

Abstract
Objective:
To determine the safety of intrathecal autologous adipose-derived mesenchymal stromal cell treatment for amyotrophic lateral sclerosis (ALS).
Methods:
Participants with ALS were enrolled and treated in this phase I dose-escalation safety trial, ranging from 1 × 107 (single dose) to 1 × 108 cells (2 monthly doses). After intrathecal treatments, participants underwent standardized follow-up, which included clinical examinations, revised ALS Functional Rating Scale (ALSFRS-R) questionnaire, blood and CSF sampling, and MRI of the neuroaxis.
Results:
Twenty-seven patients with ALS were enrolled and treated in this study. The safety profile was positive, with the most common side effects reported being temporary low back and radicular leg pain at the highest dose level. These clinical findings were associated with elevated CSF protein and nucleated cells with MRI of thickened lumbosacral nerve roots. Autopsies from 4 treated patients did not show evidence of tumor formation. Longitudinal ALSFRS-R questionnaires confirmed continued progression of disease in all treated patients.
Conclusions:
Intrathecal treatment of autologous adipose-derived mesenchymal stromal cells appears safe at the tested doses in ALS. These results warrant further exploration of efficacy in phase II trials.
Classification of evidence:
This phase I study provides Class IV evidence that in patient with ALS, intrathecal autologous adipose-derived mesenchymal stromal cell therapy is safe.
Amyotrophic lateral sclerosis (ALS) is a devastating progressive paralytic disorder due to the death of motor neurons in the brain and spinal cord. The average survival after the onset of symptoms is 3 years, and there are no adequate therapies.1 The pathomechanisms of ALS are complex and insufficiently understood.2
There has been increased interest in the potential of cell-based therapies to treat ALS. One cell type, the mesenchymal stromal cell (MSC), has been suggested as a possible therapy. MSCs secrete multiple neurotrophic growth factors and cytokines and have the ability to modulate the immune system, all of which may influence the progression of ALS.3
We have now completed a phase I dose-escalation safety clinical trial that used intrathecal delivery of adipose-derived autologous MSCs to treat advanced ALS.
METHODS
Criteria for enrollment in the study included the diagnosis of definite ALS based on El Escorial criteria, a vital capacity of >65% predicted for age and biometrics, no major medical comorbidities, and symptoms of weakness lasting >1 year but <2 years. Patients were sequentially enrolled into 5 groups of escalating MSC dose: group 1 (1 × 107, one injection), group 2 (5 × 107, one injection), group 3 (5 × 107, 2 monthly injections), group 4 (1 × 108, one injection), and group 5 (1 × 108, 2 monthly injections).
Enrolled participants underwent an open adipose biopsy of abdominal fat ≥8 weeks before MSC therapy. After isolation from the adipose biopsy, MSCs were expanded and cryopreserved in the Mayo Clinic Human Cellular Therapy Laboratory (e-Methods at Neurology.org). MSCs were suspended in lactated Ringer solution and infused intrathecally after a standard lumbar puncture. Four weekly follow-up visits took place after treatment and included review of symptom diary, vital signs, revised ALS Functional Rating Scale (ALSFRS-R) questionnaire, neurologic examination, physical examination, blood sample (weeks 1 and 3), CSF samples (weeks 1 and 4), and gadolinium-enhanced MRI of the brain and spinal cord (week 3). In participants receiving the 2 monthly treatments, the weekly follow-up visits were repeated for a total of 8 weeks. Subsequently, participants were followed up every 3 months until 2 years, death, or loss to follow-up.
RESULTS
After providing Institutional Review Board–approved informed consent, 27 patients with clinically definite ALS by El Escorial criteria were enrolled and treated in this dose-escalation safety clinical trial (table). The median age was 57 years (range 36–75 years), and the ratio of male to female patients was 15:12. The median ALSFRS-R at enrollment was 39 (range 27–47). Twenty of 27 patients had spinal-onset ALS, and 7 of 27 had bulbar onset.
Table.
Demographics and study data
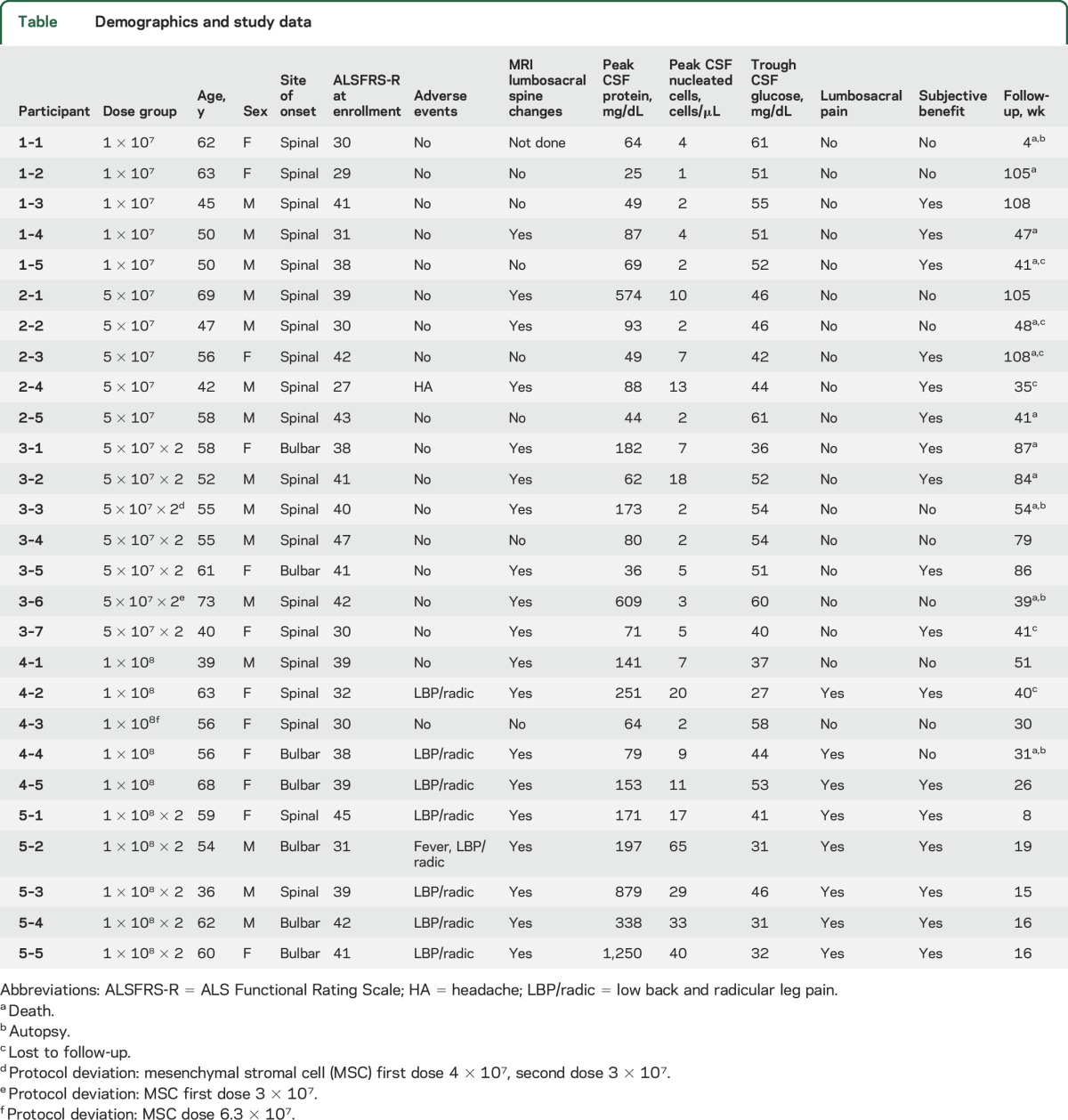
Adverse events that were considered to be possibly, probably, or definitely related to intrathecal MSCs were observed in a dose-dependent fashion. Participant 2-4 had 24 hours of headache after treatment (not position dependent). Eight of the 10 participants receiving 1 × 108 MSC dose developed low back and leg pain that began within a week of treatment, peaked in the first 3 weeks, and then resolved or became very mild (1 of 10 on visual analog scale). The pain was exacerbated by the straight leg raise test and exhibited variable intensity (2–8 of 10 on visual analog scale) and duration (range 42–77 days) among the participants. Analgesics were used during pain in 5 of 8 patients, including nonsteroidal anti-inflammatory drugs (5), gabapentin (3), and opioids (2). In participant 5-2, the pain led to postponement of the second treatment by 2 months until pain had resolved. The pain recurred on the second injection, lasting 2 months.
Although the study was not designed for efficacy, ALSFRS-R questionnaires were performed throughout the study and showed progression in all patients that did not appear, and was not reported by any patients, to be more rapid than before therapy (figure 1). Notably, 17 of 29 participants reported specific mild temporary subjective clinical improvements (typically lasting <2 months), which were reported (with overlap) as improved bulbar function (n = 8), increased limb strength (n = 5), decreased fasciculations (n = 4), decreased stiffness (n = 4), and improved energy (n = 4). Because this trial was not blinded or placebo controlled, these effects cannot be interpreted as a positive treatment response to MSC treatment.
Figure 1. ALS Functional Rating scale–revised (ALSFRS-R).
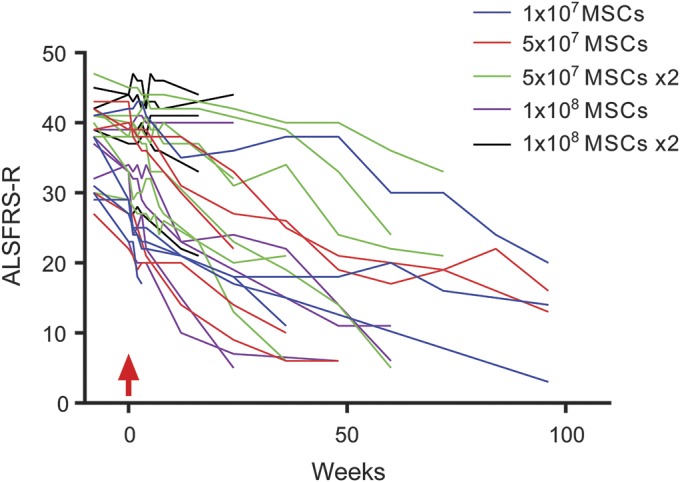
ALSFRS-R demonstrated progression during the duration of follow-up. Mesenchymal stromal cell (MSC) treatment occurred at week 0 (red arrow).
Lumbar punctures were performed at weeks 1 and 4 after intrathecal MSC treatment. Dose-dependent alterations of CSF parameters were observed. In aggregate within the different doses, CSF protein increased from baseline (mean = 56.4 mg/dL, SEM = 4.0 mg/dL) to week 1 (mean = 93.4 mg/dL, SEM = 18.7 mg/dL) and week 4 (mean = 189.2 mg/dL, SEM = 111.0 mg/dL) after intrathecal MSCs administration. There was also an increase in CSF nucleated cells (monocytes) from baseline (1.2 cells/μL, SEM 0.3 cell/μL) to week 1 (mean = 9.2 cells/μL, SEM = 2.5 cells/μL) that began to decrease by week 4 (mean = 4.7 cells/μL, SEM = 1.5 cells/μL). Glucose decreased slightly between baseline and weeks 1 and 4. No abnormal CSF oligoclonal bands or neoplastic cytology was observed in any sample. Grouped data by dose level are reported in figure 2, and individual data are included in the table.
Figure 2. MRI and CSF findings in patients receiving intrathecal mesenchymal stromal cells (MSCs).

Nodularity of lumbosacral nerve roots (red arrows) was observed in patients 3 weeks after treatment, as shown on MRIs from patient 2-1 (A, axial T2-weighted sequence; B, sagittal T2-weighted sequence; and C, sagittal gadolinium-enhanced T1-weighted sequence). (D) Elevations in CSF protein and nucleated cells were seen in a dose-dependent fashion.
MRI of the brain and spinal cord was completed, when tolerated, at week 3 after intrathecal MSCs. Abnormalities were observed in the lumbosacral nerve roots in 1 of 5 patients receiving 1 × 107, 9 of 12 receiving 5 × 107, and 9 of 10 in the 1 × 108 dose group. In these cases, the primary finding was lumbosacral nerve root thickening with clumping and gadolinium enhancement, reminiscent of arachnoiditis, which increased in severity with increasing dose (figure 2). In participant 2-1, the first participant whom we observed to have clear lumbosacral nerve root abnormalities, repeat MRI was performed at week 22 after treatment and was unchanged. CSF in participant 2-1 was also repeated at week 22 and normalized.
Four patients who died in this study underwent autopsy at a range of 31 days to 54 weeks after MSC treatment (3 of whom had abnormalities on MRI lumbosacral spine). Gross pathology did not reveal any signs of tumor formation or frank arachnoiditis in any of these cases.
DISCUSSION
Overall, intrathecal administration of MSCs in patients with ALS was safe. We observed dose-dependent changes in MRI of lumbosacral spine, CSF parameters, and temporary lumbosacral-radicular pain, all of which were tolerable. We postulate that this temporary pain relates to nerve root irritation and inflammation from MSC administration. Notably, in the 4 patients who underwent autopsies, there were no signs of tumor formation, which has been a concern with cell-based therapies. Studies are ongoing to investigate autopsy material for microscopic signs of MSC engraftment or neuroinflammation.
This phase I clinical trial of intrathecal adipose-derived MSC therapy in patients with definite ALS by El Escorial criteria corroborates other studies that have demonstrated safety of intrathecal bone marrow–derived MSCs in ALS.4–7 Although still debated, MSCs from adipose and bone marrow have similar properties, and adipose harvesting is a simpler, well-tolerated procedure. These safety data support the exploration of adipose-derived MSCs in other neurologic conditions.
Our data do not directly address the efficacy of intrathecal MSC therapy in ALS; however, we feel that the excellent safety profile justifies further investigation of this product. Notably, while we report anecdotal mild temporary subjective clinical improvements in our treatment regimen, we did not see miraculous improvements in or cessation of progression in any patients. These data may temper the sometimes irrationally exuberant enthusiasm for this therapy modality. Future studies are required to determine the efficacy and whether earlier intervention or additional MSC treatments are required to produce meaningful results. In this small phase I study, 1 × 108 MSCs were tolerated, yet this dose also caused more painful adverse effects. Our planned multidose phase II study initiates treatment at 1 × 108 MSCs but incorporates an algorithm that allows lower doses when there is protracted pain.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients with ALS and their families for volunteering for this study. The authors thank Delana Weis, Michelle Turner, Pam Vicari, Nancy Archer, and Jane Meyer for support and study coordination for this study. The authors acknowledge Drs. Michael Sarr and Todd Kellogg for surgical support and Doug Padley for supporting manufacturing of MSCs.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
revised ALS Functional Rating Scale
- MSC
mesenchymal stromal cell
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Nathan P. Staff, MD, PhD: study concept and design, obtaining funding, acquisition of data, study supervision and coordination, analysis and interpretation, drafting and revising the manuscript for content. Nicolas N. Madigan, MBBCh, PhD: acquisition of data, analysis and interpretation, revising the manuscript for content. Jonathan Morris, MD: analysis and interpretation, revising the manuscript for content. Mark Jentoft, MD, and Eric J. Sorenson, MD: acquisition of data, analysis and interpretation, revising the manuscript for content. Greg Butler, BSc: contribution of vital tools, acquisition of data, revising the manuscript for content. Dennis Gastineau, MD: study supervision and coordination, analysis and interpretation, revising the manuscript for content. Allan Dietz, PhD, and Anthony J. Windebank, MD: study concept and design, obtaining funding, acquisition of data, study supervision and coordination, analysis and interpretation, revising the manuscript for content.
STUDY FUNDING
This study was sponsored by the Mayo Clinic. The authors thank our funding partners: Jane and Judith Pape Adams Foundation, Mayo Clinic Center for Regenerative Medicine, the Department of Laboratory Pathology, and the Mayo Clinic Center for Clinical and Translational Sciences (UL1 TR000135).
DISCLOSURE
N. Staff is funded by NIH grant CA169443 and is a coinvestigator for the ALS clinical trial from BrainStorm Cell Therapeutics. N. Madigan, J. Morris, M. Jentoft, and E. Sorenson report no disclosures relevant to the manuscript. G. Butler is an inventor of technology used as a tool in this research; the technology has been licensed to a commercial entity (PLTMax; Mill Creek LifeScienes). Mayo Clinic has equity in the company, and Greg Butler and Mayo Clinic have contractual rights to receive royalties from the licensing of this technology. D. Gastineau reports no disclosures relevant to the manuscript. A. Dietz is an inventor of technology used as a tool in this research; the technology has been licensed to a commercial entity (PLTMax; Mill Creek LifeScienes). Dr. Dietz and Mayo Clinic have equity in the company and have contractual rights to receive royalties from the licensing of this technology. Dr. Dietz has governance responsibilities within this company. These conflicts have been disclosed to and are managed by the Mayo Clinic Conflict of Interest Board and are included here as directed by them. A. Windebank is a site principal investigator for the ALS clinical trial from BrainStorm Cell Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology 2002;59:280–282. [DOI] [PubMed] [Google Scholar]
- 2.Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 2013;12:310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uccelli A, Laroni A, Freedman MS. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol 2011;10:649–656. [DOI] [PubMed] [Google Scholar]
- 4.Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol 2016;73:337–44. [DOI] [PubMed] [Google Scholar]
- 5.Oh KW, Moon C, Kim HY, et al. Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Transl Med 2015;4:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazzini L, Mareschi K, Ferrero I, et al. Mesenchymal stromal cell transplantation in amyotrophic lateral sclerosis: a long-term safety study. Cytotherapy 2012;14:56–60. [DOI] [PubMed] [Google Scholar]
- 7.Karussis D, Karageorgiou C, Vaknin-Dembinsky A, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol 2010;67:1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.