

Abstract
Background
DNA damage response (DDR) defects imply genomic instability and favor tumor progression but make the cells vulnerable to the pharmacological inhibition of the DNA repairing enzymes. Targeting cellular proteins like PARPs, which cooperate and complement molecular defects of the DDR process, induces a specific lethality in DDR defective cancer cells and represents an anti-cancer strategy. Normal cells can tolerate the DNA damage generated by PARP inhibition because of an efficient homologous recombination mechanism (HR); in contrast, cancer cells with a deficient HR are unable to manage the DSBs and appear especially sensitive to the PARP inhibitors (PARPi) effects.
Main body
In this review we discuss the proof of concept for the use of PARPi in different cancer types and the success and failure of their inclusion in clinical trials.
The PARP inhibitor Olaparib [AZD2281] has been approved by the FDA for use in pretreated ovarian cancer patients with defective BRCA1/2 genes, and by the EMEA for maintenance therapy in platinum sensitive ovarian cancer patients with defective BRCA1/2 genes. BRCA mutations are now recognised as the molecular targets for PARPi sensitivity in several tumors. However, it is noteworthy that the use of PARPi has shown its efficacy also in non-BRCA related tumors. Several trials are ongoing to test different PARPi in different cancer types. Here we review the concept of BRCAness and the functional loss of proteins involved in DDR/HR mechanisms in cancer, including additional molecules that can influence the cancer cells sensitivity to PARPi. Given the complexity of the existing crosstalk between different DNA repair pathways, it is likely that a single biomarker may not be sufficient to predict the benefit of PARP inhibitors therapies. Novel general assays able to predict the DDR/HR proficiency in cancer cells and the PARPi sensitivity represent a challenge for a personalized therapy.
Conclusions
PARP inhibition is a potentially important strategy for managing a significant subset of tumors. The discovery of both germline and somatic DNA repair deficiencies in different cancer patients, together with the development of new PARP inhibitors that can kill selectively cancer cells is a potent example of targeting therapy to molecularly defined tumor subtypes.
Keywords: DNA damage response, PARP enzymes, PARP inhibitors, Cancer, BRCA1/2 and BRCAness, Clinical trials, Assays, HR proficiency and PARP activity
Methodology: sources and search terms
Literature from a range of sources, including PubMed and MEDLINE, were searched to identify recent reports regarding “DNA damage repair and PARP inhibitors” in addition to other terms relevant to this Review, including “Breast cancer and PARP”, “synthetic lethality”, “cancer and PARP inhibitors”, and “BRCAness”. The reference lists of key articles identified were also searched for additional relevant publications. The ClinicalTrials.gov database was searched using the term “PARP inhibitors” to identify relevant clinical trials.
The key points of this review are:
# The poly(ADP-ribose) polymerases (PARPs) family.
# Repair of single-strand and double-strand breaks in DNA damage.
# Homologous recombination repair (HRR) mechanisms.
# Defects in DNA Damage Response in cancer.
# BRCA1 or BRCA2 mutations.
# Synthetic lethal concept
# Molecular defects which cause the lack of homologous recombination and produce sensitivity to inhibitors of PARP activity.
# Chromosomal instability and DNA repair foci
# in vitro and ex vivo assays to predict the efficacy of PARP inhibitors.
# Success and failure of PARP inhibitors in Clinical Trials.
Background
DNA damage response (DDR) is the cellular reaction to exogenous and endogenous genotoxic injuries that may produce DNA single strand breaks (SSBs) and DNA double strand breaks (DSBs). While SSBs are repaired by mechanisms of nucleotide excision repair (NER) or base excision repair (BER), or mismatch repair (MMR), DSBs are repaired either by the mechanism of homologous recombination (HR), which utilizes the sister chromatid as a template for a correct replacement of the DNA sequence, or by the mechanism of non-homologous end joining (NHEJ), which is more prone to errors [1, 2]. The cellular choice of using HR or NHEJ is largely dependent on the phases of the cell cycle; NHEJ is present throughout the cell cycle, whereas HR predominates in the S and G2 phases, in order to ensure the high-fidelity preservation of genetic information [3]. If the repairing process does not occur correctly, the DNA injuries result in mutations and chromosomal aberrations which alter the cellular behavior and lead to cancer.
Genes that encode for enzymatic or scaffolding proteins involved in the “core” DDR activities [BER, MMR, HR and NHEJ) are: XPA-XPG, RPA, ERCC1, DNA glycosylase, APE1, DNA polymerase β/δ/ε, XRCC1, DNA ligase 1/3, DNA ligase IV, Ku70/80, RAD50/MRE11/NBS1, BRCA1, BRCA2, and RAD51 (Fig. 1) [4–9].
Fig. 1.
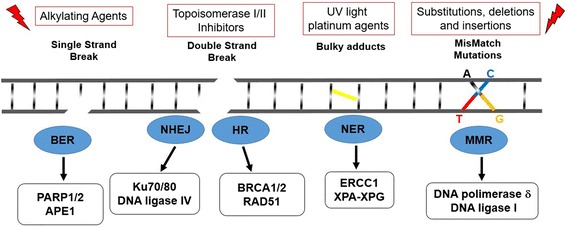
Diagram of targeted DDR pathways. In the lower part of the figure the DDR mechanisms and the related proteins involved are represented. In the upper part of the figure the targeting strategy for the corresponding defective DDR mechanisms are shown
Additionally, as a result of a computational analysis nearly 400 proteins have been identified in the regulation of the DDR processes [10–13], namely: the damage sensing kinases ATM/ATR, that activate a phosphorylation cascade signaling in response to the DSBs [14, 15]; DNA-PK, that cooperates with ATR and ATM to phosphorylate proteins involved in the DNA damage checkpoints and is required for NHEJ [16]; the kinases CHEK1 and CHEK2, that are responsible for slowing down the cell cycle progression to allow DNA repair [17]; and the nuclear phosphatase PTEN, that controls the transcription and the nuclear localization of the recombinase RAD51 [18–20]. Furthermore, ubiquitination, sumoylation, acetylation and methylation processes provide an additional layer of complexity targeting stability and efficiency of DDR proteins machinery [10, 12].
Since almost 56% of the identified 400 proteins are involved in multiple DDR pathways, a functional defect or loss of a single DDR protein may affect multiple DNA repair processes [11]. Defects in DDR seem to be positively selected in cancer cells to support the enhanced proliferation rate [21–23]. However, molecular alterations in the DNA repairing process make the cells more vulnerable to the pharmacological inhibition of the DNA repairing enzymes [24–30]. The concept of promoting the killing of cancer cells by simultaneously targeting cellular signals that cooperate and complement molecular defects to obtain cell death represents an anti-cancer strategy based on the concept of synthetic lethality (Fig. 2) [31–33].
Fig. 2.
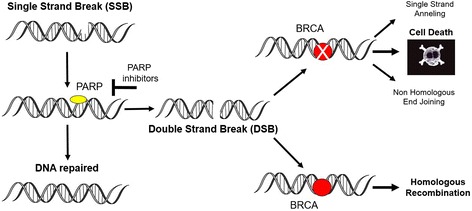
Synthetic Lethality of PARP-inhibitors in BRCA Tumors. Poly(ADP-ribose) polymerases (PARPs) repair DNA SSBs through the BER pathway. PARP inhibitors, such as olaparib, prevent repair of the SSBs, resulting in the generation of DNA DSBs. Cancer cells with a deficient homologous recombination (BRCA1/BRCA2 mutations) required for the repair of the DSBs do not compensate for the increased DNA damage caused by the inhibition of PARP enzymes and appear to be especially sensitive to treatment with these drugs
Cancer cells defective in the DSBs repair molecules involved in DDR can be targeted specifically by blocking SSBs repair by inhibiting PARP enzymes [34–41].
Main body
PARP enzymes and defects in DNA damage response in cancer
The Poly ADP-ribose polymerase (PARP) family comprises 17 members including PARP1, PARP2, PARP3, tankyrases 1 and 2 (PARP5a and 5b), all of which have been identified on the basis of their homology in the catalytic domain [42–48]. The most studied protein of the PARP family is PARP1, a nuclear protein with enzymatic and scaffolding properties, that contains an amino-terminal DNA binding domain (DBD, a central auto-modification domain (AMD), crucial for protein-protein aggregation, and a carboxyl-terminal catalytic domain (CD). The activity of the PARP1 enzyme seems to be critical in the BER but also in the HR and NHEJ mechanisms [46, 49–51]. The PARP1 enzyme transfers the first ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to the proteic residues (glutamate, aspartate and lysine) and generate an ADP-ribose unit chain (PAR), acting as a “writer” of a poly ADP-ribosylation [52, 53]. This process of “PARylation” occurs covalently on target proteins (transPARylation) and on the PARP enzyme itself (autoPARylation) producing a negative charge that alters the protein structure and function. The presence of PAR chains at the break-sites of DNA favors the non-covalent recruitment of DNA repair proteins like XRCC1, DNA ligase 3, DNA polymerase β, and the MRE11-Rad50-NBS1 (MRN) complex, for DNA resection and single strand formation, which in turn allow for RAD51 loading to initiate HR [43, 54–56].
PARP1 and PARP2 are the enzymes most extensively studied and are known to be stimulated by DNA damage, although PARP2 contributes to only 5–10% of the total PARP activity in response to DNA damage [57–60]. Tankyrases 1 and 2 are mostly involved in telomere and mitotic spindle-related functions, as well as in the regulation of Wnt signaling [61]. Overall, the PARP enzymes regulate DDR but also tumor growth and progression through transcriptional/epigenetic regulation and mRNA processing and stability [43, 62–68]. Thus, targeting PARP activity can cooperate and complement molecular defects of DDR that have been exploited therapeutically for different cancer treatments [69–72].
A wide variety of hereditary human cancers show germline alterations of genes involved in the DDR process. Germline mutations within BRCA1 carriers predispose to early onset of breast and ovarian cancer; mutations in BRCA2 genes lead to the late onset of different tumors (gastric, colon, pancreatic and melanoma) beyond breast, ovarian and prostate cancer; mutations in the ATM, NBS1, BLM, and WRN genes occur in lymphoma and leukemia; the RAD54 and CtIP genes are mutated in non-Hodgkin’s lymphoma and colon cancer; the MLH1 and MSH2 genes are mutated in hereditary non-polyposis colorectal cancer (HNPCC); the RAD51B gene is mutated in lymphoma and uterine leiomyoma and finally the RECQL4 gene is mutated in skin cancer and osteosarcoma [73–80]. Beside the DDR defects identified in hereditary human cancer, recent studies based on computational analysis have shown that almost 95% of DDR genes are mutated in coding regions at least in 15 different types of sporadic tumors [11], suggesting that mutations in DDR genes should be further investigated as possible driver mutations in cancer.
The activity of PARP inhibitors: success and failure of PARPi in Clinical Trials
The use of PARP enzyme inhibitors in cancer cells which are defective for BRCA1 and BRCA2, two proteins that localize the RAD51 recombinase to the sites of damaged DNA and promote HR repair, represent the best and most successful synthetic lethal approach in cancer therapy.
The PARP inhibitors bind the catalytic domain of the PARP protein, mostly as antagonists of the PARP cofactor β-NAD+. Because of the binding of the inhibitors, the PARP enzymes could be inhibited in the catalytic activity, with the final result of converting SSBs into DSBs and determining cell death in DSB repair deficient cells [81–88].
There are a total of nine drugs related to PARP targeting in the pipeline of drug development, some of them having a selective activity on PARP1 and PARP2, others affecting both PARP1 and PARP2 [89].
AG-014699 Rucaparib (Clovis/Pfizer), used for intravenous and oral administration, is the first inhibitor that has been introduced in clinical trials in association with chemotherapy and is now in Phase III for maintenance treatment after chemotherapy [90–92]. AZD2281 Olaparib (kuDOS/LynparzaTM, AstraZeneca), used for oral administration, is the first inhibitor identified as single antitumoral agent in cancer associated with the BRCA1 or BRCA2 mutations [93, 94]. Notably, in December 2014 Olaparib was approved by the US Food and Drug Administration (FDA) for its use in pretreated ovarian cancers with defective BRCA genes and by the European Medicines Agency (EMEA) for maintenance therapy in BRCA-mutated/platinum sensitive ovarian cancer patients. ABT-888 Veliparib [Abbvie], used for oral administration, is now in Phase III evaluation in combined therapy in advanced or recurrent solid tumors, also because of its property to reach the Central Nervous System (CNS) [95, 96]. BSI-201 Iniparib (BiPar/Sanofi), used for intravenous administration, was the first inhibitor that has entered Phase III studies and, besides the discrepancies between the phase II and III studies, has shown antitumor activity mostly in combined therapy with gemcitabine and carboplatinum [97–99]. MK-4827 Niraparib (Merck/Tesaro), used for oral administration, is a potent inhibitor of PARP1 and PARP2 and is currently being tested in Phase III clinical trials as maintenance therapy in ovarian cancer and as a treatment for breast cancer [100–102]. BMN-673 (BioMarin), used for oral administration, is more potent than Olaparib and is used in combined therapy in Phase III evaluation [103, 104]. Finally, a few drugs developed more recently like CEP-9722/8983 (Cephalon) and E7016 or E7449 (Eisai/MGI Pharma) are used for oral administration and are now being tested in Phase I toxicity studies in combined therapy [88, 105, 106]
By limiting the DDR competence, PARP inhibitors complement the mechanisms of action of chemotherapy and radiotherapy. Thus, as chemosensitizer, PARPi, has entered clinical assessment in combination with temozolomide [90, 107], DNA crosslinkers (e.g. cisplatin) [108], or cleave the sugar-phosphate backbone (e.g. bleomycin) [109]. Thus, several clinical trials with different PARP inhibitors have been conducted and are still ongoing to test the efficacy of PARPi as a single agent or in combination with radiotherapy and/or chemotherapy [https://clinicaltrials.gov/]. Clinical trials that are in progress to test PARPi efficacy have been recently summarized by Lord and Ashworth [110].
In 2005 a Phase I clinical trial assessing Olaparib as a single agent showed an objective response rate (ORR) of 47% and a disease control rate (DCR) of 63% in patients with BRCA mutations [94]. Next, Phase II studies confirmed the efficacy of Olaparib as a single agent in breast and ovarian cancer patients with BRCA mutations [111]. Significantly, the best response to Olaparib has been reported in ovarian cancer patients with BRCA mutations who have demonstrated a sensitivity to platinum treatment; patients who had a platinum-sensitive disease were Olaparib responsive with a rate of 69%, whereas those who were platinum-refractory had a response rate of 23% [111]. Thereafter, clinical trials were designed to define both the utility of Olaparib as a single agent in chemotherapy-refractory tumors, and as maintenance therapy in platinum-sensitive diseases. Carriers of BRCA mutations, responsive to previous platinum therapy, have shown better outcomes in PFS (8.4 months vs 4.8 months) with the Olaparib maintenance therapy [112–114].
Currently, Phase III clinical trials are in progress aimed at assessing the use of several PARP inhibitors (e.g. Olaparib, Niraparib, Recuparib, Veliparib, and BMN-673) in the maintenance therapy of platinum treated ovarian and breast cancer, as single agents in therapeutic settings of adjuvant and standard-of-care chemotherapy for advanced diseases. Additionally, Phase III clinical trials are ongoing to assess the efficacy of PARP inhibitors in combined treatments, in therapeutic settings of neoadjuvant strategy and in advanced diseases [https://clinicaltrials.gov/].
The positive results obtained in breast and ovarian and prostate cancer patients with BRCA mutations [111–115] have encouraged the use of PARP inhibitors in tumors with BRCA-like features (*BRCAness phenotype*). Thus, the efficacy of PARPi has been evaluated in Triple Negative Breast Tumors and High-Grade Serous Ovarian Cancer (TNBC/HGS-OVCa) that exhibited a 60-gene signature in common with BRCA mutated tumors [110, 116–121]. Moreover, clinical correlation suggested that the BRCA status conferred sensitivity to platinum chemotherapy and this can be used as a marker for HR defects to predict PARP-inhibitors sensitivity [122].
Predicting PARPi sensitivity beyond BRCA1/2 mutations and “BRCAness”
The identification of biomarkers that can predict the PARP inhibitor sensitivity of cancer cells is urgently required. Germline BRCA mutations are recognised as the molecular targets for PARPi sensitivity in breast, ovarian and prostate cancer. However, it is noteworthy that PARPi efficacy has also been reported in non-BRCA related tumors [123]. Thus, PARP inhibitors may have a utility beyond the relatively small proportion (5–10%) of cancer patients carrying BRCA mutations [102, 124].
Tumors displaying a DNA repair dysfunction, regardless of the leading genetic lesion, might exhibit a BRCA-like behavior, according to the concept of “BRCAness” [110, 116], and might therefore benefit from PARPi treatment [122, 125–128]. However, the predictive value of a “BRCAness” signature, besides BRCA1 and BRCA2 mutations, still requires prospective large-scale clinical validation before entering conventional clinical practice. Genomic alterations, gene mutations or functional loss of proteins involved in DDR mechanisms, such as ATM, ATR, CHEK1, CHEK2, DSS1, RAD51, MRE11A/NBS1, Fanconi anemia complementation group (FANC family of genes), EMSY, PALB2, XRCC2, XRCC3, or PTEN, could represent predictive markers in cancer patients to tailor a personalized treatment with PARP inhibitors [129–141].
Nevertheless, additional molecules appear to be able to influence the sensitivity to PARPi, namely the gene fusions TMPRSS2:ERG, detected in more than 50% of prostate cancers, and the EWSR1:FLI1 translocation, detected in Ewing’s sarcoma in more than 90% of patients [142, 143].
To date, a few additional proteins have been proposed as novel predictive biomarkers of PARP inhibitor sensitivity like cyclin dependent kinase CDK12, the excision repair cross-complementation group 1 ERCC1, and the proapoptotic protein CCDC6.
CDK12 attenuation in the high-grade serous ovarian cancer model [HGS-OVCa] is sufficient to confer a sensitivity to PARP1/2 inhibition [144, 145].
ERCC1 low expression in the NSCLC model is sufficient to determine a synergistic effect with PARP inhibition [146, 147].
CCDC6 loss or low expression in different cancer models impairs RAD51 foci formation, limits γH2AX foci formation by modulating the activity of the histone phosphatase PP4C and sensitizes the cancer cells to PARPi treatment [148–151].
Moreover, kinases, such as CDK5, MAPK12, PLK3, PNKP, STK22c, STK36, and deubiquitinases, such as USP1 and USP11, can produce synthetic lethal effects with PARPi in genetic perturbation screens performed following different approaches [152–157].
In Table 1 we report the altered genes which have been described so far to confer PARPi sensitivity in different in vitro and in vivo cellular models.
Table 1.
Genetic alterations that predict PARPi sensitivity
| Altered gene | PARP inhibitors | in vitro/in vivo systems | Study |
|---|---|---|---|
| BRCA2 | NU1025 and AG14361 | Chinese hamster cell V-C8 and V-C8 + B2 (BRCA2 defective and BRCA2 complemented) Human breast cancer cells MCF-7 or MDA-MB-231 (BRCA2 siRNA) |
Bryant H. et al., Nature 2005. . |
| KU0058684 and KU0058948 | Mouse ESC (lacking BRCA2 wt) HeLa (RNAi) |
Farmer H. et al, Nature 2005 McCabe N. et al., Cancer Res. 2006 |
|
| Olaparib (AZD2281) | Ovarian cancer patients ClinicalTrial NCT00753545
Prostate cancer patients ClinicalTrial, NCT01682772 |
Ledermann J. et al., N Engl J Med. 2012. Mateo J. et al., N. Engl. J. Med. 2015. |
|
| ATM | KU0058684 and KU0058948 Olaparib (AZD2281) |
HeLa (RNAi) Prostate cancer patients ClinicalTrial, NCT01682772 |
McCabe N. et al., Cancer Res. 2006. Mateo J. et al., N. Engl. J. Med. 2015. |
| ATR | KU0058684 and KU0058948 | HeLa (RNAi) |
McCabe N. et al., Cancer Res. 2006 |
| FANC A/F | KU0058684 and KU0058948 Olaparib (AZD2281) |
Mouse fibroblast (FANC KO) Prostate cancer patients ClinicalTrial, NCT01682772 |
McCabe N. et al., Cancer Res. 2006 Mateo J. et al., N. Engl. J. Med. 2015. |
| CHK2 | KU0058684 and KU0058948 Olaparib (AZD2281) |
HeLa (RNAi) Prostate cancer patients ClinicalTrial, NCT01682772 |
McCabe N. et al., Cancer Res. 2006 Mateo J. et al., N. Engl. J. Med. 2015.). |
| BRCA1 | KU0058684 and KU0058948 Olaparib (AZD2281) |
Mouse ESC (lacking BRCA2 wt) HeLa (RNAi) Ovarian cancer patients ClinicalTrial NCT00753545 Prostate cancer patients ClinicalTrial, NCT01682772 |
Farmer H. et al, Nature. 2005. McCabe N. et al., Cancer Res. 2006. Ledermann J. et al., N Engl J Med. 2012. Mateo J. et al., N. Engl. J. Med. 2015. |
| PALB2 | Olaparib (AZD2281) BMN 673 |
Human fibroblast EUFA1341 (express PALB2 mutant) Wilms tumor KT-10 cells (express truncated PALB2) |
Buisson R. etal., Nat Struct Mol Biol. 2010. Smith MA. et al., Pediatr Blood Cancer. 2015. |
| RAD51B/C | KU0058684 and KU0058948 Olaparib (AZD2281) |
HeLa (RNAi) Prostate cancer patients ClinicalTrial, NCT01682772 |
McCabe N. et al., Cancer Res. 2006. Mateo J. et al., N. Engl. J. Med. 2015. |
| RAD54 | KU0058684 and KU0058948 | Mouse ESC (lacking Rad54 wt) |
McCabe N. et al., Cancer Res. 2006. |
| ERCC1 | olaparib (AZD-2281), niraparib (MK-4827), BMN 673 olaparib (AZD-2281), veliparib (ABT-888) |
Non Small Cell Lung Cancer A549 (ERCC1 deficient clones) Non Small Cell Lung Cancer HCC827, PC9 (ERCC1 low expression) |
Postel-Vinay S. et al., Oncogene 2013. Cheng H. et al., Carcinogenesis 2013. |
| CtIP | BMN 673 KU0058948 |
Myeloid leukemia cell K562 (expressing CtIP-T) | Gaymes TJ. et al., Haematologica. 2013. |
| MRE11 | BMN 673 KU0058948 |
Hystiocitic Linphoma U937 cell (expressing MRE11-Δ57) |
Gaymes TJ. et al., Haematologica. 2013. |
| NBS1 | KU0058684 and KU0058948 | Human immortalized fibroblast | McCabe N. et al., Cancer Res. 2006. |
| DSS1 | KU0058684 and KU0058948 | HeLa (RNAi) |
McCabe N. et al., Cancer Res. 2006. |
| RPA1 | KU0058684 and KU0058948 | HeLa (RNAi) |
McCabe N. et al., Cancer Res. 2006. |
| PTEN | (KU0059436) Olaparib | Colorectal tumour cell HCT116 (truncated PTEN) endometroid adenocarcinoma cells HEC1A (truncated PTEN) Breast, prostate, melanoma, glioma and bladder cells HCC70, MDA-MB-468, PC3, RPMI-7951, A172, UM-UC3 and (PTEN deficient expression) |
Mendes-Pereira, A. et al., EMBO Mol. Med. 2009. |
| (KU0059436) Olaparib | Prostate cancer patients ClinicalTrial, NCT01682772 | Mateo J. et al., N. Engl. J. Med. 2015. | |
| ETS/ERG | (KU0059436) Olaparib MK-4827 |
Solid tumors ClinicalTrial, NCT00777582
Solid tumors ClinicalTrial, NCT00749502 |
Brenner JC. et al, Cancer Res. 2011. |
| XRCC2/XRCC3 | (3-AB) (ISQ) (NU1025) (AG14361) |
Chinese hamster ovary cell irs1/irs1SF | Bryant H. et al., Nature 2005. |
| CDK1 | AG14361 and AG014699 |
Non Small Cell Lung Cancer NCI-H1299/A549 CDK1 (RNAi) |
Johnson N. et al., Nat Med. 2011. |
| CDK12 | veliparib (ABT-888) |
Ovarian cancer OVCAR-3, OVCAR-5, OVCAR-8 CDK12 (RNAi) |
Joshi, P.M. et al., J. Biol. Chem. 2014. Bajrami,I. . et al., Cancer Res. 2014. |
| olaparib (AZD-2281) |
Ovarian cancer PEO1, OV56, COV504; OV90 cells CDK12-(RNAi) |
||
| (KU0059436) Olaparib | Prostate cancer patients ClinicalTrial, NCT01682772 | Mateo J. et al., N. Engl. J. Med. 2015. | |
| CDK5 | KU0058948 | Breast cancer CAL51 cells CDK5 (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| PLK3 | KU0058948 | Breast cancer CAL51 cells PLK3 (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| PNKP | KU0058948 | Breast cancer CAL51 cells PNKP (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| STK22C | KU0058948 | Breast cancer CAL51 cells STK22C (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| STK36 | KU0058948 | Breast cancer CAL51 cells STK36 (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| USP1 | KU0058948 | Breast cancer CAL51 cells USP1 (RNAi) |
Turner, N.C. et al., EMBO J. 2008. |
| USP11 | olaparib (AZD-2281) |
Bone osteosarcoma U2OS cell USP11 (RNAi) |
Wiltshire TD. et al., J Biol Chem. 2010. |
| CCDC6 | olaparib (AZD-2281), |
Non Small Cell Lung Cancer H1975 cells CCDC6 (RNAi) |
Morra F. et al., Int J Cancer 2015. |
The complexity of the existing crosstalk between DNA repair pathways suggests that a single biomarker may not be sufficient to predict the benefit of PARP inhibitor therapies. Therefore, DNA microarrays, real-time quantitative reverse transcriptase [qRT]-PCR, protein microarrays, mass spectrometry, immunohistochemistry and immunofluorescence assays represent powerful tools in order to identify predictive biomarkers measured at baseline or in progress of therapy in cancer patients enrolled on PARPi clinical studies.
Assays that can measure HR proficiency and PARP activity in vivo
The development of novel assays that are able to predict the HR proficiency in cancer cells represents an important challenge in the design of a personalized therapy. Several assays that can evaluate the HR proficiency in cancer cells need to be validated in prospective clinical trials for their value to predict the response to targeted therapies, Since genomic instability may represent a lifetime record of DNA repair deficiency, the genomic structural rearrangement signatures, identified in functional BRCAness, should be examined in tumor types other than ovarian and triple-negative breast cancers. [158–164].
It has been reported that the measure of telomeric allelic imbalances (NtAl), that counts the number of subtelomeric regions with allelic imbalance,in combination with the measure of loss of heterozygosity (ARD-LOH), that measures the number of regions with LOH which are larger than 15 Mband with the measure of large scale transition (LST), that counts the number of chromosomal breaks between adjacent regions of at least 10 Mb, can identify in 15 different tumor types a genomic scar signature in SNP array data and allow the selection of DNA repair-deficient cancers, to candidate cancer patients to platinum chemotherapy and PARPi [158]. On this basis Myriad Genetics used SNP profiling to develop a HR deficiency (HRD) assay which combines the mentioned three different DNA-based metrics of genomic instability [165]. The HDR assay can be performed using DNA extracted from FFPE tumor tissues and thus has been translated into clinic for perspective studies (ARIEL2 NCT#01891344)
Furthermore, a biological rationale driven (mutations in BRCA1/2) genomic instability score has been developed by integrating somatic mutations and copy number changes reported in the TCGA of 325 ovarian cancers. The identified score has been demonstrated to correlate with homozygous deletion of core HR genes in 67 HR deficient non BRCA samples compared to 152 control samples. The identified score has been also correlated with the outcome of response to treatment of platinum in ovarian cancer patients [166].
Several studies support the use of panel testing for a comprehensive analysis of mutations, expression changes of multiple genes in prospectively designed trials for the selection of patients likely to respond to platinum and PARPi. However, there is currently no gold standard method of testing for DDR cancer cell proficiency and cancer sensitivity to platinum or PARPi.
Assessing the number of nuclear foci of RAD51, FANCD2 and γH2AX may help to establish DDR cellular competence, before, during and after treatment with IR and PARP inhibitors, to predict tumor sensitivity or acquired resistance to treatments.
Therefore, the detection of RAD51 foci by IHC and IF, in ex vivo samples or in tumor biopsies during neoadjuvant therapy, may be predictive of HR defects and sensitivity to PARPi, given that PARP inhibition or loss results in an increasing RAD51 foci formation in HR intact cells [167]. This approach of RAD51 foci formation has been applied in ex vivo samples in primary ovarian cancer cultures to predict sensitivity toRucaparib, showing a negative predictive value of 100% and a positive predictive value of 93% [168]. The analysis of RAD51 foci formation in FFPE samples of breast cancer biopsied after neoadjuvant therapy also has appeared to be predictive of a response to chemotherapy [169].
The FANCD2 foci formation may be assayed to predict the sensitivity of cancer cells to cisplatin and PARPi, given that PARP inhibitors, chemotherapy and radiotherapy induce FANCD2 nuclear foci formation [170–172].
Additionally, a high-throughput screening system based on the IF of γH2AX (Rapid Automated Biodosimetry Tool) may help to screen patients sensitive to PARPi treatment, given that PARP inhibitors increase γH2AX foci in Circulating Tumor Cells [173–175]. Recently, the evaluation of RAD51 and γH2AX nuclear foci in ex vivo samples, as well as of the PARP activity, has been applied in some clinical studies [168, 176, 177].
Ongoing clinical trials with PARPi [ABT-888], either as a single agent or in combination therapy, aim to identify suitable patients for PARPi sensitivity, beside BRCA mutations, that show HR or MMR deficiency (NCT01237067, NCT02660034, NCT02576444, NCT02286687, NCT01891344, NCT02354131) by measuring γH2AX and FANCD2 foci formation in FFPE tumor samples [167, 172, 177] (NCT01017640; NCT01251874).
Tumor cells defective in the HR process might also show a compensatory induction of PARP expression. However, PARP1 protein levels do not differ between isogenic pairs of HR deficient and proficient cancer cells [178]. An enhanced PARP1 expression does not mean an enhanced PARP activity [179–182]. Since the PARP activity is associated with the levels of PAR, the detection of low levels of PAR may indicate an intrinsic low activity of PARP with a limited potential of PARP inhibitor efficacy. Therefore, the prediction of PARPi efficacy could possibly be evaluated by assays that quantify the intrinsic PARP activity in cancer cells. An Enzyme Linked Immunosorbent Assay (ELISA) may be applied for a quantitative and sensitive assessment of PAR (poly[ADP]ribose) polymer levels in tumor specimens and in peripheral blood to assess the enzyme proficiency and to predict PARPi sensitivity [176].
Conclusions
Emerging data suggest that PARP inhibition is a potentially important strategy for managing a significant subset of tumors. The discovery of both germline and somatic DNA repair deficiencies in different cancer patients, together with the development of PARP inhibitors that can kill cancer cells with these defects, is a potent example of targeting therapy to molecularly defined tumor subtypes. The assessment of genomic instability assays and of nuclear foci status, together with the levels of DNA repairing proteins may predict the outcomes of PARPi treatment in different cancer types [183–187]. In the near future, the systematic evaluation of PAR levels, γH2AX, FANC and RAD51 foci with genomic instability features in tumor biopsies before, during and after treatment might help to identify patient populations who can be classified as responders or non-responders to PARP inhibitors. However, the heterogeneity of the tumor could always limit the evaluation of specific biomarkers, especially when the analysis is performed on a small amount of tissue (tissue microarrays, TMA) by IHC or FFPE.
Acknowledgements
We are grateful to Caterina Miro and Virginia Napolitano for helpful discussion. Aniello Cerrato is grateful to CNR for the Short Term Mobility Award in 2015. Francesco Morra is grateful to Programma Garanzia Giovani Regione Campania DGR 117/2014, PIP.
Funding
This work was supported by the Associazione Italiana Ricerca sul Cancro (AIRC n. 4952 to AC), by POR Campania FSE 2007/2013 “CREME Campania Research In Experimental Medicine”, to CNR-IEOS-UOS Napoli, and by the “Ministero dell’Istruzione, dell’Università e della Ricerca” (MIUR) (PRIN 2009T5NKTB_002 to AC).
Availability of data and materials
Not applicable.
Authors’ contributions
AC designed the review and wrote the manuscript, FM helped in the design of the Table and the review; AC reviewed the manuscript and Table. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Contributor Information
Aniello Cerrato, Phone: +39-0817463605, Email: a.cerrato@ieos.cnr.it.
Angela Celetti, Phone: +39-0817463605, Email: celetti@unina.it.
References
- 1.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, Wood RD. DNA repair: from molecular mechanism to human disease. DNA Repair. 2006;5(8):986–96. doi: 10.1016/j.dnarep.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003;532(1-2):103–15. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Fortini P, Pascucci B, Parlanti E, D’Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85(11):1053–71. doi: 10.1016/j.biochi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12(2):90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining Transl. Cancer Res. 2013;2(3):130–43. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Xie Y, Xian L. Breast cancer susceptibility gene 1 [BRCA1] predict clinical outcome in platinum- and toxal-based chemotherapy in non-small-cell lung cancer [NSCLC] patients: a system review and meta-analysis. J Exp Clin Cancer Res. 2013;32:15. doi: 10.1186/1756-9966-32-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leng XF, Chen MW, Xian L, Dai L, Ma GY, Li MH. Combined analysis of mRNA expression of ERCC1 BAG-1 BRCA1 RRM1 and TUBB3 to predict prognosis in patients with non-small cell lung cancer who received adjuvant chemotherapy. J Exp Clin Cancer Res. 2012;31:25. doi: 10.1186/1756-9966-31-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowalski M, Przybylowska K, Rusin P, Olszewski J, Morawiec-Sztandera A, Bielecka-Kowalska A, Pietruszewska W, Mlynarski W, Janusz S, Majsterek I. Genetic polymorphisms in DNA base excision repair gene XRCC1 and the risk of squamous cell carcinoma of the head and neck. J Exp Clin Cancer Res. 2009;28:37. doi: 10.1186/1756-9966-28-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. 2015;15(3):166–80. doi: 10.1038/nrc3891. [DOI] [PubMed] [Google Scholar]
- 12.Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18(1):8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 13.London RE. The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst) 2015;30:90–103. doi: 10.1016/j.dnarep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou J, Qiao X, Ye H, Yang Y, Zheng X, Zhao H, Liu S. Antisense inhibition of ATM gene enhances the radiosensitivity of head and neck squamous cell carcinoma in mice. J Exp Clin Cancer Res. 2008;27:56. doi: 10.1186/1756-9966-27-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao LB, Pan XM, Sun H, Wang X, Rao L, Li LJ, Liang WB, Lv ML, Yang WZ, Zhang L. The association between ATM D1853N polymorphism and breast cancer susceptibility: a meta-analysis. J Exp Clin Cancer Res. 2010;29:117. doi: 10.1186/1756-9966-29-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 2014;17:21–9. doi: 10.1016/j.dnarep.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci. 2008;13:5016–29. doi: 10.2741/3060. [DOI] [PubMed] [Google Scholar]
- 18.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 19.McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly[ADP-ribose] polymerase inhibitors. Cancer Res. 2010;70(13):5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341(6144):395–9. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9(12):M57–60. doi: 10.1016/S0962-8924(99)01661-X. [DOI] [PubMed] [Google Scholar]
- 22.Hartman JL, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291(5506):1001–4. doi: 10.1126/science.291.5506.1001. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature Rev Cancer. 2008;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 25.Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18(1):80–6. doi: 10.1016/j.gde.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 27.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–17. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 28.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–98. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 29.Furgason JM, Bahassi M. Targeting DNA repair mechanisms in cancer. Pharmacol Ther. 2013;137(3):298–308. doi: 10.1016/j.pharmthera.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31(8):372–80. doi: 10.1016/j.tips.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 32.Brough R, Frankum JR, Costa-Cabral S, Lord CJ, Ashworth A. Searching for synthetic lethality in cancer. Curr Opin Genet Dev. 2011;21(1):34–41. doi: 10.1016/j.gde.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 33.Nijman SM, Friend SH. Cancer. Potential of the synthetic lethality principle. Science. 2013;342(6160):809–11. doi: 10.1126/science.1244669. [DOI] [PubMed] [Google Scholar]
- 34.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20(17):4704–16. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 36.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly[ADP-ribose] polymerase. Nature. 2005;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 37.Liu T, Doty B, Gibson WD. Heyer Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17(10):1260–2. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowe BP, Glazer PM. Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms. Breast Cancer Res. 2010;12(2):203. doi: 10.1186/bcr2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–93. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS, Moreno V, Mateo J, Molife LR, De Bono J, Kaye S, Lord CJ, Ashworth A. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 41.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. 2014;343(6178):1466–70. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26(8):882–93. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 43.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly[ADP-ribose]: novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 44.Hakmé A, Wong HK, Dantzer F, Schreiber V. The expanding field of poly [ADP-ribosyl]ation reactions. EMBO Rep. 2008;9(11):1094–100. doi: 10.1038/embor.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPs a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008;13:3046–82. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 46.De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84(2):137–46. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 47.Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F, Shah GM. Role of poly[ADP-ribose] polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci. 2013;110(5):1658–63. doi: 10.1073/pnas.1209507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernández-Marcelo T, Frías C, Pascua I, de Juan C, Head J, Gómez A, Hernando F, Jarabo JR, Díaz-Rubio E, Torres AJ, Rouleau M, Benito M, Iniesta P. Poly [ADP-ribose] polymerase 3 [PARP3] a potential repressor of telomerase activity. J Exp Clin Cancer Res. 2014;33:19. doi: 10.1186/1756-9966-33-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Satoh MS, Lindahl T. Role of poly[ADP-ribose] formation in DNA repair. Nature. 1992;356(6367):356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 50.Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways including DNA crosslink repair mediate alternative end joining. PLoS Genet. 2015;11(1):e1004943. doi: 10.1371/journal.pgen.1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dantzer F, Amé JC, Schreiber V, Nakamura J, Ménissier-de Murcia J, de Murcia G. Poly[ADP-ribose] polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006;409:493–510. doi: 10.1016/S0076-6879(05)09029-4. [DOI] [PubMed] [Google Scholar]
- 52.Barkauskaite E, Jankevicius G, Ahel I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol Cell. 2015;58(6):935–46. doi: 10.1016/j.molcel.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Daniels CM, Ong SE, Leung AK. The Promise of Proteomics for the Study of ADP-Ribosylation Mol. Cell. 2015;58(6):911–24. doi: 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31(19):5526–33. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28(17):2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291X(63)90024-X. [DOI] [PubMed] [Google Scholar]
- 58.Ménissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22(9):2255–63. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber A, Bai P, Murcia JM, Murcia G. PARP-1 PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair. 2004;3(8-9):1103–8. doi: 10.1016/j.dnarep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 60.Szántó M, Brunyánszki A, Kiss B, Nagy L, Gergely P, Virág L, Bai P. Poly[ADP-ribose] polymerase-2: emerging transcriptional roles of a DNA-repair protein. Cell Mol Life Sci. 2012;69(24):4079–92. doi: 10.1007/s00018-012-1003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riffell JL, Lord CJ, Ashworth A. Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov. 2012;11(12):923–36. doi: 10.1038/nrd3868. [DOI] [PubMed] [Google Scholar]
- 62.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions physiological outcomes and clinical targets. Mol Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weaver AN, Yang ES. Beyond DNA repair: additional functions of PARP-1 in cancer. Front Oncol. 2013;3:290. doi: 10.3389/fonc.2013.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Mol Aspects Med. 2013;34(6):1109–23. doi: 10.1016/j.mam.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 65.Lodhi N, Kossenkov AV, Tulin AV. Bookmarking promoters in mitotic chromatin: poly[ADP-ribose]polymerase-1 as an epigenetic mark. Nucleic Acids Res. 2014;42(11):7028–38. doi: 10.1093/nar/gku415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hottiger MO. Nuclear ADP-Ribosylation and its Role in Chromatin Plasticity Cell Differentiation and Epigenetics. Annu Rev Biochem. 2015;84:227–63. doi: 10.1146/annurev-biochem-060614-034506. [DOI] [PubMed] [Google Scholar]
- 67.Bai P. Biology of Poly[ADP-Ribose] Polymerases: The Factotums of Cell Maintenance. Mol Cell. 2015;58(6):947–58. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- 68.Bock FJ, Todorova TT, Chang P. RNA Regulation by Poly[ADP-Ribose] Polymerases. Mol Cell. 2015;58(6):959–69. doi: 10.1016/j.molcel.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Ørntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 70.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 71.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 72.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong-Brown MW, Meldrum CJ, Carpenter JE, Clarke CL, Narod SA, Jakubowska A, Rudnicka H, Lubinski J, Scott RJ. Prevalence of BRCA1 and BRCA2 germline mutations in patients with triple-negative breast cancer. Breast Cancer Res Treat. 2015;150(1):71–80. doi: 10.1007/s10549-015-3293-7. [DOI] [PubMed] [Google Scholar]
- 74.Salo-Mullen EE, O’Reilly EM, Kelsen DP, Ashraf AM, Lowery MA, Yu KH, Reidy DL, Epstein AS, Lincoln A, Saldia A, Jacobs LM, Rau-Murthy R, Zhang L, Kurtz RC, Saltz L, Offit K, Robson ME, Stadler ZK. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015;121(24):4382–8. doi: 10.1002/cncr.29664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maia S, Cardoso M, Paulo P, Pinheiro M, Pinto P, Santos C, Pinto C, Peixoto A, Henrique R, Teixeira MR. The role of germline mutations in the BRCA1/2 and mismatch repair genes in men ascertained for early-onset and/or familial prostate cancer. Fam Cancer. 2016;15(1):111–21. doi: 10.1007/s10689-015-9832-x. [DOI] [PubMed] [Google Scholar]
- 76.Bielorai B, Fisher T, Waldman D, Lerenthal Y, Nissenkorn A, Tohami T, Marek D, Amariglio N, Toren A. Acute lymphoblastic leukemia in early childhood as the presenting sign of ataxia-telangiectasia variant. Pediatr Hematol Oncol. 2013;30(6):574–82. doi: 10.3109/08880018.2013.777949. [DOI] [PubMed] [Google Scholar]
- 77.Varon R, Reis A, Henze G, von Einsiedel HG, Sperling K, Seeger K. Mutations in the Nijmegen Breakage Syndrome gene (NBS1) in childhood acute lymphoblastic leukemia (ALL) Cancer Res. 2001;61(9):3570–2. [PubMed] [Google Scholar]
- 78.Arora H, Chacon AH, Choudhary S, McLeod MP, Meshkov L, Nouri K, Izakovic J. Bloom syndrome. Int J Dermatol. 2014;53(7):798–802. doi: 10.1111/ijd.12408. [DOI] [PubMed] [Google Scholar]
- 79.Ponti G, Castellsagué E, Ruini C, Percesepe A, Tomasi A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin Genet. 2015;87(6):507–16. doi: 10.1111/cge.12529. [DOI] [PubMed] [Google Scholar]
- 80.Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22(8):898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 81.Zaremba T, Curtin NJ. PARP inhibitor development for systemic cancer targeting. Anticancer Agents Med Chem. 2007;7(5):515–23. doi: 10.2174/187152007781668715. [DOI] [PubMed] [Google Scholar]
- 82.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8(4):363–9. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 83.Sandhu SK, Yap TA, de Bono JS. Poly[ADP-ribose] polymerase inhibitors in cancer treatment: a clinical perspective. Eur J Cancer. 2010;46(1):9–20. doi: 10.1016/j.ejca.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 84.Kummar S, Chen A, Parchment RE, Kinders RJ, Ji J, Tomaszewski JE, Doroshow JH. Advances in using PARP inhibitors to treat cancer. BMC Med. 2012;10:25. doi: 10.1186/1741-7015-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murai SY, Huang BB, Das A, Renaud Y, Zhang JH, Doroshow J, Ji S, Takeda Y. Pommier Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas. 2015;81(1):5–9. doi: 10.1016/j.maturitas.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 87.Steffen JD, Tholey RM, Langelier MF, Planck JL, Schiewer MJ, Lal S, Bildzukewicz NA, Yeo CJ, Knudsen KE, Brody JR, Pascal JM. Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res. 2014;74(1):31–7. doi: 10.1158/0008-5472.CAN-13-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–43. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H. Phase I study of the poly [ADP-ribose] polymerase inhibitor AG014699 in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–23. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drew Y, Ledermann JA, Jones A, et al. Phase II trial of the poly[ADP-ribose]polymerase [PARP] inhibitor AG-014699 in BRCA 1 and 2 mutated advanced ovarian and/or locally advanced or metastatic breast cancer. J Clin Oncol. 2011;29(Suppl):3104. [Google Scholar]
- 92.Kristeleit RBH, Lorusso P, Patel M, et al. Phase 1 study of continuous oral rucaparib: analysis of patient subgroup with ovarian/peritoneal cancer. J Clin Oncol. 2013;31(Suppl):2585. [Google Scholar]
- 93.Fong P, Boss D, Carden C, Roelvink M, De Greve J, Gourley C. AZD2281 [KU-0059436] a PARP [poly ADP-ribose polymerase] inhibitor with single agent anticancer activity in patients with BRCA deficient ovarian cancer: results from a phase I study. J Clin Oncol. 2008;26(15S):Abstract 551. [Google Scholar]
- 94.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly[ADP-ribose] polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 95.Mendeleyev J, Kirsten E, Hakam A, Buki KG, Kun E. Potential chemotherapeutic activity of 4-iodo-3-nitrobenzamide. Metabolic reduction to the 3-nitroso derivative and induction of cell death in tumor cells in culture. Biochem Pharmacol. 1995;50(5):705–14. doi: 10.1016/0006-2952(95)00189-7. [DOI] [PubMed] [Google Scholar]
- 96.Huggins-Puhalla SL, Beumer JH, Appleman LJ, et al. A phase I study of chronically dosed singleagent veliparib [ABT-888] in patients [pts] with either BRCA 1/2-mutated cancer [BRCA+] platinumrefractory ovarian cancer or basal-like breast cancer [BRCA-wt] J Clin Oncol. 2012;30(Suppl):3054. [Google Scholar]
- 97.O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M, Richards P, Specht JM, Yardley DA, Carlson RW, Finn RS, Charpentier E, Garcia-Ribas I, Winer EP. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32(34):3840–7. doi: 10.1200/JCO.2014.55.2984. [DOI] [PubMed] [Google Scholar]
- 98.Mateo J, Ong M, Tan DS, Gonzalez MA, de Bono JS. Appraising iniparib the PARP inhibitor that never was—What must we learn? Nat Rev Clin Oncol. 2013;10(12):688–96. doi: 10.1038/nrclinonc.2013.177. [DOI] [PubMed] [Google Scholar]
- 99.Sinha G. Downfall of iniparib: a PARP inhibitor that doesn’t inhibit PARP after all. J Natl Cancer Inst. 2014;106(1):djt447. doi: 10.1093/jnci/djt447. [DOI] [PubMed] [Google Scholar]
- 100.Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: A Poly[ADP-ribose] Polymerase [PARP] Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J Med Chem. 2015;58(8):3302–14. doi: 10.1021/jm5018237. [DOI] [PubMed] [Google Scholar]
- 101.Sandhu SK, Wenham RM, Wilding G, McFadden M, Sun L, Toniatti C, Stroh M, Carpenter CL, De Bono JS, Schelman WR. First-in human trial of a poly[ADP-ribose] polymerase [PARP] inhibitor MK-4827 in advanced cancer patients [pts] with antitumor activity in BRCA-deficient and sporadic ovarian cancers. J Clin Oncol. 2010;28(15s):Abstract 3001. [Google Scholar]
- 102.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM. The poly[ADP-ribose] polymerase inhibitor niraparib [MK4827] in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–92. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 103.De Bono JS, Mina LA, Gonzalez-Hernandez M, et al. First-in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumors. J Clin Oncol. 2013;31(Suppl):2580. [Google Scholar]
- 104.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord J, Post LE, Ashworth A. BMN 673 a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–15. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miknyoczki S, Chang H, Grobelny J, Pritchard S, Worrell C, McGann N, Ator M, Husten J, Deibold J, Hudkins R, Zulli A, Parchment R, Ruggeri B. The selective poly [ADP-ribose] polymerase-1 [2] inhibitor CEP-8983 increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol Cancer Ther. 2007;6(8):2290–302. doi: 10.1158/1535-7163.MCT-07-0062. [DOI] [PubMed] [Google Scholar]
- 106.Russo AL, Kwon H-C, Burgan WE, Carter D, Beam K, Weizheng X, Zhang J, Slusher BS, Chakravarti A, Tofilon PJ, Camphausen K. In vitro and in vivo radiosensitization of glioblastoma cells by the poly [ADP-ribose] polymerase inhibitor E7016. Clin Cancer Res. 2009;15(2):607–12. doi: 10.1158/1078-0432.CCR-08-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action repair and resistance. Curr Mol Pharmacol. 2012;5(1):102–14. doi: 10.2174/1874467211205010102. [DOI] [PubMed] [Google Scholar]
- 108.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 109.Liu LV, Bell CB, 3rd, Wong SD, Wilson SA, Kwak Y, Chow MS, Zhao J, Hodgson KO, Hedman B, Solomon EI. Definition of the intermediates and mechanism of the anticancer drug bleomycin using nuclear resonance vibrational spectroscopy and related methods. Proc Natl Acad Sci. 2010;107(52):22419–24. doi: 10.1073/pnas.1016323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 111.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J. Oral poly[ADP-ribose] polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 112.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB. Poly[ADP]-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 113.Elit L, Hirte H. Palliative systemic therapy for women with recurrent epithelial ovarian cancer: current options. Onco Targets Ther. 2013;6:107–18. doi: 10.2147/OTT.S30238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 115.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A’Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373(18):1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 117.Holstege H, Horlings HM, Velds A, Langerød A, Børresen-Dale AL, van deVijver MJ, Nederlof PM, Jonkers J. BRCA1-mutated and basal-like breast cancers have similar aCGH profiles and a high incidence of protein truncating TP53 mutations. BMC Cancer. 2010;10:654. doi: 10.1186/1471-2407-10-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, Carey LA. Poly[ADP-ribose] polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res. 2010;16(19):4702–10. doi: 10.1158/1078-0432.CCR-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C. Iniparib plus chemotherapy in metastatic triplenegativebreast cancer. N Engl J Med. 2011;364(3):205–14. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 120.Kim A, Ueda Y, Naka T, Enomoto T. Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res. 2012;31:14. doi: 10.1186/1756-9966-31-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eckstein N. Platinum resistance in breast and ovarian cancer cell lines. J Exp Clin Cancer Res. 2011;30:91. doi: 10.1186/1756-9966-30-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Levine DA, Cannistra SA. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2 multicentre open-label non-randomised study. Lancet Oncol. 2011;12(9):852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 124.De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. 2014;16(1):43–72. doi: 10.1593/neo.131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–9. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 126.Olopade OI, Wei M. FANCF methylation contributes to chemoselectivity in ovarian cancer. Cancer Cell. 2003;3(5):417–20. doi: 10.1016/S1535-6108(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 127.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly[ADP ribose] polymerase inhibition. Cancer Res. 2006;66(16):8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 128.Rodriguez AA, Makris A, Wu MF, Rimawi M, Froehlich A, Dave B, Hilsenbeck SG, Chamness GC, Lewis MT, Dobrolecki LE, Jain D, Sahoo S, Osborne CK, Chang JC. DNA repair signature is associated with anthracycline response in triple negative breast cancer patients. Breast Cancer Res Treat. 2010;123(1):189–96. doi: 10.1007/s10549-010-0983-z. [DOI] [PubMed] [Google Scholar]
- 129.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, Newell DR, D’Andrea AD, Curtin NJ, Wong KK, Shapiro GI. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17(7):875–82. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A, Chelliah R, Curtin N, Farzaneh F, Shall S, Mufti GJ. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica. 2013;98(9):1397–406. doi: 10.3324/haematol.2012.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smith MA, Hampton OA, Reynolds CP, Kang MH, Maris JM, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Wu J, Kurmasheva RT, Wheeler DA, Houghton PJ. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer. 2015;62(1):91–8. doi: 10.1002/pbc.25201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6-7):315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A, Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53–75. doi: 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 134.Tentori L, Ricci-Vitiani L, Muzi A, Ciccarone F, Pelacchi F, Calabrese R, Runci D, Pallini R, Caiafa P, Graziani G. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer. 2014;14:151. doi: 10.1186/1471-2407-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buisson R, Dion-Côté AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17(10):1247–54. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, Gentileschi MP, Barilà D, Soddu S. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013;32:95. doi: 10.1186/1756-9966-32-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, Kazakoff S, Quek K, Wilhelm-Benartzi C, Curry E, Leong HS, Australian Ovarian Cancer Study Group. Hamilton A, Mileshkin L, Au-Yeung G, Kennedy C, Hung J, Chiew YE, Harnett P, Friedlander M, Quinn M, Pyman J, Cordner S, O’Brien P, Leditschke J, Young G, Strachan K, Waring P, Azar W, Mitchell C, Traficante N, Hendley J, Thorne H, Shackleton M, Miller DK, Arnau GM, Tothill RW, Holloway TP, Semple T, Harliwong I, Nourse C, Nourbakhsh E, Manning S, Idrisoglu S, Bruxner TJ, Christ AN, Poudel B, Holmes O, Anderson M, Leonard C, Lonie A, Hall N, Wood S, Taylor DF, Xu Q, Fink JL, Waddell N, Drapkin R, Stronach E, Gabra H, Brown R, Jewell A, Nagaraj SH, Markham E, Wilson PJ, Ellul J, McNally O, Doyle MA, Vedururu R, Stewart C, Lengyel E, Pearson JV, Waddell N, deFazio A, Grimmond SM, Bowtell DD. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–94. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 138.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 140.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grützmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, Australian Pancreatic Cancer Genome Initiative. Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM, Whole genomes redefine the mutational landscape of pancreatic cancer Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, Palanisamy N, Siddiqui J, Yan W, Cao X, Mehra R, Sabolch A, Basrur V, Lonigro RJ, Yang J, Tomlins SA, Maher CA, Elenitoba-Johnson KS, Hussain M, Navone NM, Pienta KJ, Varambally S, Feng FY, Chinnaiyan AM. Mechanistic rationale for inhibition of poly[ADP-ribose] polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19(5):664–78. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Brenner JC, Feng FY, Han S, Patel S, Goyal S, Bou-Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA, Chinnaiyan AM. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012;72(7):1608–13. doi: 10.1158/0008-5472.CAN-11-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12 a kinase that promotes homologous recombination repair and resistance to cisplatin and poly[ADP-ribose] polymerase inhibitors. J Biol Chem. 2014;289(13):9247–53. doi: 10.1074/jbc.M114.551143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, Chen L, Kozarewa I, Assiotis I, Fenwick K, Natrajan R, Lord CJ, Ashworth A. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–97. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cheng H, Zhang Z, Borczuk A, Powell CA, Balajee AS, Lieberman HB, Halmos B. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1-low non-small cell lung cancer cells. Carcinogenesis. 2013;34(4):739–49. doi: 10.1093/carcin/bgs393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Postel-Vinay S, Bajrami I, Friboulet L, Elliott R, Fontebasso Y, Dorvault N, Olaussen KA, André F, Soria JC, Lord CJ, Ashworth A. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32(47):5377–87. doi: 10.1038/onc.2013.311. [DOI] [PubMed] [Google Scholar]
- 148.Merolla F, Luise C, Muller MT, Pacelli R, Fusco A, Celetti A. Loss of CCDC6 the first identified RET partner gene affects pH2AX S139 levels and accelerates mitotic entry upon DNA damage. PLoS ONE. 2012;7(5):e36177. doi: 10.1371/journal.pone.0036177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morra F, Luise C, Visconti R, Staibano S, Merolla F, Ilardi G, Guggino G, Paladino S, Sarnataro D, Franco R, Monaco R, Zitomarino F, Pacelli R, Monaco G, Rocco G, Cerrato A, Linardopoulos S, Muller MT, Celetti A. New therapeutic perspectives in CCDC6 deficient lung cancer cells. Int J Cancer. 2015;136(9):2146–57. doi: 10.1002/ijc.29263. [DOI] [PubMed] [Google Scholar]
- 150.Morra F, Luise C, Merolla F, Poser I, Visconti R, Ilardi G, Paladino S, Inuzuka H, Guggino G, Monaco R, Colecchia D, Monaco G, Cerrato A, Chiariello M, Denning K, Claudio PP, Staibano S, Celetti A. FBXW7 and USP7 regulate CCDC6 turnover during the cell cycle and affect cancer drugs susceptibility in NSCLC. Oncotarget. 2015;6(14):12697–709. doi: 10.18632/oncotarget.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Malapelle U, Morra F, Ilardi G, Visconti R, Merolla F, Cerrato A, Napolitano V, Monaco R, Guggino G, Monaco G, Staibano S, Troncone G, Celetti A. USP7 inhibitors downregulating CCDC6 sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer. 2016; doi: 10.1016/j.lungcan. [DOI] [PubMed]
- 152.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stec E, Locco L, Szymanski S, Bartz SR, Toniatti C, Needham RH, Palmieri A, Carleton M, Cleary MA, Jackson AL, Linsley PS, Strulovici B, Ferrer M, Santini F. A multiplexed siRNA screening strategy to identify genes in the PARP pathway. J Biomol Screen. 2012;17(10):1316–28. doi: 10.1177/1087057112453071. [DOI] [PubMed] [Google Scholar]
- 154.Neijenhuis S, Bajrami I, Miller R, Lord CJ, Ashworth A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair (Amst) 2013;12(6):394–402. doi: 10.1016/j.dnarep.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 155.Wiltshire TD, Lovejoy CA, Wang T, Xia F, O’Connor MJ, Cortez D. Sensitivity to poly[ADP-ribose] polymerase [PARP] inhibition identifies ubiquitin-specific peptidase 11 [USP11] as a regulator of DNA double-strand break repair. J Biol Chem. 2010;285(19):14565–71. doi: 10.1074/jbc.M110.104745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2008;7(12):2010–9. doi: 10.1016/j.dnarep.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 157.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27(9):1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marquard AM, Eklund AC, Joshi T, Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi Z, Birkbak NJ. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3:9. doi: 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, Bowman-Colin C, Li Y, Greene-Colozzi A, Iglehart JD, Tung N, Ryan PD, Garber JE, Silver DP, Szallasi Z, Richardson AL. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2(4):366–75. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, Tran TV, Williams D, Iliev D, Jammulapati S, FitzGerald LM, Krivak T, DeLoia JA, Gutin A, Mills GB, Lanchbury JS. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–82. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, Houdayer C, Sastre-Garau X, Vincent-Salomon A, Stoppa-Lyonnet D, Stern MH. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–62. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 162.Baumbusch LO, Helland Å, Wang Y, Liestøl K, Schaner ME, Holm R, Etemadmoghadam D, Alsop K, Brown P, Australian Ovarian Cancer Study Group. Mitchell G, Fereday S, DeFazio A, Bowtell DD, Kristensen GB, Lingjærde OC, Børresen-Dale AL. High levels of genomic aberrations in serous ovarian cancers are associated with better survival. PLoS ONE. 2013;8(1):e54356. doi: 10.1371/journal.pone.0054356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Donnell RL O, McCormick A, Mukhopadhyay A, Woodhouse LC, Moat M, Grundy A, Dixon M, Kaufman A, Soohoo S, Elattar A, Curtin NJ, Edmondson RJ. The use of ovarian cancer cells from patients undergoing surgery to generate primary cultures capable of undergoing functional analysis. PLoS ONE. 2014;9(6):e90604. doi: 10.1371/journal.pone.0090604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Patterson MJ, Sutton RE, Forrest I, Sharrock R, Lane M, Kaufmann A, O’Donnell R, Edmondson RJ, Wilson BT, Curtin NJ. Assessing the function of homologous recombination DNA repair in malignant pleural effusion (MPE) samples. Br J Cancer. 2014;111(1):94–100. doi: 10.1038/bjc.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lim D, Ngeow J. Evaluation of the methods to identify patients who may benefit from PARP inhibitor use. Endocr Relat Cancer. 2016;23(6):R267–85. doi: 10.1530/ERC-16-0116. [DOI] [PubMed] [Google Scholar]
- 166.Zhang S, Yuan Y, Hao D. A genomic instability score in discriminating nonequivalent outcomes of BRCA1/2 mutations and in predicting outcomes of ovarian cancer treated with platinum-based chemotherapy. PLoS ONE. 2014;9(12):e113169. doi: 10.1371/journal.pone.0113169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, Parchment RE. Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly [ADP-ribose] polymerase in tumor biopsies of mouse xenografts. Clin Cancer Res. 2008;14(21):6877–85. doi: 10.1158/1078-0432.CCR-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle S, Los G, Hostomsky Z, Edmondson RJ, Curtin NJ. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly[ADP-ribose] polymerase inhibitors. Clin Cancer Res. 2010;16(8):2344–51. doi: 10.1158/1078-0432.CCR-09-2758. [DOI] [PubMed] [Google Scholar]
- 169.Graeser MK, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis-Filho J, Dowsett M, Ashworth A, Turner N. A marker of homologous recombination predicts pathological complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16(24):6159–68. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249–62. doi: 10.1016/S1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 171.Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24(13):5850–62. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Duan W, Gao L, Zhao W, Leon M, Sadee W, Webb A, Resnick K, Wu X, Ramaswamy B, Cohn DE, Shapiro C, Andreassen PR, Otterson GA, Villalona-Calero MA. Assessment of FANCD2 nuclear foci formation in paraffin-embedded tumors: a potential patient-enrichment strategy for treatment with DNA interstrand crosslinking agents. Transl Res. 2013;161(3):156–64. doi: 10.1016/j.trsl.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, Parchment RE, Doroshow JH, Pommier Y. Histone {gamma}H2AX and Poly[ADP-Ribose] as Clinical Pharmacodynamic Biomarkers. Clin Cancer Res. 2010;16(18):4532–42. doi: 10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Avondoglio D, Scott T, Kil WJ, Sproull M, Tofilon PJ, Camphausen K. High throughput evaluation of gamma-H2AX. Radiat Oncol. 2009;4:31. doi: 10.1186/1748-717X-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Garty G, Chen Y, Salerno A, Turner H, Zhang J, Lyulko O, Bertucci A, Xu Y, Wang H, Simaan N, Randers-Pehrson G, Yao YL, Amundson SA, Brenner DJ. The RABIT: a rapid automated biodosimetry tool for radiological triage. Health Phys. 2010;98(2):209–17. doi: 10.1097/HP.0b013e3181ab3cb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu X, Palma J, Kinders R, Shi Y, Donawho C, Ellis PA, Rodriguez LE, Colon-Lopez M, Saltarelli M, LeBlond D, Lin CT, Frost DJ, Luo Y, Giranda VL. An enzyme-linked immunosorbentpoly[ADP-ribose] polymerase biomarker assay for clinical trials of PARP inhibitors. Anal Biochem. 2008;381(2):240–7. doi: 10.1016/j.ab.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 177.Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, Ji J, Monks A, Low JA, Chen A, Murgo AJ, Collins J, Steinberg SM, Eliopoulos H, Giranda VL, Gordon G, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH. Phase 0 clinical trial of the poly [ADP-ribose] polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27(16):2705–11. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res. 2009;7(8):1304–9. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, Issaeva N, Sleeth K, Sharma RA, Helleday T. Poly[ADP-ribose] polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70(13):5389–98. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 180.Brustmann H. Poly[adenosine diphosphate-ribose] polymerase expression in serous ovarian carcinoma: correlation with p53 MIB-1 and outcome. Int J Gynecol Pathol. 2007;26(2):147–53. doi: 10.1097/01.pgp.0000235064.93182.ec. [DOI] [PubMed] [Google Scholar]
- 181.Zaremba T, Ketzer P, Cole M, Coulthard S, Plummer ER, Curtin NJ. Poly[ADP-ribose] polymerase-1 polymorphisms expression and activity in selected human tumour cell lines. Br J Cancer. 2009;101(2):256–62. doi: 10.1038/sj.bjc.6605166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Goncalves A, Finetti P, Sabatier R, Gilabert M, Adelaide J, Borg JP, Chaffanet M, Viens P, Birnbaum D, Bertucci F. Poly[-ADP- ribose] polymerase-1 mRNA expression in human breast cancer: a meta-analysis. Breast Cancer Res Treat. 2010;127(1):273–81. doi: 10.1007/s10549-010-1199-y. [DOI] [PubMed] [Google Scholar]
- 183.Bepler G, Begum M, Simon GR. Molecular analysis-based treatment strategies for nonsmall cell lung cancer. Cancer Control. 2008;15(2):130–9. doi: 10.1177/107327480801500205. [DOI] [PubMed] [Google Scholar]
- 184.Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, Tursz T, Le Chevalier T, Soria JC. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355(10):983–91. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 185.Sinicrope F, Foster NR, Sargent DJ, Thibodeau SN, Smyrk TC, O’Connell MJ. Model-based prediction of defective DNA mismatch repair using clinicopathological variables in sporadic colon cancer patients. Cancer. 2010;116(7):1691–8. doi: 10.1002/cncr.24913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang LE, Hu Z, Sturgis EM, Spitz MR, Strom SS, Amos CI, Guo Z, Qiao Y, Gillenwater AM, Myers JN, Clayman GL, Weber RS, El-Naggar AK, Mao L, Lippman SM, Hong WK, Wei Q. Reduced DNA repair capacity for removing tobacco carcinogen- induced DNA adducts contributes to risk of head and neck cancer but not tumor characteristics. Clin Cancer Res. 2010;16(2):764–74. doi: 10.1158/1078-0432.CCR-09-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Asakawa H, Koizumi H, Koike A, Takahashi M, Wu W, Iwase H, Fukuda M, Ohta T. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010;12(2):R17. doi: 10.1186/bcr2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.