

Abstract
Background
Cultivated flax (Linum usitatissimum L.) is widely used for production of textile, food, chemical and pharmaceutical products. However, various stresses decrease flax production. Search for genes, which are involved in stress response, is necessary for breeding of adaptive cultivars. Imbalanced concentration of nutrient elements in soil decrease flax yields and also results in heritable changes in some flax lines. The appearance of Linum Insertion Sequence 1 (LIS-1) is the most studied modification. However, LIS-1 function is still unclear.
Results
High-throughput sequencing of transcriptome of flax plants grown under normal (N), phosphate deficient (P), and nutrient excess (NPK) conditions was carried out using Illumina platform. The assembly of transcriptome was performed, and a total of 34924, 33797, and 33698 unique transcripts for N, P, and NPK sequencing libraries were identified, respectively. We have not revealed any LIS-1 derived mRNA in our sequencing data. The analysis of high-throughput sequencing data allowed us to identify genes with potentially differential expression under imbalanced nutrition. For further investigation with qPCR, 15 genes were chosen and their expression levels were evaluated in the extended sampling of 31 flax plants. Significant expression alterations were revealed for genes encoding WRKY and JAZ protein families under P and NPK conditions. Moreover, the alterations of WRKY family genes differed depending on LIS-1 presence in flax plant genome. Besides, we revealed slight and LIS-1 independent mRNA level changes of KRP2 and ING1 genes, which are adjacent to LIS-1, under nutrition stress.
Conclusions
Differentially expressed genes were identified in flax plants, which were grown under phosphate deficiency and excess nutrition, on the basis of high-throughput sequencing and qPCR data. We showed that WRKY and JAS gene families participate in flax response to imbalanced nutrient content in soil. Besides, we have not identified any mRNA, which could be derived from LIS-1, in our transcriptome sequencing data. Expression of LIS-1 flanking genes, ING1 and KRP2, was suggested not to be nutrient stress-induced. Obtained results provide new insights into edaphic stress response in flax and the role of LIS-1 in these process.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-016-0927-9) contains supplementary material, which is available to authorized users.
Keywords: Linum usitatissimum, Flax, Edaphic stress, Phosphate deficiency, Excess nutrition, LIS-1, WRKY, JAZ, qPCR, High-throughput sequencing
Background
Cultivated flax (Linum usitatissimum L.) is widely used for production of textile, chemicals, food, and pharmaceutical products. However, various biotic and abiotic stresses decrease flax oil and fiber yield. Therefore, cultivation of resistant to unfavorable environment L. usitatissimum varieties is required for obtaining of high and stable yield. Understanding of mechanisms of flax stress resistance is necessary for breeding of cultivars, which are tolerant to stress conditions. Search for genes, which are involved in stress response, is important for progress in genetics of tolerance and genomics-assisted breeding of flax. Edaphic stresses, including deficient or excessive nutrition, significantly limit the production of flax [1], and is of particular interest for investigation. Moreover, imbalanced concentration of nutrient elements in soil result in heritable phenotype and genotype changes in some flax lines, which were called plastic [2–9]. Offspring of plastic line, which stably inherited the changes, were termed genotrophs [7]. The most famous flax plastic line is ‘Stormont cirrus’ and the most studied genetic modification is Linum Insertion Sequence 1 (LIS-1). LIS-1 is a 5.7-kb sequence that appear into a particular site of flax genome under nutrient deficiency, however, LIS-1 function is still unclear [2–4, 10, 11].
In the present work, we investigated the impact of soil nutrient stress on flax plants and compared the responses to nutrient deficiency or excess in line ‘Stormont cirrus’ with and without LIS-1 and non-plastic cultivar ‘Bethune’. High-throughput sequencing and quantitative RCR (qPCR) were used for gene expression evaluation in flax plants and allowed us to identify genes, which are likely involved in response to unfavorable nutrient content in soil.
Methods
Plant material
L. usitatissimum plants of cultivar ‘Bethune’ and line ‘Stormont Cirrus’ with and without LIS-1 were grown under normal (N), phosphate deficiency (P) and excess nutrition (NPK) conditions as described earlier [11]. Plant material was obtained from nine plants of ‘Bethune’ (three plants were grown under N conditions, 3 – under P, and 3 – under NPK), ten plants of ‘Stormont Cirrus’ with stable presence of LIS-1 (3 – N, 3 – P, and 4 – NPK), and 12 plants of ‘Stormont Cirrus’ with stable absence of LIS-1 (4 – N, 4 – P, 4 – NPK). Upper leaves from individual plants after 6 weeks of growth were collected and immediately frozen in liquid nitrogen. Plant samples were stored at −70 °C. Total DNA was isolated using chloroform:isoamyl alcohol extraction as described earlier [12]. Total RNA was extracted using RNA MicroPrep kit (Zymo Research, USA). The LIS-1 status of each individual plant was tested by PCR with primers of Chen et al. [3] and agarose gel electrophoresis as described earlier [11].
Transcriptome sequencing
CDNA library preparation was performed using TruSeq RNA SamplePrep (Illumina, USA). Poly-A containing mRNA fraction was isolated using poly-T oligoattached magnetic beads. Pooled samples were used to prepare three libraries for line ‘Stormont Cirrus’ grown under N, P, and NPK conditions. Library quality was evaluated using Agilent 2100 Bioanalyzer (Agilent Technologies, USA), then libraries were sequenced on Illumina GAIIx platform. Raw reads were trimmed and filtered with trimmomatic [13]. The assembly of transcriptome was performed using SOAPdenovo-Trans transcriptome assembler. Assembled transcripts were mapped to L. usitatissimum ‘reference’ transcriptome database of JGI Genome portal with blastn (PhytozomeV11: Lusitatissimum_200_v1.0.transcript.fa.gz; [14]). If several assembled transcripts were mapped to one ‘reference’ transcript, then we merged them into one transcript and filled gaps from the ‘reference’ sequence. Next we transferred annotations from ‘reference’ dataset to the assembled transcripts. For the further analysis, only mapped transcripts were used that made assembly more reliable.
To assess differential expression, reads were aligned to the assembled transcripts with bowtie2 [15] and read count per transcript values were calculated with rsem [16]. Library normalization was performed with total number of reads. The comparison of mRNA levels in N, P, and NPK libraries was carried out using fold change (normalized read count in NPK or P/normalized read count in N) and p-value (χ 2 test) parameters [17].
Gene set enrichment analysis with Gene Ontology data was performed using Goseq for genes with the highest fold changes (http://bioconductor.org/packages/release/bioc/html/goseq.html).
For identification of mRNAs derived from LIS-1 or its 5′ and 3′ flanking sequences, obtained Illumina transcriptomic reads were aligned to GenBank accessions AF104351.1, AF300798.1, and AF300797.1 using blastn [18].
QPCR analysis
PCR reactions were performed using a 7500 Real-Time PCR System (Applied Biosystems, USA) in 20 μl reaction mix containing 1× 2-FRT PCR mix (Amplisens, Russia), 250 nM of dNTPs mix (Fermentas, Lithuania), 350 nM of forward and reverse primers, 2 U of TaqF polymerase (Amplisens), 1× EvaGreen (Biotium, USA), and cDNA. The following amplification program was used: 95 °C for 15 min; 50 cycles of 95 °C for 15 s, 60–62 °C for 60 s. In total, nine flax plants of cultivar ‘Bethune’, ten plants of line ‘Stormont Cirrus’ with LIS-1 and 12 plants without LIS-1, which were grown under N (ten plants), P (10 plants), and NPK (11 plants) conditions, were studied. Expression of 15 genes was evaluated with primers, which were selected using NCBI Primer Blast [19] preferably with amplicon spanning exon-exon junction (see Additional file 1: Table S1). For qPCR data analysis, reference genes ETIF3H and ETIF3E [20] were used. All calculations were performed using the program ATG (Analysis of Transcription of Genes) [21]. The ΔC t eff values, which are directly proportional to the expression levels, were calculated according to the formula [11]:
where Ct, replicate-averaged threshold cycle; E, efficiency of reaction for each pair of primers. Each qPCR reaction was repeated three times. Amplification efficiency was 95 % or higher, threshold cycle varied from 23 to 34 for all primer pairs (Additional file 1: Table S1).
Kruskal–Wallis and Mann-Whitney tests were used for assessment of statistical significance of revealed expression alterations. P ≤ 0.05 were considered statistically significant.
Results
High-throughput sequencing of N, P, and NPK flax plant libraries
Three transcriptome libraries were constructed from flax plants grown under normal (N), phosphate deficient (P), and nutrient excess (NPK) conditions. High-throughput sequencing using GAIIx Illumina platform generated a total 10.6, 13.6, and 18.6 million reads (75 nucleotides in length) for N, P, and NPK conditions, respectively (Sequence Read Archive – SRP083007). All three libraries demonstrated very similar statistics: GC content was equal to 47–48 %, average quality per read was about 38, PCR sequence duplication level varied from 15 to 17 %. After mapping to the reference L. usitatissimum transcriptome (JGI Genome portal), joining hits and filling gaps, 34924, 33797, and 33698 unique transcripts were identified for N, P, and NPK libraries, respectively. The maximum transcript length was 14619 nucleotides.
Gene expression analysis on the basis of high-throughput sequencing data
The number of reads aligned to each assembled transcript was determined and normalized per 10 million reads. For the visualization of expression analysis results, the diagrams were constructed (Fig. 1). Gene Ontology enrichment analysis was performed for top 30 differentially expressed genes. Both under P and NPK conditions, expression of the majority of genes from the top 30 was decreased. Growing of flax plants under phosphate deficiency resulted mainly in the decrease of expression of transcription factors (p = 2.4*10−7). Thus, transcription factors could play an important role in flax adaptation to nutrition stress that is in concordance with data on the role of transcription factors in plant tolerance to abiotic stresses [22]. Under P conditions, expression alterations of genes, which participate in histone acetyltransferase activity (p = 2.9*10−5), sequence-specific DNA binding (p = 8.0*10−5), asparagine biosynthetic process (p = 2.1*10−3), protein binding process (p = 4.8*10−3), zinc ion binding (p = 0.03), and methyltransferase activity (p = 0.07) was also revealed. Under NPK conditions, differentially expression was observed for genes, which participate in histone acetyltransferase activity (p = 4.6*10−4), iron ion binding (p = 4.6*10−4), transcription cofactor activity (p = 4.6*10−4), oxidoreductase activity (p = 2.6*10−3), protein binding (p = 3.4*10−3), ion channel activity (p = 0.02), etc.
Fig. 1.
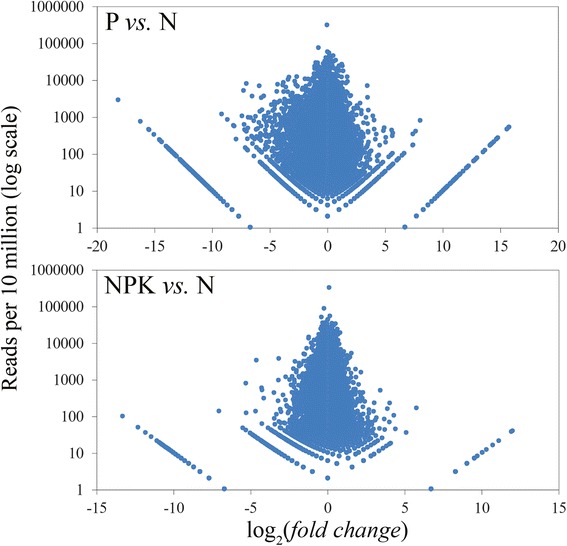
Relative and absolute expression levels of genes in plants grown under phosphate deficiency (P vs. N) or excess nutrition (NPK vs. N). High-throughput sequencing data. Each point represents the data for one gene. Y-axis – the sum of normalized reads in normal (N) and nutrient stress (P or NPK) conditions
LIS-1 derived RNAs
For identification of mRNAs derived from LIS-1, its 5′ and 3′ flanking sequences, obtained reads were aligned to GenBank accession AF104351.1, AF300797.1, and AF300798.1, respectively. For LIS-1 5′ and 3′ flanking sequences, coincidence was revealed for regions, which are corresponded to exons of Inhibitor of growth 1 (ING1) and Kip-related cyclin-dependent kinase inhibitor 2 (KRP2) genes (Fig. 2). Expression analysis on the basis of our high-throughput sequencing data revealed no difference in expression level of KRP2 gene under N, P, and NPK conditions. A slight increase of expression was observed for ING1 gene under P conditions. For LIS-1, we identified only few read k-mers, which coincided with short regions of the sequence. Moreover, matched k-mers were identified in both sample with LIS-1 and samples without LIS-1. Therefore, we concluded that no reads were derived from LIS-1.
Fig. 2.
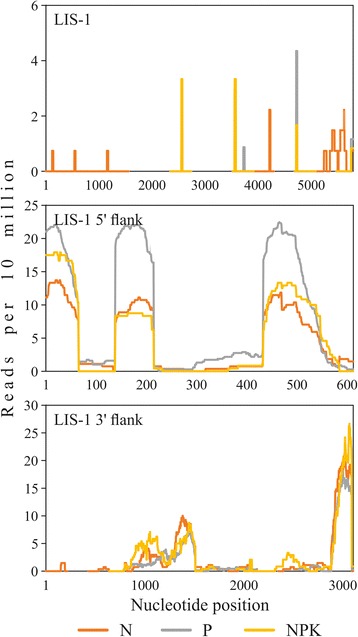
The coverage of LIS-1, LIS-1 5′ and 3′ flanking sequences. High-throughput sequencing data. N – normal conditions, P – phosphate deficiency, NPK – excess nutrition. GenBank accessions: LIS-1 – AF104351.1, LIS-1 5′ flank – AF300797.1, LIS-1 3′ flank – AF300798.1
QPCR analysis of gene expression in flax plants grown under imbalanced nutrition
We used high-throughput sequencing data for three pooled plant samples grown under different nutrition conditions for preliminary evaluation of gene expression. For reliable evaluation of expression alterations, we performed qPCR analysis in the extended sampling (ten flax plants grown under N, 11 – under P, and 10 – under NPK conditions). On the basis of high-throughput sequencing data, 20 genes with up- or down-regulation under nutrition stress were chosen. Twenty pairs of primers for qPCR were designed and tested, but only 13 of them showed high efficiency and specificity. QPCR analysis was performed for plants of line ‘Stormont cirrus’ with and without LIS-1 and cultivar ‘Bethune’ to evaluate the expression of genes encoding the following proteins: WRKY DNA-binding protein 70 (WRKY70); WRKY DNA-binding protein 33 (WRKY33); WRKY DNA-binding protein 40 (WRKY40); JAZ8/TIFY5A jasmonate-zim-domain protein 8 (JAZ8); JAS1, JAZ10, TIFY9 jasmonate-zim-domain protein 10 (JAZ10); Putative nuclease HARBI1 (HARBI1); S-adenosyl-L-methionine-dependent methyltransferase (SAM MTase); Neutral invertase (A/N-Invs); CCR4-associated factor 1 homolog 11 (CAF1-11); BTB and TAZ domain protein 4 (BT4); MLP-like protein 423 (MLP423); Ethylene-responsive transcription factor ERF (ERF); Myb domain protein 41 (MYB41). Besides, expression of ING1 and KRP2 genes, which are located in LIS-1 flanking sequences, was evaluated.
Significant expression alterations were revealed for genes encoding WRKY DNA-binding protein family and genes encoding JAZ jasmonate-zim-domain protein family in ‘Stormont cirrus’ plants with and without LIS-1 as well as in ‘Bethune’ plants grown under different nutrient conditions (Fig. 3).
Fig. 3.

Expression of five genes in plants of cultivar ‘Bethune’ and line ‘Stormont Cirrus’ with (+) and without (-) LIS-1 grown under phosphate deficiency (P), excess nutrition (NPK), or normal (N) conditions. QPCR data. Rectangles correspond to the ranges containing 50 % of the values (between the 25th and 75th percentiles); the bars are the maximum and minimum ΔC t eff values
For JAZ gene family, the reaction on different nutrition conditions was similar in the majority of groups of samples – expression was increased under P compared to NPK conditions. Median of for JAZ10 was −3.4 under P and −5.1 under NPK in ‘Stormont cirrus’ with LIS-1; −3.5 under P and −5.5 under NPK in ‘Stormont cirrus’ without LIS-1; −3.7 under P and −5.6 under NPK in ‘Bethune’. Median of for JAZ8 was −5.9 under P and −5.7 under NPK in ‘Stormont cirrus’ with LIS-1; −4.6 under P and −7.2 under NPK in ‘Stormont cirrus’ without LIS-1; −3.7 under P and −7.4 under NPK in ‘Bethune’. JAZ family proteins are repressors of jasmonic acid, which regulates many aspects of plant growth, development and stress responses [23]. Our results suggest that JAZ family proteins are involved in response to deficient and excessive nutrition in flax.
For WRKY gene family, expression was also increased under P compared to NPK conditions, but only in genotypes without LIS-1. Median of for WRKY70 was −0.4 under P and −2.3 under NPK in ‘Stormont cirrus’ without LIS-1; −1.9 under P and −4.3 under NPK in ‘Bethune’. Median of for WRKY33 was −0.7 under P and −2.9 under NPK in ‘Stormont cirrus’ without LIS-1; −1.5 under P and −2.7 under NPK in ‘Bethune’. Median of for WRKY40 was −0.5 under P and −2.9 under NPK in ‘Stormont cirrus’ without LIS-1; −1.0 under P and −3.4 under NPK in ‘Bethune’. For ‘Stormont cirrus’ with LIS-1 the trend was reverse: median of for WRKY70 was −3.6 under P and −2.8 under NPK; for WRKY33 – –2.0 under P and −1.4 under NPK; for WRKY40 – −2.0 under P and −1.3 under NPK. Proteins of WRKY family are transcription factors, which are involved in many processes in plants including stress response [24]. On the basis of our data, we can speculate that the genes of this family participate in flax response to imbalanced nutrition. It is worth noting that expression alterations of WRKY family genes differed depending on LIS-1 presence in flax plant genome.
For the other examined genes, statistically significant expression alterations were revealed only for particular groups of samples (Additional file 2: Figure S1). Expression was altered in ‘Stormont cirrus’ with LIS-1 for genes encoding Putative nuclease HARBI1 (median of was −5.2 under P, −3.8 under NPK, and −2.8 under N), MLP423 (−4.2 under P, −2.9 under NPK, and −4.8 under N), ING1 (−3.4 under P and −2.4 under NPK), A/N-Invs (−1.5 under P and 0.2 under NPK), BT4 (−4.2 under P and −3.0 under NPK); in ‘Stormont cirrus’ without LIS-1 – for genes encoding ING1 (−4.2 under P and −3.2 under N), BT4 (−4.6 under P and −3.1 under N), KRP2 (−1.8 under P, −2.4 under NPK, and −2.0 under N), and ERF (−3.4 under P, −7.2 under NPK, and −5.7 under N); in ‘Bethune’ – only for MYB41 (−1.8 under P, −3.7 under NPK, and −2.6 under N). For genes encoding CAF1-11 and SAM MTase, significant expression alterations were not revealed. Thus, the majority of expression alterations of listed above genes under imbalanced nutrition were observed for plastic line ‘Stormont cirrus’, while in non-plastic cultivar ‘Bethune’ only one gene was differentially expressed.
Discussion
The role of LIS-1 in response to imbalanced nutrition
High-throughput sequencing of three cDNA libraries, which were prepared from flax plants grown under different nutrition conditions, was performed. Transcriptome assembly enabled identification of 34924, 33797, and 33698 unique transcripts for N, P, and NPK libraries, respectively. For recognition of LIS-1 derived reads, we chose Blast due to its tolerance to mismatches and long insertions/deletions since one cannot exclude high polymorphism of LIS-1 between genotypes. As a result, we have not revealed any LIS-1 derived mRNA in our sequencing data. Earlier, it was reported that LIS-1 sequence did not contain large open reading frames [4]. In our previous study, we have not identified any LIS-1 transcribed microRNA as well [11]. In the present work, we revealed statistically significant expression alterations of KRP2 and ING1 genes, which are adjacent to LIS-1, under imbalanced nutrition conditions. However, the degree of changes was not high and correlation between LIS-1 status and expression alterations was not observed. For ING1 gene, a slight increase of expression was revealed under P conditions in all groups of samples: ‘Stormont cirrus’ with LIS-1 – median : −3.4 in P and −2.8 in N; ‘Stormont cirrus’ without LIS-1 – median : −4.2 in P and −3.2 in N; ‘Bethune’ – median : −3.8 in P and −2.9 in N. Expression level of KRP2 gene varied slightly under different nutrition conditions (median was in the range from 1.4 to 2.4 in all groups of samples). Therefore, we can conclude that LIS-1 presence most likely does not result in expression alterations of its flanking genes under nutrient stress.
Evaluation of the gene expression alterations under P and NPK condition
For our study, we used seeds of plastic line ‘Stormont cirrus’ obtained from plants with stable status of LIS-1: with LIS-1 and without LIS-1. Also, we used non-plastic cultivar ‘Bethune’ in which LIS-1 never occurs. Flax plants were grown under N, P, and NPK conditions and qPCR analysis was performed to assess the expression of genes, which were chosen on the basis of high-throughput sequencing data, in extended sampling of 31 plants. Alike expression alterations were revealed for studied genes of JAZ family (JAZ8 and JAZ10) under N, P, and NPK conditions for the majority of groups: expression increased under P conditions and decreased under NPK compared to N (only for ‘Stormont cirrus’ with LIS-1, expression of JAZ8 was almost the same under P and NPK conditions). JAZ family proteins are repressors of phytohormone jasmonates, which play important role in plant growth, development and stress tolerance [23, 25–27]. Also, the interaction of ubiquitination process and jasmonate signaling was revealed [28]. However, little is known about the role of JAZ proteins in response to nutrient stress. Differential expression of TIFY family genes under phosphate deficiency was revealed in common bean after 25 days of growth: some of TIFY family genes were induced and others were repressed. Participation of TIFY family genes in Phaseolus vulgaris response and adaptation to P-starvation was assumed [29]. The involvement of JAZ proteins in nutrient deficiency response was revealed in rice and chickpea: differential expression was shown under macro and micronutrients deficiency; most of chickpea and rice JAZ genes were up-regulated after 7 days and down-regulated after 15 days of phosphate deficiency [30]. In the present work in flax plants after 6 weeks of growth under phosphate deficiency, JAZ genes were up-regulated, while under excess nutrition, JAZ genes were down-regulated. Obtained results indicate the involvement of JAZ proteins in flax response to imbalanced nutrition. Our data make a contribution to the knowledge on the role of JAZ proteins in abiotic stress response in plants.
Significant expression alterations in flax plants grown under imbalanced nutrition were revealed for genes of WRKY family (WRKY33, WRKY40, and WRKY70). WRKY gene family, one of the largest families of transcription factors in plants, participates in diverse biological processes, including response to biotic and abiotic stresses [31]. Numerous studies on WRKY protein role were performed for different plant species [31, 32]. Deficiency of nutrition elements or their imbalance in soil is a serious problem for agricultural production, so, plant response to this stress is intensively studied. It was shown that WRKY family genes are involved in tolerance to phosphate starvation in rice: overexpression of WRKY74 gene increase tolerance to phosphate starvation, while lines with low WRKY74 level were sensitive to this stress [33]. In Arabidopsis, WRKY42 regulates phosphate homeostasis: WRKY42 overexpressing lines were more sensitive to phosphate deficiency [34]. However, overexpression of WRKY45 in Arabidopsis increased phosphate uptake [35]. For flax, the involvement of WRKY family genes only in saline-alkaline stress response was known [36, 37]. In the present work, we revealed significant expression decrease of WRKY33, WRKY40, and WRKY70 under NPK compared to P conditions for flax genotypes without LIS-1 (‘Stormont cirrus’ without LIS-1 and ‘Bethune’). However, for ‘Stormont cirrus’ with LIS-1, expression increase or retention of studied WRKY family genes was found under NPK compared to P conditions. It is known that flax small genotrophs with LIS-1 are more adaptive to nutrient deficiency in comparison with lines without LIS-1 [2]. In the present study, we showed that in more adaptive to P conditions ‘Stormont cirrus’ genotypes (‘Stormont cirrus’ with LIS-1), expression of WRKY family genes was up-regulated in contrast to less adaptive ones (‘Stormont cirrus’ without LIS-1). In ‘Bethune’ cultivar, expression alterations of WRKY family genes were similar to ‘Stormont cirrus’ without LIS-1. We assumed that WRKY family genes are involved in nutrition stress response in flax and their overexpression is a feature of less adaptive to phosphate deficiency lines and cultivars.
Conclusions
Search for genes, which are involved in flax response to imbalanced nutrition, was performed. Differentially expressed genes were identified in flax plants, which were grown under phosphate deficiency and excess nutrition, on the basis of high-throughput sequencing and qPCR data. Expression of WRKY and JAS gene families altered in all examined groups of flax plants that allowed us to assume the important role of these genes in flax response to nutrition stress. Moreover, the expression level of WRKY family genes was different in responsive and adaptive to phosphate deficiency genotypes, which differed by LIS-1 presence. Besides, we have not identified any mRNA, which could be derived from LIS-1, in our transcriptome sequencing data. Expression alterations of LIS-1 flanking genes, ING1 and KRP2, were slight and did not correlate with LIS-1 presence in flax plant genome pointing that these genes are not nutrient stress-induced in flax. These data provide new insights into edaphic stress response in flax and the role of LIS-1 in this process.
Acknowledgments
The authors thank Lomonosov Moscow State University for the help with plant growing and All-Russian Research Institute for Flax for the selection and provision of seeds. This work was performed using the equipment of “Genome” center of Engelhardt Institute of Molecular Biology.
Declarations
This article has been published as part of BMC Plant Biology Volume 16 Supplement 3, 2016: Selected articles from BGRS\SB-2016: plant biology. The full contents of the supplement are available online at http://bmcplantbiol.biomedcentral.com/articles/supplements/volume-16-supplement-3.
Funding
This work and the publications costs were financially supported by the Russian Science Foundation, grant 16-16-00114.
Availability of data and materials
The datasets generated during the current study are available in the Sequence Read Archive – SRP083007, https://www.ncbi.nlm.nih.gov/sra/SRP083007.
Authors’ contributions
AD, TR, NB, and NM conceived and designed the work; AVK, NKo, ASS, AAK, AVS, AFS, NKi, TR, OY, NB, and NM performed the experiments; AD, GK, NKo, MB, OM, NB, and NM analyzed the data; AD, GK, and NM wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
Primer sequences, amplification efficiencies and threshold cycles (Ct) for genes examined in the study. (DOCX 17 kb)
Expression of ten genes in plants of cultivar ‘Bethune’ and line ‘Stormont Cirrus’ with (+) and without (-) LIS-1 grown under phosphate deficiency (P), excess nutrition (NPK), or normal (N) conditions. QPCR data. Rectangles correspond to the ranges containing 50 % of the values (between the 25th and 75th percentiles); the bars are the maximum and minimum ΔC t eff values. (TIF 873 kb)
Contributor Information
Alexey A. Dmitriev, Email: Alex_245@mail.ru
Anna V. Kudryavtseva, Email: rhizamoeba@mail.ru
George S. Krasnov, Email: gskrasnov@mail.ru
Nadezhda V. Koroban, Email: nosy57@gmail.com
Anna S. Speranskaya, Email: hannadt@mail.ru
Anastasia A. Krinitsina, Email: ankrina@gmail.com
Maxim S. Belenikin, Email: molecular.modeler@gmail.com
Anastasiya V. Snezhkina, Email: leftger@rambler.ru
Asiya F. Sadritdinova, Email: aska2228@mail.ru
Natalya V. Kishlyan, Email: natalya-kishlyan@yandex.ru
Tatiana A. Rozhmina, Email: vniil@mail.ru
Olga Yu. Yurkevich, Email: olikys@gmail.com
Olga V. Muravenko, Email: olgmur1@yandex.ru
Nadezhda L. Bolsheva, Email: nlbolsheva@mail.ru
Nataliya V. Melnikova, Email: mnv-4529264@yandex.ru
References
- 1.Muir AD, Westcott ND. Flax: the genus Linum. London: Taylor & Francis; 2003. [Google Scholar]
- 2.Bickel CL, Lukacs M, Cullis CA. The loci controlling plasticity in flax. Res Rep Biol. 2012;3:1–11. doi: 10.2147/RRB.S27198. [DOI] [Google Scholar]
- 3.Chen Y, Lowenfeld R, Cullis CA. An environmentally induced adaptive (?) insertion event in flax. Int J Genet Mol Biol. 2009;1(3):38–47. [Google Scholar]
- 4.Chen Y, Schneeberger RG, Cullis CA. A site-specific insertion sequence in flax genotrophs induced by environment. New Phytol. 2005;167(1):171–80. doi: 10.1111/j.1469-8137.2005.01398.x. [DOI] [PubMed] [Google Scholar]
- 5.Cullis CA, Charlton L. The induction of ribosomal DNA changes in flax. Plant Sci Lett. 1981;20:213–7. doi: 10.1016/0304-4211(81)90264-9. [DOI] [Google Scholar]
- 6.Durrant A. Environmental conditioning of flax. Nature. 1958;181:928–9. doi: 10.1038/181928a0. [DOI] [Google Scholar]
- 7.Durrant A. The environmental induction of heritable change in Linum. Heredity. 1962;17:27–61. doi: 10.1038/hdy.1962.2. [DOI] [Google Scholar]
- 8.Evans GM, Durrant A, Rees H. Associated nuclear changes in the induction of flax genotrophs. Nature. 1966;212:697–9. doi: 10.1038/212697a0. [DOI] [Google Scholar]
- 9.Tyson H, Fieldes MA. Molecular-weight and net charge of peroxidase isozymes in F1 hybrids between L and S flax genotrophs. Biochem Genet. 1982;20:919–27. doi: 10.1007/BF00484069. [DOI] [PubMed] [Google Scholar]
- 10.Bastaki NK, Cullis CA. Floral-dip transformation of flax (Linum usitatissimum) to generate transgenic progenies with a high transformation rate. J Vis Exp. 2014(94). doi: 10.3791/52189. [DOI] [PMC free article] [PubMed]
- 11.Melnikova NV, Dmitriev AA, Belenikin MS, Koroban NV, Speranskaya AS, Krinitsina AA, et al. Identification, expression analysis, and target prediction of flax genotroph MicroRNAs under normal and nutrient stress conditions. Front Plant Sci. 2016;7:399. doi: 10.3389/fpls.2016.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melnikova NV, Kudryavtseva AV, Zelenin AV, Lakunina VA, Yurkevich OY, Speranskaya AS, et al. Retrotransposon-based molecular markers for analysis of genetic diversity within the Genus Linum. Biomed Res Int. 2014;2014:231589. doi: 10.1155/2014/231589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012;72(3):461–73. doi: 10.1111/j.1365-313X.2012.05093.x. [DOI] [PubMed] [Google Scholar]
- 15.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melnikova NV, Dmitriev AA, Belenikin MS, Speranskaya AS, Krinitsina AA, Rachinskaia OA, et al. Excess fertilizer responsive miRNAs revealed in Linum usitatissimum L. Biochimie. 2015;109:36–41. doi: 10.1016/j.biochi.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1-2):203–14. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 19.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huis R, Hawkins S, Neutelings G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.) BMC Plant Biol. 2010;10:71. doi: 10.1186/1471-2229-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krasnov GS, Oparina NY, Dmitriev AA, Kudryavtseva AV, Anedchenko EA, Kondrat’eva TT, et al. RPN1, a new reference gene for quantitative data normalization in lung and kidney cancer. Mol Biol. 2011;45(2):211–20. doi: 10.1134/S0026893311020129. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Wang H, Shao H, Tang X. Recent advances in utilizing transcription factors to improve plant abiotic stress tolerance by transgenic technology. Front Plant Sci. 2016;7:67. doi: 10.3389/fpls.2016.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santner A, Estelle M. The JAZ proteins link jasmonate perception with transcriptional changes. Plant Cell. 2007;19(12):3839–42. doi: 10.1105/tpc.107.056960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5:1. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pauwels L, Goossens A. The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell. 2011;23(9):3089–100. doi: 10.1105/tpc.111.089300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazan K. Diverse roles of jasmonates and ethylene in abiotic stress tolerance. Trends Plant Sci. 2015;20(4):219–29. doi: 10.1016/j.tplants.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Goossens J, Fernandez-Calvo P, Schweizer F, Goossens A. Jasmonates: signal transduction components and their roles in environmental stress responses. Plant Mol Biol. 2016;91:673–89. doi: 10.1007/s11103-016-0480-9. [DOI] [PubMed] [Google Scholar]
- 28.Thines B, Katsir L, Melotto M, Niu Y, Mandaokar A, Liu G, et al. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature. 2007;448(7154):661–5. doi: 10.1038/nature05960. [DOI] [PubMed] [Google Scholar]
- 29.Aparicio-Fabre R, Guillen G, Loredo M, Arellano J, Valdes-Lopez O, Ramirez M, et al. Common bean (Phaseolus vulgaris L.) PvTIFY orchestrates global changes in transcript profile response to jasmonate and phosphorus deficiency. BMC Plant Biol. 2013;13:26. doi: 10.1186/1471-2229-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh AP, Pandey BK, Deveshwar P, Narnoliya L, Parida SK, Giri J. JAZ repressors: potential involvement in nutrients deficiency response in rice and chickpea. Front Plant Sci. 2015;6:975. doi: 10.3389/fpls.2015.00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bakshi M, Oelmuller R. WRKY transcription factors: Jack of many trades in plants. Plant Signal Behav. 2014;9(2):e27700. doi: 10.4161/psb.27700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Song Y, Li S, Zhang L, Zou C, Yu D. The role of WRKY transcription factors in plant abiotic stresses. Biochim Biophys Acta. 2012;1819(2):120–8. doi: 10.1016/j.bbagrm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Dai X, Wang Y, Zhang WH. OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. J Exp Bot. 2016;67(3):947–60. doi: 10.1093/jxb/erv515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su T, Xu Q, Zhang FC, Chen Y, Li LQ, Wu WH, et al. WRKY42 modulates phosphate homeostasis through regulating phosphate translocation and acquisition in Arabidopsis. Plant Physiol. 2015;167(4):1579–91. doi: 10.1104/pp.114.253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, Xu Q, Kong YH, Chen Y, Duan JY, Wu WH, et al. Arabidopsis WRKY45 transcription factor activates PHOSPHATE TRANSPORTER1;1 expression in response to phosphate starvation. Plant Physiol. 2014;164(4):2020–9. doi: 10.1104/pp.113.235077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Y, Wu G, Yuan H, Cheng L, Zhao D, Huang W, et al. Identification and characterization of miRNAs and targets in flax (Linum usitatissimum) under saline, alkaline, and saline-alkaline stresses. BMC Plant Biol. 2016;16(1):124. doi: 10.1186/s12870-016-0808-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Y, Huang W, Chen H, Wu G, Yuan H, Song X, et al. Identification of differentially expressed genes in flax (Linum usitatissimum L.) under saline-alkaline stress by digital gene expression. Gene. 2014;549(1):113–22. doi: 10.1016/j.gene.2014.07.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the current study are available in the Sequence Read Archive – SRP083007, https://www.ncbi.nlm.nih.gov/sra/SRP083007.