

Summary
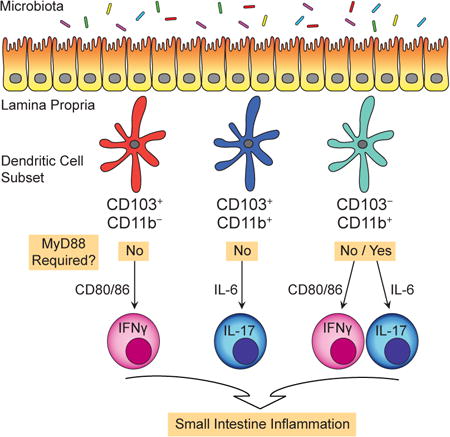
Normal dynamics between microbiota and dendritic cells (DCs) support modest numbers of T cells, yet these do not cause inflammation. The DCs that induce inflammatory T cells and the signals that drive this process remains unclear. Here we demonstrate that small intestine DCs lacking the signaling attenuator A20 induce inflammatory T cells and that the signals perceived and antigen presenting cell (APC) functions are unique for different DC subsets. Thus, while CD103+CD11b− DCs exclusively instruct IFNγ+ T cells, CD103+CD11b+ DCs exclusively instruct IL-17+ T cells. Surprisingly, APC functions of both DC subsets are upregulated in a MyD88-independent fashion. In contrast, CD103−CD11b+ DCs instruct both IFNγ+ and IL-17+ T cells and only the IL-17-inducing APC functions require MyD88. In disease pathogenesis both CD103− CD11b+ and CD103+CD11b+ DCs expand pathologic Th17 cells. Thus, in disease pathogenesis specific DCs instruct specific inflammatory T cells.
Graphical abstract
Introduction
The mucosal immune system must co-exist with a large and diverse number of intestine-resident microorganisms, collectively referred to as the intestinal microbiota. It is now clear that intestinal microbiota can potently influence the composition and function of immune cells, including mucosal T cells (Honda & Littman 2012). While microbiota-reactive T cells naturally exist during health, an increase in abundance, stimulation, or function of these T cells is thought to contribute to the development of inflammatory bowel disease (IBD).
Dendritic cells (DCs) are specialized antigen presenting cells (APCs) that sense microbes, activate naïve T cells, and regulate immunity (Steinman 2012; Hammer & Ma 2013). DCs of intestine lamina propria (LP) were thought to uniformly express CD103, however with recent lineage verification of a CD103− DC population (Cerovic et al. 2013; Scott et al. 2014), three bona fide DC subsets are now recognized: CD103+CD11b−, CD103+CD11b+ and CD103− CD11b+ DCs. All three express Zbtb46, require FLT3 ligand, and migrate to lymph nodes (Bogunovic et al. 2009; Varol et al. 2009; Cerovic et al. 2013; Scott et al. 2014). In lymph nodes, and possibly within intestine, DCs instruct T cell responses (Geem et al. 2014; Goto et al. 2014; Semmrich et al. 2012; Bekiaris et al. 2014). Normal dynamics between microbiota and DCs support modest numbers of IFNγ-producing, T helper 1 (Th1) and IL-17-producing (Th17) CD4 T cells, although these do not cause inflammation. The DC subsets and APC functions that give rise to inflammatory T cells, particularly those that cause IBD, remain poorly understood.
APC functions of DCs are upregulated by inflammatory signals and MyD88 signals, which include toll-like receptor (TLR), IL-1 and IL-18 pathways are considered most potent (Hou et al. 2008; Spörri & Reis e Sousa 2005; Iwasaki & Medzhitov 2010). MyD88 signals are triggered by microbiota and in certain settings MyD88 signals drive inflammation in colon (Hoshi et al. 2012; Feng et al. 2010). Exactly how MyD88 signals triggered by microbiota drive inflammation or control DC functions is unclear. One difficulty is that in health, intestinal DCs and their migratory counterparts in lymph nodes have limited APC function (Persson et al. 2013; Fujimoto et al. 2011). Exogenous inflammatory stimuli are required to enhance APC functions and these may alter DCs in non-physiologic ways. The physiologic signals that control APC functions are a topic of great interest because these signals likely control how DCs instruct T cells and also, the three DC subsets of intestine may each instruct different T cells, particularly Th17 cells (Persson et al. 2013; Schlitzer et al. 2013; Bekiaris et al. 2014).
We previously reported DCs lacking A20 (gene name Tnfaip3), an intracellular suppressor of multiple signaling pathways (Lee et al. 2000; Boone et al. 2004; Hitotsumatsu et al. 2008), are hyper-responsive to physiologic MyD88 signals and physiologic signals that are MyD88-independent (Hammer et al. 2011). Although both signals enhanced DC-stimulation of T cells in mice with DC-specific, Cd11c-cre, conditional knockout of A20 (A20cko mice), DC-expansion of T cells in lymph nodes occurred in an exclusively MyD88-dependent fashion (Hammer et al. 2011). While pathologic potential of MyD88 is known, the physiologic settings wherein MyD88-independent signals have potent effect on DCs and whether such signals can be pathologic are unknown. In IBD, human genetic studies link A20 to Crohn's disease and ulcerative colitis (Wellcome Trust Case Control Consortium et al. 2007; Graham et al. 2008; 1000 Genomes Project Consortium et al. 2012), and in IBD, DC-intrinsic signaling pathways, MyD88-dependent and perhaps also MyD88-independent, could be pathologic.
Here we used A20cko mice to determine how intestinal DCs regulate T cells and intestinal homeostasis. We additionally used mice with DC-specific loss of both A20 and MyD88 (A20/Myd88cko mice) to determine the roles of physiologic MyD88 signals. We show that A20cko mice develop inflammation of small intestine that requires microbiota, DCs and T cells.
Surprisingly, DC-expansion of inflammatory T cells can occur in a MyD88-independent fashion. Without the need for exogenous stimuli, all three intestinal DC subsets in A20cko mice have remarkable ability to activate naïve T cells and instruct cytokine production. We further show that in small intestine, CD103−CD11b+ and CD103+CD11b+ DCs instruct and expand pathologic Th17 cells, indicating that in disease pathogenesis, specific DC subsets instruct specific types of inflammatory T cells.
Results
DCs drive spontaneous inflammation in small intestine, in a MyD88-independent fashion
Inflammatory bowel disease involves dynamics between microbe and host and we reported that A20cko mice develop inflammation in colon (Hammer et al. 2011). Colon anatomy, physiology, and cellular composition are distinct from small intestine and inflammation does not affect both organs by default. We thus tested whether DC-expression of A20 was required for small intestine homeostasis. Remarkably, small intestine of A20cko mice was inflamed, which resulted in increased organ weight and thickening of the intestinal wall (Fig. 1a, S1a). Small intestine inflammation occurred within ten weeks of age and preceded colitis (Fig. S1a), which we reported is most prominent in mice six months or older (Hammer et al. 2011). Inflammation did not occur in co-housed, littermate control, A20wt mice (see methods for co-housing). Given disease severity we hypothesized that MyD88, which is known to drive lymphadenopathy in A20cko mice (Hammer et al. 2011), was required for small intestine inflammation. In contrast to our predictions, A20/Myd88cko mice also demonstrated severe small intestine inflammation and increased organ weight (Fig. 1a, S1a). Genomic qPCR confirmed >95% deletion of A20 and MyD88 genes in DCs of small intestine lamina propria (SI-LP) (Fig. S1b). These genes were deleted to a lesser extent in SI-LP macrophages, a fraction of which are known to undergo gene recombination in Cd11c-cre mice (Fig. S1b). Inflammation in A20/Myd88cko mice was due to loss of A20, and not loss of MyD88, since co-housed A20-wild type mice (Myd88cko) had no inflammation (Fig. 1a).
Figure 1. A20cko and A20/Myd88cko mice develop spontaneous small intestine inflammation that shares histologic features resembling Crohn's disease.
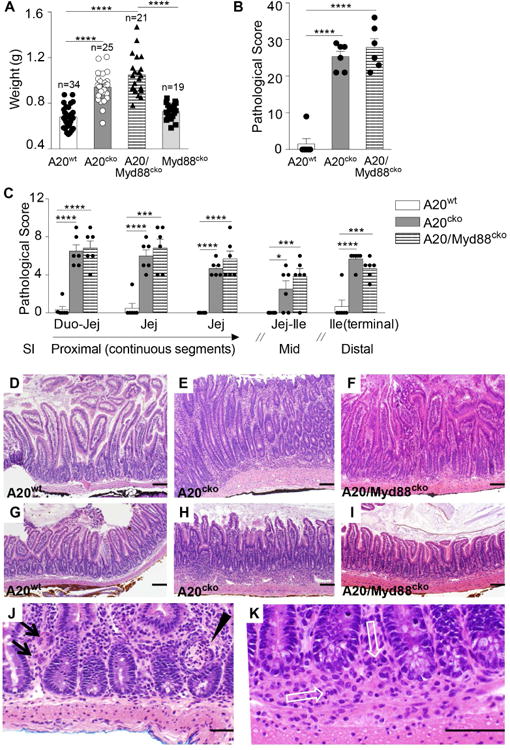
(a) Organ weights of the small intestine from A20wt, A20cko, A20/Myd88cko and Myd88cko mice (n = 19-34 mice of each genotype, aged 9-12 weeks). (b-c) Five segments of small intestine (SI) were dissected from mice of the indicated genotype, ages 14-20 weeks and evaluated for pathology in a blinded fashion. Segments of SI were from proximal and middle (mid) regions, and also terminal ileum, as described in methods. Abbreviations: Duodenum (Duo), Jejunum (Jej), Ileum (Ile). The combined score of all SI regions evaluated (b), and the individual score of each evaluated region (c). (d-k) Hematoxylin and eosin stained SI segments of proximal jejunum (d-f) and the terminal ileum (g-i) are shown at the same magnification for co-housed 14 week-old A20wt (d, g), A20cko (e, h) and A20/Myd88cko (f, i) mice. Inflammatory infiltrates are evident in the LP of A20cko and A20/Myd88cko mice, particularly beneath the crypts. These extend focally into the submucosa. (j) Examples of multinucleated giant cells characteristic of granulomatous inflammation (black arrows) and crypt abscesses (black arrowhead) in the proximal jejunum of a 10 week-old A20cko mouse. (k) Example of a cluster of epithelioid macrophages (granuloma) with neutrophils (white open arrows) in the LP and submucosa of the terminal ileum of a 14 week-old A20/Myd88cko mouse. Thick black bar = 100μm. Error bars represent mean ± SEM, *, P <0.05, ***, P <0.001, ****, P <0.0001 (unpaired student's t-test). See also Figure S1.
Histologic examination showed small intestine organ weight of A20cko mice and A20/Myd88cko mice was increased due to inflammation and cellular infiltration, the extent and severity of which was similar in both genotypes (Fig. 1b). Inflammation was generally present in all segments of small intestine examined, but of greater extent (involving >60% of the area) in segments containing duodenum/proximal jejunum and terminal ileum (Fig. 1c). Inflammatory infiltrates in proximal jejunum (Fig. 1d-f) and in terminal ileum (Fig. 1g-i) typically expanded the LP, particularly beneath crypts. Foci of inflammatory infiltrates commonly extended into submucosa or were transmural (Fig. 1e,f and h,i). Inflammatory infiltrate was primarily composed of mononuclear cells, admixed with varying numbers of neutrophils. Foci of epithelioid macrophages (granulomas) were frequent, often accompanied by multinucleated giant cells (Fig. 1j,k). Crypt abscesses were also observed, particularly in proximal jejunum (Fig. 1j). Thus, both A20cko and A20/Myd88cko mice develop spontaneous small intestine inflammation sharing histologic features with Crohn's disease. In these analyses we found little to no pathologic inflammation in colon despite extensive inflammation involving small intestine, suggesting the physiologic factors driving pathogenesis were unique in these two organs. The persistence of small intestine inflammation in A20/Myd88cko mice further suggests that physiologic signals other than MyD88 can drive pathologic inflammation of small intestine.
DCs and T cells together drive small intestine inflammation in a MyD88-independent fashion
Different DC subsets have distinct developmental requirements and cellular responses to microbial products. To determine the role of A20 in each DC subset of SI-LP, we analyzed the three DC subsets using a panel of markers verified to discriminate CD14+ macrophages from CD24+ DCs (Tamoutounour et al. 2012; Scott et al. 2014). Using this standard approach we could unambiguously distinguish SI-LP macrophages and DCs, despite inflammation in A20cko and A20/Myd88cko mice (Fig. 2a, S1c).
Figure 2. In A20cko and A20/Myd88cko mice, intestinal DCs are phenotypically mature and expand pathological mucosal T cells that cause small intestine inflammation.
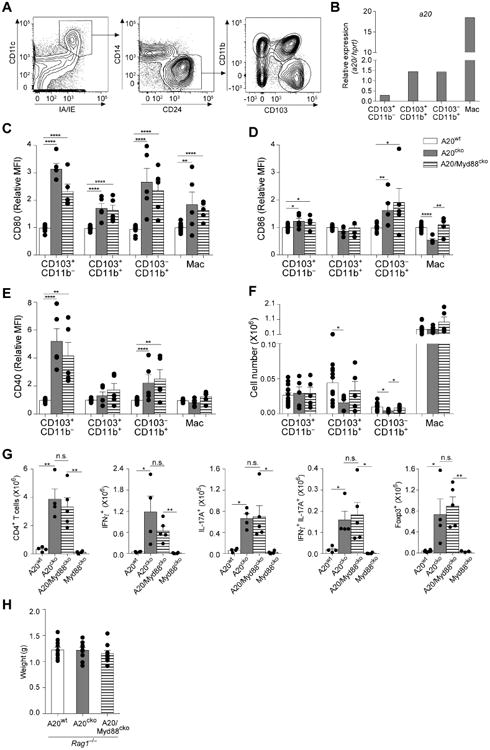
(a) Representative flow cytometry gating strategy in A20wt mice to identify the three bona fide DC populations of SI-LP. (b) Expression of A20 mRNA by the indicated SI-LP DC subset or macrophages from wild-type mice, relative to hprt. (c-f) Cell surface expression of maturation markers CD80 (c), CD86 (d) CD40 (e) and total cell number (f) of SI-LP DCs and macrophages. Cellular expression of maturation markers is represented as mean fluorescence intensity (MFI) relative to that of the same population in wild-type (WT) mice. (g) Cell number of SI-LP CD4+ T cells, IFNγ+, IL-17+, IFNγ+IL-17+ and Foxp3+CD4+ T cells from mice of the indicated genotype. Data in (c-g) was combined from at least 3 independent experiments with mice between 10-14 weeks of age, including at least one mouse of each genotype per experiment. Each dot represents one mouse. (h) Organ weights of small intestine from Rag1-/- mice of the indicated genotypes (n = 9-11 mice of each genotype, aged 12-14 weeks). Error bars represent mean ± SEM, *, P <0.05, **, P <0.01, ****, P <0.0001 (unpaired student's t-test). See also Figure S2.
In control A20wt mice, A20 mRNA was abundant in SI-LP macrophages, CD103+CD11b+, and CD103−CD11b+ DCs (Fig. 2b). CD103+CD11b− DCs expressed little A20 mRNA, although upon deletion of A20 in A20cko mice, CD80 expression was increased 3 fold, suggesting that low level expression of A20 was physiologically relevant. CD80 was also upregulated by the other DCs and by macrophages (Fig. 2c). While all SI-LP DCs in A20cko mice upregulated CD80, and all failed to express A20 mRNA (Fig. S1d), other maturation markers were upregulated differently by each subset. Only CD103−CD11b+ DCs highly upregulated CD86, whereas CD40, a classical maturation marker, was most dramatically upregulated on CD103+CD11b− DCs (Fig. 2d,e). Neither CD86 nor CD40 was upregulated by CD103+CD11b+ DCs or macrophages. In bone-marrow derived DCs, which receive no maturation stimulus, loss of A20 did not cause spontaneous expression of maturation markers (Fig. S1e). These data suggest that although all SI-LP DCs normally express A20, each has subset-specific characteristics that result in distinct maturation phenotypes in A20cko mice. Similar phenotypic changes occurred in A20/Myd88cko mice, indicating that MyD88 was not required (Fig. 2c-e).
Despite these phenotypic changes neither DCs or macrophages were numerically expanded (Fig. 2f). By contrast, Th1, Th17, and IL-17+IFNγ+ CD4 T cells were expanded more than 10-fold in A20cko mice (Fig. 2g). Foxp3+ T cells were also expanded, suggesting that expansion of CD4 T cells was not due to deficiency in numbers of regulatory T cells (Fig. 2g). Consistent with our observation that inflammation of small intestine was driven in a MyD88-independent fashion, CD4 T cells were also expanded more than 10-fold in A20/Myd88cko mice (Fig. 2g). These data suggest that in small intestine, physiologic signals that are MyD88-independent can induce DC maturation and T cell expansion.
Given that SI-LP T cells were expanded we considered that T cells may be required for inflammation. To test this, we crossed A20cko and A20/Myd88cko mice to Rag1-/- mice. In contrast to Rag1-sufficient mice, small intestine organ weight of Rag1-/- A20cko and Rag1-/- A20/Myd88cko was normal and there was no inflammation (Fig. 2h, Fig. S2a,b). From these data we conclude that SI-LP DCs in A20cko and A20/Myd88cko mice expand pathologic T cells that cause small intestine inflammation. Although T cells caused inflammation, they were not required for DC maturation, suggesting that this was not secondary to inflammation (Fig. S2c,d). Collectively, the persistence of DC maturation and expansion of pathologic T cells in A20/Myd88cko mice suggests that in small intestine, MyD88-independent signals can potently induce pathological, T cell-mediated inflammation.
Microbiota trigger MyD88-independent pathways to induce small intestine inflammation
Microbiota are well known to trigger MyD88 signals that expand pathologic T cells, however, these observations were made in colon (Hoshi et al. 2012; Feng et al. 2010). We hypothesized that microbiota can also trigger MyD88-independent signals to induce pathologic T cells in small intestine. To test this in A20cko and A20/Myd88cko mice, we added antibiotics to drinking water of lactating dams and to their offspring until 10 weeks of age. Antibiotics decreased stool 16S DNA by a minimum factor of 104 (Fig. S3a). Separate cohorts of age-matched mice provided water alone were analyzed together with antibiotic-treated mice as untreated, genotype-matched controls. In contrast to mice provided water alone, organ weight of small intestine in antibiotic-treated A20cko and A20/Myd88cko mice was similar to that of antibiotic-treated A20wt mice (Fig. 3a). Moreover, antibiotics prevented inflammation (Fig. S3b,c). These data indicate that small intestine inflammation in A20cko and A20/Myd88cko mice required microbiota.
Figure 3. Microbiota are required for small intestinal inflammation and expansion of pathological mucosal T cells in A20cko and A20/Myd88cko mice.
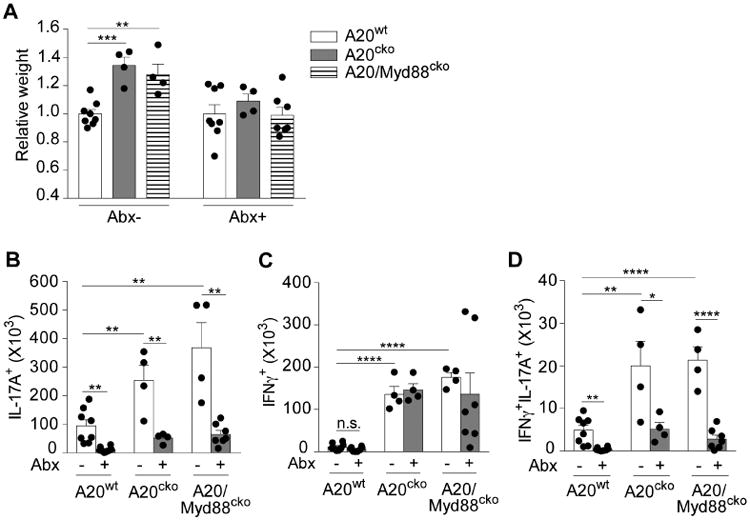
(a) Organ weights of small intestine from mice of the indicated genotypes, treated with or without broad-spectrum antibiotics (Abx) for 9-10 weeks. Mice from these treatment groups were analyzed for cell number of SI-LP IL-17+ (b), IFNγ+ (c) and IFNγ+IL-17+ CD4 T cells (d). Each dot represents one mouse. Results are combined from 4 independent experiments. Error bars show mean ± SEM, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001 (unpaired student's t-test). See also Figure S3.
Microbiota-reactive T cells exist in health but do not cause inflammation (Honda & Littman 2012). Thus, antibiotics given to A20wt mice reduced Th17 cells 8-fold (Fig. 3b). Th17 cells, which expanded and were pathologic in A20cko mice, were reduced 5-fold by antibiotics, indicating that microbiota were required. Importantly, antibiotics also reduced Th17 cells in A20/Myd88cko mice, a 7-fold decrease compared to A20/Myd88cko mice given water alone (Fig. 3b). Collectively, these data indicate that expansion of pathologic Th17 cells and small intestine inflammation in A20cko and A20/Myd88cko mice were both driven in a microbiota-dependent fashion. Additionally, in small intestine, microbiota-triggered signals that are independent of MyD88 can potently induce inflammatory Th17 cells.
Th1 cells are also present in the healthy intestine, although microbiota-reactive Th1 cells are poorly defined. Although A20cko and A20/Myd88cko mice had abundant Th1 cells, antibiotics did not reduce these, although there was a downward trend in A20/Myd88cko mice (Fig. 3c). Unlike Th1, antibiotics reduced IFNγ+IL-17+ T cells in A20cko and A20/Myd88cko mice 5-fold (Fig. 3d), suggesting that these IFNγ-producing T cells were expanded in a microbiota-dependent fashion and were linked to small intestine inflammation in A20cko and A20/Myd88cko mice. Collectively, these data support a pathologic role for signals independent of MyD88 in dynamics between microbiota, DCs and T cells in small intestine.
CD103+CD11b− DCs and CD103+CD11b+ DCs have biased APC functions that are upregulated in a MyD88-independent fashion
In the absence of exogenous stimulus, SI-LP DCs from WT mice have limited ability to instruct naïve T cells to produce IFNγ or IL-17 (Fujimoto et al. 2011; Persson et al. 2013). Because these T cell populations expanded and caused small intestine inflammation in A20cko mice, we hypothesized that SI-LP DCs in these mice have enhanced and pathologic APC functions. Additionally, because T cells expanded similarly in A20cko and A20/Myd88cko mice we reasoned that physiologic signals independent of MyD88 were sufficient to induce pathologic DC functions in one or more DC subsets. To test these hypotheses, we sorted each DC subset and assayed APC function in co-cultures with naïve OT-II T cells + endotoxin-free Ova-peptide. Importantly, no exogenous stimuli were added.
We first analyzed CD103+CD11b− DCs. When isolated from A20wt mice, CD103+CD11b− DCs instructed less than 0.3% of all T cells to produce either IFNγ or IL-17 (Fig. 4a). In stark contrast, CD103+CD11b− DCs from A20cko mice induced a robust population of IFNγ+ T cells, more than 250-fold that of wild-type (Fig. 4a,b). CD103+CD11b− DCs from A20/Myd88cko mice had similarly robust ability to instruct IFNγ+ T cells, indicating that MyD88 signals were not required to enhance APC function (Fig. 4a,b). Interestingly, although IFNγ-inducing APC functions were markedly enhanced in CD103+CD11b− DCs from A20cko and A20/Myd88cko mice, these same DCs had little ability to induce IL-17+ T cells (Fig. 4a,b). Thus, while CD103+CD11b− DCs from A20cko and A20/Myd88cko mice could instruct as many as 40% of all T cells to produce IFNγ, populations of IL-17+ T cells were consistently less than 0.3%. The strong bias of CD103+CD11b− DCs towards IFNγ-inducing APC functions were further supported by ELISA assay of cytokine in DC–T cell co-culture supernatant. While IFNγ protein in CD103+CD11b− co-cultures from A20cko and A20/Myd88cko mice was increased more than 100-fold compared to control, IL-17 protein remained low and was statistically similar to A20wt co-cultures (Fig. 4c). These data suggest that CD103+CD11b− DCs are strongly biased to instruct inflammatory Th1 cells.
Figure 4. Each SI-LP DC subset possesses unique APC functions, and differential requirements for MyD88 signals.
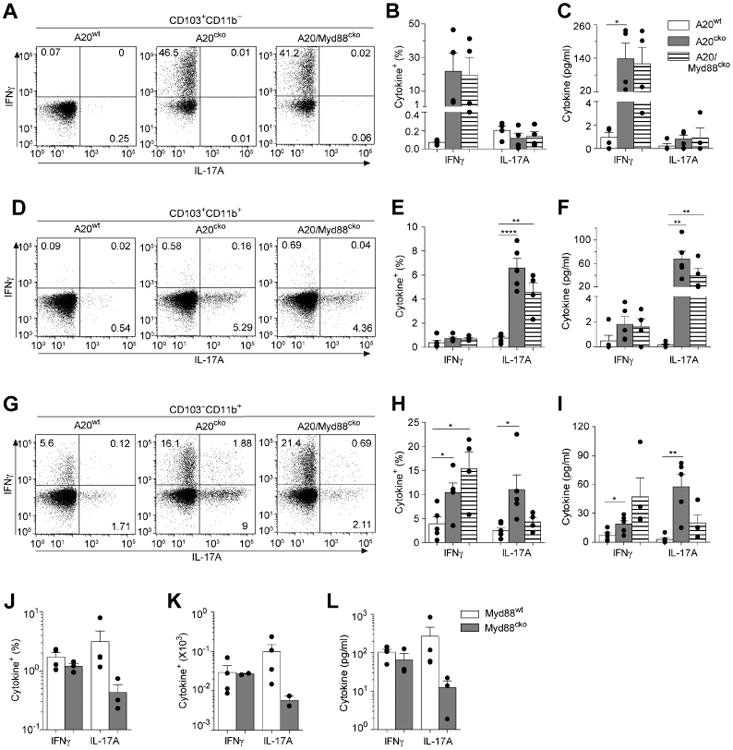
SI-LP CD103+CD11b− DCs were isolated from mice of the indicated genotype and co-cultured 1:1 with naïve OT-II T cells for 4 days. (a) Representative flow plots of IFNγ+ and IL-17+ T cells in each CD103+CD11b− DC–T cell co-culture. The percentages of IFNγ+ or IL-17+ CD4 T cells (b) and ELISA quantification of IFNγ and IL-17 protein (c) in co-cultures of CD103+CD11b− DCs from A20wt, A20cko and A20/Myd88cko mice. OT-II co-cultures with CD103+CD11b+ DCs (d-f) or CD103−CD11b+ DCs (g-i) were assayed as above. Data are combined from 3 independent experiments. Each dot represents one experiment, or replicates within an experiment, using DCs pooled from at least 3 mice of each genotype, and including all 3 genotypes for each experiment. (j-k) CD103−CD11b+ DCs isolated from co-housed Myd88wt and Myd88cko mice were co-cultured with naïve OT-II T cells, as above. The percentages (j) and total cell number (k) of IFNγ+ or IL-17+ T cells, or IFNγ or IL-17A protein in culture supernatant (l) assayed from two independent experiments, with at least 5 mice per genotype, and including both genotypes for each experiment. Each dot represents replicates within an experiment. Error bars represent mean ± SEM, *, P < 0.05, **, P < 0.01, ****, P < 0.0001, (unpaired student's t-test). See also Figure S4.
In mirror image, CD103+CD11b+ DCs isolated from A20cko mice preferentially instructed IL-17+ T cells (Fig. 4d,e). Compared to A20wt, IL-17-inducing APC functions of CD103+CD11b+ DCs from A20cko mice were enhanced more than 5-fold and remained elevated in CD103+CD11b+ DCs from A20/Myd88cko mice (Fig. 4d,e). By contrast, in these same co-cultures we detected very few IFNγ+ T cells. Enhanced and biased instruction of IL-17+ T cells was also reflected in supernatants of CD103+CD11b+ DC co-cultures from A20cko and A20/Myd88cko mice. In these co-cultures, the abundance of IL-17 protein was increased more than 300-fold compared to controls (Fig. 4f). IFNγ protein was modestly increased and may have been produced by IFNγ+IL-17+ T cells, since these were induced at much higher frequencies in both A20cko and A20/Myd88cko CD103+CD11b+ DC co-cultures (Fig S4a). These data suggest that CD103+CD11b+ DCs are heavily biased to instruct inflammatory Th17 cells.
In these experiments we found increased IFNγ+ or IL-17+ Tcells in DC–T cell co-cultures correlated with reduced Foxp3+ T cells (Fig S4b). We also tested APC functions of SI-LP macrophages from A20cko and A20/Myd88cko mice and found few IFNγ+ or IL-17+ T cells, less than 1,000-fold that of DCs from the same mice. (Fig. S4c). Less than one IFNγ+ or IL-17+ T cell was generated by each macrophage and the total number of T cells remaining in macrophage co-cultures was significantly less than DC co-cultures. These data suggest that SI-LP macrophages in A20cko and A20/Myd88cko mice have little ability to instruct naïve CD4 T cells to produce cytokines and that these APC functions were most potently upregulated by DCs. Alternatively, although most SI-LP macrophages recombined the floxed A20 allele and as a bulk population some cytokine genes were upregulated (Fig. S1b, S4d), it is possible that the remaining A20-sufficient macrophages suppress naïve T cell activation, as previously described (Denning et al. 2007). Nevertheless, in contrast to macrophages, it is clear that SI-LP CD103+CD11b− and CD103+CD11b+ DCs from A20cko and A20/Myd88cko mice DCs can potently activate naïve T cells and instruct inflammatory cytokine production, with surprisingly biased instruction for IFNγ or IL-17. It appears that the physiologic signals that can upregulate APC functions of CD103+CD11b+ and CD103+CD11b− DCs are independent of MyD88.
IL-17-inducing APC functions of CD103−CD11b+ DCs require MyD88
CD103−CD11b+ DCs are the least abundant SI-LP DC subset and little information exists on this population (Scott et al. 2014; Cerovic et al. 2013). When we tested APC functions of CD103− CD11b+ DCs from wild-type mice we routinely detected 2% or more cytokine-producing T cells, which was a much higher percentage compared to CD103+ DC co-cultures (Fig. 4g,h). Wild-type CD103−CD11b+ DCs instructed IFNγ+ or IL-17+ T cells with equal efficiency and were the most capable DC subset for instruction of IL-17+IFNγ+ T cells (Fig. S4a). Our findings confirm that in health, CD103−CD11b+ DCs possess dual APC functions (Scott et al. 2014). Interestingly, both IFNγ- and IL-17-inducing APC functions were enhanced in CD103−CD11b+ DCs from A20cko mice (Fig. 4g,h). Thus, loss of A20 did not appear to introduce bias to APC functions, but instead upregulated the dual IFNγ- and IL-17-inducing APC functions that were readily apparent in A20wt DCs. When we tested whether APC functions of CD103−CD11b+ DCs required MyD88, we were surprised to find that MyD88 was selectively required to induce IL-17+ T cells (Fig. 4g,h). Thus, while CD103−CD11b+ DC co-cultures from A20cko and A20/Myd88cko mice had similar percentages of IFNγ+ T cells and IFNγ protein, IL-17+ T cells and IL-17 protein were reduced 3-fold in A20/Myd88cko DC co-cultures (Fig. 4g-i). Collectively, these data suggest that while IFNγ-inducing APC functions were upregulated in a MyD88-independent fashion in CD103−CD11b+ DCs, instruction of IL-17+ T cells required MyD88.
Given these results we reasoned that MyD88 should also play a role in APC functions of A20wt, CD103−CD11b+ DCs. We tested this by performing a side-by-side analysis of SI-LP DCs from A20wt/Myd88wt (Myd88wt) and littermate A20wt/Myd88cko mice (Myd88cko). CD103−CD11b+ DCs from Myd88wt and Myd88cko mice were equal for expansion of IFNγ+ T cells and co-cultures contained similar amounts of IFNγ protein (Fig. 4j-l). However, in the absence of MyD88, IL-17+ T cells were reduced 5-fold. Loss of APC functions due to MyD88 deficiency was specific to CD103−CD11b+ DCs since we found no differences in APC functions of CD103+ DCs from Myd88wt or Myd88cko mice (Fig. S4e-g). These data suggest that among SI-LP DCs, physiologic MyD88 signals have most potent influence on APC functions of CD103−CD11b+ DCs, and in these DCs, MyD88 selectively upregulates IL-17-inducing APC functions. Because CD103− CD11b+ DCs from A20wt and A20cko mice both upregulate IL-17-inducing APC functions in a MyD88-dependent fashion, the link between MyD88 and CD103−CD11b+ DC-instruction of IL-17+ T cells appears to be required in health and in inflammation.
Distinct mechanisms enhance IFNγ- or IL-17-inducing APC functions of SI-LP DCs
Our results above suggested that APC functions of SI-LP DCs were enhanced in A20cko mice and that each DC subset possessed unique, subset-specific APC functions. In intestinal DCs, it is not known what mechanisms enhance DC-instruction of T cells, or whether APC functions of IFNγ-inducing and IL-17-inducing DCs can be enhanced by a common mechanism. To test if the different DC subsets use similar or distinct mechanisms to instruct IFNγ+ or IL-17+ T cells, we added blocking antibodies to DC–T cell co-cultures. We first tested requirements for CD80/86, since all three SI-LP DCs in A20cko mice upregulated one or both molecules (Fig. 2).
CD103+CD11b− DCs from A20cko mice have enhanced and biased abilities to instruct IFNγ+ T cells (Fig. 4). In co-cultures with control antibody, numbers of IFNγ+ T cells were increased >25 fold compared to control DCs (Fig. 5a). By contrast, upon addition of CD80/86 blocking antibodies, IFNγ+ T cells were essentially abolished (Fig. 5a). Numerically, CD80/86 blocking resulted in 20-fold fewer IFNγ+ T cells in co-cultures of CD103+CD11b− DCs from A20cko mice. Comparatively the total T cell number in these co-cultures was at most reduced 2.5-fold (Fig. S5), suggesting that blocking CD80/86 most significantly blocked instruction of IFNγ+ T cells, APC functions which were otherwise enhanced in CD103+CD11b− DCs DCs from A20cko mice. From these data we conclude that enhanced IFNγ-inducing APC functions of CD103+CD11b− DCs from A20cko mice require DC-expression of CD80/86 costimulatory molecules.
Figure 5. Enhanced APC functions of IFNγ-inducing DCs require CD80/86 and IL-17-inducing DCs require IL-6.
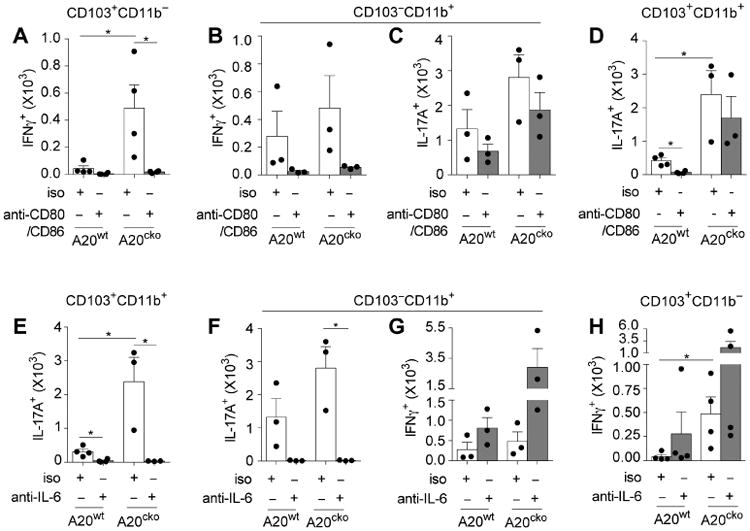
Number of IFNγ+ T cells induced in co-cultures with SI-LP CD103+CD11b− DCs (a) or CD103− CD11b+ DCs (b) from A20wt and A20cko mice as in Fig. 4, in the presence of antibodies blocking CD80 and CD86 (anti-CD80/86) or isotype control (iso). (c) Numbers of IL-17+ T cells induced in CD103−CD11b+ DC co-cultures from (b) were assayed in parallel. Number of IL-17+ T cells induced in CD103+CD11b+ DC co-cultures in the presence of anti-CD80/86 (d) or IL-6 blocking antibody (e). Number of IL-17+ T cells induced in co-cultures with CD103-CD11b+ DCs (f), in the presence of IL-6 blocking antibody or isotype control. (g) The number of IFNγ+ T cells induced in CD103−CD11b+ DC co-cultures from (f) was assayed in parallel. (h) Numbers of IFNγ+ T cells induced in CD103+CD11b- DC co-cultures in the presence of IL-6 blocking antibody or isotype control. Results were combined from 3 independent experiments, each including pooled DCs from at least 3 mice of each genotype. Error bars represent mean ± SEM. See also Figure S5.
IFNγ-inducing APC functions were also enhanced in CD103−CD11b+ DCs from A20cko mice (Fig. 4). CD80/86 blocking markedly decreased the number of IFNγ+ T cells expanded by CD103− CD11b+ DCs, similar to results with CD103+CD11b− DCs (Fig. 5b). Thus, upregulation of CD80/86 by SI-LP DCs can enhance instruction of IFNγ+ T cells. These data also suggest that despite their subset-specific differences, cellular mechanisms that enhance APC functions are shared by CD103+CD11b− and CD103−CD11b+ DCs. Notably, between these two, only CD103− CD11b+ DCs possess both IFNγ- and IL-17-inducing APC functions (Fig. 4). Thus, by using CD103−CD11b+ DCs we could test whether CD80/86 expression, on the same DC, was required to instruct IFNγ+ T cells and also IL-17+ T cells. Surprisingly, while CD80/86 blocking antibodies in CD103−CD11b+ DC co-cultures reduced numbers of IFNγ+ T cells 9-fold, the numbers of IL-17+ T cells changed very little (1.6-fold) (Fig. 5b,c). These data suggest that cellular mechanisms that enhance IL-17-inducing APC functions of CD103−CD11b+ DCs do not require CD80/86 and are thus distinct from mechanisms that instruct IFNγ+ T cells. In support of this conclusion CD103+CD11b+ DCs from A20cko mice, which have enhanced and biased abilities to instruct IL-17+ T cells (Fig. 4), were not inhibited by CD80/86 blocking antibodies (Fig. 5d). Thus, IL-17-inducing APC functions upregulated by CD103+CD11b+ and CD103−CD11b+ DCs of A20cko mice did not require CD80/86. Collectively, these data indicate that physiologic signals that enhance IFNγ-inducing APC functions of SI-LP DCs do so in part by upregulating CD80/86, but this does not enhance IL-17-inducing APC functions.
Several cytokines are suggested to play a role in instruction or survival of IL-17+ T cells, including IL-6 (Denning et al. 2011; Persson et al. 2013; Hu et al. 2011). IL-6 can also support IFNγ+ T cells, particularly in inflammation (Dejean et al. 2009; Nish et al. 2014). To test whether IL-6 enhances IFNγ-inducing and/or IL-17-inducing functions of SI-LP DCs, we added IL-6 blocking antibodies to DC–T cell co-cultures. CD103+CD11b+ DCs from A20cko mice have enhanced and biased ability to instruct IL-17+ T cells (Fig. 4), and essentially all IL-17+ T cells were abolished upon addition of IL-6 blocking antibody (Fig. 5e). IL-6 blockade also abolished IL-17+ T cells from co-cultures of CD103−CD11b+ DCs (Fig. 5f), indicating that IL-6 was required for APC functions of both CD103+CD11b+ and CD103−CD11b+ DCs. Again, because CD103− CD11b+ DCs possessed dual APC functions, we tested whether IL-6 expressed by those DCs enhanced both IL-17- and IFNγ-inducing APC functions. In marked contrast to IL-17+ T cells, IL-6 blockade in CD103−CD11b+ DC co-cultures did not inhibit expansion of IFNγ+ T cells (Fig. 5g). Consistent with this result, IL-6 blockade did not impair expansion of IFNγ+ T cells by CD103+CD11b− DCs (Fig. 5h). These data suggest that IL-6 is selectively required to enhance IL-17-inducing APC functions of SI-LP DCs.
IL-1β and TNFα could also influence DC-instruction of T cells (Shaw et al. 2012), but blocking antibodies to these cytokines did not impair APC functions of any DC subset (data not shown). Our results suggest that among SI-LP DCs, upregulation of IFNγ-inducing APC functions correlate with upregulation of CD80/86, whereas IL-17-inducing APC functions require upregulation of IL-6. These molecules were similarly required by SI-LP DCs from A20/Myd88cko mice (data not shown). High expression of CD80/86 and IL-6 by SI-LP DCs of A20cko and A20/Myd88cko mice likely contributes to T cell-mediated inflammation of small intestine.
SI-LP DCs have subset-specific requirements for MyD88 to drive cytokine gene expression
Our results above indicated that only specific DCs upregulated IL-17-inducing APC functions and these required IL-6. The physiologic signals that induce IL-6 and whether inducible expression of IL-6 is exclusive to IL-17-inducing DC subsets are not known. To investigate these questions we first quantified IL-6 mRNA in CD103+CD11b− DCs. Because CD103+CD11b− DCs failed to instruct IL-17+ T cells (Fig. 4), we reasoned that these DCs failed to upregulate IL-6. Surprisingly, CD103+CD11b− DCs from A20cko mice and also A20/Myd88cko mice expressed high IL-6 mRNA, upregulated 4-fold compared to controls (Fig. 6a). These results stood out to us for two reasons. First, because these DCs did not instruct IL-17+ T cells these data suggest that upregulation of IL-6 by CD103+CD11b− DCs was insufficient to upregulate IL-17-inducing APC functions. Thus, high expression of IL-6 is not exclusive to IL-17-inducing SI-LP DCs. Second, these data indicate that in CD103+CD11b− DCs, IL-6 can be upregulated in a MyD88-independent fashion.
Figure 6. CD103+CD11b− DCs uniquely upregulate IL-6, IL-23a and IL-1β mRNA in a MyD88-independent fashion, yet these DCs do not upregulate IL-17-inducing APC functions.
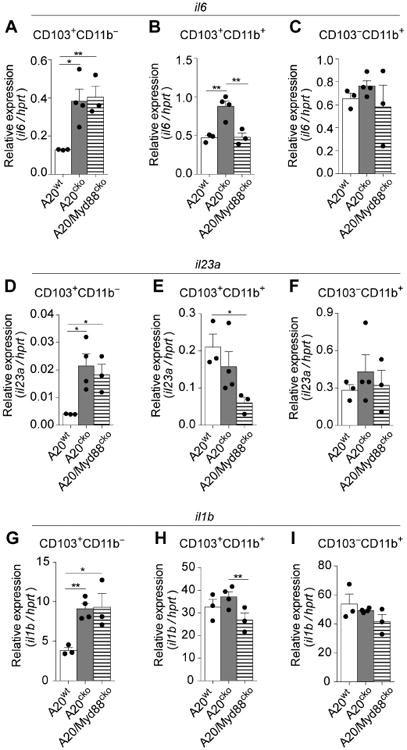
Abundance of IL-6 mRNA expressed by SI-LP CD103+CD11b− DCs (a), CD103+CD11b+ DCs (b), or CD103−CD11b+ DCs (c), from mice of the indicated genotype. Expression of IL-23(p19) (d-f), and IL-1β mRNA (g-i) were assayed in parallel. Results are relative to hprt and are combined from 3 independent experiments, including at least 2 mice of each genotype per experiment. Error bars represent mean ± SEM. See also Figure S7.
We next analyzed CD103+CD11b+ DCs. These DCs from A20cko and A20/Myd88cko mice have enhanced ability to instruct IL-17+ T cells (Fig. 4). Because IL-17-inducing APC functions of CD103+CD11b+ DCs do not require MyD88 we reasoned that IL-6 mRNA would be upregulated in DCs of both A20cko and A20/Myd88cko mice. However, while CD103+CD11b+ DCs from A20cko mice indeed expressed high levels of IL-6, DCs from A20/Myd88cko mice did not, indicating that physiologic MyD88 signals were required (Fig. 6b). Moreover, IL-6 mRNA in CD103+CD11b+ DCs from A20/Myd88cko mice was identical to A20wt, suggesting high IL-6 (significantly above WT) is likely not the only factor that enhances IL-17-inducing APC functions. Accordingly, IL-6 mRNA above wild-type was seemingly not required to enhance IL-17-inducing APC functions of CD103−CD11b+ DCs, since IL-6 was similarly expressed by CD103−CD11b+ DCs from A20wt, A20cko and A20/Myd88cko mice (Fig. 6c). We also assayed IL-6 protein in DC–T cell co-culture supernatants and found IL-6 protein was abundant in co-cultures of DCs from A20cko and A20/Myd88cko mice; however, this IL-6 protein may be secondary to T cell activation (Fig. S6). Thus, when analyzed directly ex vivo, our data suggest that IL-17-inducing APC functions of SI-LP DCs can be enhanced without significantly increased expression of IL-6 mRNA. Additionally, physiologic signals in A20cko mice induce high IL-6 mRNA in both CD103+CD11b− and CD103+CD11b+ DCs, and of these two, only CD103+CD11b+ DCs upregulate IL-6 in a MyD88-dependent fashion.
Given these results we also analyzed IL-23a(p19) and IL-1β mRNA, which are commonly induced in a MyD88-dependent fashion, although the physiologic signals that drive expression are unknown. Surprisingly, upregulation of IL-23a and IL-1β mRNA was exclusive to CD103+CD11b− DCs, and both cytokines were upregulated in a MyD88-independent fashion in this DC subset (Fig. 6d-i). IL-12a(p35) and IL-12b(p40) mRNA were not upregulated, indicating that gene expression was not generally “promiscuous” in CD103+CD11b− DCs from A20cko or A20/Myd88cko mice (Fig. S7). From these data we conclude that physiologic signals that are independent of MyD88 can induce IL-23a, IL-1β and IL-6 mRNA in CD103+CD11b− DCs.
By contrast, these cytokines appeared to be induced in a MyD88-dependent fashion in CD103+CD11b+ DCs (Fig. 6b,e,h). CD103+CD11b+ DCs from A20/Myd88cko mice actually expressed less IL-23a than did A20wt, suggesting that physiologic MyD88 signals were critical (Fig. 6b,e). MyD88 was also required in SI-LP macrophages of A20cko mice, which upregulated only IL-23a and IL-6 (Fig. S4d). None of the cytokine genes tested were upregulated in CD103− CD11b+ DCs (Fig. 6c,f,i and Fig. S7). Although cytokine expression may be enhanced post-transcriptionally, at the mRNA level, our findings suggest requirements for MyD88 are unique for each cytokine and for each DC subset. Like cytokine mRNA, each DC subset has a unique maturation phenotype and together, these subset-specific factors likely enhance and bias APC functions.
CD103+CD11b+ and CD103−CD11b+ DCs instruct inflammatory Th17 cells in disease pathogenesis of small intestine
In health, CD103+CD11b+ DCs support Th17 cells, although these do not cause inflammation (Persson et al. 2013; Schlitzer et al. 2013). In pathogenesis, it is not known whether specific inflammatory T cells require specific DC subsets. To test whether CD103+CD11b+ DCs were required to instruct inflammatory Th17 cells we generated A20cko mice that lacked CD103+CD11b+ DCs by crossing to the DC-ablation strain, Langerin-DTA mice (Kaplan et al. 2005; Welty et al. 2013). In Langerin-DTA mice the human Langerin promoter drives intracellular expression of diphtheria toxin, and this transgene is known to ablate Langerhans cells and also SI-LP CD103+CD11b+ DCs. We analyzed co-housed, Langerin-DTA transgene-positive A20cko mice (A20cko-DTA+) and littermate control, transgene-negative, A20cko-DTA− mice at 9-12 weeks of age.
In A20cko-DTA+ mice CD103+CD11b+ DCs were numerically reduced to just 7% that of co-housed littermate A20cko-DTA− mice (Fig. 7a,b). CD103+CD11b+ DCs in A20wt-DTA+ mice were ablated with even higher efficiency (98%). Although previous studies reported the Langerin-DTA transgene did not ablate other SI-LP DCs (Welty et al. 2013; Panea et al. 2015), we found numbers of CD103−CD11b+ DCs in both A20wt-DTA+ mice and A20cko-DTA+ mice were markedly reduced (Fig. 7a,b). CD103−CD11b+ DCs were on average reduced 2-fold in A20cko-DTA+ mice and as much as 8-fold in A20wt-DTA+ mice. Thus, the two DC subsets with potent IL-17-inducing APC functions in DC–T cell co-cultures were depleted in A20cko-DTA+ mice. These mice were not deficient in CD103+CD11b− DCs or macrophages (Fig. 7b).
Figure 7. In small intestine inflammation CD103+CD11b+ DCs and CD103−CD11b+ DCs expand inflammatory Th17 cells.
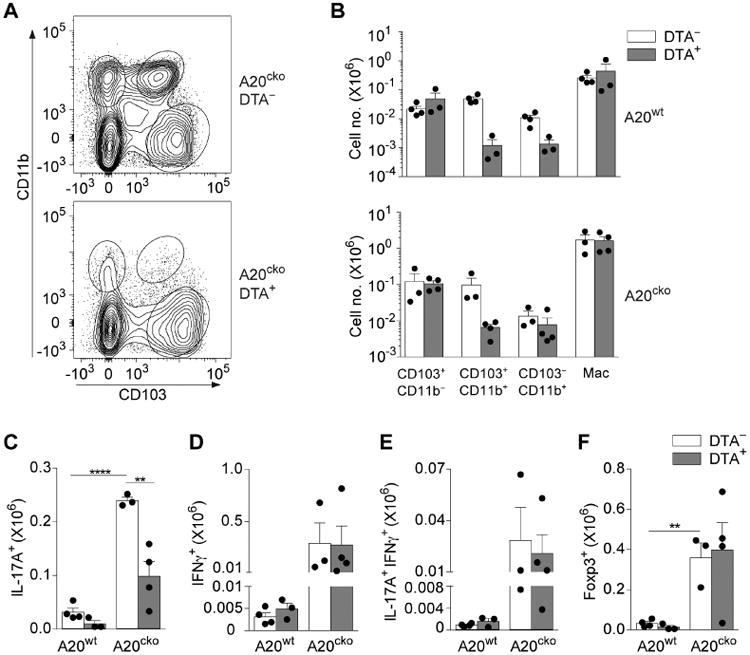
(a) Representative flow plot of SI-LP DCs in co-housed, littermate A20cko-DTA− mice and A20cko-DTA+ mice at 9-12 weeks of age. (b) Absolute number of each SI-LP DC subset in A20wt or A20cko mice, either DTA− or DTA+. (c-f) Absolute number of SI-LP CD4 T cells, either IL-17+ (c), IFNγ+ (d), IL-17+IFNγ+ (e) or Foxp3+ CD4 T cells (f) in co-housed A20wt or A20cko mice, additionally either DTA− or DTA+. Results were combined from 3 independent experiments including at least one mouse of each genotype. Each dot represents a mouse. Error bars represent mean ± SEM. *, P < 0.05, **, P < 0.01, ****, P < 0.0001, (unpaired student's t-test).
Compared to A20wt-DTA− mice, littermate A20wt-DTA+ mice had fewer SI-LP Th17 cells (Fig. 7c). This is consistent with previous reports and supports a specialized role for CD103+CD11b+ DCs and also CD103−CD11b+ DCs in expansion of mucosal Th17 cells during health. In inflamed small intestine of A20cko-DTA− mice numbers of mucosal Th17 cells were markedly expanded (Fig. 7c). By contrast, in co-housed, littermate A20cko-DTA+ mice, Th17 expansion was significantly attenuated. On average, Th17 cells in A20cko-DTA+ mice were just one-third that of A20cko-DTA− mice, suggesting that inflammatory Th17 cells in A20cko mice required CD103+CD11b+ DCs and CD103−CD11b+ DCs (Fig. 7c). Unlike Th17 cells, Th1 cells were expanded >55-fold in both A20cko-DTA− and DTA+ mice (Fig. 7d). Additionally, both mice had expanded numbers of IL-17+IFNγ+ and Foxp3+ T cells (Fig. 7e,f). The persistence of these SI-LP T cells, yet marked reduction of Th17 cells in A20cko-DTA+ mice indicates that APC functions of CD103+CD11b+ DCs, and also CD103−CD11b+ DCs, are required to instruct and expand inflammatory Th17 cells in the pathogenesis of small intestine inflammation sharing histologic features with Crohn's Disease. Thus, in the context of pathologic inflammation, SI-LP DCs have subset-specific APC functions that define clear relationships between specific DCs and specific pathologic T cells.
Discussion
Dendritic cells are quintessential APCs, yet how APC functions are controlled by microbiota is poorly understood. Our goals were to understand the physiologic signals perceived by intestinal DCs and the roles of these signals in DC functions and intestinal homeostasis. We showed that APC functions of SI-LP DCs can be potently upregulated by MyD88-independent signals and, in a MyD88-independent fashion, microbiota, DCs and T cells can together drive inflammation of small intestine that histologically resembles Crohn's Disease. The pathologic potential of MyD88-independent signals in DCs is upregulated in SI-LP DCs lacking A20, an intracellular suppressor of MyD88-dependent and –independent signals (Lee et al. 2000; Boone et al. 2004; Hitotsumatsu et al. 2008; Hammer et al. 2011). Although A20-suppression is also active in DCs outside of the intestine, it is interesting that in non-intestinal DCs, APC functions that expand T cells in A20cko mice require MyD88 (Hammer et al. 2011). By contrast, T cells in small intestine are expanded in a MyD88-independent fashion and we show microbiota drive this expansion. Although it is possible that microbiota stimulate DCs during the isolation procedure, enhanced APC functions of SI-LP DCs are well supported in vivo, in which inflammation and expansion of SI-LP T cells occurs in both A20cko and A20/Myd88cko mice. Moreover, we identified two DC subsets in A20cko mice that instruct IL-17+ T cells and ablation of these DCs significantly diminished Th17 cells in vivo. That each SI-LP DC subset of A20cko and A20/Myd88cko mice upregulates distinct cytokines, costimulatory molecules and APC functions strongly suggest that these phenotypic and functional changes are triggered in vivo, within the intestine microenvironment. Phenotypic and functional changes of SI-LP macrophages do not mimic those of DCs and our findings and the work of others on A20-floxed Lyz2-cre mice (macrophage deletion), would suggest that loss of A20-expression in macrophages is insufficient to drive inflammation in small intestine (Matmati et al. 2011). Collectively, our data highlight pathologic roles for microbiota-triggered signals, independent of MyD88, in DC-expansion of microbiota-reactive T cells and IBD pathogenesis.
IBD has two main subtypes: ulcerative colitis which is limited to colon and Crohn's disease, which can affect any part of the gastrointestinal tract and commonly involves small intestine. Small intestine inflammation also occurs in TNFΔARE mice, which overexpress TNF (Kontoyiannis et al. 1999), and SAMP1/YitFc mice (Pizarro et al. 2011). Both develop ileitis, although the pathologic signals are not defined. In some facilities IL-10-/- mice develop inflammation of both colon and small intestine and evidence suggests that different microbiota drive pathogenesis in each intestinal organ (Kühn et al. 1993; Kim et al. 2005; Moran et al. 2009; Burich et al. 2001). Further studies with A20cko and A20/Myd88cko mice, which develop ileitis as well as inflammation of jejunum and other regions of small intestine, will yield insight into how region-specific microbiota influence disease pathogenesis.
By depletion of CD103+CD11b+ and CD103−CD11b+ DCs in A20cko-DTA+ mice we show that these DCs instruct pathologic Th17 cells. Although previous studies with Langerin-DTA mice reported no loss of CD103−CD11b+ DCs (Welty et al. 2013; Panea et al. 2015), this subset is heterogeneous and numbers may differ between mouse facilities (Cerovic et al. 2013; Scott et al. 2014). Other possibilities for these disparate findings may be procedural differences in LP preparation or strategies for flow cytometry gating. We found SI-LP DCs equally express CD24 and CD26 yet, CD14 and CD64 are slightly higher on CD103−CD11b+ DCs. Because these markers discriminate DCs and macrophages, special care may be needed to ensure proper gating of CD103−CD11b+ DCs.
Understanding the functions of CD103−CD11b+ DCs is of particular interest, since our data indicate that these DCs instruct inflammatory IL-17+ T cells, particularly downstream of MyD88 signals. We now decribe how APC functions of intestinal DCs are altered by physiologic MyD88 signals. No verified methods exist to selectively ablate CD103−CD11b+ DCs and we do not know whether these DCs, or CD103+CD11b+ DCs, are dominant for expansion of inflammatory Th17 cells in A20cko mice. However, CD103+CD11b+ DCs are absent in A20cko-DTA+ mice, while ∼50% of CD103−CD11b+ DCs remain. Thus, the Th17 cells that persist in A20cko-DTA+ mice, which are expanded 10-fold compared to A20wt-DTA+ mice, are likely instructed by the remaining CD103−CD11b+ DCs. These DCs must also expand IL-17+IFNγ+ T cells, since these persist in A20cko-DTA+ mice. IL-17+IFNγ+ and Th17 cells may be a spectrum of one population and our results with A20cko-DTA+ mice may suggest that DCs induce RUNX or other regulators of Th17 identity (Reis et al. 2013; Wang et al. 2014; Harbour et al. 2015). Alternatively, loss of Th17, yet expansion of IL-17+IFNγ+ T cells in A20cko-DTA+ mice may indicate that these are two separate populations, the latter of which requires CD103−CD11b+ DCs. Macrophages could further expand IL-17+ T cells (Shaw et al. 2012; Panea et al. 2015; Seo et al. 2015). However, because macrophage numbers are normal in A20cko-DTA+ mice, our data suggest macrophages have limited roles in expansion of inflammatory Th17 cells in disease pathogenesis of small intestine.
Th1 cells also expanded in disease pathogenesis and our data suggest that CD103−CD11b+ DCs and CD103+CD11b− DCs instruct IFNγ+ T cells. While our manuscript was under review two studies found that CD103+CD11b− DCs expand SI-LP Th1 cells during health, which supports our finding of functional specialization of these DCs for IFNγ+ T cells (Luda et al. 2016; Ohta et al. 2016). IFNγ-inducing APC functions of CD103+CD11b− DCs are also required for pathogen immunity and cytoprotection of intestinal epithelial cells (Mashayekhi et al. 2011; Muzaki et al. 2015). Whether these functions are shared by CD103−CD11b+ DCs is unknown.
How do DCs acquire subset-specific APC functions? What factors reinforce APC functions that are biased? Conversely, what factors awaken dual APC functions in CD103−CD11b+ DCs? Interaction with species of microbiota, expression of pattern recognition receptors, signaling molecules, or signal-induced gene transcription, could be unique for each DC subset. Indeed, we found different DCs required different signals to induce IL-6. DC-expression of costimulatory molecules, cytokines, and dynamics of the immune synapse collaborate to instruct T cell cytokine and understanding how microbiota control these dynamics in different DC subsets is fundamental to understand functional-specialization of intestinal DCs.
Experimental Procedures
Mice
A20-floxed, Myd88-floxed, Cd11c-cre and human Langerin-DTA transgenic mice have been described (Kaplan et al. 2005; Caton et al. 2007; Hou et al. 2008; Tavares et al. 2010). All mice were purchased from Jackson Laboratories or re-derived by embryo transfer into a maximum barrier facility at Duke University. Routine screening of maximum barrier facilities confirmed exclusion of Helicobacter, Pastuerella, and Norovirus. For all breedings littermate, Cd11c-cre-negative mice were co-housed with experimental mice and used as control, A20wt mice. Animal protocols were approved by Duke University Institutional Animal Care and Use Committee.
Antibiotics
Drinking water containing ampicillin (1g/L), vancomycin (500mg/L), neomycin sulfate (1g/L) and metronidazole (1g/L) was provided on d18-19 of gestation or to dams within 2 days after delivery. Weaned pups received antibiotics for an additional 6-7 weeks. Stool DNA was prepared using PowerSoil® (Mo bio) and 16S DNA quantified using the following primers: ACTCCTACGGGAGGCAGCAGT, GTATTACCGCGGCTGCTGGCAC (Bergström et al. 2012).
Intestinal lamina propria preparation
Small intestine was flushed with HBSS/10mM HEPES and Peyer's patches removed. Intestine was cut longitudinally and washed thoroughly with HBSS/10mM HEPES to remove lumen contents. Organ weight was recorded after brief wicking of liquid. Intestine was then cut into 0.5cm pieces and processed according to methods specified by the lamina propria dissociation kit and GentleMACS™ Dissociator (Miltenyi), with modifications as described in Supplemental Experimental Procedures.
Flow cytometry, cell sorting and DC-T cell co-cultures
SI-LP cells were stained with Live/dead® fixable dead cell staining (Thermo Fisher) and immune populations gated as follows: CD4 T cells (CD45+TCRβ+CD4+), DCs (CD45+IA/IE+CD11c+CD24+CD14-), and Macrophages (CD45+IA/IE+CD11b+CD24-CD14+CD103-). For cytokine stains SI-LP cells were stimulated for 4h with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of brefeldin A. Cells were analyzed on a BD Canto or sorted with Astrios (Beckman Coulter).
Sorted SI-LP macrophages were gated as IA/IEhiCD14+CD11b+CD103−. To sort SI-LP DCs, cells were first enriched by CD11c microbeads and an AutoMACS (Miltenyi), or by Nycoprep centrifugation (Axis-Shield). DCs were gated as IA/IEhiCD11c+CD26+CD14− and then subsetted by CD103 and CD11b expression. For co-cultures, negatively enriched (EasySep™) OT-II T cells were sorted for TCRβ+CD4+CD62LhiCD44lowCD25−. These naïve OT-II T cells were co-cultured for 4 days with DCs or macrophages at a 1:1 ratio in a 96-well round-bottom plate. Co-cultures also included endotoxin free, OVA-peptide (5μg/ml, JPT) and TGFβ (1ng/ml, peprotech). Within a given experiment each well contained an equivalent number of DCs (of an individual DC subset) or macrophages from mice of each genotype. Some experiments included 10μg/mL of the following antibodies: Rat IgG (RTK2071), hamster IgG (G235-2356), anti-CD80 (16-10A1), anti-CD86 (GL1) or anti-IL-6 (MP5-20F3). IL-17A, IFNγ and IL-6 in supernatants were assayed by ELISA (Biolegend). Cytokine+ T cells were assayed by 4h re-stimulation with plate-bound anti-CD3 (5μg/ml) and anti-CD28 (5μg/ml). Results from ELISA and T cell numbers were normalized to 1,000 input naïve OT-II cells.
Quantitative PCR
Described in Supplemental Experimental Procedures
Histology
Five 2 cm segments of small intestine were collected in accordance with defined anatomic locations (Treuting & Dintzis 2012). The first segment contained a small portion of lower stomach and the pylorus-adjacent intestine, which includes duodenum and proximal jejunum. This was followed immediately after by two contiguous segments of proximal jejunum. The fourth segment contained middle small intestine (jejunum-ileum) and the last was adjacent to the ileocecal junction (terminal ileum). Segments were color-coded, fixed in Carnoy's, and sections stained with hematoxylin and eosin. Slides were coded to blind the pathologist and each segment scored using a previously described scoring system (Hale et al. 2005), with modifications as described in Supplemental Experimental Procedures.
Statistical analysis
Student's t-test (GraphPad Prism) was used for statistical analysis.
Supplementary Material
Acknowledgments
The authors thank Donald Cook for critique of the manuscript and scientific input, Garnett Kelsoe, Michael Krangel, Qi-Jing Li and Lawrence David for reagents and equipment, Timothy Denning, Susan Tonkonogy, Siqi Liu, Liang Chen and Aspen Reese for technical advice, Lynn Martinek and J Michael Cook for cell sorting. Support for Fernanda P. Benzatti was provided by CAPES/Brazil grant 9693/14-09. This work was funded by grants awarded to Gianna E. Hammer: Pilot/Feasibility funds from the Center of Gastrointestinal Biology and Disease grant P30 DK034987, foundation awards from PSC partners seeking a cure, and American Gastroenterological Association. Gianna E. Hammer is a Pew Biomedical Scholar and is supported by Pew Charitable Trusts.
Footnotes
Author contributions: Conceptualization, G.E.H. and J.L.; Investigation, J.L., H.H., F.P.B., J.J.Z., N.Y., L.P.H. and G.E.H.; Resources, J.L., H.H., A.B.K., J.J.Z., A.M. and G.E.H.; Writing – Original Draft, G.E.H. and J.L.; Writing – Review and Editing, G.E.H., L.P.H. and J.L.; Funding Acquisition, Visualization and Supervision, G.E.H.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunological reviews. 2014;260(1):86–101. doi: 10.1111/imr.12194. [DOI] [PubMed] [Google Scholar]
- Bergström A, et al. Introducing GUt low-density array (GULDA): a validated approach for qPCR-based intestinal microbial community analysis. FEMS microbiology letters. 2012;337(1):38–47. doi: 10.1111/1574-6968.12004. [DOI] [PubMed] [Google Scholar]
- Bogunovic M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31(3):513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nature immunology. 2004;5(10):1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- Burich A, et al. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. American journal of physiology Gastrointestinal and liver physiology. 2001;281(3):G764–78. doi: 10.1152/ajpgi.2001.281.3.G764. [DOI] [PubMed] [Google Scholar]
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. The Journal of experimental medicine. 2007;204(7):1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerovic V, et al. Intestinal CD103(-) dendritic cells migrate in lymph and prime effector T cells. Mucosal immunology. 2013;6(1):104–113. doi: 10.1038/mi.2012.53. [DOI] [PubMed] [Google Scholar]
- Dejean AS, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nature immunology. 2009;10(5):504–513. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. Journal of immunology (Baltimore, Md : 1950) 2011;187(2):733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, et al. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nature immunology. 2007;8(10):1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- Feng T, et al. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. Journal of Experimental Medicine. 2010;207(6):1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto K, et al. A new subset of CD103+CD8alpha+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces Th1 response and CTL activity. Journal of immunology (Baltimore, Md : 1950) 2011;186(11):6287–6295. doi: 10.4049/jimmunol.1004036. [DOI] [PubMed] [Google Scholar]
- Geem D, et al. Specific microbiota-induced intestinal Th17 differentiation requires MHC class II but not GALT and mesenteric lymph nodes. Journal of immunology (Baltimore, Md : 1950) 2014;193(1):431–438. doi: 10.4049/jimmunol.1303167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40(4):594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nature genetics. 2008;40(9):1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale LP, Gottfried MR, Swidsinski A. Piroxicam treatment of IL-10-deficient mice enhances colonic epithelial apoptosis and mucosal exposure to intestinal bacteria. Inflammatory bowel diseases. 2005;11(12):1060–1069. doi: 10.1097/01.mib.0000187582.90423.bc. [DOI] [PubMed] [Google Scholar]
- Hammer GE, Ma A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annual review of immunology. 2013;31:743–791. doi: 10.1146/annurev-immunol-020711-074929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer GE, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nature immunology. 2011;12(12):1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour SN, et al. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(22):7061–7066. doi: 10.1073/pnas.1415675112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitotsumatsu O, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28(3):381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annual review of immunology. 2012;30:759–795. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nature communications. 2012;3:1120. doi: 10.1038/ncomms2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29(2):272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, et al. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity. 2011;35(6):1010–1022. doi: 10.1016/j.immuni.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (New York, NY) 2010;327(5963):291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH, et al. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23(6):611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Kim SC, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128(4):891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Kühn R, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science (New York, NY) 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luda KM, et al. IRF8 Transcription-Factor-Dependent Classical Dendritic Cells Are Essential for Intestinal T Cell Homeostasis. Immunity. 2016;44(4):860–874. doi: 10.1016/j.immuni.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Mashayekhi M, et al. CD8α(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35(2):249–259. doi: 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nature genetics. 2011;43(9):908–912. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- Moran JP, et al. Bifidobacterium animalis causes extensive duodenitis and mild colonic inflammation in monoassociated interleukin-10-deficient mice. Inflammatory bowel diseases. 2009;15(7):1022–1031. doi: 10.1002/ibd.20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzaki ARBM, et al. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-γ-induced anti-inflammatory response in epithelial cells. Mucosal immunology. 2015 doi: 10.1038/mi.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nish SA, et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. In: Taniguchi T, editor. eLife. Vol. 3. 2014. p. e01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, et al. Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Scientific reports. 2016;6:23505. doi: 10.1038/srep23505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panea C, et al. Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell reports. 2015;12(8):1314–1324. doi: 10.1016/j.celrep.2015.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson EK, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. 2013;38(5):958–969. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Pizarro TT, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn's disease-like ileitis. Inflammatory bowel diseases. 2011;17(12):2566–2584. doi: 10.1002/ibd.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis BS, et al. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nature immunology. 2013;14(3):271–280. doi: 10.1038/ni.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38(5):970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CL, et al. CCR2(+)CD103(-) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal immunology. 2014 doi: 10.1038/mi.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmrich M, et al. Directed antigen targeting in vivo identifies a role for CD103+ dendritic cells in both tolerogenic and immunogenic T-cell responses. Mucosal immunology. 2012;5(2):150–160. doi: 10.1038/mi.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SU, et al. Distinct Commensals Induce Interleukin-1β via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunity. 2015;42(4):744–755. doi: 10.1016/j.immuni.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, et al. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. Journal of Experimental Medicine. 2012;209(2):251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spörri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nature immunology. 2005;6(2):163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- Steinman RM. Decisions about dendritic cells: past, present, and future. Annual review of immunology. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- Tamoutounour S, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. European journal of immunology. 2012;42(12):3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- Tavares RM, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33(2):181–191. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuting PM, Dintzis SM. Comparative Anatomy and Histology. Academic Press; 2012. [Google Scholar]
- Varol C, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31(3):502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity. 2014;40(3):355–366. doi: 10.1016/j.immuni.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nature genetics. 2007;39(11):1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welty NE, et al. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. Journal of Experimental Medicine. 2013;210(10):2011–2024. doi: 10.1084/jem.20130728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.