

Abstract
Off-target binding of hydrophobic drugs can lead to unwanted side effects, either through specific or nonspecific binding to unintended membrane protein targets; however, distinguishing the binding of drugs to membrane proteins from that of detergents, lipids and cofactors is challenging. Here we use high-resolution mass spectrometry to study the effects of HIV protease inhibitors on the human zinc metalloprotease ZMPSTE24. This intramembrane protease plays a major role in converting prelamin A to mature lamin A. We monitored proteolysis of farnesylated prelamin A peptide by ZMPSTE24 and unexpectedly found retention of the C-terminal peptide product with the enzyme. We also resolved binding of zinc, lipids, and HIV protease inhibitors and showed that drug binding blocked prelamin A peptide cleavage and conferred stability to ZMPSTE24. Our results not only have relevance for the progeria-like side effects of certain HIV protease inhibitor drugs but also highlight new approaches for documenting off-target drug binding.
Graphical abstract
Introduction
Intramembrane proteases such as rhomboids, presenilin, and signal peptide peptidase are intriguing because of their ability to cleave peptide bonds within the lipid bilayer (reviewed in1). Understanding the catalytic mechanism and substrate-binding properties of these proteases poses technical challenges because of the hydrophobicity of the proteases themselves as well as of their substrates. To study the function of intramembrane proteases, these proteins are extracted and purified in detergents to keep the protein in solution and in an active state. Mass spectrometry of membrane proteins offers a powerful means for studying the behaviour of these proteins, primarily because this approach offers the potential to deconvolute a subset of potential ligand-binding moieties2,3, including lipids, substrates, metal ions, and drugs. Simultaneous binding of these factors has not been demonstrated previously using mass spectrometry due to the limited resolution available in earlier experiments. To evaluate the capacity of mass spectrometry to monitor off-target binding of drugs and to uncover mechanistic implications of this binding, we have investigated the interaction between HIV protease inhibitors and the human integral membrane metalloprotease ZMPSTE24.
ZMPSTE24 is a recently characterised member of the intramembrane class of proteases whose catalytic site lies within the plane of the lipid bilayer1,4,5. The X-ray structure of human ZMPSTE24 was solved at 3.4 Å, revealing a seven transmembrane α-helical barrel structure surrounding a voluminous water-filled, intramembrane chamber, capped by a zinc metalloprotease domain6. The catalytic site, including the zinc ion, faces into the chamber. Analogous features were identified in the yeast orthologue Ste24p7. Remarkably, the intramembrane cavities in ZMPSTE24 and Ste24p are large enough to accommodate a 10-kDa protein or ~1000 water molecules. Whether lipids might be housed within this chamber is not known. Human ZMPSTE24 is an endoplasmic reticulum/nuclear membrane protease that has dual functions in the maturation and processing of prelamin A to lamin A. First, ZMPSTE24 is capable of cleaving the last three residues (SIM) from prelamin A’s carboxyl-terminal CaaX motif (where C is cysteine, a is generally an aliphatic amino acid, and X is any residue). This CaaX-cleavage step is also performed by another ER membrane protease, RCE18,9. In a second and unique function, ZMPSTE24 mediates the final step of lamin A biogenesis, clipping off the last 15 amino acid residues of prelamin A, including its C-terminal farnesylcysteine10. This step releases mature lamin A, which is one of the principal protein components of the nuclear lamina.
Defective ZMPSTE24-mediated processing of prelamin A causes progeroid syndromes with clinical phenotypes resembling those of physiologic aging, for example thin skin, partial lipodystrophy, osteoporosis, and atherosclerotic coronary disease. The classic premature aging disorder of children, Hutchinson-Gilford progeria syndrome, is caused by a splicing mutation that results in an internal deletion of 50 amino acids within the carboxyl terminus of prelamin A; this deletion eliminates the ZMPSTE24 cleavage site in prelamin A and thereby blocks eliminates the endoproteolytic cleavage step that would ordinarily release mature lamin A11. ZMPSTE24 null mutations that completely block ZMPSTE24 activity result in restrictive dermopathy, a severe neonatal progeroid disorder characterized by a complete blockade of lamin A biogenesis and a striking accumulation of farnesyl–prelamin A12. Partial loss-of-function ZMPSTE24 mutations that do not fully block lamin A biogenesis lead to a moderate accumulation of farnesyl–prelamin A and a less severe progeroid disorder called mandibuloacral dysplasia13–15.
Interestingly, several HIV protease inhibitors (e.g., lopinavir, ritonavir, amprenavir) but not others (e.g., darunavir) block ZMPSTE24 activity in cultured fibroblasts and lead to an impressive accumulation of farnesyl–prelamin A. Futhermore, biochemical studies showed these inhibitors blocked the enzymatic activity of purified yeast Ste24p16–18. These were surprising findings, as the HIV proteases are aspartyl proteases, whereas ZMPSTE24 and Ste24p are zinc metalloproteases with a distinct mechanism of catalysis. It is noteworthy that long-term therapy with certain HIV protease inhibitors, including lopinavir/ritonavir (Kaletra), has been associated with aging-like phenotypes similar to those in patients with progeroid syndromes (partial lipodystrophy, atherosclerotic disease, osteoporosis)16,19,20. Indeed, when human or mouse fibroblasts are incubated with therapeutic concentrations of lopinavir, the amount of prelamin A accumulation is nearly as great as that in fibroblasts from patients with debilitating progeroid disorders13,21. At this point, however, the extent to which the progeria-like side effects of HIV protease inhibitors can be attributed to ZMPSTE24 inhibition is controversial, in part because other drugs within in commonly prescribed antiretroviral drug cocktails may also contribute to the observed side effects22. Importantly, while the ability of some HIV- protease inhibitors to block prelamin A processing in cultured cells is well established16,17 until the present work there has been no direct evidence showing that HIV protease inhibitors bind to human ZMPSTE24 at the molecular level.
In this study, we develop and apply a high-resolution mass spectrometry approach23 adapted for membrane proteins24 to understand the capacity of human ZMPSTE24 to cleave prelamin A and to better characterize the metal-, lipid-, and drug-binding properties of the membrane protein. Our studies monitor the proteolytic processing of prelamin A and investigate whether the products of the cleavage reaction remain within ZMPSTE24’s intramembrane chamber. We also probe the ability of HIV protease inhibitors to bind to ZMPSTE24 and inhibit the processing of prelamin A. We show that some HIV protease inhibitors, but not others, bind ZMPSTE24, prevent the cleavage of prelamin A, and lead to increased stability of the enzyme, as judged by resistance to unfolding.
Results
Mass spectra of ZMSTE24 reveal binding of zinc and phospholipids
ZMPSTE24 was expressed in Sf9 cells and purified in the detergent octyl glucose neopentyl glycol (OGNG) with cholesteryl hemisuccinate (CHS) as described6 ZMPSTE24 was introduced into the Q Exactive Orbitrap mass spectrometer under conditions optimised for the preservation of noncovalent interactions23,25 and adapted for transmission of membrane proteins24 (Fig. 1). The charge state series confirms the mass of the protein and reveals two additional charge state series corresponding to the association of lipids co-purifying with the enzyme. Expansion of the predominant charge state (16+) exposes the presence of a small population of apo ZMPSTE24 (with a mass difference of 62 Da, consistent with an absence of a zinc ion) (Fig. 1 inset). The catalytic Zn2+ ion is essential for binding and cleavage of prelamin A peptides and its presence in the majority of the ZMPSTE24 molecules implies that the Zn2+ coordination site is maintained.
Figure 1.
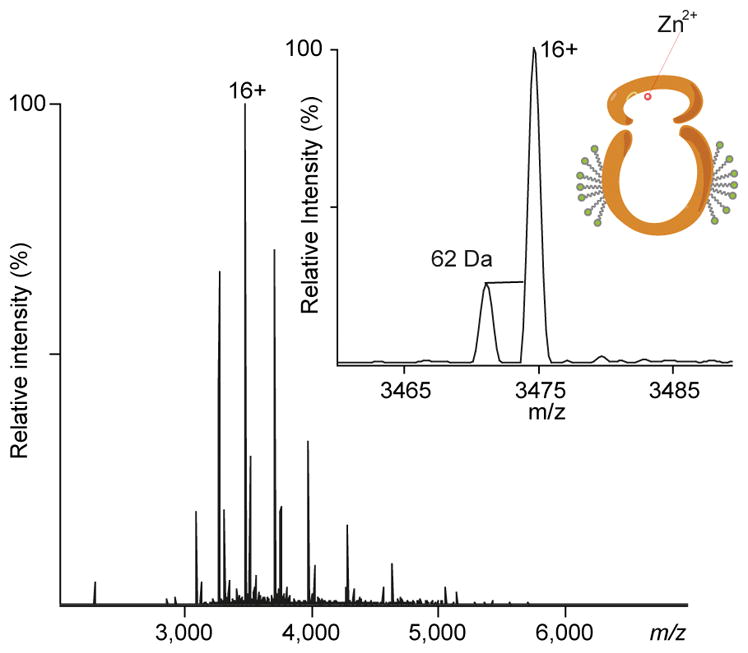
Mass spectrum of ZMPSTE24 reveals zinc binding. (A) High-resolution Orbitrap QExactive mass spectrum of ZMPSTE24 released from a detergent (octyl glucose neopentyl glycol, OGNG) micelle. Adduct peaks at higher m/z are due to binding of lipids (POPG and cholesterol hemisuccinate) that co-purify with the protein. Inset: holo and apo-metallo ZMPSTE24 can be resolved (masses of 55579 Da and 55517 Da, respectively), confirming that Zn2+ is bound to the majority of the protein population.
The interaction of ZMPSTE24 with substrate peptides
We first used western blots of SDS-polyacrylamide gels to verify prelamin A accumulation in a human fibroblast cell line treated with either a protein farnesyltransferase inhibitor (ABT-100, 0.5 μM) or lopinavir (20 μM). The blots were incubated with a goat IgG against human lamin A/C or a monoclonal antibody against the C-terminus of prelamin A. These studies showed that the protein farnesyltransferase inhibitor caused an accumulation of nonfarnesylated prelamin A, whereas lopinavir led to an accumulation of farnesyl–prelamin A (Supplementary Fig. 1) and validate our previous findings16,17.
To investigate the interactions of the protein ZMPSTE24 with prelamin A at the molecular level, ZMPSTE24 in OGNG/CHS micelles was incubated with equimolar amounts of a 26-mer farnesylated synthetic peptide corresponding to the C terminus of prelamin A (Supplementary Fig. 2). The synthetic peptide mimics the prelamin A substrate in mammalian cells, except that the synthetic peptide terminates with a farnesylcysteine rather than a farnesylcysteine methyl ester. Mass spectra were acquired before adding the synthetic peptide and at intervals for a period of 1 h (Fig. 2). After a 2-min incubation at room temperature, the mass spectrum shows two charge states series—assigned to the ZMPSTE24 protein and the 26-mer prelamin A peptide bound to ZMPSTE24. Although partial collapse of the large intramembrane chamber in the gas phase cannot be ruled out, the survival of this large noncovalently bound ZMPSTE24 peptide at collision energies required to completely release the OGNG micelle implies that the sequestration within the chamber protects the peptide from dissociation.
Figure 2.

Mass spectra of ZMPSTE24 allow the cleavage of the prelamin peptide to be monitred in real-time. ZMPSTE24 was preprared in OGNG micelles and incubated with with an equimolar concentration of the peptide the 26-mer prelamin A peptide. Mass spectra were recorded on a Q-Tof mass spectrometer at different time intervals as indicated. Apo protein charge states are labelled (brown triangles). Binding of the full-length 26-mer prelamin A peptide is observed within 2 min (blue peaks). The corresponding 15-mer peptide cleavage product is formed with time (green peaks). The majority of the peptide was cleaved within ~12 min, and the cleaved peptide product remains associated with ZMPSTE24. The schematic for ZMPSTE24 depicts the full-length 26-mer prelamin A peptide and its farnesyl lipid tail (red). Right hand panels show the presence of 26-mer peptide and the cleavage product (1016 for the triply charged ion), indicating that the majority of the substrate peptide was hydrolysed within ~12 min to form the doubly charged product ion at 903 m/z.
After a 2-min incubation period of the peptide with ZMPSTE24, peaks were also observed that correspond to the binding of ZMPSTE24 to the proteolytically processed peptide product (i.e., a farnesylated 15-mer peptide released from the 26-mer peptide). Surprisingly, the wild-type ZMPSTE24 and the farnesylated 15-mer peptide product formed a stable complex, with the 15-mer peptide remaining bound to ZMPSTE24 for time periods ranging from 1 to 24 h. In a control experiment, we performed studies with a catalytically inactive mutant ZMPSTE24 protein (ZMPSTE24-E336A) and found no interaction between ZMPSTE24-E336A and the prelamin A peptide without the C-terminus. It is also possible to follow ZMPSTE24-mediated cleavage of the prelamin A peptide by studying the release of low-molecular weight peptide products following activation in the gas phase. The full-length peptide substrate and its product peptide were assigned as triply- and doubly-charged species (m/z 1016 and 903, respectively), corresponding to masses of 3046 Da and 1806 Da, respectively (Supplementary Fig. 3). After ~12 min, the peak corresponding to substrate peptide had significantly diminished and there was no further increase in the intensity of the product at 60 min. We interpret these findings as showing complete endoproteolytic processing of the peptide and conclude that the cleavage reaction is nearly complete by ~12 min.
To define the impact of protein farnesylation on protease activity, ZMPSTE24 was incubated with a 14-mer synthetic peptide spanning the second cleavage site for prelamin A (Supplementary Fig. 2). This nonfarnesylated peptide was incubated in an equimolar ratio with ZMPSTE24 at a concentration of 15 μM. Hydrolysis was monitored in real time as above, and a digested peptide fragment was detected at low intensity at m/z 525 after ~10 min (Supplementary Fig. 4). Approximately 35% of the nonfarnesylated peptide was hydrolysed after 1 h. The slow product formation with the nonfarnesylated peptide stands in contrast to results with the farnesylated 26-mer peptide, where the endoproteolytic cleavage reaction was complete within ~15 min. Moreover, binding of the 10-mer cleavage product within the ZMPSTE24 chamber could not be detected in the mass spectrum at the concentrations tested (data not shown). Together, these observations confirm the importance of protein farnesylation for efficient prelamin A processing and for binding of the cleaved peptide within the ZMP cavity.
Drug binding to ZMPSTE24
To explore the binding of drugs to ZMPSTE24, we added lopinavir (15 μM) to form an equimolar solution of ZMPSTE24 in C8E4. Several adduct peaks were observed and resolved using the high-resolution Q Exactive Orbitrap mass spectrometer. Peaks were assigned to association with cholesterol hemisuccinate (CHS) (present in the purification buffer), OGNG, and lopinavir (Supplementary Fig. 5). At the very same drug concentrations and with the same experimental conditions, binding of amprenavir and ritonavir were observed, albeit in lower amounts (i.e., lower peak intensities for drug-bound states). Interestingly, the higher molecular mass of ritonavir made it possible to confirm binding to the apo form of ZMPSTE24 as well as to the Zn2+-bound enzyme. Another HIV protease inhibitor, darunavir, which shares some structural features with lopinavir and ritonavir, showed no binding to ZMPSTE24 at the same 15 μM concentration used for the other drugs. Our results establish an order of affinity (based on peak height and identical mass spectrometry procedures and identical solution concentrations) with lopinavir > ritonavir > amprenavir > darunavir.
In order to probe further the different binding affinities of ZMPSTE24 to the HIV protease inhibitors we performed a competitive drug-binding assay. Three inhibitors (lopinavir, ritonavir, darunavir) were mixed at equimolar concentrations (7 μM) and added to a solution of ZMPSTE24. Low concentrations of DMSO are required to retain solubility of these drugs. The final concentration of each drug in the 1:1:1 solution was 7 μM. The resulting mass spectrum reveals peaks at low m/z consistent with the masses of each drug (Figure 3). At the high m/z region, binding to ZMPSTE24 of lopinavir and ritonavir, but not darunavir, is observed; in addition, adduct peaks were assigned to CHS and OGNG. Interestingly the intensities of the peaks assigned to ZMPSTE24–drug conjugates showed similar trends to the intensity profiles observed when the drugs were added separately to ZMPSTE24 (Supplementary Fig. 5), with lopinavir forming a higher population of protein drug conjugate than ritonavir.
Figure 3.

Mass spectra recorded after competitive binding of HIV protease inhibitor drugs to ZMPSTE24. An Orbitrap mass spectrum shows the interaction between ZMPSTE24 and 1:1:1 solition of lopinavir, ritonavir, and darunavir (main panel). Left inset: expansion of the low m/z region of the spectrum confirms the presence of all three inhibitors in solution. Right inset: expansion of the mass spectrum from m/z 5020 – 5135 reveals binding of CHS, OGNG, lopinavir, and ritonavir to the 11+ charge state of ZMPSTE24. No peak was observed for binding of darunavir under these competitive binding conditions. Charge state series are labelled: yellow hexagon, zinc-bound ZMPSTE24; dark blue circle, ZMPSTE24·CHS; green circle, ZMPSTE24·OGNG; black circle, ZMPSTE24·lopinavir; red circle, ZMPSTE24·ritonavir.
To obtain relative binding affinities of the HIV-PIs, we determined their solution phase dissociation constants with established mass spectrometry methods3,26. We varied the concentration of lopinavir and ritonavir in intervals of 2.5 μM from 4 μM to 20 μM (and used a fixed concertation of ZMPSTE24, 15 μM). The mass spectra clearly show an increase in drug binding to ZMPSTE24 with increasing concentrations of each HIV-PI (Supplementary Fig. 6). The solubility of the drugs limited the upper range of the concentrations that could be tested, but measurements at low μM concentrations were carried out in triplicate and were reproducible (Supplementary Fig. 7). The KD values obtained by this method were 25 ± 1.2 and 30 ± 1.3 μM for lopinavir and ritonavir, respectively.
ZMPSTE24 activity is blocked by HIV protease inhibitors
Having documented binding of HIV-PIs to ZMPSTE24, the next question was whether this binding affected the catalytic activity of ZMPSTE24 against the 26-mer prelamin A peptide (Fig. 2). After first establishing that ZMPSTE24 retains catalytic activity in the presence of the detergent used for drug-binding experiments (C8E4), we tested whether it is possible to form a ZMPSTE24–drug–prelamin A peptide ternary complex. In our initial studies, we added lopinavir to ZMPSTE24 before attempting to detect the ZMPSTE24–lopinavir–peptide complex. However, after adding lopinavir to the enzyme, we could detect binding of lopinavir to ZMPSTE24, but we were unable to detect binding of the prelamin A peptide to ZMPSTE24. Moreover, the mass spectrum recorded at low m/z values indicated that the peptide remained intact and did not undergo cleavage. In contrast, when ZMPSTE24 was incubated in the presence of darunavir, we detected proteolysis of the prelamin A peptide at low m/z. This is direct confirmation that darunavir—in contrast to lopinavir—does not bind to ZMPSTE24 and block its activity.
Given that HIV-PIs such as lopinavir prevent the binding of prelamin A peptide and block the endoproteolytic cleavage reaction, they likely bind to the active site of the enzyme within ZMPSTE24’s large intramembrane chamber. We suspected that this binding event might confer stability to ZMPSTE24 and increase its resistance to protein unfolding in the gas phase. We used ion-mobility mass spectrometry (IM-MS)27–29, a technique that measures the rotational collision cross section, to compare the stability of ZMPSTE24–drug conjugates with the apo form. IM-MS has been used previously to assess the effects of lipid binding to membrane proteins30,31, and in these studies, the ligand-bound protein complex was compared with the apo protein in the same spectrum. Considering first the unfolding trajectory of the Zn-bound protein, we note that it transitions from a compact native-like state at low collision energies (~70 V, 11+ charge state) to a relatively long-lived intermediate and an unfolded states with arrival times of 3.6 ms and 5.3 ms at 70 and 140 V, respectively (Supplementary Fig. 8). Selecting the ritonavir-bound protein ions and subjecting these to increasing collision energy altered the corresponding unfolding transitions. Notably, the onset of the intermediate state occurred at higher activation voltages for the ritonavir-bound state than for the Zn-bound protein (~80 V). Similarly, when lopinavir was added to ZMPSTE24 and the lopinavir-bound peak subjected to collisional activation, the transition to the intermediate state occurred at higher collision energies (~85 V), consistent with stabilisation of the protein–drug conjugate and increased resistance to unfolding with respect to the zinc-bound form. We conclude that both ritonavir and lopinavir stabilise ZMPSTE24 and that the effect is greater for lopinavir, consistent with the more favourable KD value determined for lopinavir binding to the enzyme.
Discussion
In the current study, we showed the utility of novel mass spectrometry approaches to study the intramembrane metalloprotease ZMPSTE24. We monitored and resolved the binding of metal ions, lipids, drugs, and detergents, and investigated the processing of a farnesylated 26-mer peptide prelamin A substrate. We examined the binding and cleavage of the peptide substrate, as well as the fate of the 15-mer farnesylated peptide cleavage product. That a population of the cleaved peptide can be retained within the intramembrane chamber of ZMPSTE24 for at least 24 h implies that there is a tight association between ZMPSTE24 and the cleavage product. However, we cannot exclude the possibility that the cleaved farnesylated 15-mer peptide product is released from ZMPSTE24 in vivo in the presence of a lipid bilayer or is released in the presence of specific protein binding partners. In any case, our observation of persistent binding of the cleaved peptide to ZMPSTE24 in vitro is intriguing and indicates an unexpected stability of the peptide–ZMPSTE24 complex. We speculate that the sequestration of the peptide product within the ZMPSTE24 chamber could serve to protect the cell from toxic effects of the farnesylated peptide. Presumably, the peptide is eventually released from the intramembrane chamber into an ER luminal compartment for disposal.
It is widely assumed that protein farnesylation facilitates the association of prelamin A with nuclear membranes and assists in the delivery of mature lamin A to the nuclear lamina32, but recent studies suggest that this effect may be rather modest in vivo. Genetically modified mice that synthesize mature lamin A directly (bypassing prelamin A synthesis and prelamin A processing steps) have no obvious abnormality in the targeting of lamin A to the nuclear rim33. However, the fact that prelamin A farnesylation and prelamin A proteoltyic processing has been conserved during evolution strongly suggests that it must be functionally important. At this point, the “physiologic rationale” for prelamin A farnesylation is incompletely understood, but the current studies have uncovered a novel biochemical insight into the importance of prelamin A farnesylation. We show that protein farnesylation is important for efficient prelamin A processing by ZMPSTE24. The absence of farnesyl lipid anchor on prelamin A caused markedly reduced binding of a prelamin A peptide to ZMPSTE24 as well as reduced proteolysis. The fact that the absence of farnesylation retards but does not fully block ZMPSTE24-mediated prelamin A proteolysis probably explains a longstanding mystery in prelamin A processing. For years, it has been unclear why high doses of potent protein farnesyltransferase inhibitors (FTIs) were incapable of fully inhibiting the biogenesis of mature lamin A from prelamin A34. Similarly, it was unexpected that a knockout of protein farnesyltransferase (or a knockout of both protein farnesyltransferase and geranylgeranyltransferase-I) in the liver did not completely abolish lamin A biogenesis from prelamin A35. Our current studies provide a plausible explanation—that ZMPSTE24-mediated prelamin A cleavage persists, albeit at a lower level, when prelamin A farnesylation is absent.
Having established conditions to monitor ZMPSTE24-mediated prelamin A processing, we examined the effects of HIV protease inhibitors on this process. We documented, for the first time, direct binding of HIV-PIs (lopinavir, ritonavir, amprenavir) to ZMPSTE24 and showed that each of those drugs blocked prelamin A proteolysis. We also showed that binding of these drugs confers stability upon ZMPSTE24 as evidenced by resistance to unfolding. Interestingly, darunavir did not bind to ZMPSTE24, or interfere with proteolysis. Our KD values are of the same order as previously reported IC50 values for lopinavir and ritonavir17, and our Western Blot studies on a human fibroblast cell line support the inhibition of prelamin A processing by lopinavir. Of note, our findings are also consistent with earlier studies showing that lopinavir is much more potent than darunavir in causing an accumulation of farnesyl–prelamin A in cultured fibroblasts17.
In our studies, we tested HIV protease inhibitor drugs over a range of concentrations from 4 μM to 20 μM, which is similar to the therapeutic concentrations of the drugs36. Our biochemical findings are therefore consistent with the ability of similar concentrations of HIV-PIs to inhibit prelamin A processing in cultured cells as shown here and reported previously16,17. However, we would point out that HIV-PIs are highly protein-bound in plasma; thus, it is possible that the concentration of unbound (bioavailable) drug in the cells of HIV patients may be 30–100 fold lower37,38 than the 4 - 20 μM concentration used in our experiments. It would be highly desirable to define the effective concentration of drug in the nuclear membranes where ZMPSTE24 resides. ZMPSTE24 protein concentrations are also extremely relevant to the ability of HIV-PIs to block prelamin A processing16,17, and for that reason it would also be desirable to know the exact concentration of ZMPSTE24 protein in different tissues. At this point, there is conflicting information on the impact of HIV-PIs on prelamin A processing in vivo. Western blot studies show an accumulation of prelamin A in adipose tissue of HIV patients treated with an HIV-PI39, suggesting that HIV-PIs do inhibit ZMPSTE24 processing, but a more recent study did not find a significant effect of HIV-PIs on prelamin A processing in peripheral blood mononuclear cells20. We would argue that the biochemical findings in the current studies—that HIV-PIs bind directly to ZMPSTE24 and block proteolysis, provide an impetus to pursue additional studies to define the impact of the drugs on prelamin A processing in different tissues in vivo.
In conclusion, the mass spectrometry approaches that we applied in this study extend earlier studies of metal binding and drugs to soluble protein targets40–44 and provide direct evidence for the binding of HIV protease inhibitors to the membrane protein ZMPSTE24, with and without zinc binding, and in the presence of detergent and lipid adducts. This is not only the first direct evidence of drug binding to ZMPSTE24 but our results are corroborated by the resistance to unfolding conferred by these binding events. Our findings are also important because it was intuitively surprising that these drugs, which were designed as aspartyl protease inhibitors, would bind to a zinc metalloprotease. We strongly suspect that these off-target binding events are common with frequently used pharmaceutical agents. We propose that the methodologies applied here will therefore be widely employed for probing these unwanted associations in other membrane proteins where the potential for off-target hydrophobic interactions is predicted to be high45.
Methods
Protein purification
ZMPSTE24 was overexpressed and purified as described by Quigley et al6. Briefly, full-length ZMPSTE24 was expressed in Sf9 cells with an pFB-CT10HF-LIC vector containing a ZMPSTE24 open reading frame with a C-terminal tobacco etch virus (TEV) cleavage site, a 10× His tag, and a FLAG tag. The final protein was purified in OGNG detergent supplemented with 0.018% CHS.
Peptides
Synthetic peptides corresponding to the C-terminal sequences from of prelamin A (consisting of 26 or 14 amino acids) were synthesised by a commercial supplier (Peptide Protein Research Ltd). The C-terminal cysteine of the 26- mer peptide was farnesylated. A stock 20 mM solution of peptides in DMSO was prepared and diluted to 100 μM in a 200 mM ammonium acetate buffer supplemented with 0.16% OGNG.
Human immunodeficiency virus protease inhibitors (HIV-PIs)
Lopinavir, ritonavir, amprenavir, and darunavir were purchased Sigma or Cambridge Biosciences in powder form. The drugs were solubilised in DMSO at a stock concertation of 5 mM concentration and then further diluted to 100 μM in 200 mM ammonium acetate supplemented with 0.5% C8E4.
Mass spectrometry
QExactive
High-resolution mass spectrometry was performed using a modified QExactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) optimised for transmission of high m/z ions (Heck Nat Methods) and analysis of membrane proteins24. Modifications included a lower frequency RF applied to the quadrupole, higher pressures in the HCD cell, and modified electronics for extended frequency range detection on the Orbitrap. Typically the following instrument conditions were applied: The capillary voltage was set at 1.2 kV, and the ion transfer capillary was set to 30 °C in order to maintain ‘soft’ evaporation conditions. The S-lens RF potential was set to 100 V for better transmission of protein-detergent micelles. The quadrupole was set to from 2,000 to 10,000 m/z range.
For OGNG solubilised ZMPSTE24 protein complexes, collisional activation was applied by adjusting the voltage applied in the HCD cell to 100 V. For C8E4 solubilised ZMPSTE24–drug binding experiments, no activation was applied in the HCD cell in order to preserve drug binding. Argon pressure in the HCD cell was set at 1.29 × 10−9 mbar. The C-trap entrance lens was set to 6.0 V. The automatic gain control was set to 1 × 106 with a maximum inject time of 100 ms. Scans were collected from 2,000 to 8,000 m/z and from 500 to 800 m/z for protein complexes and drug molecules, respectively. For spectra recorded for drug molecules at low mass-to-charge ratios, the pressure in the HCD cell was decreased. The resolution of the instrument was set to 17,500 at m/z=200 (a transient time of 64 ms) with 1 microscan or 10 microscans summed into a single scan to increase signal-to-noise ratios. The noise level parameter was set to a default value 4.64. Calibration of the instrument was performed with cesium-iodide clusters up to 11,340 m/z.
QToF and Synapt G1
Solutions of ZMPSTE24 and various peptides were introduced into a modified QToF mass spectrometer using gold-coated glass needles, and spectra were recorded under the following conditions: capillary, cone, and collision cell voltages of 1.4 kV, 50 V, and 150 V, respectively. The backing pressure was set to 4.9mBar. Drift time measurements of C8E4-solubilised ZMPSTE24 with or without drugs were performed on a Synapt G1 mass spectrometer under the following conditions: capillary, cone and transfer voltages of 1.4 kV, 10 V, and 10 V, respectively. The flow rate of Argon was set at 8 ml/min in the collision cell and nitrogen gas was admitted at a flow rate of 22 ml/min into the ion-mobility cell, producing pressures of 7.2–7.3 × 10−2 mbar and 0.578 mbar, respectively. The backing pressure was set at 4.39 mbar.
Peptide binding experiments
Purified ZMPSTE24 was buffer exchanged to 200 mM ammonium acetate supplemented with 0.16% OGNG and 0.018% CHS using micro biospin columns (BioRad). Peptide solutions were added to buffer exchanged protein solutions to yield a final peptide concentration of 15 μM.
Drug binding experiments
ZMPSTE24 was buffer exchanged into 200 mM ammonium acetate supplemented with 0.5% C8E4 detergent. The stock solution of HIV-PI drugs were added to buffer exchanged ZMPSTE24 protein either separately, at concentrations from 2 to 18 μM, or in a 1:1:1 mixture of lopinavir, ritonavir and darunavir prepared by mixing equimolar concentrations to yield a final concentration of 7 μM for each drug.
Western blots
Cell extracts from a human fibroblast cell line that had been exposed for three days to a protein farnesyltransferase inhibitor (ABT-100, 0.5 μM) or lopinavir (20 μM) for 3 days were size-fractionated on 4–12% polyacrylamide-SDS gels. The size-fractionated proteins were then transferred to sheets of nitrocellulose membrane for western blotting. The blots were incubated with a goat IgG against human lamin A/C (Santa Cruz) or a monoclonal antibody (7G11) against the C terminus of prelamin A. Binding of the primary antibodies was detected with IRDye-conjugated secondary antibodies, followed by scanning of the blots with an Odyssey Infrared Imaging System (Li-Cor Biosciences).
Supplementary Material
Figure 4.

Collision-induced unfolding reveals differences in the unfolding trajectories of free ZMPSTE24 compared with its drug-bound states. (A) Mass spectrum and arrival times of ZMPSTE24 acquired under collisional activation (80 V) (upper panel) in the presence of ritonavir in C8E4. The arrival time distribution of the 11+ charge state is consistent with partial unfolding of the enzyme, whereas charge states 10+ and 9+ remain compact, consistent with native-like structure in the gas phase. Peaks assigned to binding of the drug are labelled with black circles. The 11+ charge states of apo and the ZMPSTE24 ritonavir complex, selected for collision induced unfolding measurements, are shown in the ion mobility data (white circle). Native and intermediate states are apparent at this voltage (80 V). (B) Comparison of the collision-induced unfolding from 70 V to 110 V of the 11+ charge state of Zn2+-bound ZMPSTE24 with unfolding in the presence of ritonavir or lopinavir. The transition from compact folded state to the first intermediate state is highlighted (white arrows). This transition occurs at a higher voltage for lopinavir-bound ZMPSTE24 than for the ritonavir-bound ZMPSTE24.
Acknowledgments
This work is supported by program grants from the Medical Research Council (98101), the National Institutes of Health (NIH), and an ERC Advanced Investigator Award IMPRESS (26851). C.V.R. is a Royal Society Research Professor. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. S.M. was funded for this work by NIH grant R01 GM041223; S.G.Y. was supported by NIH grants AG035626-10 and HL126551. We thank Shipeng Wang and Berenice Rotty for purification of three of the protein samples used in this study.
Footnotes
Author Contributions
S.M. and C.V.R designed the research with assistance from A.Q. and E.P.C. A.Q. and E.P.C. purified ZMPSTE24. S.M. and J.G. carried out the mass spectrometry experiments. J.M. initiated preliminary mass spectrometry experiment. S.M (Susan Michaelis) helped with new reagents. S.G.Y performed the western blot analysis. S.M. and C.V.R. wrote the manuscript with contributions from all authors.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Wolfe MS, Kopan R. Intramembrane proteolysis: theme and variations. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- 2.Marcoux J, et al. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopper JT, Robinson CV. Mass spectrometry quantifies protein interactions--from molecular chaperones to membrane porins. Angew Chem Int Ed Engl. 2014;53:14002–14015. doi: 10.1002/anie.201403741. [DOI] [PubMed] [Google Scholar]
- 4.Freeman M. The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu Rev Cell Dev Biol. 2014;30:235–254. doi: 10.1146/annurev-cellbio-100913-012944. [DOI] [PubMed] [Google Scholar]
- 5.Avci D, Lemberg MK. Clipping or Extracting: Two Ways to Membrane Protein Degradation. Trends Cell Biol. 2015;25:611–622. doi: 10.1016/j.tcb.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Quigley A, et al. The structural basis of ZMPSTE24-dependent laminopathies. Science. 2013;339:1604–1607. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- 7.Pryor EE, Jr, et al. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339:1600–1604. doi: 10.1126/science.1232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria--new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Manolaridis I, et al. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504:301–305. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies BS, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Ann Rev Genomics& Human Gen. 2009;10:153–174. doi: 10.1146/annurev-genom-082908-150150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eriksson M, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moulson CL, et al. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum Mutat. 2007;28:882–889. doi: 10.1002/humu.20536. [DOI] [PubMed] [Google Scholar]
- 13.Shackleton S, et al. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet. 2005;42:e36. doi: 10.1136/jmg.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmad Z, Zackai E, Medne L, Garg A. Early onset mandibuloacral dysplasia due to compound heterozygous mutations in ZMPSTE24. Am J Med Genet A. 2010;152A:2703–2710. doi: 10.1002/ajmg.a.33664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrowman J, Wiley PA, Hudon-Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet. 2012;21:4084–4093. doi: 10.1093/hmg/dds233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coffinier C, et al. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci USA. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coffinier C, et al. A potent HIV protease inhibitor, darunavir, does not inhibit ZMPSTE24 or lead to an accumulation of farnesyl-prelamin A in cells. J Biol Chem. 2008;283:9797–9804. doi: 10.1074/jbc.M709629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hudon SE, et al. HIV-protease inhibitors block the enzymatic activity of purified Ste24p. Biochem Biophys Res Commun. 2008;374:365–368. doi: 10.1016/j.bbrc.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrowman J, Michaelis S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol Chem. 2009;390:761–773. doi: 10.1515/BC.2009.080. [DOI] [PubMed] [Google Scholar]
- 20.Perrin S, et al. HIV protease inhibitors do not cause the accumulation of prelamin A in PBMCs from patients receiving first line therapy: the ANRS EP45 “aging” study. PLoS One. 2012;7:e53035. doi: 10.1371/journal.pone.0053035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 22.Torres RA, Lewis W. Aging and HIV/AIDS: pathogenetic role of therapeutic side effects. Lab Invest. 2014;94:120–128. doi: 10.1038/labinvest.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJ. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Methods. 2012;9:1084–1086. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- 24.Gault J, et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat Methods. 2016 doi: 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belov ME, et al. From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- 26.Erba EB, Zenobi R. Mass spectrometric studies of dissociation constants of noncovalent complexes. Annu Rep Prog Chem, Sect C. 2011;107:199–228. doi: 10.1039/C1PC90006D. [DOI] [Google Scholar]
- 27.Clemmer DE, Hudgins RR, Jarrold MF. Naked Protein Conformations: Cytochrome c in the Gas Phase. J Am Chem Soc. 1995;117:10141–10142. doi: 10.1021/ja00145a037. [DOI] [Google Scholar]
- 28.Gill AC, Jennings KR, Wyttenbach T, Bowers MT. Conformations of biopolymers in the gas phase: a new mass spectrometric method. Int J of Mass Spectrom. 2000;195–196:685–697. doi.org/10.1016/S1387-3806(99)00256-0. [Google Scholar]
- 29.Ruotolo BT, et al. Ion mobility-mass spectrometry reveals long-lived, unfolded intermediates in the dissociation of protein complexes. Angew Chem Int Ed Engl. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]
- 30.Laganowsky A, et al. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allison TM, et al. Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nat Commun. 2015;6:8551. doi: 10.1038/ncomms9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silvius JR, l’Heureux F. Fluorimetric evaluation of the affinities of isoprenylated peptides for lipid bilayers. Biochemistry. 1994;33:3014–3022. doi: 10.1021/bi00176a034. [DOI] [PubMed] [Google Scholar]
- 33.Coffinier C, et al. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem. 2010;285:20818–20826. doi: 10.1074/jbc.M110.128835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toth JI, et al. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SH, et al. Severe hepatocellular disease in mice lacking one or both CaaX prenyltransferases. J Lipid Res. 2012;53:77–86. doi: 10.1194/jlr.M021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z. Second generation HIV protease inhibitors against resistant virus. Exp Opin Drug disc. 2008;3:775–786. doi: 10.1517/17460441.3.7.775. [DOI] [PubMed] [Google Scholar]
- 37.Boffito M, et al. Protein binding in antiretroviral therapies. AIDS Res Hum Retroviruses. 2003;19:825–835. doi: 10.1089/088922203769232629. [DOI] [PubMed] [Google Scholar]
- 38.Solas C, et al. Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003;47:238–243. doi: 10.1128/AAC.47.1.238-243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caron M, et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14:1759–1767. doi: 10.1038/sj.cdd.4402197. [DOI] [PubMed] [Google Scholar]
- 40.Loo JA, DeJohn DE, Du P, Stevenson TI, Ogorzalek Loo RR. Application of mass spectrometry for target identification and characterization. Med Res Rev. 1999;19:307–319. doi: 10.1002/(sici)1098-1128(199907)19:4<307::aid-med4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Harvey SR, et al. Small-molecule inhibition of c-MYC:MAX leucine zipper formation is revealed by ion mobility mass spectrometry. J Am Chem Soc. 2012;134:19384–19392. doi: 10.1021/ja306519h. [DOI] [PubMed] [Google Scholar]
- 42.Pacholarz KJ, Garlish RA, Taylor RJ, Barran PE. Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery. Chem Soc Rev. 2012;41:4335–4355. doi: 10.1039/c2cs35035a. [DOI] [PubMed] [Google Scholar]
- 43.Maple HJ, et al. Application of the Exactive Plus EMR for automated protein-ligand screening by non-covalent mass spectrometry. Rapid Commun Mass Spectrom. 2014;28:1561–1568. doi: 10.1002/rcm.6925. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs AD, et al. Resolution of Stepwise Cooperativities of Copper Binding by the Homotetrameric Copper-Sensitive Operon Repressor (CsoR): Impact on Structure and Stability. Angew Chem Int Ed Engl. 2015;54:12795–12799. doi: 10.1002/anie.201506349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kell DB, Dobson PD, Bilsland E, Oliver SG. The promiscuous binding of pharmaceutical drugs and their transporter-mediated uptake into cells: what we (need to) know and how we can do so. Drug Discov Today. 2013;18:218–239. doi: 10.1016/j.drudis.2012.11.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.