

 A metallomics approach can be used to probe the mechanisms by which the co-administration of sulfur-containing ‘chemoprotective agents’ can modulate the metabolism of cisplatin in the bloodstream.
A metallomics approach can be used to probe the mechanisms by which the co-administration of sulfur-containing ‘chemoprotective agents’ can modulate the metabolism of cisplatin in the bloodstream.
Abstract
Numerous in vivo studies have shown that the severe toxic side-effects of intravenously administered cisplatin can be significantly reduced by the co-administration of sulfur-containing ‘chemoprotective agents’. Using a metallomics approach, a likely biochemical basis for these potentially useful observations was only recently uncovered and appears to involve the reaction of chemoprotective agents with cisplatin-derived Pt-species in human plasma to form novel platinum–sulfur complexes (PSC's). We here reveal aspects of the structure of two PSC's and establish the identification of an optimal chemoprotective agent to ameliorate the toxic side-effects of cisplatin, while leaving its antineoplastic activity largely intact, as a feasible research strategy to transform cisplatin into a safer and more effective anticancer drug.
Introduction
The serendipitous discovery of the antiproliferative effects of cis-diaminedichloroplatinum(ii) or cisplatin [CP] on E. coli cells in the 1960s combined with its approval by the FDA in 1978 heralded the era of platinum-based chemotherapy.1 Despite the subsequent FDA approval of second- and third-generation platinum-based anticancer drugs, such as carboplatin in 1989 and oxaliplatin in 2002,2 CP – which is intravenously administered either alone or in combination with other anticancer drugs – remains one of the most effective anticancer drugs that is used worldwide owing to its broad spectrum of activity towards a variety of cancers, including testicular, ovarian, head and neck, colorectal, bladder, cervical and lung cancer as well as melanoma and lymphomas.3 Extensive studies into the metabolism of CP have revealed that it constitutes a prodrug and it is commonly believed that the binding of the CP-derived monoaqua hydrolysis product [(NH3)2PtCl(H2O)]+ to DNA represents its likely mode of action.3c Given the complexity of the intracellular biochemistry of CP,3a,4 however, further studies are needed to rule out whether other biomolecular mechanisms may also contribute to its overall activity.
Severe toxic side-effects of CP
In contrast to so-called ‘molecularly targeted’ anticancer drugs which target a single pathway to kill cancer cells, CP represents a ‘shotgun’ cytotoxin which offers two major advantages: it is active against many different cell types in a tumour and its utilization is conceptually less susceptible to the development of resistance (although it does occur).5 Shotgun cytotoxins, however, also exhibit a dark side. With regard to CP, its therapeutic use and efficacy is inherently limited by the severe toxic side-effects that this metal-based drug exhibits on several non-proliferating cell types,4c which often results in life-long impacts on the quality of life of patients.6 For example, 30 to 60% of patients suffer from nephrotoxicity,7 more than 60% of pediatric patients develop bilateral and permanent hearing loss8 and up to 90% of patients develop neurotoxicity.3b Although nephrotoxicity in patients can be somewhat ameliorated by increased hydration or the administration of mannitol,9 no approved procedures exist to completely eliminate ototoxicity; the latter therefore constitutes a primary dose limiting factor.10 Likewise, there are currently no established clinical procedures to reduce or ameliorate the neurotoxicity of this otherwise very effective anticancer drug.11
Strategies to ameliorate the severe toxic side-effects of Pt-based anticancer drugs
Owing to the efficiency of CP and its inherent limitations, considerable research efforts are directed to improve the tumor selectivity of Pt-based anticancer drugs. This fundamental challenge can be addressed either by synthesizing novel Pt-compounds3c,12 or by improving the delivery of established or newly synthesized Pt-based drugs to the tumor by drug-delivery vehicles.13 The third principle approach – the one that is of focal interest in the context of this mini-review – aims to reduce the CP-induced severe toxic side-effects by the co-administration of small-molecular-weight ‘chemoprotective agents’, while leaving the anticancer effect of CP largely intact.14 Compared to the first two approaches, the latter approach is potentially more cost effective since it aims to selectively reduce the severe toxic side-effects of an already FDA-approved Pt-drug with a chemoprotective agent that may – ideally – already be approved by the FDA (i.e. costly drug approval processes are avoided altogether).
Reducing CP-induced toxic side-effects by ‘chemoprotection’
Numerous studies with animal models or patients have demonstrated that chemoprotective agents, such as sodium thiosulfate (STS),15 N-acetyl-l-cysteine (NAC),15a,16 amifostine,15c,17 sodium diethyldithiocarbamate,10,15c,18 d-methionine,9,19 l-methionine,19a,20 l-glutathione (GSH),21 cimetidine,22 sodium salicylate,23 l-carnosine,24 2,3-dimercapto-1-propanesulfonic acid,25 and procaineamide hydrochloride26 can significantly reduce some of the severe toxic side-effects of CP. While amifostine is the only chemoprotective agent that has been specifically approved by the FDA for CP therapy,27 STS and NAC have been approved for the treatment of cyanide poisoning and for acetaminophen overdose, while d-methionine is currently undergoing a phase 3 clinical trial to reduce noise-induced hearing loss.6
From a biochemical point of view, the perhaps surprising lack of understanding as to the mode of action of the aforementioned phenomenological observations at a molecular level (i.e. the effect of chemoprotective agents on the metabolism of CP) must be attributed to the complexity of biological systems (e.g. the bloodstream) and a lack of appropriate tools to gain insight. In this context, one is reminded of F. Dyson's notion that “New directions in science are launched by new tools much more often than by new concepts. The effect of a tool driven revolution is to discover new things that need to be explained.” To this end, we have developed an instrumental metallomics tool28 that allowed us to better understand the underlying chemistry in order to ultimately translate this straightforward approach to achieve actual benefits for cancer patients. In brief, this metallomics tool allows to directly analyze blood plasma for essentially all contained Pt-species by using size-exclusion chromatography coupled on-line to an inductively coupled plasma atomic emission spectrometer. This principle approach dramatically reduces the complexity that is inherently associated with the analysis of blood plasma into all of its constituents because only the sub-population of Pt-species is detected. It is useful to discuss some key issues that pertain to this chemoprotection approach in more detail.
Considerations pertaining to the mechanistic aspects of the ‘chemoprotection’ approach
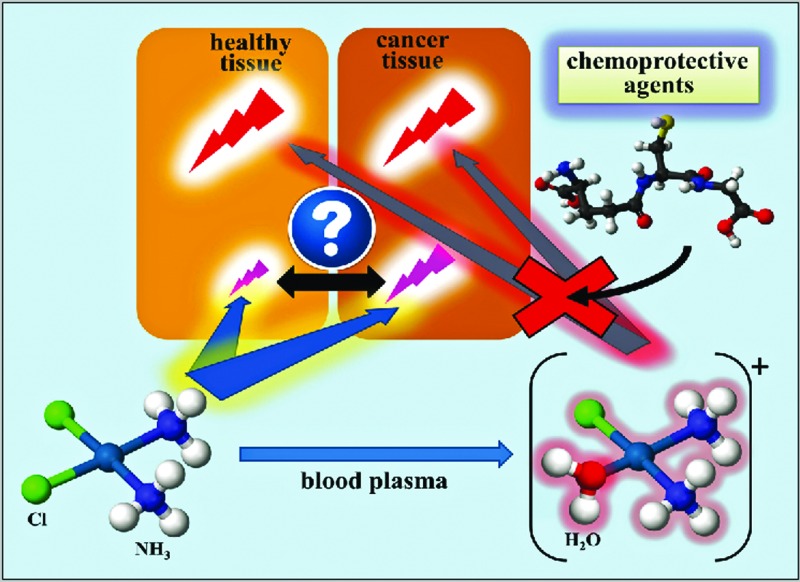
The bloodstream represents the first biological compartment where chemical reactions between intravenously administered CP and a chemoprotective agent may ensue. Although relevant chemistry between CP (and its metabolites) and a chemoprotective agent may also occur in internal organs,29 it is very difficult to tune the metabolism of CP therein and it will therefore not be further discussed. Conceptually, the intravenous administration to a patient with an appropriate chemoprotective agent should allow one to modulate the metabolism of CP in the bloodstream, which will ultimately determine which Pt-species are left in the blood circulation to subsequently interact with healthy (unintended) and tumor tissue cells (intended) (Fig. 1). In order to evaluate the potential of this ‘chemoprotection approach’, one needs to first understand the metabolism of CP in the bloodstream itself28b before one can probe the effect that chemoprotective agents may exert on its metabolism (Fig. 1) as well as other blood constituents, such as erythrocytes as well as plasma proteins and metalloproteins.30
Fig. 1. Conceptual depiction of the chemical reactions that occur between cisplatin (CP) and chemoprotective agents in human plasma in vitro as well as the repercussions that these Pt-species will exert in the whole organism in vivo. CPHP refers to all CP-derived hydrolysis products and PP refers to plasma proteins.

It is commonly believed that intravenously administered CP does not hydrolyze in the bloodstream, owing to the comparatively high concentration of Cl– (∼100 mM) in human blood plasma. After the addition of CP to human plasma in vitro, however, highly toxic CP-derived hydrolysis products (CPHP)31 and plasma protein bound Pt-species (Pt-PP) were present within as little as 5 min,28b,32 while the majority of Pt was still present as the parent drug. All chemoprotective agents that we have investigated – STS, NAC, d-methionine and GSH – affected the metabolism of CP in plasma by producing additional Pt-peaks that did not match the retention time of the Pt-peaks that were detected when only CP was added to plasma (in this case Pt-peaks corresponding to CP, CPHP's and Pt-PP's were detected).30,33 The additional Pt-peaks were therefore assigned to novel Pt-containing sulfur complexes or PSC's in blood plasma.30,33 The mechanism of formation of these PSC's likely involves the reaction of the highly reactive monoaqua hydrolysis product of CP – [(NH3)2PtCl(H2O)]+ – with each chemoprotective agent.33b Based on this demonstrated ‘tunability’ of the metabolism of CP with chemoprotective agents in blood plasma, it is now possible to discuss potential advantages and disadvantages of this principle approach to possibly improve Pt-based anticancer drugs that are currently in use.
The advantages of a ‘chemoprotection approach’ are that the identification of an inexpensive and safe chemoprotective agent that can neutralize highly toxic CPHP's31 should allow one to reduce the severe toxic side effects of CP while minimally affecting the efficacy of the parent drug CP (Fig. 1).32 There are three potential disadvantages that have to be considered in the context of this principle approach, (a) the chemoprotective agent itself may exert adverse toxic effects by affecting the integrity of endogenous plasma metalloproteins, such as transferrin, ferritin and Zn bound to human serum albumin (which would result in potential toxicity),30 (b) the chemoprotective agent may decrease the plasma lifetime of the parent CP (which would decrease the efficacy of the latter) and/or (c) the PSC may not be effectively excreted, which in turn could result in possible organ toxicity (e.g. if the PSC traverses the blood–brain barrier).
Tuning the metabolism of CP with an optimal chemoprotective agent
In the context of translating this chemoprotection approach30,32,33 into practical benefits for patients (e.g. reduction of ototoxicity, neurotoxicity and/or nephrotoxicity), it is important to clearly distinguish between information that can be obtained from in vitro studies (e.g. using human plasma) and information that can only be derived from in vivo studies (e.g. using an appropriate animal model). In vitro studies are crucial to establish whether the mode of action of a chemoprotective agent involves the formation of a complex with CPHP's in plasma. If this is the case, the structure of the formed PSC can either be elucidated by X-ray absorption spectroscopy (Fig. 2)34 and/or mass spectrometry (Fig. 3).6 In vitro studies can also establish an effective molar ratio between the chemoprotective agent and CP which will preclude the formation of free CPHP's in plasma and minimize a chemoprotective agent-induced perturbation of endogenous plasma metalloproteins.30,33b Furthermore, the antitumour activity of the formed PSC's and their acute (IC50) as well as their potential long-term toxicity can be established by assessing toxicogenomic endpoints35 in cell culture experiments using appropriate cell lines. After the completion of these in vitro studies, in vivo studies with an appropriate animal model are absolutely necessary to further optimize the molar ratio between the chemoprotective agent and CP, their order of injection as well as the time lag between the administration of the two drugs.36 In addition, in vivo studies are required to detect a potentially adverse organ accumulation of a formed PSC (e.g. in the brain) and to assess the clearance of formed PSC complexes from the bloodstream to the kidneys and ultimately the urine.
Fig. 2. X-ray absorption spectroscopy-derived structure of the [Pt(S2O3)4]6– that was isolated from PBS-buffer (STS : CP = 400 : 1). (A) Shows the Pt LIII EXAFS oscillations and (B) the Pt–S phase corrected Fourier transforms, for data (red lines), together with the best fit (blue lines). Best fits indicated four equivalent Pt–S at distance R of 2.31 Å, with σ 2, the mean square deviation in R, of 0.0031 Å2. The inset shows the energy minimized geometry optimized density functional theory structure for [Pt(S2O3)4]6– constrained to S 4 point group symmetry to assist with convergence. The computed Pt–S bond-lengths were 2.33 Å, in excellent agreement with the EXAFS results, and both are in agreement with the Pt–S distance of the sole reported crystal structure of a terminal thiosulfate bound to a Pt(ii) ion.34 .

Fig. 3. A LC-ESI-MS spectrum (negative mode) obtained when PBS-buffer was spiked with CP, incubated at 37 °C for 10 min and then spiked with NAC and analyzed 50 min later.6 NAC : CP molar ratio 400 : 1, CP: cisplatin, NAC: N-acetylcysteine.

Future outlook
The elucidation of the mechanisms by which chemoprotective agents affect the metabolism of CP in the bloodstream is absolutely critical in the context of developing a clinical treatment protocol that can be employed to significantly reduce the toxic side-effects of CP in patients while maintaining its antitumor efficacy. This task is now within reach since state-of-the-art metallomics tools can be applied in conjunction with animal studies.30,33a,b In principle, the approach of selectively inactivating the toxic hydrolysis products of CP in the bloodstream by an optimal chemoprotective agent, while maximizing the lifetime of the active anticancer prodrug CP in the blood circulation would effectively transform this anticancer drug into a much safer one. This overall strategy may also allow one to improve the therapeutic potential of CP (as well as other toxic metal-based anticancer drugs) by escalating the dose that is administered to a patient. This would be particularly useful as this would allow one to treat even those patients in which the tumor has developed drug resistance and where the CP dose can often not be further increased because of the inherent severe toxic side-effects of CP.
Acknowledgments
MS is a Fellow in the Canadian Institutes of Health Research Training grant in Health Research Using Synchrotron Techniques (CIHR-THRUST). AN is supported by a research grant from the Alberta's Children's Hospital Foundation. GNG and IJP are supported by Canada Research Chairs, with research at the University of Saskatchewan further supported by NSERC, CIHR, SHRF and the Government of Saskatchewan. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory is supported by the U.S. DOE, Office of Science, OBES under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) including P41GM103393. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Biographies

Melani Sooriyaarachchi
Melani Sooriyaarachchi was born in Sri Lanka and received her BSc (Hons) in Chemistry from the University of Colombo in 2007. She started her graduate studies at the University of Calgary in 2008 under the supervision of Dr J. Gailer where she completed her MSc (2011) and subsequently PhD (2015). Her research was focused on studying the metabolism of various clinically relevant metal-based anticancer drugs in human plasma, when used individually or in combination. She was a CIHR-THRUST fellow during 2011–2015. Currently she is a chemistry instructor at the Southern Alberta Institute of Technology.

Graham N. George
Graham George was educated at King's College London (BSc, 1979) and the University of Sussex (DPhil, 1983). After postdoctoral fellowships at the University of Sussex and at Exxon Research & Engineering Co. in New Jersey, USA, he continued at Exxon as a Principal Investigator until moving to the Stanford Synchrotron Radiation Laboratory in 1992. In 2003 took up the position of full professor and Tier 1 Canada Research Chair of X-ray Absorption Spectroscopy at the University of Saskatchewan. His research directions include a career-long interest in X-ray spectroscopy and the development of new methods for understanding the roles and mechanisms of metals in biology. He is married to fellow professor and Canada Research Chair Ingrid Pickering, and they have three children.

Ingrid J. Pickering
Ingrid Pickering received her BA in Natural Sciences from the University of Cambridge (UK) in 1986 and following this used Physical Chemistry to study heterogeneous catalysis at the Royal Institution and received a PhD from Imperial College London (UK) in 1990. After two years as a postdoctoral fellow with Exxon Research and Engineering Co. (NJ, USA), she moved to the Stanford Synchrotron Radiation Laboratory (CA, USA) in 1992. She came to Canada and her present position at the University of Saskatchewan in 2003. Ingrid is currently Professor and Tier 1 Canada Research Chair in Molecular Environment Science at the University of Saskatchewan. Her research investigates the roles of metals and other elements in biological systems from the environment to human health.

Aru Narendran
Aru Narendran is a full professor in the departments of pediatrics and oncology, Faculty of Medicine, University of Calgary. His primary research and clinical interests focus on the development of new agents and novel therapeutics for refractory pediatric malignancies. He is the recipient of a number of awards including the Odile Schweisguth International Prize for pediatric oncology research and the young investigator award from the Children's oncology group (COG). Currently, he directs the POETIC pre-clinical and drug discovery laboratory with the mandate to identify agents and regimens for early phase clinical trials for currently difficult to treat pediatric cancer patients.

Jürgen Gailer
Jürgen Gailer received his MSc and PhD from the University of Graz/Austria in 1997. As an Erwin Schroedinger fellow he moved to the Department of Molecular and Cellular Biology of the University of Arizona (Tucson/USA) and subsequently transferred to the Department of Nutritional Sciences as a research associate. In 2001 he moved to the GSF National Research Center for Environment and Health (Munich/Germany), where he was an Alexander von Humboldt fellow until 2002. In 2003 he became team leader in biopharmaceutical production at Boehringer IngelheimAustria/Vienna and joined the Department of Chemistry of the University of Calgary in 2004 where he is an associate professor and has a co-appointment in the Environmental Science Program. His research aims to develop and apply metallomics methods to better understand the role that non-essential metals and metal-based drugs play in biology.
References
- Klein A. V. and Hambley T. W., in Ligand Design in Medicinal Inorganic Chemistry, ed. T. Storr, John Wiley & Sons, Ltd, 1st edn, 2014, pp. 9–45. [Google Scholar]
- Kelland L. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- (a) Tan C.-P., Lu Y.-Y., Ji L.-N., Mao Z.-W. Metallomics. 2014;6:978–995. doi: 10.1039/c3mt00225j. [DOI] [PubMed] [Google Scholar]; (b) Maccio A., Madeddu C. Expert Opin. Pharmacother. 2013;14:1839–1857. doi: 10.1517/14656566.2013.813934. [DOI] [PubMed] [Google Scholar]; (c) Pizarro A. M., Sadler P. J. Biochimie. 2009;91:1198–1211. doi: 10.1016/j.biochi.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Gibson D. Dalton Trans. 2009:10681–10689. doi: 10.1039/b918871c. [DOI] [PubMed] [Google Scholar]; (b) Casini A., Reedijk J. Chem. Sci. 2012;3:3135–3144. [Google Scholar]; (c) Sancho-Martinez S. M., Prieto-Garcia L., Prieto M., Lopez-Novoa J. M., Lopez-Hernandez F. J. Pharmacol. Ther. 2012;136:35–55. doi: 10.1016/j.pharmthera.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Hambley T. W. Dalton Trans. 2007:4929–4937. doi: 10.1039/b706075k. [DOI] [PubMed] [Google Scholar]
- Sooriyaarachchi M., Ameliorating the toxic side-effects of cisplatin by systematically modulating its metabolism in human plasma with chemoprotective agents, PhD thesis, University of Calgary, April 2015. [Google Scholar]
- (a) Pabla N., Dong Z. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]; (b) Sahni V., Choudhury D., Ahmed Z. Nat. Rev. Nephrol. 2009;5:450–462. doi: 10.1038/nrneph.2009.97. [DOI] [PubMed] [Google Scholar]
- Brock P. R., Knight K. R., Freyer D. R., Campbell K. C. M., Steyger P. S., Blakely B. W., Rassekh S. R., Chang K. W., Fliogor B. J., Rajput K., Sullivan M., Neuwelt E. A. J. Clin. Oncol. 2012;30:2408–2417. doi: 10.1200/JCO.2011.39.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. C. M., Rybak L. P., Meech R. P., Hughes L. Hear. Res. 1996;102:90–98. doi: 10.1016/s0378-5955(96)00152-9. [DOI] [PubMed] [Google Scholar]
- Berry J. M., Jacobs C., Sikic B. I., Halsey J., Borch R. F. J. Clin. Oncol. 1990;8:1585–1590. doi: 10.1200/JCO.1990.8.9.1585. [DOI] [PubMed] [Google Scholar]
- Wolf S., Barton D., Kottschade L., Grothey A., Loprinzi C. Eur. J. Cancer. 2008;44:1507–1515. doi: 10.1016/j.ejca.2008.04.018. [DOI] [PubMed] [Google Scholar]
- (a) Harper B. W., Krause-Heuer A. M., Grant M. P., Manohar M., Garbutcheon-Singh K. B., Aldrich-Wright J. R. Chem. – Eur. J. 2010;16:7064–7077. doi: 10.1002/chem.201000148. [DOI] [PubMed] [Google Scholar]; (b) Bruijnincx P. C. A., Sadler P. J. Curr. Opin. Chem. Biol. 2008;12:197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Liao L., Liu J., Dreaden E. C., Morton S. W., Shopsowitz K. E., Hammond P. T., Johnson J. A. J. Am. Chem. Soc. 2014;136:5896–5899. doi: 10.1021/ja502011g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krause-Heuer A. M., Wheate N. J., Tilby M. J., Pearson D. G., Ottley C. J., Aldrich-Wright J. R. Inorg. Chem. 2008;47:6880–6888. doi: 10.1021/ic800467c. [DOI] [PubMed] [Google Scholar]; (c) Kratz F., Muller I. A., Ryppa C., Warnecke A. ChemMedChem. 2008;3:20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]; (d) Plumb J. A., Venugopal B., Oun R., Gomez-Roman N., Kawazoe Y., Venkataramanan N. S., Wheate N. J. Metallomics. 2012;4:561–567. doi: 10.1039/c2mt20054f. [DOI] [PubMed] [Google Scholar]
- Wang X., Guo Z. Anti-Cancer Agents Med. Chem. 2007;7:19–34. doi: 10.2174/187152007779314062. [DOI] [PubMed] [Google Scholar]
- (a) Dickey D. T., Wu Y. J., Muldoon L. L., Neuwelt E. A. J. Pharmacol. Exp. Ther. 2005;314:1052–1058. doi: 10.1124/jpet.105.087601. [DOI] [PubMed] [Google Scholar]; (b) Saito T., Zhang Z. J., Manabe Y., Ohtsubo T., Saito H. Eur. Arch. Oto-Rhino-Laryngol. 1997;254:281–286. doi: 10.1007/BF02905989. [DOI] [PubMed] [Google Scholar]; (c) Gandara D. R., Wiebe V. J., Perez E. A., Makuch R. W., deGregorio M. W. Crit. Rev. Oncol. Hematol. 1990;10:353–365. doi: 10.1016/1040-8428(90)90010-p. [DOI] [PubMed] [Google Scholar]; (d) Erdlenbruch B., Pekrun A., Schiffman H., Witt O., Lakomek M. Med. Pediatr. Oncol. 2002;38:349–352. doi: 10.1002/mpo.1343. [DOI] [PubMed] [Google Scholar]; (e) Nagai N., Hotta K., Yamamura H., Ogata H. Cancer Chemother. Pharmacol. 1995;36:404–410. doi: 10.1007/BF00686189. [DOI] [PubMed] [Google Scholar]
- (a) Muldoon L. L., Walker-Rosenfeld S. L., Halce C., Purcell S. E., Bennett L. C., Neuwelt E. A. J. Pharmacol. Exp. Ther. 2001;296:797–805. [PubMed] [Google Scholar]; (b) Wu Y. J., Muldoon L. L., Neuwelt E. A. J. Pharmacol. Exp. Ther. 2005;312:424–431. doi: 10.1124/jpet.104.075119. [DOI] [PubMed] [Google Scholar]; (c) Dickey D. T., Muldoon L. L., Doolittle N. D., Peterson D. R., Kraemer D. F., Neuwelt E. A. Cancer Chemother. Pharmacol. 2008;62:235–241. doi: 10.1007/s00280-007-0597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Treskes M., van der Vijgh W. J. F. Cancer Chemother. Pharmacol. 1993;33:93–106. doi: 10.1007/BF00685326. [DOI] [PubMed] [Google Scholar]; (b) Glover D., Grabelsky S., Fox K., Weiler C., Cannon L., Glick J. Int. J. Radiat. Oncol., Biol., Phys. 1989;16:1201–1204. doi: 10.1016/0360-3016(89)90283-6. [DOI] [PubMed] [Google Scholar]
- (a) Rybak L. P., Ravi R., Somani S. M. Toxicol. Sci. 1995;26:293–300. doi: 10.1006/faat.1995.1100. [DOI] [PubMed] [Google Scholar]; (b) Borch R. F., Pleasants M. E. Proc. Natl. Acad. Sci. U. S. A. 1979;76:6611–6614. doi: 10.1073/pnas.76.12.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Church M. W., Kaltenbach J. A., Blakely B. W., Burgio D. L. Hear. Res. 1995;86:195–203. doi: 10.1016/0378-5955(95)00066-d. [DOI] [PubMed] [Google Scholar]
- (a) Jones M. M., Basinger M. Anticancer Res. 1989;9:1937–1942. [PubMed] [Google Scholar]; (b) Cloven N. G., Re A., McHale M. T., Burger R. A., Disaia P. J., Rose G. S., Campbell K. C. M., Fan H. Anticancer Res. 2000;20:4205–4210. [PubMed] [Google Scholar]; (c) Ekborn A., Laurell G., Johnström P., Wallin I., Eksborg S., Ehrsson H. Hear. Res. 2002;165:53–61. doi: 10.1016/s0378-5955(02)00277-0. [DOI] [PubMed] [Google Scholar]; (d) Campbell K. C. M., Meech R. P., Rybak L. P., Hughes L. F. Hear. Res. 1999;138:13–28. doi: 10.1016/s0378-5955(99)00142-2. [DOI] [PubMed] [Google Scholar]; (e) Korver K. D., Rybak L. P., Whitworth C., Campbell K. M. Otolaryngol.–Head Neck Surg. 2002;126:683–689. doi: 10.1067/mhn.2002.125299. [DOI] [PubMed] [Google Scholar]
- Basinger M. A., Jones M. M., Holscher M. A. Toxicol. Appl. Pharmacol. 1990;103:1–15. doi: 10.1016/0041-008x(90)90257-u. [DOI] [PubMed] [Google Scholar]
- (a) Hamers F., Brakkee J. H., Cavalletti E., Tedeschi M., Marmonti L., Pezzoni G., Neijt J. P., Gispen W. H. Cancer Res. 1993;53:544–549. [PubMed] [Google Scholar]; (b) Zunino F., Pratesi G., Micheloni A., Cavalletti E., Sala F., Tofanetti O. Chem.-Biol. Interact. 1989;70:89–101. doi: 10.1016/0009-2797(89)90065-3. [DOI] [PubMed] [Google Scholar]; (c) Re F., Bohm S., Oriana S., Battista Spatti G., Zunino F. Cancer Chemother. Pharmacol. 1990;25:355–360. doi: 10.1007/BF00686237. [DOI] [PubMed] [Google Scholar]
- Katsuda H., Yamashita M., Katsura H., Yu J., Waki Y., Nagata N., Sai Y., Miyamoto K. Biol. Pharm. Bull. 2010;33:1867–1871. doi: 10.1248/bpb.33.1867. [DOI] [PubMed] [Google Scholar]
- (a) Hyppolito M. A., de Oliveira J. A. A., Rossato M. Eur. Arch. Oto-Rhino-Laryngol. 2006;263:798–803. doi: 10.1007/s00405-006-0070-6. [DOI] [PubMed] [Google Scholar]; (b) Li G. M., Sha S. H., Zotova E., Arezzo J., Van de Water T., Schacht J. Lab. Invest. 2002;82:585–596. doi: 10.1038/labinvest.3780453. [DOI] [PubMed] [Google Scholar]
- Fouad A. A., Morsy M. A., Gomaa W. Environ. Toxicol. Pharmacol. 2008;25:292–297. doi: 10.1016/j.etap.2007.10.026. [DOI] [PubMed] [Google Scholar]
- Sato T., Okubo M., Sawaki K., Maehashi H., Kawaguchi M. J. Pharmacol. Sci. 2010;112:361–368. doi: 10.1254/jphs.09323fp. [DOI] [PubMed] [Google Scholar]
- (a) Viale M., Zhang J. X., Pastrone M., Mariggio M. A., Esposito M., Lindup W. E. J. Pharmacol. Exp. Ther. 2000;293:829–836. [PubMed] [Google Scholar]; (b) Ognio E., Chiavarina B., Peterka M., Mariggio M. A., Viale M. Chem.-Biol. Interact. 2006;164:232–240. doi: 10.1016/j.cbi.2006.10.001. [DOI] [PubMed] [Google Scholar]; (c) Zicca A., Cafaggi S., Mariggi M. A., Vannozzi M. O., Ottone V., Bocchini V., Caviglioli G., Viale M. Eur. J. Pharmacol. 2002;442:265–272. doi: 10.1016/s0014-2999(02)01537-6. [DOI] [PubMed] [Google Scholar]; (d) Fenoglio C., Boncompagni E., Chiavarina B., Cafaggi S., Cilli M., Viale M. Anticancer Res. 2005;25:4123–4128. [PubMed] [Google Scholar]
- Gula A., Ren L., Zhou Z., Lu D., Wang S. Int. J. Pharmacol. 2013;453:441–447. doi: 10.1016/j.ijpharm.2013.06.019. [DOI] [PubMed] [Google Scholar]
- (a) Manley S. A., Byrns S., Lyon A. W., Brown P., Gailer J. J. Biol. Inorg. Chem. 2009;14:61–74. doi: 10.1007/s00775-008-0424-1. [DOI] [PubMed] [Google Scholar]; (b) Sooriyaarachchi M., Narendran A., Gailer J. Metallomics. 2011;3:49–55. doi: 10.1039/c0mt00058b. [DOI] [PubMed] [Google Scholar]
- Schmitt N. C., Rubel E. W., Nathanson N. M. J. Neurosci. 2009;29:3843–3851. doi: 10.1523/JNEUROSCI.5842-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sooriyaarachchi M., Narendran A., Gailer J. Metallomics. 2013;5:197–207. doi: 10.1039/c3mt00012e. [DOI] [PubMed] [Google Scholar]
- Daley-Yates P. T., McBrien D. C. H. Biochem. Pharmacol. 1984;33:3063–3070. doi: 10.1016/0006-2952(84)90610-5. [DOI] [PubMed] [Google Scholar]
- Morris T. T., Ruan Y., Lewis V. A., Narendran A., Gailer J. Metallomics. 2014;6:2034–2041. doi: 10.1039/c4mt00220b. [DOI] [PubMed] [Google Scholar]
- (a) Sooriyaarachchi M., Narendran A., Gailer J. Metallomics. 2012;4:960–967. doi: 10.1039/c2mt20076g. [DOI] [PubMed] [Google Scholar]; (b) Sooriyaarachchi M., Narendran A., White W. H., Gailer J. Metallomics. 2014;6:532–541. doi: 10.1039/c3mt00238a. [DOI] [PubMed] [Google Scholar]; (c) Sooriyaarachchi M., Gibson M. A., Lima B. d. S., Gailer J. Can. J. Chem. 2016;94:360–366. [Google Scholar]
- Sooriyaarachchi M., Gailer J., Dolgova N. V., Pickering I. J., George G. N. J. Inorg. Biochem. 2016 doi: 10.1016/j.jinorgbio.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H., Fostel J. M., Aruga C., Bauer D., Deng S., Dickinson D., Le Fevre A. C., Fornace A. J. J., Gu Y., Hoflack J. C., Shiiyama M., Smith R., Snyder R. D., Spire C., Tanaka G., Aubrecht J. Toxicol. Sci. 2009;110:341–352. doi: 10.1093/toxsci/kfp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon L. L., Pagel M. A., Kroll R. A., Brummett R. E., Doolittle N. D., Zuhowski E. G., Egorin M. J., Neuwelt E. A. Clin. Cancer Res. 2000;6:309–315. [PubMed] [Google Scholar]