ACE-2 activity correlated inversely with ACE-1 activity (r = −0.453, P < 0.0001), and the ratio of ACE-1 to ACE-2 was significantly elevated in AD (P < 0.0001). Finally, we show that the ratio of Ang II to Ang (1–7) (a proxy measure of ACE-2 activity indicating conversion of Ang II to Ang (1–7)) is reduced in AD.
Conclusions
Together, our findings indicate that ACE-2 activity is reduced in AD and is an important regulator of the central classical ACE-1/Ang II/AT1R axis of RAS, and also that dysregulation of this pathway likely plays a significant role in the pathogenesis of AD.
Electronic supplementary material
The online version of this article (doi:10.1186/s13195-016-0217-7) contains supplementary material, which is available to authorized users.
Keywords: Angiotensin-converting enzyme-2, Renin-angiotensin system, Angiotensin-converting enzyme-1, Angiotensin II, Alzheimer’s disease
Background
Genetic, clinical and epidemiological data as well as experimental cell and animal studies all support a role for the renin-angiotensin system (RAS) in the pathogenesis of Alzheimer’s disease (AD) [1]. Many of the pro-inflammatory, anti-cholinergic and vasopressor actions of RAS associated with the pathogenesis of AD are mediated by angiotensin II (Ang II) signalling through the angiotensin II type 1 receptor (AT1R), commonly referred to as the classical axis (reviewed in [1]). Intracerebroventricular infusion of Ang II increased both amyloid-β (Aβ) (via increased amyloidogenic processing of amyloid precursor protein [APP]) [2] and tau pathology, and also reduced cognitive performance [3], in aged normal rats. We have previously reported that angiotensin-converting enzyme-1 (ACE-1), the rate-limiting enzyme in the production of angiotensin II (Ang II), is increased in AD in human brain tissue [4, 5]. Angiotensin II type 1 receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACEIs) reduce the amount of AD-like pathology and improve cognitive performance in most but not all mouse models of AD [6–11]. Translation of these treatments in AD is also supported in secondary outcomes of clinical trials of various ARBs and ACEIs, as well as in epidemiological studies where the prevalence of AD was reduced [12–16]. Last, the ACE-1 indel polymorphism (rs1799752) is a genetic risk factor for sporadic AD [17]. This finding has previously been supported by several meta-analyses [18–22] but not by recent genome-wide association studies.
ACE-2 is a zinc metallopeptidase which shares 42% sequence homology within the ACE-1 catalytic region [23, 24]. The ACE-2 metalloprotease is expressed mostly as a transmembrane protein, but it also exists in an active soluble truncated form [24]. It is expressed predominantly in endothelial and arterial smooth muscle cells throughout the body [25], but it is also expressed in non-vascular cells within the brain, including neuronal cell bodies [26] and astroglial cells [27]. Upon its discovery, ACE-2 was shown to generate angiotensin 1–7 (Ang (1-7)) from Ang II, and, to a lesser extent, angiotensin 1–9 (Ang (1-9)) from Ang I [23, 24, 28]. Emerging data suggest that ACE-2-mediated conversion of Ang II to Ang (1–7) and subsequent activation of the Mas receptor by Ang (1–7) (comprising the ACE-2/Ang (1-7) /Mas axis) oppose the local actions of the classical RAS pathway in both the periphery (reviewed in [29]) and brain (reviewed in [30–33]). In experimental animal studies, ACE-2 regulates blood pressure by counteracting the effects of the classical axis. A reduction in ACE-2 expression has been implicated in cardiac and renal pathologies (reviewed in [30]) associated with chronic hypertension. Activation of brain ACE-2 has been shown to be neuroprotective in animal models of ischaemic stroke [34, 35].
Previous studies have suggested a link between reduced activity of the ACE-2/Ang (1–7)/Mas axis and neurodegenerative conditions, including multiple sclerosis [36]. A recent study provided the first clues of an association with AD and reported reduced serum ACE-2 activity in patients with AD compared with control subjects [37]. Notably, this study also identified that ACE-2 converts Aβ43 (an early deposited and highly amyloidogenic form of Aβ that seeds plaque formation [38]) to Aβ42, which in turn is cleaved by ACE-1 to less toxic Aβ40 and Aβ41 species [37]. Ang (1–7) levels were also reduced in a mouse model of sporadic AD in association with hyperphosphorylation of tau [39].
In the present study, we investigated the expression and distribution of ACE-2 in relation to AD pathology and the classical RAS axis in human post-mortem brain tissue. We show, for the first time to our knowledge, that ACE-2 activity is reduced in human post-mortem brain tissue in AD in relation to Aβ and tau pathology, and also that ACE-2 correlates inversely with ACE-1 activity. We also show that the ratio of Ang II to Ang (1–7) (a proxy measure of ACE-2 activity) was increased in AD, indicating reduced conversion of Ang II to Ang (1–7). Together, these data indicate that the ACE-2/Ang (1–7)/Mas axis is dysregulated in AD and that loss of function of this regulatory arm of RAS may contribute, at least in part, to overactivation of the classical RAS axis associated with AD pathogenesis.
Methods
Case selection
Brain tissue was obtained from the South West Dementia Brain Bank, University of Bristol, UK, with local research ethics committee approval (National Research Ethics Service 08/H0106/28 + 5). Tissue was dissected from the mid-frontal cortex (Brodmann area 9) in 90 cases of AD and 59 age-matched controls. Brains had been subjected to detailed neuropathological assessment according to the National Institute on Aging-Alzheimer’s Association guidelines [40], and AD pathology was a sufficient explanation for the dementia in these cases. Control brains were from people who had no history of dementia, had been extensively assessed neuropathologically, and had few or absent neuritic plaques, Braak tangle stage III or less, and no other neuropathological abnormalities. The demographic data for these cases are presented in Table 1, and the Medical Research Council UK Brain Banks Network (MRC UK-BBN) database identifiers are shown in Additional file 1: Table S1.
Table 1.
Demographics of the study cohort
| Control (n = 59) | AD (n = 90) | |
|---|---|---|
| Age, years, mean ± SD | 78.5 ± 10.1 | 78.5 ± 9.7 |
| Sex, F/M | 22/37 | 55/35 |
| PM delay, h, mean ± SD) | 43.8 ± 36.4 | 45.2 ± 25.1 |
AD Alzheimer’s disease, PM Post-mortem
Previous measurements of ACE-1 activity, measured by fluorogenic activity assay, were available for all cases [4, 41]. Total soluble (Nonidet P-40-extracted) and insoluble (6 M guanidine hydrochloride-extracted) Aβ levels were measured previously by sandwich enzyme-linked immunosorbent assay (ELISA) [42], and cerebral amyloid angiopathy (CAA) severity, which was graded semi-quantitatively on a 4-point scale by a method adapted from that of Olichney et al. [43], had previously been reported [44]. Phosphorylated tau (p-tau) load (area fraction of cerebral cortex immunopositive for p-tau) had been measured for all cases, as previously reported [45, 46]. ACE1 genotype data for the Alu 237-bp insertion(I)/deletion(D) (indel) polymorphism (rs1799752) in intron 16 of the ACE1 gene were previously reported [5, 41]. Last, all cases had previously been apolipoprotein E (APOE)-genotyped [44, 47] by a polymerase chain reaction method [48].
Brain tissue
The right cerebral cortex had been fixed in 10% formalin for a minimum of 3 weeks before the tissue was processed and paraffin blocks were taken for pathological assessment. The left cerebral hemisphere had been sliced and frozen at −80 °C until used for biochemical assessment. For each case, 200 mg of dissected frozen brain tissue was homogenised in a Precellys homogeniser (Stretton Scientific, Stretton, UK) as previously described [4, 5]. The samples were centrifuged at 13,000 rpm, and the clarified supernatants were aliquoted and stored at −80 °C until required. Total protein was measured using the Total Protein kit (Sigma-Aldrich, Poole, UK) following the manufacturer’s guidelines. All brain tissue was obtained within 72 h after death.
ACE-2 activity assay
ACE-2 activity was measured in brain tissue using the SensoLyte® 390 ACE2 activity assay kit (catalogue number AS-72086; AnaSpec, Fremont, CA, USA). The assay was performed in black, flat-bottomed, non-binding, 96-well Nunc FluoroNunc plates (Fisher Scientific, Loughborough, UK) following the manufacturer’s guidelines with minor modifications. Brain tissue homogenates were prepared in assay buffer provided in the kit, to which 0.05% Triton X-100 was added. Samples were centrifuged at 13,000 rpm for 15 minutes at 4 °C, and supernatants were removed and stored at −80 °C until used. Supernatants were diluted 1:100 in the proprietary ACE-2 assay buffer and incubated for 10 minutes at room temperature prior to addition of the ACE-2-specific fluorescence resonance energy transfer (FRET) peptide and then incubated for 30 minutes in the dark. Cleavage of the ACE-2 FRET peptide was measured using a BMG FLUOstar OPTIMA microplate reader (BMG Labtech, Aylesbury, UK) at an excitation/emission wavelength of 330/390 nm. ACE-2 activity was interpolated from a serial dilution of 7-methoxycoumarin-4-yl-acetyl (Mca) fluorescence reference standard, and measurements for each case were repeated in duplicate.
To confirm the specificity of the commercial ACE-2 assay kit, we measured ACE-2 activity in a subset of samples (ten controls and ten AD) for which we had previously measured ACE-2 activity as outlined above. The assay was performed in black, flat-bottomed, non-binding, 96-well Nunc FluoroNunc plates. Recombinant human ACE-2 (440-6 ng/ml) (R&D Systems, Cambridge, UK) and brain tissue supernatants (diluted 1:20) were diluted in assay buffer (75 mM Tris, 1 M NaCl, pH 7.5) and pre-incubated with an ACE-2 specific inhibitor, MLN4760 (10 μM) (Calbiochem, Nottingham, UK) or assay buffer alone for 10 minutes at 37 °C. An ACE-2 fluorogenic peptide Mca-APK(Dnp) (Enzo Life Sciences, Exeter, UK) was then added, and the reaction was incubated at 37 °C for 30 minutes in the dark. Fluorescence was read at an excitation/emission wavelength of 330/405 nm using a BMG FLUOstar OPTIMA microplate reader. ACE-2-specific activity was calculated after subtracting fluorescence in the presence of MLN-4760 from the uninhibited sample. We observed a very strong correlation between the independent measurements of ACE-2 in the presence of MLN4760 (10 μM) and with the kit, indicating the specificity of the ACE-2 assay kit (Additional file 2: Figure S1).
Angiotensin II sandwich ELISA
Ang II levels were measured in brain tissue homogenates extracted in 1% SDS lysis buffer (100 μM NaCl, 10 mM Tris, pH 6, 1 μM phenylmethylsulphonylfluoride, 1 μg/ml aprotinin [Sigma-Aldrich] and 1% SDS in distilled water) using a commercially available sandwich ELISA kit (Abcam, Cambridge, UK) following the manufacturer’s guidelines. In brief, recombinant human Ang II or brain tissue supernatants (diluted 1:2 in PBS) were added in duplicate to wells that had been pre-coated with an Ang II-specific capture antibody and incubated for 2 h at room temperature. After a wash step, the wells were incubated for 2 h with biotinylated anti-Ang II antibody at room temperature. The plate was again washed, followed by a 30-minute incubation with streptavidin/HRP. After a final wash, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added for 20 minutes, and the absorbance at 450 nm was read using a FLUOstar OPTIMA plate reader. The concentration of Ang II was interpolated from a serial dilution of recombinant Ang II (1000–62.5 pg/ml) and measured in duplicate for each case.
Angiotensin (1–7) direct ELISA
Ang (1–7) levels were measured in human brain tissue homogenates in 1% SDS lysis buffer (see above) using an in-house direct ELISA kit. Recombinant human Ang (1–7) (Enzo Life Sciences) or human brain tissue homogenates (diluted 1:40 In PBS) were incubated for 2 h in a clear, high binding capacity Nunc MaxiSorp plate (Thermo Fisher Scientific, Waltham, MA, USA) at 26 °C with shaking. The wells were washed five times in PBS with 0.05% Tween-20 and blocked for 1 h in PBS:1% bovine serum albumin (Sigma-Aldrich). After another five washes, the wells were incubated with biotinylated anti-human Ang 1–7 (2 μg/ml in PBS) (Cloud-Clone, Wuhan, China) for 2 h at 26 °C with shaking, followed by a further wash step. Streptavidin/HRP (1:200) in PBS/0.01% Tween-20 was added to each well, which was incubated at room temperature for 20 minutes in the dark. TMB substrate (R&D Systems) was added after a further wash and left to develop in the dark for 20 minutes. Absorbance at 450 nm was read following the addition of 2 N sulphuric acid (‘stop’ solution) using a FLUOstar OPTIMA plate reader. Ang (1–7) concentration was interpolated from a standard curve generated by serially diluting recombinant human Ang (1–7) (5000–78.125 pg/ml). The assay showed minimal cross-reactivity with a number of closely related peptides, including Ang I, Ang II and Ang III.
ACE-2 immunoperoxidase labelling
Formalin-fixed, paraffin-embedded tissue sections (7 μm) were cut and de-waxed prior to immunohistochemistry. Sections were pre-treated in trisodium citrate buffer (9 mM), pH 6, and microwaved for 5 minutes, left to stand for 5 minutes, and boiled for a further 5 minutes before being left to stand for 15 minutes at room temperature. Sections were then rinsed thoroughly and covered in horse serum blocking solution, rinsed again, and incubated overnight at room temperature with anti-ACE-2 antibody (0.05 μg/ml, ab15348; Abcam). Bound antibody was visualised using a biotinylated universal antibody followed by VECTASTAIN Elite ABC avidin-biotin complex kit (Vector Laboratories, Peterborough, UK) and a reaction with 0.01% H2O2. Specificity of the antibody was assessed by pre-adsorption of the ACE-2 antibody with a 250-fold molar excess of recombinant human ACE-2 protein (R&D Systems).
Statistical analysis
Unpaired two-tailed t tests or analysis of variance (ANOVA) with Bonferroni’s post hoc analysis was used for comparisons between groups, and Pearson’s test was used to assess linear correlation with SPSS version 16 (SPSS, Chicago, IL, USA) and GraphPad Prism version 6 (GraphPad Software, La Jolla, CA, USA) software. P values <0.05 were considered statistically significant.
Results
ACE-2 enzyme activity is reduced in Alzheimer’s disease in association with increasing Aβ load and tau pathology
ACE-2 activity was significantly reduced by approximately 50% in the mid-frontal cortex in AD compared with age-matched controls (P < 0.0001) (Fig. 1a). ACE-2 varied according to disease severity when the controls and AD cases were grouped and stratified into the following Braak tangle stage groups: 0–II, III–IV, and V-VI (P < 0.0001 by ANOVA). Post hoc analysis using the Bonferroni correction for multiple comparisons revealed that ACE-2 activity was significantly reduced in Braak tangle stages V–VI compared with stages 0–II (P < 0.0001) and stages III–IV (P < 0.05) (Fig. 1b). No difference was observed between Braak stages 0–II and stages III–IV.
Fig. 1.
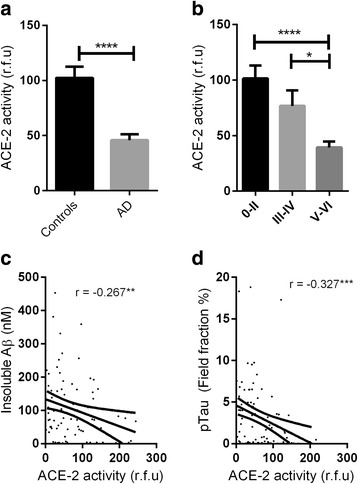
Angiotensin-converting enzyme 2 (ACE-2) activity is reduced in Alzheimer’s disease (AD). a Bar chart showing reduced ACE-2 activity in the mid-frontal cortex in AD (n = 90) compared with age-matched controls (n = 59) (P < 0.0001). b Bar chart showing reduced ACE-2 activity in relation to disease severity when all cases were combined and grouped according to Braak stage (0–II, II–IV, and V–VI) (P < 0.0001). Post hoc analysis revealed that ACE-2 activity was reduced in Braak tangle stages V–VI compared with stages 0–II and III-IV (P < 0.0001 and P < 0.05 respectively) and in Braak tangle stages III–IV compared with stages 0–II, but the difference was not statistically different. The bars indicate the mean value and SEM. c and d Scatterplots showing that ACE-2 activity was inversely correlated with insoluble amyloid-β (Aβ) load (measured by enzyme-linked immunosorbent assay) (r = −0.267, P < 0.01) and phosphorylated tau (p-tau) load (measured by field fraction analysis) (r = −0.327, P < 0.001). The solid inner line indicates the best-fit linear regression and the outer lines the 95% confidence intervals. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. rfu Relative fluorescence units
In a combined AD and control cohort, ACE-2 activity correlated inversely with total insoluble Aβ levels (r = −0.267, P < 0.01) (Fig. 1c) but not with soluble Aβ (data not shown). ACE-2 correlated inversely with β-secretase activity (r = −0.277 P < 0.001) (Additional file 3: Figure S2). ACE-2 correlated inversely with p-tau load (r = 0.327, P < 0.01) (Fig. 1d).
ACE-2 activity is reduced in relation to APOE and ACE1 polymorphisms and CAA severity
ACE-2 activity was significantly lower in individuals possessing an APOE ε4 allele, an established genetic risk factor for sporadic AD [49], than in those without (P < 0.05) (Fig. 2a). ACE-2 activity also differed significantly between ACE1 (rs1799752) indel genotypes (P < 0.05), with individuals who were homozygous II for ACE-1 (previously associated with increased risk for AD [17]) having the lowest ACE-2 activity, although post hoc analysis revealed that this did not reach statistical significance (Fig. 2b).
Fig. 2.
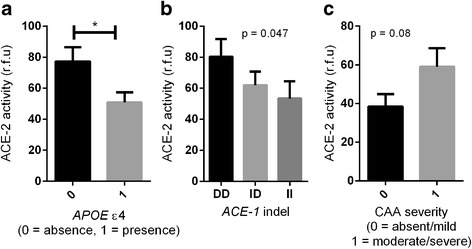
Angiotensin-converting enzyme 2 (ACE-2) activity is reduced in association with apolipoprotein E (APOE) ε4 and ACE1 (rs1799752) indel polymorphism and increased in cerebral amyloid angiopathy (CAA). a Bar chart showing reduced ACE-2 activity in individuals with an APOE ε4 allele (P < 0.05). b Bar chart showing that ACE-2 activity varied according to ACE1 indel polymorphism (P < 0.05), with a trend towards reduced ACE-2 activity in ACE-1 II homozygotes. c Bar chart showing elevated ACE-2 activity in moderate to severe CAA compared with absent to mild CAA, approaching significance (P = 0.08). The bars indicate the mean value and SEM. *P < 0.05. rfu Relative fluorescence units
We assessed ACE-2 activity in relation to CAA severity and found, as for ACE-1 activity [4], a tendency, although not significant, towards increased ACE-2 activity in cases with moderate to severe CAA compared with absent to mild CAA (P = 0.08) (Fig. 2c).
ACE-2 is inversely correlated with ACE-1, and the ratio of ACE-1 to ACE-2 is increased in Alzheimer’s disease
ACE-2 activity correlated inversely with ACE-1 activity in a combined AD and control cohort (r = −0.453, P > 0.0001) (Fig. 3a). The same pattern was observed and remained statistically significant when the control (r = −0.390, P < 0.05) and AD (r = −0.257, P < 0.05) groups were analysed separately.
Fig. 3.
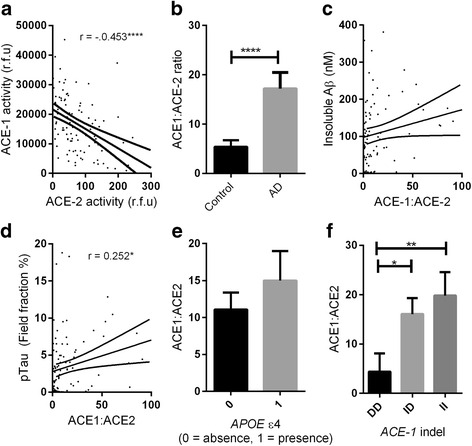
Angiotensin-converting enzyme 2 (ACE-2) activity is inversely correlated with ACE-1 activity, and the ACE-1/ACE-2 ratio is increased, in Alzheimer’s disease (AD). a Scatterplot showing a strong inverse relationship between ACE-1 and ACE-2 activity in mid-frontal cortex (r = −.453, P < 0.0001). The inner solid line indicates the best-fit linear regression and the outer lines the 95% confidence intervals. Each dot represents an individual brain. b Bar chart showing elevated ACE-1/ACE-2 ratio in AD (P < 0.0001). c and d Scatterplots showing positive correlation between the ACE-1/ACE-2 ratio and insoluble amyloid-β (Aβ) load (r = 0.199, P = 0.059) and p-tau load (r = 0.252, P < 0.05). e Bar chart showing a trend towards increased ACE-1/ACE-2 ratio in individuals who possessed an apolipoprotein E (APOE) ε4 allele. f Bar chart showing lower ACE:ACE-2 ratio in individuals who were homozygous DD for the ACE1 (rs1799752) indel polymorphism compared with II (P < 0.01) and ID (P < 0.05). The bars indicate the mean value and SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. rfu Relative fluorescence units
Previous reports have suggested the ratio of ACE-1 to ACE-2 is a good proxy measure for the activation status of classical and regulatory RAS pathways [33]. With this in mind, we calculated the ACE-1/ACE-2 ratio for all cases and found that it was significantly increased in AD compared with controls (P > 0.0001) (Fig. 3b). The ACE-1/ACE-2 ratio also correlated positively with insoluble Aβ level, approaching significance (r = 0.199, P = 0.059) (Fig. 3c), and significantly with p-tau (r = 0.252, P < 0.05) (Fig. 3d). The ACE-1/ACE-2 ratio was increased in individuals possessing an APOE ε4 allele, approaching significance (P = 0.093) (Fig. 3e), and differed significantly according to ACE1 (rs1799752) indel polymorphism (P < 0.01). Post hoc analysis revealed that the ratio was significantly higher in individuals with ACE1 II (AD risk factor) than in DD (P < 0.01) and in ID than in DD (P < 0.05) (Fig. 3f).
Ang II/Ang (1-7) ratio is increased in AD
Ang II levels were significantly increased in mid-frontal cortex in AD compared with age-matched controls (P < 0.0001) (Fig. 4a), whereas Ang (1–7) levels were unchanged (Fig. 4b). We calculated the Ang II/Ang (1–7) ratio (as a proxy indicator of ACE-2 activity) and found that the Ang II/Ang (1–7) ratio was significantly increased in AD (P > 0.001) (Fig. 4c). These data indicate that the conversion of Ang II to Ang (1–7) is likely to be reduced in AD because of lower ACE-2 activity.
Fig. 4.
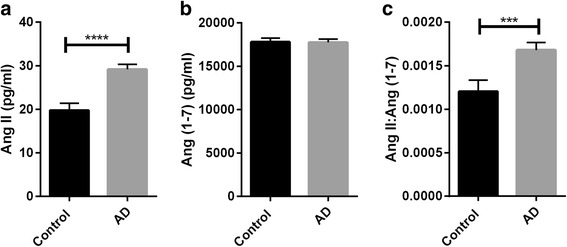
The ratio of angiotensin II (Ang II) to angiotensin (1–7) (Ang (1-7)) (a proxy measure of ACE-2 activity) is increased, indicating reduced conversion of Ang II to Ang (1–7) in Alzheimer’s disease (AD). Bar charts showing a elevated Ang II levels in AD and b unchanged Ang (1–7) levels in AD compared with age-matched controls in mid-fontal cortex. c Bar chart showing the Ang II/Ang (1–7) ratio was significantly increased in AD (P < 0.001). The bars indicate the mean value and SEM. ***P < 0.001, ****P < 0.0001
ACE-2 expression in human brain tissue
ACE-2 was localised primarily to capillaries but also had a perivascular distribution around larger arterioles (Fig. 5a). ACE-2 labelled non-vascular cells that strongly resembled astrocytes (Fig. 5b and c). Labelling was not observed with pre-adsorption of the ACE-2 antibody with recombinant human ACE-2, demonstrating specificity of the antibody (Fig. 5d).
Fig. 5.
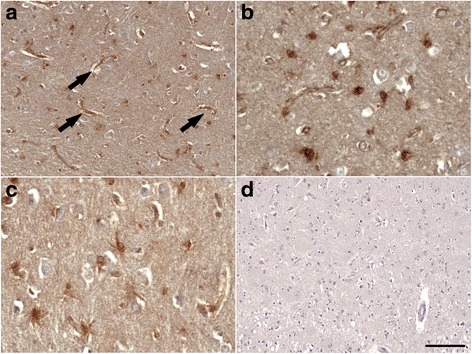
Angiotensin-converting enzyme 2 (ACE-2) expression in mid-frontal cortex in Alzheimer’s disease. a and b ACE-2 displayed strong capillary labelling (black arrows) and abundant perivascular labelling of larger arterioles (scale bar = 100 μm). Shown in b at higher magnification (scale bar = 50 μm). b and c ACE-2 was present in astrocytes (scale bar = 50 μm). d Pre-adsorption of ACE-2 antibody with recombinant human ACE-2 abolished labelling, confirming antibody specificity (scale bar = 100 μm)
Discussion
In the present study, we show that ACE-2 activity is reduced in post-mortem brain tissue in AD in association with increased Aβ and tau pathology. The reduction in ACE-2 was more pronounced in individuals carrying an APOE ε4 allele and in those who were homozygous II for the ACE1 (rs1799752) indel polymorphism (both of which are suggested genetic risk factors for AD [17]). ACE-2 activity correlated inversely with ACE-1 activity (which we have previously shown to be increased in AD [4, 5]), and the ACE-1/ACE-2 ratio was higher in AD. Together, these data strongly suggest that reduced ACE-2 activity within the brain contributes to AD pathogenesis and is associated with increased activation of the central classical RAS axis.
The brain has its own intrinsic RAS [50–52], and we have shown in our previous studies that ACE-1, the rate-limiting enzyme in the production of Ang II, is overactive in AD [4, 5]. It is widely accepted that Ang II-mediated signalling via AT1R (commonly termed the classical axis) is overactive in AD and is associated with AD pathogenesis (reviewed in [1]). This view has been supported in various animal studies in which infusion of Ang II resulted in elevated plaque and tau pathology and significant cognitive impairment [2, 3]. Secondary observations in clinical trials and epidemiological studies have provided further evidence that RAS-targeting drugs that either block the production of Ang II or prevent AT1R-mediated signalling reduce the prevalence of AD [12–16], while cognitive performance is improved and pathology reduced, in animal models of AD [6–11]. Until recently, the prevailing view of the RAS in AD has been oversimplified because it has failed to consider the contribution of the other downstream RAS regulatory pathways within the brain.
In this study, we found reduced brain ACE-2 activity in AD, which supports a recent study showing lower peripheral serum ACE-2 levels in AD [37]. ACE-2 activity correlated inversely with parenchymal Aβ load and increased p-tau levels. We also observed a strong inverse relationship between ACE-2 and β-secretase activity, suggesting that ACE-2 may contribute in some way to regulating the amyloidogenic processing of APP. There are several possible mechanisms that link reduced ACE-2 activity to the pathogenesis of AD. Firstly, lower ACE-2 activity will, via a lower conversion of Ang II to Ang (1–7), result in elevated Ang II levels (as we have shown in this study). An increase in Ang II/Ang (1–7) ratio has commonly been reported in other chronic conditions associated with overactivation of the central axis [53]. Secondly, ACE-2 is primarily responsible for generating Ang (1–7) from Ang II [24, 54, 55], and subsequent Ang (1–7) activation of the Mas receptor counter-regulates the detrimental effects of the classical (ACE-1/Ang II/AT1R) axis [56–58] and has been linked with enhancing learning and memory processing [59, 60]. Lastly, ACE-2 has recently been shown to convert Aβ43, a highly amyloidogenic form of Aβ that seeds plaque formation [38], to Aβ42, which in turn is cleaved by ACE-1 to Aβ40 or, to a lesser extent, Aβ41, which have reduced toxicity [37]. Lower ACE-2 activity in AD may therefore promote the early deposition of Aβ43 and prevent downstream cleavage of Aβ42 by ACE-1.Together, these data suggest a putative protective role of the ACE-2/Ang (1–7)/Mas pathway, not only against the development of pathology but also against the decline in cognitive function, that is lost in AD.
Our findings indicate that the balance between the classical (ACE-1/Ang II/AT1R) axis and regulatory (ACE-2/Ang (1–7)/Mas) axis of RAS is disturbed in AD, as previously shown in various mouse models of cardiovascular disease [33] and diabetic nephropathy [53]. ACE-2 activity is reduced in AD and is inversely correlated with increasing ACE-1 activity, and the ACE-1/ACE-2 ratio is increased in AD in association with disease pathology. These findings support commonly observed traits in cardiac and renal pathologies showing that dysregulation of the ACE-2/Ang (1–7)/Mas pathway, including reduced ACE-2 activity, is associated with sustained hypertension mediated by overactivation of the classical axis (reviewed in [30, 61]). Despite the ratio of Ang II to Ang (1–7) (a proxy measure of ACE-2 activity) being increased in AD (i.e., reduced conversion of Ang II to Ang (1-7)), we did not observe an overall reduction in total Ang (1–7) in AD. This is inconsistent with a recent report showing reduced serum Ang (1–7) levels, rather than reduced ACE-2 activity, in senescence-accelerated mouse prone 8, a mouse model of sporadic AD (involving overexpression of APP). The authors observed that Ang (1–7) levels correlated inversely with Ang II and p-tau levels [39]. The reason for the discrepant findings between human and mouse brain tissue is unclear; however, both studies indicate that the ACE-2/Ang (1–7)/Mas pathway is dysregulated in AD and that further work is required to determine the exact contribution of each component of the pathway in AD.
Activation of the ACE-2/Ang (1–7)/Mas pathway, by inducing ACE-2 activity, or infusion of Ang (1–7) or a Mas receptor agonist, is protective in various experimental animal models of cardiovascular disease and is associated with a reduction of the classical RAS pathway (reviewed in [32, 61]). Neuronal overexpression of brain ACE-2 is also neuroprotective in a chronic hypertension mouse model (transgenic for renin and angiotensinogen that overproduces Ang II) following experimental induction of ischaemic stroke [34, 35, 62]. These protective effects were partially reversed in the presence of a Mas receptor antagonist, demonstrating the specificity of the ACE-2/Ang (1–7)/Mas pathway, and they have been shown to be mediated by counter-regulating the effects of Ang II-mediated reactive oxygen species production [63]. In AD, there is growing recognition that re-positioning of brain-penetrating ARBs and ACEIs may have clinical benefits in AD [64]. In addition to reducing the central pool of Ang II, ARBs and ACEIs might also exert their protective effects by preventing AT1R-mediated reduction in ACE-2 activity [65] that can be reversed by ARBs [27, 66–69]. ACE-2 activation is also associated with reduced ACE-1 activity [70] and with down-regulation of Ang II levels and AT1R expression [27, 65, 71–73]. These studies suggest that activation of ACE-2 may exert protective effects in AD above and beyond dampening RAS activation that the use of ACEIs and ARBs currently allow.
Lastly, we explored the distribution of ACE-2 within the mid-frontal and temporal cortices and found it to be localised predominantly within endothelial cells and smooth muscle cells of cerebral arteries, as previously reported [25]. Interestingly, as for ACE-1, we also observed extensive perivascular ACE-2 expression and found that ACE-2 activity was increased in individuals with moderate to severe CAA, as has previously been shown for ACE-1 [4]. We speculate that the sequential cleavage of Aβ43, first by ACE-2, and the subsequent cleavage of Aβ42 to Aβ40 (the predominant species in CAA [74]) by ACE-1, provides a potential mechanistic link with CAA. Further studies are required to determine the relationship between ACE-2 and CAA severity.
Conclusions
These data indicate that reduced activity of the ACE-2/Ang (1–7)/Mas axis is strongly linked to overactivity of the classical RAS pathway and with AD-related pathology.
Acknowledgements
We acknowledge Professor Seth Love, University of Bristol, for his academic input and neuropathological assessment.
Funding
This work was supported by Alzheimer’s Research UK (ART-PG2011-1). The South West Dementia Brain Bank is part of the Brains for Dementia Research program, jointly funded by Alzheimer’s Research UK and the Alzheimer’s Society, and is supported by Bristol Research into Alzheimer’s and Care of the Elderly (BRACE) and the Medical Research Council.
Availability of data and material
All data within the article is linked to the MRC UK-BBN by a unique numeric MRC UK-BBN identifier (Additional file 2: Figure S1). There is no risk of disclosure of personal information, because all of the information held within the database has been anonymised.
Authors’ contributions
JSM carried out the angiotensin-II measurements and validated the ACE-2 activity measurements, performed the statistical analysis and was primarily responsible for drafting and finalizing the manuscript. SW carried out the ACE-2 activity measurements, performed statistical analysis and helped to draft the manuscript. NAM carried out the angiotensin (1–7) measurements, performed statistical analysis and helped draft the manuscript. LEP carried out the ACE-2 immunolabelling and analysis and revised the manuscript. PGK conceived and was responsible for overall planning and design of the study, and helped to revise and finalize the manuscript. All authors read and approved the final manuscript.
Authors’ information
All authors are members of the Dementia Research Group, Clinical Neurosciences, School of Clinical Sciences, University of Bristol, Bristol, UK.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The use of human brain tissue for this study was approved by the management committee of the South West Dementia Brain Bank (Human Tissue Authority licence number 12273) under the terms of Bristol Research Ethics Committee approval of the brain bank (reference 08/H0106/28 + 5). All participants provided consent to post-mortem removal of whole brain and CSF and the retention of these for use in research. Consent included access to the donor’s medical records to collect information on past medical history relevant to the donation, but that in all publications this information would be anonymised.
Abbreviations
- ACE
Angiotensin-converting enzyme
- ACEI
Angiotensin-converting enzyme inhibitors
- AD
Alzheimer’s disease
- Ang (1–7)
Angiotensin (1–7) peptide
- Ang (1–9)
Angiotensin (1–9) peptide
- Ang II
Angiotensin II peptide
- ANOVA
Analysis of variance
- APOE
Apolipoprotein E
- APP
Amyloid precursor protein
- ARB
Angiotensin II type 1 receptor blocker
- AT1R
Angiotensin II type 1 receptor
- Aβ
Amyloid-β
- CAA
Cerebral amyloid angiopathy
- D/D ACE-1 (rs1799752)
Deletion/deletion polymorphism
- ELISA
Enzyme-linked immunosorbent assay
- FRET
Fluorescence resonance energy transfer
- I/D ACE-1 (rs1799752)
Insertion/deletion polymorphism
- I/I ACE-1 (rs1799752)
Insertion/insertion polymorphism
- Mca
7-Methoxycoumarin-4-yl-acetyl
- MRC
Medical Research Council
- MRC UK-BBN
Medical Research Council UK Brain Banks Network
- p-tau
Phosphorylated tau
- PM
Post-mortem
- RAS
Renin-angiotensin system
- rfu
Relative fluorescence units
- TMB
3,3′,5,5′-Tetramethylbenzidine
Additional files
MRC identifiers for all cases. (DOC 80 kb)
Scatterplot showing a strong positive correlation between two independent measures of ACE-2 activity in brain tissue samples. ACE-2 was measured using either a commercially available ACE-2 activity assay kit (SensoLyte® 390) or an ACE-2 fluorogenic peptide substrate (Mca-APK[Dnp]) in the presence of a selective ACE-2 inhibitor, MLN4760 (10 μM). The solid inner line indicates the best-fit linear regression, and the outer lines the 95% confidence intervals. Each point represents a separate brain. ****P < 0.0001. (TIF 26 kb)
Scatterplot showing an inverse relationship between ACE-2 activity and BACE-1 activity in a combined Alzheimer’s disease and age-matched control cohort. ACE-2 activity was measured using the SensoLyte® 390 ACE-2 activity assay kit, and BACE-1 activity was measured using the β-secretase specific fluorogenic substrate (Mca-SEVNLDAEFRK[Dnp]RR-NH2). The inner solid line indicates the best-fit linear regression, and the outer lines the 95% confidence intervals. Each point represents a separate brain. ***P < 0.001. (TIF 25 kb)
Contributor Information
Patrick Gavin Kehoe, Phone: +44 (0) 117 4147821, Email: Patrick.Kehoe@bristol.ac.uk.
Steffenny Wong, Email: sw0485@bristol.ac.uk.
Noura AL Mulhim, Email: na15725@bristol.ac.uk.
Laura Elyse Palmer, Email: laura.e.palmer@bristol.ac.uk.
J. Scott Miners, Phone: +44 (0) 117 4147818, Email: Scot.Miners@bristol.ac.uk.
References
- 1.Kehoe PG, Miners S, Love S. Angiotensins in Alzheimer’s disease - friend or foe? Trends Neurosci. 2009;32:619–28. doi: 10.1016/j.tins.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Zhu D, Shi J, Zhang Y, Wang B, Liu W, Chen Z, et al. Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLoS One. 2011;6:e16037. doi: 10.1371/journal.pone.0016037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian M, Zhu D, Xie W, Shi J. Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett. 2012;586:3737–45. doi: 10.1016/j.febslet.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Miners JS, Ashby E, Van Helmond Z, Chalmers KA, Palmer LE, Love S, et al. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2008;34:181–93. doi: 10.1111/j.1365-2990.2007.00885.x. [DOI] [PubMed] [Google Scholar]
- 5.Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E, et al. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res. 2009;1:163–77. [PMC free article] [PubMed] [Google Scholar]
- 6.Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, et al. Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. 2010;13:195–201. doi: 10.1089/rej.2009.0944. [DOI] [PubMed] [Google Scholar]
- 7.Dong YF, Kataoka K, Tokutomi Y, Nako H, Nakamura T, Toyama K, et al. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J. 2011;25:2911–20. doi: 10.1096/fj.11-182873. [DOI] [PubMed] [Google Scholar]
- 8.Ongali B, Nicolakakis N, Tong XK, Aboulkassim T, Papadopoulos P, Rosa-Neto P, et al. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis. 2014;68:126–36. doi: 10.1016/j.nbd.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, et al. Cognitive deficit in amyloid-β-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-γ activation. Hypertension. 2009;54:782–7. doi: 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, et al. Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada K, Uchida S, Takahashi S, Takayama M, Nagata Y, Suzuki N, et al. Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer’s disease. Brain Res. 2010;1352:176–86. doi: 10.1016/j.brainres.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis. 2011;26:699–708. doi: 10.3233/JAD-2011-110347. [DOI] [PubMed] [Google Scholar]
- 13.Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, et al. Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet. 1998;352:1347–51. doi: 10.1016/S0140-6736(98)03086-4. [DOI] [PubMed] [Google Scholar]
- 14.Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzourio C, Anderson C, Chapman N, Woodward M, Neal B, MacMahon S, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069–75. doi: 10.1001/archinte.163.9.1069. [DOI] [PubMed] [Google Scholar]
- 16.Kume K, Hanyu H, Sakurai H, Takada Y, Onuma T, Iwamoto T. Effects of telmisartan on cognition and regional cerebral blood flow in hypertensive patients with Alzheimer’s disease. Geriatr Gerontol Int. 2012;12:207–14. doi: 10.1111/j.1447-0594.2011.00746.x. [DOI] [PubMed] [Google Scholar]
- 17.Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C, et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet. 1999;21:71–2. doi: 10.1038/5009. [DOI] [PubMed] [Google Scholar]
- 18.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 19.Elkins JS, Douglas VC, Johnston SC. Alzheimer disease risk and genetic variation in ACE: a meta-analysis. Neurology. 2004;62:363–8. doi: 10.1212/01.WNL.0000106823.72493.FF. [DOI] [PubMed] [Google Scholar]
- 20.Kehoe PG, Katzov H, Feuk L, Bennet AM, Johansson B, Wiman B, et al. Haplotypes extending across ACE are associated with Alzheimer’s disease. Hum Mol Genet. 2003;12:859–67. doi: 10.1093/hmg/ddg094. [DOI] [PubMed] [Google Scholar]
- 21.Lehmann DJ, Cortina-Borja M, Warden DR, Smith AD, Sleegers K, Prince JA, et al. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am J Epidemiol. 2005;162:305–17. doi: 10.1093/aje/kwi202. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 23.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 24.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 25.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: a first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292:R373–81. doi: 10.1152/ajpregu.00292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher PE, Chappell MC, Ferrario CM, Tallant EA. Distinct roles for ANG II and ANG-(1–7) in the regulation of angiotensin-converting enzyme 2 in rat astrocytes. Am J Physiol Cell Physiol. 2006;290:C420–6. doi: 10.1152/ajpcell.00409.2004. [DOI] [PubMed] [Google Scholar]
- 28.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 29.Xia H, Lazartigues E. Angiotensin-converting enzyme 2: central regulator for cardiovascular function. Curr Hypertens Rep. 2010;12:170–5. doi: 10.1007/s11906-010-0105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107:1482–94. doi: 10.1111/j.1471-4159.2008.05723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu P, Sriramula S, Lazartigues E. ACE2/ANG-(1–7)/Mas pathway in the brain: the axis of good. Am J Physiol Regul Integr Comp Physiol. 2011;300:R804–17. doi: 10.1152/ajpregu.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang T, Gao L, Lu J, Zhang YD. ACE2-Ang-(1–7)-Mas axis in brain: a potential target for prevention and treatment of ischemic stroke. Curr Neuropharmacol. 2013;11:209–17. doi: 10.2174/1570159X11311020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos RAS, Ferreira AJ, Silva AC S e. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1–7)-Mas axis. Exp Physiol. 2008;93:519–27. doi: 10.1113/expphysiol.2008.042002. [DOI] [PubMed] [Google Scholar]
- 34.Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, et al. Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96:1084–96. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y. Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br J Pharmacol. 2012;167:1520–32. doi: 10.1111/j.1476-5381.2012.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawajiri M, Mogi M, Higaki N, Matsuoka T, Ohyagi Y, Tsukuda K, et al. Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult Scler. 2009;15:262–5. doi: 10.1177/1352458508097923. [DOI] [PubMed] [Google Scholar]
- 37.Liu S, Liu J, Miura Y, Tanabe C, Maeda T, Terayama Y, et al. Conversion of Aβ43 to Aβ40 by the successive action of angiotensin-converting enzyme 2 and angiotensin-converting enzyme. J Neurosci Res. 2014;92:1178–86. doi: 10.1002/jnr.23404. [DOI] [PubMed] [Google Scholar]
- 38.Zou K, Liu J, Watanabe A, Hiraga S, Liu S, Tanabe C, et al. Aβ43 is the earliest-depositing Aβ species in APP transgenic mouse brain and is converted to Aβ41 by two active domains of ACE. Am J Pathol. 2013;182:2322–31. doi: 10.1016/j.ajpath.2013.01.053. [DOI] [PubMed] [Google Scholar]
- 39.Jiang T, Zhang YD, Zhou JS, Zhu XC, Tian YY, Zhao HD, et al. Angiotensin-(1–7) is reduced and inversely correlates with tau hyperphosphorylation in animal models of Alzheimer’s disease. Mol Neurobiol. 2016;53:2489–97. doi: 10.1007/s12035-015-9260-9. [DOI] [PubMed] [Google Scholar]
- 40.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miners JS, van Helmond Z, Raiker M, Love S, Kehoe PG. ACE variants and association with brain Aβ levels in Alzheimer’s disease. Am J Transl Res. 2010;3:73–80. [PMC free article] [PubMed] [Google Scholar]
- 42.van Helmond Z, Miners JS, Kehoe PG, Love S. Higher soluble amyloid β concentration in frontal cortex of young adults than in normal elderly or Alzheimer’s disease. Brain Pathol. 2010;20:787–93. doi: 10.1111/j.1750-3639.2010.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, et al. The apolipoprotein E ε4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology. 1996;47:190–6. doi: 10.1212/WNL.47.1.190. [DOI] [PubMed] [Google Scholar]
- 44.Chalmers K, Wilcock GK, Love S. APOE ε4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A β protein. Neuropathol Appl Neurobiol. 2003;29:231–8. doi: 10.1046/j.1365-2990.2003.00457.x. [DOI] [PubMed] [Google Scholar]
- 45.Ballard CG, Chalmers KA, Todd C, McKeith IG, O’Brien JT, Wilcock G, et al. Cholinesterase inhibitors reduce cortical Aβ in dementia with Lewy bodies. Neurology. 2007;68:1726–9. doi: 10.1212/01.wnl.0000261920.03297.64. [DOI] [PubMed] [Google Scholar]
- 46.Chalmers KA, Wilcock GK, Vinters HV, Perry EK, Perry R, Ballard CG, et al. Cholinesterase inhibitors may increase phosphorylated tau in Alzheimer’s disease. J Neurol. 2009;256:717–20. doi: 10.1007/s00415-009-5000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love S, Nicoll JA, Hughes A, Wilcock GK. APOE and cerebral amyloid angiopathy in the elderly. Neuroreport. 2003;14:1535–6. doi: 10.1097/00001756-200308060-00027. [DOI] [PubMed] [Google Scholar]
- 48.Wenham PR, Price WH, Blandell G. Apolipoprotein E genotyping by one-stage PCR. Lancet. 1991;337:1158–9. doi: 10.1016/0140-6736(91)92823-K. [DOI] [PubMed] [Google Scholar]
- 49.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E ε4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–9. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 50.Wright JW, Harding JW. Brain renin-angiotensin—a new look at an old system. Prog Neurobiol. 2011;95:49–67. doi: 10.1016/j.pneurobio.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Wright JW, Harding JW. The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch. 2013;465:133–51. doi: 10.1007/s00424-012-1102-2. [DOI] [PubMed] [Google Scholar]
- 52.Phillips MI, de Oliveira EM. Brain renin angiotensin in disease. J Mol Med. 2008;86:715–22. doi: 10.1007/s00109-008-0331-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Padda RS, Shi Y, Lo CS, Zhang SL, Chan JS. Angiotensin-(1–7): a novel peptide to treat hypertension and nephropathy in diabetes? J Diabetes Metab. 2015;6:615. doi: 10.4172/2155-6156.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elased KM, Cunha TS, Marcondes FK, Morris M. Brain angiotensin-converting enzymes: role of angiotensin-converting enzyme 2 in processing angiotensin II in mice. Exp Physiol. 2008;93:665–75. doi: 10.1113/expphysiol.2007.040311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 56.Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1–7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289:H2281–90. doi: 10.1152/ajpheart.00618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Santos RA, Campagnole-Santos MJ, Andrade SP. Angiotensin-(1–7): an update. Regul Pept. 2000;91:45–62. doi: 10.1016/S0167-0115(00)00138-5. [DOI] [PubMed] [Google Scholar]
- 58.Santos RAS, Silva AC S e, Maric C, Silva DMR, Machado RP, de Buhr I, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–63. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hellner K, Walther T, Schubert M, Albrecht D. Angiotensin-(1–7) enhances LTP in the hippocampus through the G-protein-coupled receptor Mas. Mol Cell Neurosci. 2005;29:427–35. doi: 10.1016/j.mcn.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 60.Lazaroni TL, Raslan AC, Fontes WR, de Oliveira ML, Bader M, Alenina N, et al. Angiotensin-(1–7)/Mas axis integrity is required for the expression of object recognition memory. Neurobiol Learn Mem. 2012;97:113–23. doi: 10.1016/j.nlm.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 61.Feng Y, Xia H, Santos RA, Speth R, Lazartigues E. Angiotensin-converting enzyme 2: a new target for neurogenic hypertension. Exp Physiol. 2010;95:601–6. doi: 10.1113/expphysiol.2009.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J, Zhao Y, Chen S, Wang J, Xiao X, Ma X, et al. Neuronal over-expression of ACE2 protects brain from ischemia-induced damage. Neuropharmacology. 2014;79:550–8. doi: 10.1016/j.neuropharm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng J, Li G, Chen S, Bihl J, Buck J, Zhu Y, et al. Activation of the ACE2/Ang-(1–7)/Mas pathway reduces oxygen-glucose deprivation-induced tissue swelling, ROS production, and cell death in mouse brain with angiotensin II overproduction. Neuroscience. 2014;273:39–51. doi: 10.1016/j.neuroscience.2014.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashby EL, Kehoe PG. Current status of renin-aldosterone angiotensin system-targeting anti-hypertensive drugs as therapeutic options for Alzheimer’s disease. Expert Opin Investig Drugs. 2013;22:1229–42. doi: 10.1517/13543784.2013.812631. [DOI] [PubMed] [Google Scholar]
- 65.Xia H, Feng Y, Obr TD, Hickman PJ, Lazartigues E. Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension. 2009;53:210–6. doi: 10.1161/HYPERTENSIONAHA.108.123844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chappel MC, Ferrario CM. ACE and ACE2: their role to balance the expression of angiotensin II and angiotensin-(1–7) Kidney Int. 2006;70:8–10. doi: 10.1038/sj.ki.5000321. [DOI] [PubMed] [Google Scholar]
- 67.Ferrario CM, Varagic J. The ANG-(1–7)/ACE2/Mas axis in the regulation of nephron function. Am J Physiol Renal Physiol. 2010;298:F1297–305. A published erratum appears in Am J Physiol Renal Physiol. 2010;299:F1515. doi: 10.1152/ajprenal.zh2-6109-corr.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Igase M, Kohara K, Nagai T, Miki T, Ferrario CM. Increased expression of angiotensin converting enzyme 2 in conjunction with reduction of neointima by angiotensin II type 1 receptor blockade. Hypertens Res. 2008;31:553–9. doi: 10.1291/hypres.31.553. [DOI] [PubMed] [Google Scholar]
- 69.Iwanami J, Mogi M, Tsukuda K, Wang XL, Nakaoka H, Ohshima K, et al. Role of angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis in the hypotensive effect of azilsartan. Hypertens Res. 2014;37:616–20. doi: 10.1038/hr.2014.49. [DOI] [PubMed] [Google Scholar]
- 70.Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res. 2011;92:401–8. doi: 10.1093/cvr/cvr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol. 2010;108:923–32. doi: 10.1152/japplphysiol.00840.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiao L, Gao L, Lazartigues E, Zucker IH. Brain-selective overexpression of angiotensin-converting enzyme 2 attenuates sympathetic nerve activity and enhances baroreflex function in chronic heart failure. Hypertension. 2011;58:1057–65. doi: 10.1161/HYPERTENSIONAHA.111.176636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, et al. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res. 2008;102:729–36. doi: 10.1161/CIRCRESAHA.107.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, et al. Amyloid β protein (Aβ) in Alzheimer’s disease brain: biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43) J Biol Chem. 1995;270:7013–6. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]