

SUMMARY
Nucleotide excision repair (NER) is an evolutionarily conserved mechanism that processes helix-destabilizing and/or -distorting DNA lesions, such as UV-induced photoproducts. Here, we investigate the dynamic protein-DNA interactions during the damage recognition step using single-molecule fluorescence microscopy. Quantum dot-labeled Rad4-Rad23 (yeast XPC-RAD23B ortholog) forms nonmotile complexes or conducts a one-dimensional search via either random diffusion or constrained motion. Atomic force microcopy analysis of Rad4 with the β-hairpin domain 3 (BHD3) deleted reveals that this motif is non-essential for damage-specific binding and DNA bending. Furthermore, we find that deletion of seven residues in the tip of β-hairpin in BHD3 increases Rad4-Rad23 constrained motion at the expense of stable binding at sites of DNA lesions, without diminishing cellular UV resistance or photoproduct repair in vivo. These results suggest a distinct intermediate in the damage recognition process during NER, allowing dynamic DNA damage detection at a distance.
Keywords: Rad4, Rad23, XPC, nucleotide excision repair, xeroderma pigmentosum, single particle tracking, dynamic DNA damage recognition, DNA tightrope assay, quantum dots
INTRODUCTION
Cellular metabolism and environmental factors continually damage DNA, threatening genome integrity. Unrepaired DNA damage results in increased mutations and cell death, contributing to a number of human diseases, including cancer (Hoeijmakers, 2001). Several DNA repair pathways have evolved to identify and remove different lesions. Nucleotide excision repair (NER) is a highly conserved repair mechanism that recognizes and repairs a variety of helix-destabilizing/distorting DNA lesions, including: UV-induced 6-4 pyrimidine-pyrimidone photoproducts (6-4PPs) and cyclobutane pyrimidine dimers (CPDs), as well as bulky chemical adducts resulting from cisplatin or aromatic hydrocarbons (Friedberg et al., 2006; Gillet and Scharer, 2006). Mutations in NER genes cause several human diseases including xeroderma pigmentosum (XP). Patients with this disorder can be assigned to one of eight different complementation groups (A–G, V), are highly sensitive to sunlight, and exhibit an increased skin cancer risk (DiGiovanna and Kraemer, 2012).
Mammalian NER involves nearly 30 different proteins, recruited in a highly orchestrated manner (Aboussekhra et al., 1995). Two different sub-pathways exist for the initiation of NER: global genome (GG-) and transcription-coupled (TC-) repair (Gillet and Scharer, 2006; Hanawalt and Spivak, 2008; Scharer, 2013; Vermeulen and Fousteri, 2013). In GG-NER, the XPC-RAD23B-CETN2 complex initially recognizes various NER substrates including 6-4PPs, while detection of the less distorting CPDs is first accomplished by UV-DDB, which then hands it off to XPC (Scharer, 2013). In TC-NER, lesions are first identified by stalled RNA polymerases at damage sites before recruitment of other TC-NER specific factors (Vermeulen and Fousteri, 2013). GG- and TC-NER converge when TFIIH verifies the presence of actual chemical base damage using the ATPase activity of XPB and the helicase activity of XPD (Coin et al., 2007; Kuper et al., 2014; Sugasawa et al., 2009). Subsequent recruitment of XPA, RPA, XPG, and XPF-ERCC1 allows incision complex assembly, in which structure-specific endonucleases XPF-ERCC1 and XPG make incisions 5′ and 3′ to the lesion in the damaged strand, respectively. The resulting gap is filled by DNA pol δ/ε/κ, RFC, and PCNA, before the newly synthesized repair patch is sealed by DNA ligase I (Scharer, 2013).
The Saccharomyces cerevisiae Rad4-Rad23 complex is orthologous to human XPC-RAD23B, sharing structural and functional similarities (Legerski and Peterson, 1992; Masutani et al., 1994). An X-ray crystal structure of Rad4-Rad23 bound to a 24-base pair duplex DNA harboring a CPD-containing mismatch lesion reveals that Rad4 consists of a transglutaminase homology domain (TGD) and three β-hairpin domains (BHD 1 – 3, Figure 1A) (Min and Pavletich, 2007). β-hairpin 3 of BHD3 (hereafter β-hairpin 3) is inserted into the DNA double helix at the lesion site. While the BHD2-BHD3 groove holds the two nucleotides displaced from the undamaged strand opposite the lesion, the CPD is flipped out of the helix and away from the protein. Finally the DNA duplex shows a kink of ~42°, similar to that resulting from binding of XPC-RAD23B to a cholesterol moiety in DNA, as measured by scanning force microscopy (Janicijevic et al., 2003). Because of the lack of direct contact with the lesion by Rad4, Rad4 (XPC) is proposed to indirectly recognize locally destabilized duplex DNA by probing the two strands’ propensity to open, which allows insertion of β-hairpin 3 (Min and Pavletich, 2007). This hypothesis provides a working model for how Rad4 (XPC) recognizes chemically and structurally diverse DNA damage in vitro, such as a cholesterol-modified nucleotide, 6-4PP, cisplatin 1,3-d(GTG) intrastrand adduct, C8-dG acetylaminofluorene, and 5R-thymine glycol (Brown et al., 2010; Hey et al., 2002; Jansen et al., 1998; Kusumoto et al., 2001; Sugasawa et al., 2002; Yeo et al., 2012). Previous studies on domain deletions and mutated XPC constructs employing bulk biochemical binding assays and a fluorescence-based cellular method suggest a two-stage damage recognition model. In this model, XPC uses the BHD1/BHD2/β-turn interface to conduct a sliding search for DNA damage, followed by the more energetically costly β-hairpin insertion (Camenisch et al., 2009). This model is consistent with the kinetic gating mechanism of damage recognition, proposed by Min and co-workers (Chen et al., 2015).
Figure 1. Rad4-Rad23 Crystal Structure, Experimental Schematics, and WT on UV-irradiated λ-DNA.
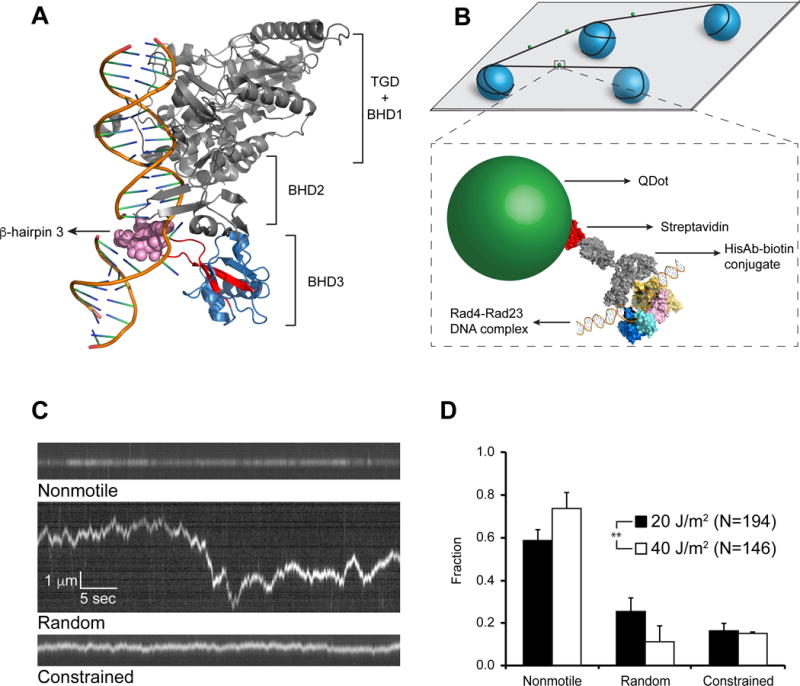
(A) Co-crystal of Rad4-Rad23 with CPD-mismatch-containing DNA (PDB ID: 2QSG). TGD domain and β-hairpin domains 1 and 2 of Rad4, as well as Rad23, are shown in gray, β-hairpin domain 3 in blue, with β-hairpin 3 in red. Residues 599–605 are shown as pink spheres.
(B) Schematics of flow cell and protein conjugation strategy. Top: 5 μm diameter poly-L-lysine coated silica beads (blue) are deposited on polyethylene glycol treated coverslip (gray). DNA (black) is elongated and strung up across beads by flow. Bottom: His-tagged Rad4-Rad23 (yellow, pink, cyan, and blue) is labeled with streptavidin (red)-coated quantum dot (SAQD, green) through a His-antibody (His-Ab)-biotin conjugate (gray). See also Figure S1.
(C) Representative kymographs depicting top: nonmotile (see also Movie S1), middle: random diffusion (see also Movie S2), and bottom: constrained motion (see also Movie S3) particles. Scale bars in middle panel apply to all three kymographs.
(D) Bar graph of fractions of each observed motion type on λ-DNA irradiated with 20 J/m2 (black bars) or 40J/m2 (white bars) UV light (p = 0.0026, χ2 test). All bar graph data in this study are represented as weighted means ± weighted standard deviations over four to five independent experimental days. (Statistical significance *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001, ****: p ≤ 0.0001)
From the perspective of protein-DNA interactions, DNA damage recognition provides a unique example of the ‘speed-stability paradox’: a protein searching for target DNA sites needs to accomplish overall fast searching through a relatively smooth diffusion energy landscape while also achieving stable formation of a protein-DNA complex at the target site (deep local energy well) (Slutsky and Mirny, 2004; Tafvizi et al., 2011). We used a single-molecule DNA tightrope assay (Kad et al., 2010) and atomic force microscopy (AFM) (Ghodke et al., 2014; Yeh et al., 2012) to (1) directly test the two-stage damage recognition model; (2) visualize how Rad4 searches for DNA damage; and (3) explore the specific role of BHD3. Using different DNA lesions and protein variants, we provide a model for how Rad4 utilizes different structural domains to achieve damage recognition in a dynamic process. Rad4 first undergoes a fast initial quality check on DNA for damage detection through random diffusion and DNA bending by BHD1 and BHD2. Initial damage encounter likely triggers a protein conformational change such that it enters a more rigorous damage recognition mode characterized by constrained motion with a steeper energy landscape. This constrained motion, or ‘recognition-at-a-distance,’ helps to reconcile the apparent lack of specificity of Rad4 for CPDs in vitro (Guzder et al., 1998) with its essential role of CPD repair in vivo (Verhage et al., 1994). Finally, in a lesion and sequence specific manner, damage recognition is achieved, leading to nonmotile long-lived Rad4-DNA complexes.
RESULTS
Rad4-Rad23 Utilizes a Combination of 3D and 1D Approaches to Search for Damage on DNA
To directly visualize the Rad4-Rad23 search process, we performed single-molecule tightrope assays (Ghodke et al., 2014; Kad et al., 2010) using N-terminally histidine-tagged Rad4 labeled with streptavidin-conjugated quantum-dots (SAQD) through biotinylated anti-histidine-tag antibody (HisAb) (Figure 1B and S1). The wildtype (WT) Rad4-Rad23 used here is essentially the same as that in crystal structures (Figure 1A), spanning all four DNA-interacting domains of Rad4 (His-scRad4 101-632) and all Rad23 domains except for an internal UBA1 domain (Rad23 1-398_Δ135-299). This WT complex exhibits DNA binding behavior similar to the full-length Rad4-Rad23 complex in electrophoretic mobility shift assays (EMSAs) (Min and Pavletich, 2007). DNA tightropes were suspended between 5 μm poly-L-lysine coated silica beads deposited randomly on a PEGylated coverslip via hydrodynamic flow using a syringe pump. SAQD-labeled Rad4-Rad23 was injected into the flow cell in the presence of DNA tightropes and observations were started immediately after flow was stopped. For experiments performed with UV-irradiated λ-DNA containing on average one photoproduct per 2,200 bp, we detected consistent binding of Rad4-Rad23 to DNA throughout the flow cell. Over a period of ~2 hours, both motile and nonmotile complexes were observed. While some particles dissociated, we saw few arrivals during recording. This indicates that Rad4-Rad23 has a rapid on-rate through the initial 3D diffusion process, followed by 1D diffusion on DNA.
Closer examination of kymographs obtained from single particle tracking of labeled Rad4-Rad23 on DNA tightropes revealed three distinct classes of protein complex movement: nonmotile (Figure 1C top and Movie S1), random diffusion (Figure 1C middle and Movie S2), and constrained motion (Figure 1C bottom and Movie S3). Nonmotile particles showed no discernable movement along the trajectory of DNA (three pixels, ~500 bp, see Supplemental Experimental Procedures) throughout the five-minute recording window. Randomly diffusing particles exhibited increasing displacement from starting positions over time; approximately 5 kbp or larger. Finally, constrained particles oscillated around certain positions on DNA and appeared restricted within ~1–2 kbp in total end-to-end displacement. On UV-irradiated λ-DNA (at 20 J/m2), 59 ± 5% of all observed WT Rad4-Rad23 particles (N = 194) were nonmotile, 25 ± 6% diffused randomly, and 16 ± 4% underwent constrained motion (Figure 1D).
Sliding is the Main Component of Observed 1D Diffusion of Rad4-Rad23
Among the various modes of possible protein-DNA interactions, both sliding and hopping along DNA are perceived as 1D diffusion. However, their mechanisms are fundamentally different. Proteins undergoing 1D sliding maintain contacts with DNA continuously through a corkscrew motion along the helical path. In contrast, hopping involves microscopic dissociation from and rebinding to the same piece of DNA, without macro-dissociating into solution (Halford and Marko, 2004; Kad and Van Houten, 2012). Therefore, diffusion by sliding should be relatively insensitive to changes in salt concentrations, while hopping particles are expected to show an increase in their diffusion coefficients (D) as the distances between hops grow under higher salt conditions (Berg et al., 1981; Blainey et al., 2006). Thus to differentiate between the two mechanisms, we repeated experiments with WT proteins on 20 J/m2 UV-irradiated λ-DNA at 100 mM and 150 mM NaCl (N = 172 and 169, respectively), compared with 75 mM NaCl. Across three salt conditions, nonmotile fractions remained similar (~60%, Figure 2A). Additionly, distributions of log10D (see Supplemental Experimental Procedures) of over 90% of all motile particles, random and constrained, were normal with similar means and standard deviations in all three salt conditions (Figure 2B–D). The apparent insensitivity of diffusion coefficients to higher ionic strengths therefore indicates that under these salt concentrations (75–150 mM NaCl), the majority (>90%) of all motile Rad4-Rad23 slide as they 1D diffuse along DNA. Interestingly, we also observed that at 150 mM NaCl, the number of particles possessing the highest diffusion coefficients (D ~ 0.1 μm2/s) increased by ~10% of the total (N = 65, Figure 2D) compared to results seen in 75 and 100 mM NaCl.
Figure 2. Behavior of WT Rad4-Rad23 on UV-irradiated λ-DNA at Different Salt Concentrations.
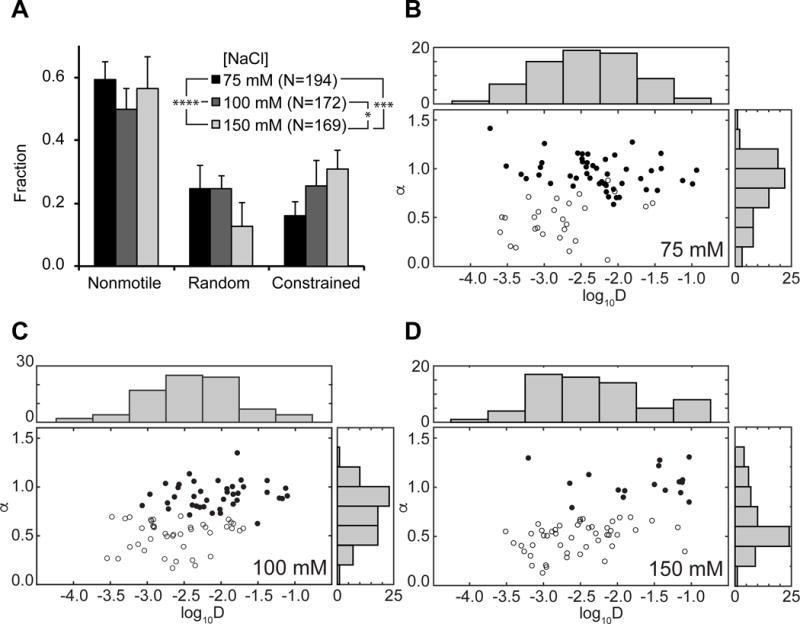
(A) Distributions of observed motion types at different salt concentrations. Data at 75 mM NaCl reproduced from Figure 1D (p = 0.0005, χ2 test).
(B – D) Anomalous diffusion exponent (α) vs. diffusion coefficient (log10D) plotted for random (filled circles) and constrained (empty circles) particles at (B) 75 mM, (C) 100 mM, and (D) 150 mM NaCl. Distributions of diffusion coefficients log10D and anomalous diffusion exponents α are plotted above and to the right of each scatter plot, respectively.
To investigate the diffusion nature in more detail, we analyzed the anomalous diffusion exponent α for all motile particles (see Supplemental Experimental Procedures). α is expected to be ~ 1 for randomly diffusing particles and < 1 for constrained particles. These analyses show that the fraction of particles undergoing constrained motion increased with more physiological salt concentrations (Figure 2A). Increasing ionic strength to 150 mM NaCl resulted in an increased population that exhibited α< 1 (Figure 2D). Finally, comparing relationships between anomalous diffusion exponent α and diffusion coefficient D across three salt concentrations (Figure 2B–D), the fast diffusers (D ~ 0.1 μm2/s) seen at 150 mM NaCl appear to have arisen from the random diffusion population (Figure 2D). Taken together, these data support the hypothesis that particles undergoing constrained motion are indeed sliding on DNA; their behavior remained relatively unchanged when challenged with higher salt. On the other hand, we cannot exclude the possibility that some randomly diffusing particles may undergo hopping at higher salt concentrations.
Rad4-Rad23 Exhibits Lesion-Specific Damage Recognition
It is interesting to note that ~60% of WT Rad4 observed on UV-irradiated λ-DNA were nonmotile (Figure 1D, black bars). Increasing the UV dose two-fold increased the percentage of nonmotile particles and decreased random movers (Figure 1D, white bars). UV irradiation induces a mixture of CPDs and 6-4PPs at about a 3:1 ratio (Friedberg et al., 2006). Rad4-Rad23 binds poorly to CPDs in vitro, while possessing an order of magnitude higher affinity toward the helix-distorting 6-4PPs (Guzder et al., 1998). We thus characterized the Rad4-Rad23 behavior on DNA substrates that harbor one type of DNA lesion in the same repeating sequence context. To this end, we made long DNA-damage arrays by tandemly ligating multiple linearized plasmids, each contained either one CPD or one fluorescein-modified deoxythymidine (Fl-dT) per 2,030 bp, as previously described (Ghodke et al., 2014). Rad4 binds tightly to Fl-dT (Krasikova et al., 2013), making it a model substrate with high specificity (Figure S2A). As expected, SAQD-labeled Rad4-Rad23 formed arrays of nonmotile complexes when introduced into flow cells in the presence of Fl-dT DNA tightropes (Figure 3A–B and Movie S4), with inter-particle spacing being integer-multiples of 2 kbp (Figure S3). Overall, 80 ± 18% of all Rad4-Rad23 particles on Fl-dT DNA were nonmotile, while random and constrained movers represented 12 ± 11% and 8 ± 12%, respectively (Figure 3E, N = 211). In comparison, the nonmotile Rad4-Rad23 population was reduced by 2-fold to 42 ± 10% on CPD-containing DNA damage arrays (N = 106). The fraction of random movers on CPD substrates remained at 14 ± 6%, whereas that of constrained particles increased more than five-fold to 44 ± 4% compared to Fl-dT (Figure 3C–E and Movie S5). In contrast, Rad4-Rad23 behavior on undamaged DNA was statistically different from that on CPD arrays (Figure 3E, p = 0.0187, χ2 test) and important differences were noted in motion types: more particles (26 ± 12%) diffused randomly and less (27 ± 8%) underwent constrained motion on undamaged DNA as compared to CPDs (Figure 3E). These results suggest that constrained motion is directly due to recognition of CPDs. Furthermore, only 6% of all partiles on DNA bound at two positions on undamaged DNA tightropes; over 55% and 41% of particles bound to Fl-dT- and CPD-containing substrates at regular intervals consistent with inter-lesion distance of 2 kbp (Figure S3). The presence of Rad4-Rad23 arrays on CPD substrates and the lack thereof on undamaged DNA argue that, in our tightrope setup, the protein senses the relatively minor helical distortion caused by this lesion.
Figure 3. Lesion-Dependent Motions in Damage Recognition by Rad4-Rad23.
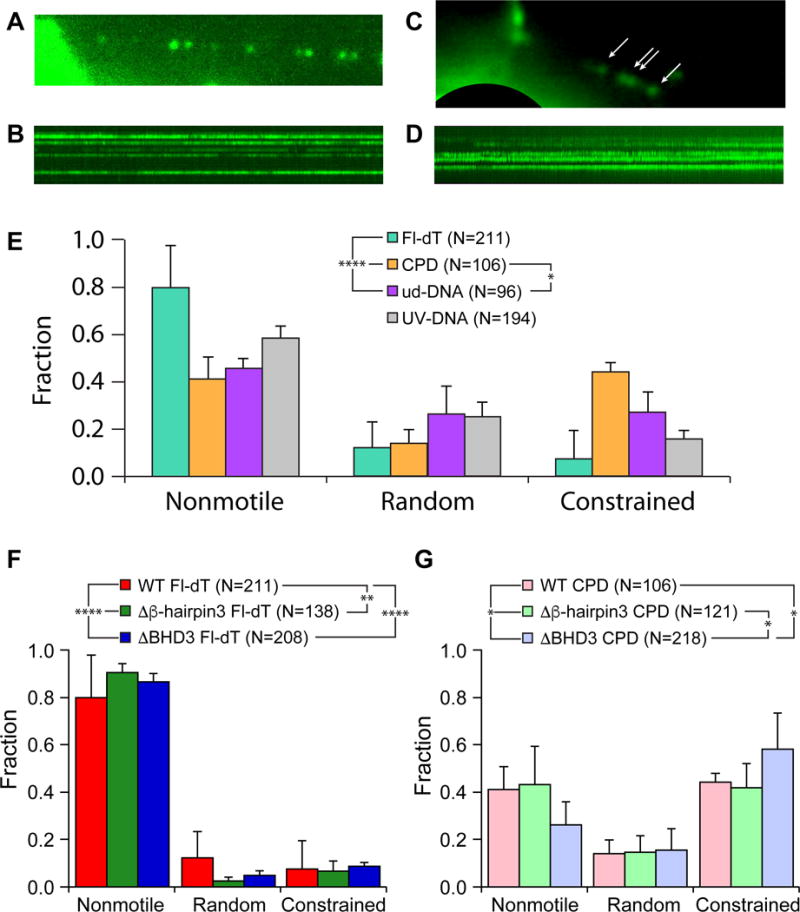
(A) Single frame of quantum dot-labeled Rad4-Rad23 assembled in an array on Fl-dT-containing DNA. See also Movie S4.
(B) Kymograph of Rad4-Rad23 particles assembled on Fl-dT array shown in (A).
(C) Single frame of quantum dot-labeled Rad4-Rad23 assembled in an array on CPD-containing DNA. Rad4-Rad23 particles are indicated by white arrows. See also Movie S5.
(D) Kymograph of Rad4-Rad23 particles assembled on CPD array shown in (C).
(E) Distributions of motion types of WT Rad4-Rad23 observed on DNA damage arrays show lesion-dependent behavior (Fl-dT – green, CPD – orange, undamaged DNA – purple, UV-irradiated λ-DNA – gray). See also Figure S2 and S3.
(F) and (G) Distributions of motion types of WT, Δβ-hairpin3, and ΔBHD3 observed on DNA damage arrays containing sites of Fl-dT (WT – red, Δβ-hairpin3 – green, ΔBHD3 – blue), and CPD (WT – pink, Δβ-hairpin3 – mint, ΔBHD3 – lavender), respectively. WT data reproduced from Figure 3E.
Truncations in the β-hairpin Domain 3 (BHD3) of Rad4 Increase Constrained Motion
Co-crystal structure of Rad4-Rad23 bound to model DNA lesions show that the tip of β-hairpin 3 of Rad4 inserts into the DNA duplex at the lesion site, suggesting that this structural component is important for DNA damage recognition (Figure 1A). Previous EMSAs have also demonstrated that deletions of the β-hairpin tip (Δβ-hairpin3, His-scRad4 101-632_Δ599-606) or β-hairpin domain 3 (ΔBHD3, His-scRad4 101-540) abolished lesion-specific binding of Rad4-Rad23 (Chen et al., 2015; Min and Pavletich, 2007). We further investigated the role of the β-hairpin 3 motif in Rad4’s lesion recognition by examining the diffusion behavior of Δβ-hairpin3 (β-hairpin 3 tip deletion, His-scRad4 101-632_Δ599-606) and ΔBHD3 (β-hairpin 3 domain deletion, His-scRad4 101-540) (Figure 1A) on UV-irradiated λ-DNA. Both mutants showed a decrease in nonmotile particles (30–40%) compared to WT (~60%, Figure 4A). An overall upward trend in the random diffusion population also corresponded to the increasing loss of residues within BHD3 in these two mutants (25% for WT, 33% for Δβ-hairpin3, and 39% for ΔBHD3; Figure 4A). Interestingly, the fraction of Δβ-hairpin3 undergoing constrained motion is almost double of WT or ΔBHD3.
Figure 4. Motion and Dissociation Kinetics of Rad4 WT and β-hairpin 3 Mutants on UV-irradiated λ-DNA.
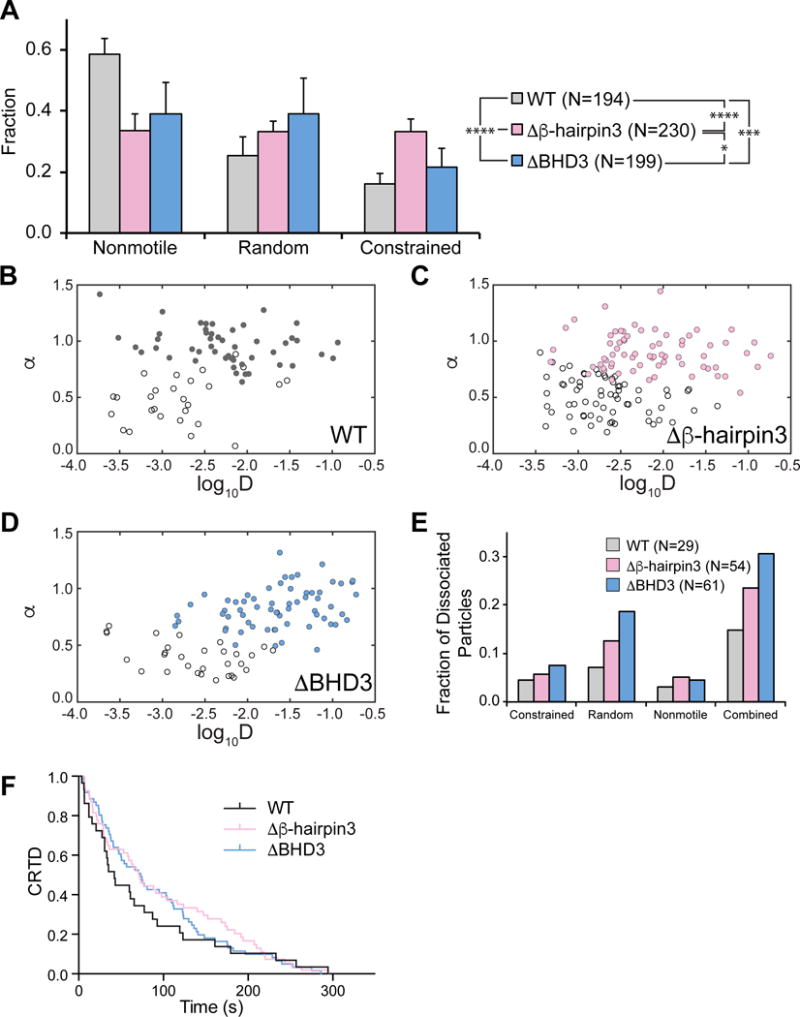
(A) Distributions of observed motion types from Rad4 WT and mutants.
(B – D) Anomalous diffusion exponent (α) vs. diffusion coefficient (log10D) plotted for random (filled circles) and constrained (empty circles) particles of WT (B), Δβ-hairpin3 (C), and ΔHD3 (D). See also Figure S4.
(E) Dissociating particles as fractions of total particles observed increase with larger deletions in Rad4 BHD3 sequence.
(F) Cumulative residence time distribution (CRTD) plot of lifetimes of Rad4 WT and mutants that dissociated during observation. See also Figure S5.
χ2 analysis of WT and both mutants shows that distributions of motion types were indeed affected by deletions of the damage-sensing β-hairpin 3 (p < 0.0001, Figure 4A). Histograms of diffusion coefficients and anomalous diffusion exponents from all motile particles also show that while the Δβ-hairpin3 diffusion coefficient was similar to WT, deletion of the full domain (ΔBHD3) caused ~25% of proteins to diffuse significantly faster (Figure S4). Finally, as compared to WT, both Δβ-hairpin3 and ΔBHD3 mutants appear to be more prone to dissociation (15%, 24%, 31%, respectively, Figure 4E). However, at low particle counts, neither dissociation kinetics nor mean lifetimes of the dissociating proteins were significantly different across three protein variants, shown by Mantel-Cox log-rank test of survival curves (Figure 4F, p > 0.5) and one-way ANOVA of off-rates obtained from single-exponential fitting of lifetime histograms (Figure S5, p > 0.2). Further analysis of these protein variants on DNA damage arrays revealed distinct behavior of the deletion mutants compared to WT on Fl-dT substrates (Figure 3F, p < 0.01, χ2 test). While Δβ-hairpin3 behaved similarly to WT on CPD substrates, ΔBHD3 showed significant increase in constrained motion (Figure 3G, p < 0.05, χ2 test).
Rad4 Variant Lacking β-hairpin Domain 3 (ΔBHD3) is Capable of Specific Binding and DNA Bending to Fl-dT-Containing DNA Fragments
The co-crystal of DNA-bound Rad4-Rad23 indicates that DNA binding by WT causes a kink in the DNA of about 42° (Figure 1A) (Min and Pavletich, 2007). Having shown that the ΔBHD3 variant forms stable complexes on UV-irradiated λ-DNA tightropes and binds to Fl-dT-containing a short DNA fragment in fluorescence anisotropy experiments (Figure S2 and Table S1), we asked if DNA bending is also a feature of binding by this mutant. Using AFM we studied the bending of a 538-bp dsDNA fragment that containes a Fl-dT lesion at 30% of the contour length from one end. Naked DNA was bent by only 4 ± 32° (N = 245, Figure S6), whereas we observed that WT specifically bound at 32 ± 13% contour length (N = 335, Figure 5A) bent the DNA 43 ± 24° (N = 189, Figure 5B); remarkably similar to specifically bound ΔBHD3 (31 ± 10% contour length, N = 148, Figure 5C) which bent DNA 36.5 ± 29.1° (N = 101, Figure 5D). Lastly, consistent with our previous report on WT protein binding to undamaged DNA fragments (Chen et al., 2015), both WT and ΔBHD3 induced bending in DNA even when not specifically bound to the Fl-dT lesion (white bars, Figure 5B and D).
Figure 5. Specific Binding and DNA Bending by WT and ΔBHD3.
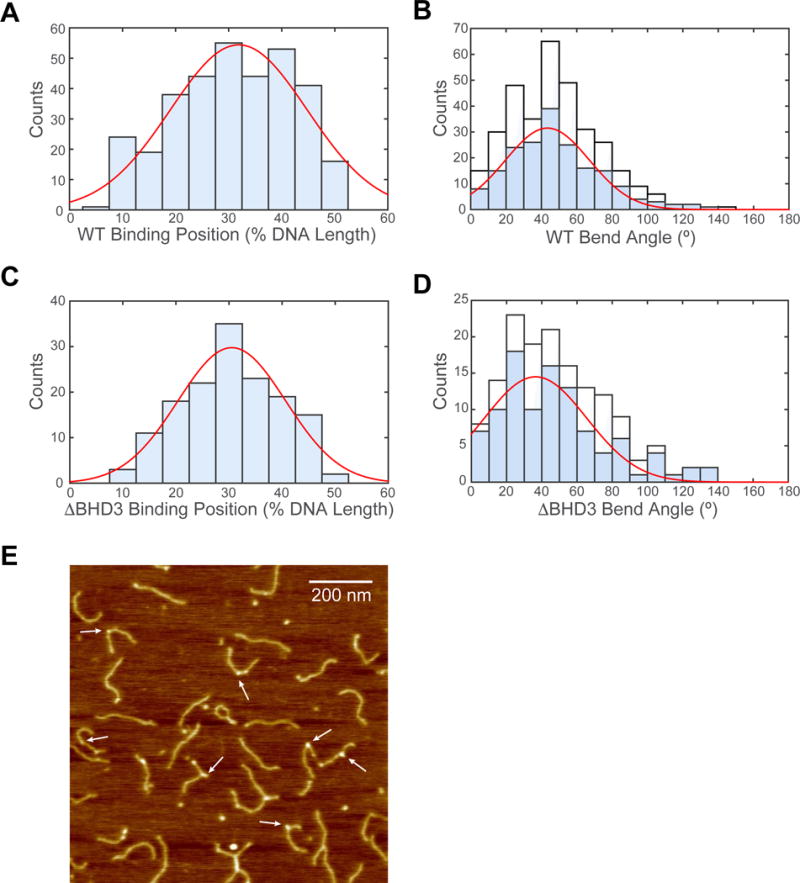
(A) Histogram and Gaussian fitting (red curve) of WT binding positions (32 ± 13%, N = 335) on DNA fragment in terms of percentage of total contour length measured from one end.
(B) Histogram of DNA bend angles at all internal WT binding sites (white, N = 335). Histogram (blue) and Gaussian fitting (red curve) of DNA bend angles (43 ± 24°, N = 189) at WT proteins specifically bound between 20% and 40%. See also Figure S6.
(C) Histogram and Gaussian fitting (red curve) of ΔBHD3 binding positions (31 ± 10%, N = 148) on DNA fragment in terms of percentage of total contour length measured from one end.
(D) Histogram of DNA bend angles at all internal ΔBHD3 binding sites (white). Histogram (blue) and Gaussian fitting (red curve) of DNA bend angles (37 ± 29°, N = 101) at ΔBHD3 specifically bound between 20% and 40%.
(E) Representative AFM image of ΔBHD3 bound to Fl-dT-containing DNA fragments. White arrows highlight representative binding events scored in data analysis. See also Figure S6.
Deletions of C-terminal Regions in Rad4 Confer Varying Degrees of UV Sensitivity and Repair in Yeast
Since the seven amino acid deletion in β-hairpin 3 resulted in more constrained motion on UV-irradiated DNA, we next tested whether this variant promotes efficient repair in vivo. We thus conducted UV survival and DNA repair assays on yeast strains carrying different Rad4 mutants with deletions and truncations around β-hairpin 3 within the RAD4 locus (Figure 1A and Figure 6A). Notably, deletion of the seven amino acid tip of β-hairpin 3 (rad4 Δ599–605) showed WT levels of UV resistance (Figure 6B, compare pink and black lines). Removal of the entire β-hairpin 3 from stem to tip (Figure 6B, rad4 Δ590–615, red) led to increased UV sensitivity comparable to deleting the entire β-hairpin domain 3 (Figure 6B, rad4 Δ541–632, blue). However, both of these constructs were considerably more UV resistant than the rad4Δ strain. Finally, cells with truncation from BHD3 to the C-terminus (Figure 6B, rad4 Δ541-Cterm, green) are as UV sensitive as the rad4Δ strain (Figure 6B, rad4 Δ, orange). Protein expression levels of FLAG-tagged Rad4 WT and mutants were probed with α-FLAG antibody (Figure 6C). Western blotting data suggest that removal of the entire β-hairpin 3 destabilized the protein and could contribute to some of the observed UV sensitivity due to reduced Rad4 protein levels.
Figure 6. UV Survival and Rates of CPD Removal of Yeast Carrying Different Rad4 Variants.
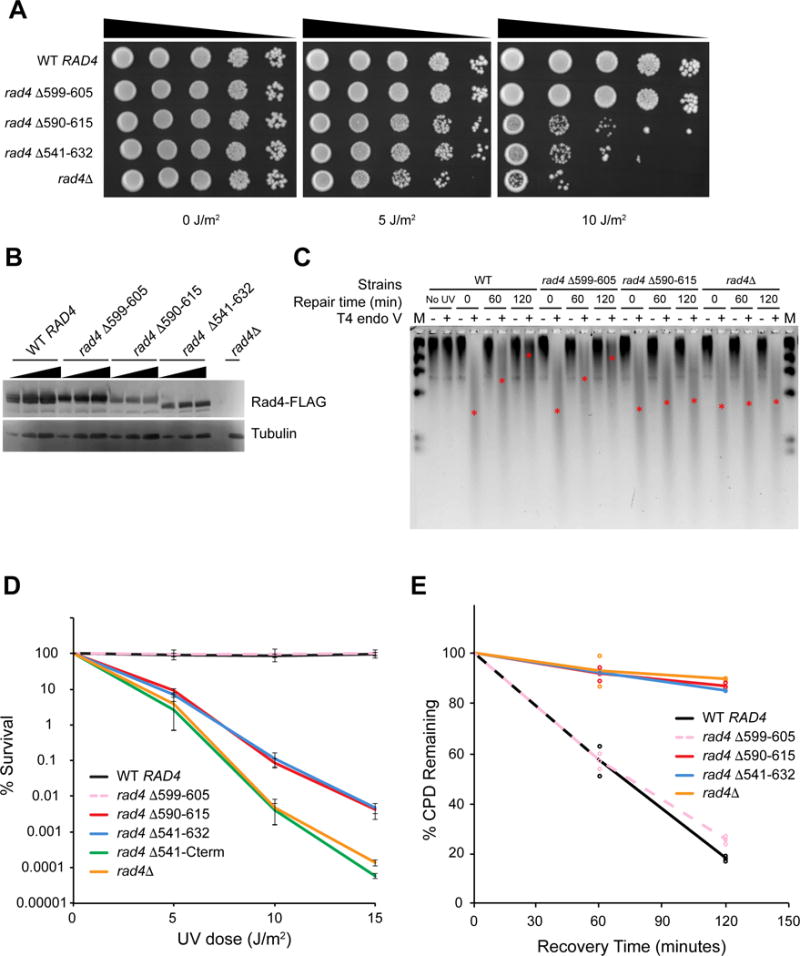
(A) Serial dilutions of yeast cells (BY4742) expressing different 3×FLAG-tagged Rad4 variants on YPD plates, 72 hours after UV irradiation.
(B) Expression levels of 3×FLAG-tagged Rad4 variants detected with anti-FLAG antibody.
(C) Genomic DNA of yeast cells after UV irradiation and recovery digested with T4 endo V, separated on alkaline agarose gel, and detected with SYBR Gold. Approximate positions of the ensemble average size of DNA in each lane are denoted with red asterisks (*). DNA marker (M, λ DNA-HindIII) was loaded in the left- and right-most lanes.
(D) Quantitative UV-survival of yeast cells (BY4741) expressing different untagged Rad4 variants. WT RAD4 – black, rad4 Δ599–605 (Δβ-hairpin3) – pink dashed, rad4 Δ590–615 – red, rad4 Δ541–632 (ΔBHD3) – blue, rad4 Δ541-cterm – green, rad4Δ – orange.
(E) Quantitative rates of CPD removal of yeast cells (BY4741) expressing different untagged Rad4 variants, determined by T4 endo V digestion. Color scheme same as in Figure 6D. See also Figure S7.
To investigate whether the mutant lacking the seven amino acid tip of β-hairpin 3 (Δβ-hairpin3, rad4 Δ599–605), which undergoes more constrained motion on UV-irradiated λ-DNA, also showed WT levels of photoproduct repair, we used two different experimental approaches: 1) T4 phage pyrimidine dimer glycosylase (Endo V) incisions on genomic DNA (Figure 6C and E), and 2) antibody slot blots of total genomic DNA for CPD and 6-4 photoproducts (Figure S7). These data indicate that loss of seven amino acids from the tip of β-hairpin 3 did not affect the rates of CPD or 6-4 photoproducts repair. However, consistent with UV survival data, larger deletions of β-hairpin domain 3 resulted in loss of photoproduct removal.
DISCUSSION
Here, we employed single-molecule methods to uncover the dynamic nature of the Rad4-Rad23 damage recognition process. We found that Rad4-Rad23 forms stable protein-DNA complexes or slides on DNA one-dimensionally to search for damage. In addition to random 1D diffusion, we showed that some Rad4-Rad23 molecules exhibited constrained motion (~1–2 kbp) around damage sites in a lesion-dependent manner. This apparent subdiffusive behavior was also influenced by deletions made in the β-hairpin domain 3 of Rad4. Surprisingly, AFM experiments revealed that Rad4-Rad23 lacking β-hairpin domain 3 (ΔBHD3) binds specifically to Fl-dT, while inducing a bend in DNA similar to WT binding, suggesting that BHD3 is not directly involved in initial damage detection or DNA bending at Fl-dT modified sites. Furthermore, we demonstrated that Rad4 mutant Δβ-hairpin3 lacking seven amino acids (FERGSTV) at the tip of the β-hairpin 3 caused a 2-fold increase in constrained motion on DNA tightropes, while maintaining WT levels of UV resistance as well as CPD and 6,4-photoproduct removal in yeast. This work examining long range motions of Rad4/Rad23 on DNA strongly supports a model in which Rad4 uses constrained motion around CPD sites as this “recognition-at-a-distance” mechanism allows efficient repair.
Alternative Damage Recognition Mechanism for Sub-Optimal Substrates through Constrained Motion by Rad4-Rad23
Our working model for Rad4-Rad23 damage recognition suggests that constrained motion represents an intermediate interrogation step. UV-irradiation induces structurally distinct lesions that are differentially bound by Rad4-Rad23 (Guzder et al., 1998). CPDs are less distorting to the DNA helix than 6-4PPs (Kim et al., 1995) and Fl-dT likely intercalates between DNA base pairs (Jaciuk et al., 2011). Robust stable and specific binding of Rad4-Rad23 to Fl-dT-containing DNA damage arrays (Figure 3E) is consistent with our fluorescence anisotropy measurements (Figure S2 and Table S1), as well as previous reports (Krasikova et al., 2013). In contrast, the subdiffusive population of WT Rad4-Rad23 on CPD-containing damage arrays increased 5-fold compared to Fl-dT at the expense of stably bound particles (Figure 3E). This observation substantiates the idea that in addition to stable binding at specific lesions; Rad4-Rad23 can effectively convey damage recognition through constrained motion around damage sites, particularly at the ‘sub-optimal’ weakly distorting CPDs.
Observation of distinct constrained motion of Rad4-Rad23 around CPD sites (Movie S5) may also help reconcile the discrepancy in reported roles of Rad4 in CPD removal, where Rad4 is incapable of recognizing CPDs in vitro (Guzder et al., 1998), yet indispensable for removal of thymine dimers in vivo (Verhage et al., 1994). Past biochemical studies characterizing binding of Rad4-Rad23 utilized short (~100 bp) damage-containing DNA fragments (Guzder et al., 1998; Krasikova et al., 2013). Since the protein exhibits oscillatory motion of ~1–2 kbp around CPD sites on naked DNA, one plausible explanation for Rad4-Rad23’s apparent lack of specificity towards CPD is that Rad4-Rad23 may dissociate from ends of such short DNA fragments in vitro. Our data from experiments on both UV survival as well as CPD and 6-4PP repair kinetics support a model in which Rad4 recruits downstream repair factors while undergoing constrained motion on genomic DNA. Since eukaryotic DNA is organized into chromatin with one nucleosome every 147 bp (Luger, 2003), and only 1–2 nucleosomes are removed during NER (Nag and Smerdon, 2009), this constrained motion would be of significantly shorter ranges than measured on DNA tightropes, making site-specific recruitment of downstream proteins even more efficient.
BHD3-Independent DNA Bending as an Initial Quality Check by Rad4
Our data on DNA bending by specifically bound WT Rad4-Rad23 (Figure 5B, blue bars) are consistent with the crystal structure (Min and Pavletich, 2007), as well as a previous study on XPC binding to cholesterol damage (Janicijevic et al., 2003). We also observed bending in DNA produced by non-specifically bound WT proteins (Figure 5B, white bars), consistent with our previous report (Chen et al., 2015). β-hairpin 3, seen inserted between DNA strands at site of lesion in the crystal structure (Figure 1A), has been hypothesized to be crucial for damage recognition (Min and Pavletich, 2007). Remarkably, we have shown that Rad4 lacking the entire β-hairpin domain 3 (ΔBHD3) was capable of specific binding to the Fl-dT lesion (Figure 5C) and induced a bend in DNA comparable to that caused by WT protein both at damage sites (Figure 5D, blue bars) and on non-specific undamaged sequences (Figure 5D, white bars). These data suggest that Rad4 checks the integrity of DNA using β-hairpin domains 1 and 2. This initial recognition is thus independent of the energetically costly insertion of β-hairpin 3 into DNA. Since β-hairpin insertion is slow and rate-limiting when binding to mismatch DNA (Chen et al., 2015), DNA bending may serve as a rapid initial quality check on a much faster timescale, e.g. during linear diffusion on DNA where the residence time at each base pair is ~10 μs (see Supplemental Experimental Procedures). At strongly helix-distorting lesions such as a Fl-dT or 6-4PP, bending/twisting of DNA could lead to spontaneous base-flipping (Su et al., 2005) and trapping of Rad4-Rad23 in an energy minimum such that robust protein-DNA complexes are formed (Chen et al., 2015; Velmurugu et al., 2016). In contrast, at damage sites with minimal helical distortions, where β-hairpin 3 insertion and subsequent protein-DNA complex stabilization are less attainable, TFIIH and Rad14 (XPA) may be relied on more heavily, as in the recently proposed ‘tripartite DNA lesion recognition and verification’ process (Li et al., 2015). Increased involvement of downstream NER factors would also help explain the slower repair rate of CPDs (Mitchell et al., 1985). Finally, in addition to damage recognition by Rad4-Rad23, the Rad7-Rad16 complex, known to be essential for dimer removal in silenced genes and again contribute to ~20–30% of CPD repair in the non-transcribed strands of active genes (Verhage et al., 1994), have also been implicated in some cases to function as a putative damage sensor (Guzder et al., 1997; Lettieri et al., 2008).
A Dynamic Multi-Step Damage Recognition Model
Subdiffusion of macromolecules in biological systems has been observed previously (Dunn et al., 2011; Ghodke et al., 2014; Gorman et al., 2007; Hofling and Franosch, 2013; Hughes et al., 2013; Lin et al., 2014). Rad4 showed increased constrained motion at physiological salt concentrations (Figure 2), which could be due to the faborable hydrophobic interactions between aromatic side-chains (F556, F597, and F599) and DNA bases at elevated ionic strengths. A recent molecular dynamics simulations study has identified that F556, F597, and F599 form a Phe ‘flipping path’ in BHD3, facilitating β-hairpin 3 insertion (Mu et al., 2015). Because the correct orientation of F599 during base flipping was essential in allowing complete insertion of the hairpin, loss of F599 in Δβ-hairpin3 may impede or abort the insertion process, resulting in increased constrained motion rather than formation of stable complexes. The hypothesis that Rad4-Rad23 undergoing constrained motion remains ‘repair competent’ is corroborated by the finding that yeast carrying the protein variant lacking seven amino acids at the tip of β-hairpin 3 (Δβ-hairpin3) are as UV-resistant as WT (Figure 6A) and show the same rates of CPD and 6-4 photoproduct removal (Figure 6E and S7). Any decrease in recognition and repair due to reduced levels of stable binding of Δβ-hairpin3 is apparently compensated by this ‘recognition-at-a-distance’ achieved through subdiffusion of the protein around the lesion. We thus envision Rad4-Rad23 as a first responder to arrive at the scene of an accident, able to direct other emergency workers to the site without being directly on the scene. Indeed, ‘recognition-at-a-distance’ may be applicable to a wide range of proteins that need to achieve target binding and signal for downstream processes, such as those involved in replication and transcription, during which ‘molecular traffic jams’ could occur (Finkelstein and Greene, 2013). This mechanism would allow weakly interacting or sub-optimal target sites to be recognized and acted upon, while reducing potential steric hindrance or target site occlusion problems between the tightly bound recognition proteins and subsequent factors that need access to the targets. Mismatch repair proteins that dissociate from mismatched bases in an ATP-dependent manner to recruit the next proteins may also fall into this general category (Gorman et al., 2007).
The balance in maintaining speed and specificity to target search and recognition by DNA binding proteins has been subject to both theoretical and experimental studies (Tafvizi et al., 2011). In a previously established two-state model, a protein is considered to have two conformations, one that allows rapid diffusion on a smooth energy landscape, and the other that binds to target with a rugged landscape required for high specificity (Slutsky and Mirny, 2004). Similar to the previously proposed conformational proofreading mechanism (Ghodke et al., 2014; Savir and Tlusty, 2007), our data on diffusion of Rad4 WT and mutants support a dynamic model with multiple intermediate states that utilize different structural domains of the protein to achieve efficient damage recognition (Figure 7). We calculate that the energy barrier to free diffusion is ~1.60 kBT and ~0.37 kBT for WT undergoing constrained motion and random diffusion, respectively (see Supplemental Experimental Procedures). DNA bending and other interactions between β-hairpin domain 2 and DNA likely contribute to the ruggedness of energy landscape during subdiffusion. Base flipping and stabilization of flipped-out bases following β-hairpin 3 insertion, which amounts to ~5.7 kBT (Chen et al., 2015; Mu et al., 2015), make further contributions towards and exceeding the theoretical minimum energy difference requirement at target sites (~5.7 kBT for yeast genome, see Supplemental Experimental Procedures) (Goffeau et al., 1996; Slutsky and Mirny, 2004). Overall, assuming WT diffusional behavior is observed in a yeast cell nucleus containing ~1.2 × 107bp of genomic DNA and ~870 copies of Rad4, we can estimate the shortest possible time needed to search the genome from the typical range of motion of a Rad4-Rad23 molecule and its measured average lifetime. Such calculation yields a lower limit of genome search time of roughly 2 – 3 minutes (see Supplemental Experimental Procedures).
Figure 7. Working Model for Dynamic Lesion Recognition by Rad4-Rad23.
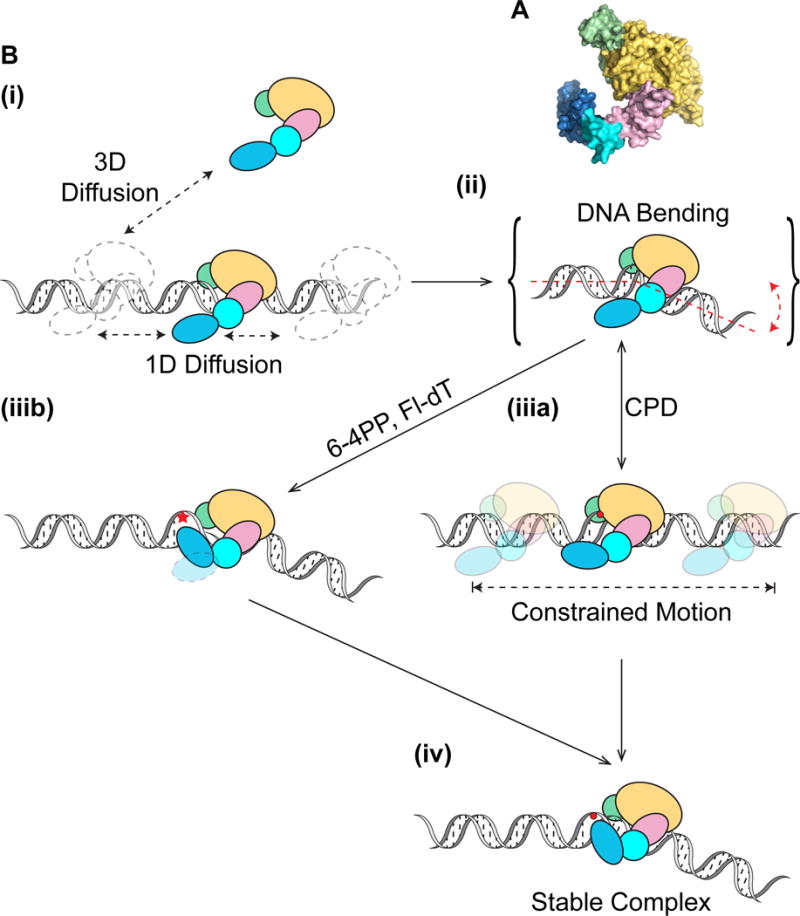
(A) Domains of Rad4-Rad23 (PDB: 2QSF) color-coded as shown in (B): TGD – yellow, BHD1 – pink, BHD2 – cyan, BHD3 – blue, Rad23 – green.
(B) Rad4-Rad23 scans DNA through 3D or 1D diffusion (i) and tests integrity of DNA via bending/twisting during 1D diffusion on DNA (ii). Depending on the type of damage encountered, Rad4-Rad23 can either undergo constrained motion around lesion due to lack of β-hairpin 3 insertion (iiia), or alternatively rapidly forms stable protein-DNA complex at site of lesion with β-hairpin 3 inserted for stabilization in a twist-open action (Velmurugu et al., 2016) (iiib). While it is possible that the DNA in (iiia) is bent, for simplicity this is not shown. Extra time spent probing the lesion, afforded by constrained motion of Rad4-Rad23, could also lead to stable binding of the protein at sites that require larger base opening/flipping energies (iv).
Our working model of recognition suggests that, Rad4-Rad23 scans DNA for lesion through a combination of 3D and 1D diffusion (Figure 7B(i)). During diffusion, the integrity of DNA is being checked through bending or twisting (Velmurugu et al., 2016), involving β-hairpin domains 1 and 2 in the search process (Figure 7B(ii)). While diffusing one dimensionally on DNA, a protein conformational change could be triggered by lesion encounter, which may allow Rad4 to enter a binding state of stronger interactions with DNA, resulting in the subdiffusion of Rad4-Rad23 on DNA. Both DNA bending/twisting and protein conformational changes could contribute to shorter regions of interrogation by the protein (Figure 7B(iiia)). Alternatively, spontaneous helix opening and base flipping, which are energetically linked to DNA bending, may follow at sites of severely helix-destabilizing lesions, facilitating immediate β-hairpin 3 insertion and leading to stable binding (Figure 7B(iiib)). Otherwise, BHD3 continues to interact with and probe DNA as the protein undergoes constrained motion. As the subdiffusive protein has limited range, β-hairpin 3 insertion is afforded more opportunities to proceed, therefore leading to recognition of difficult targets with slower base-flipping rates, consistent with the previously published kinetic gating mechanism of Rad4-Rad23 damage recognition (Chen et al., 2015; Velmurugu et al., 2016). Both recognition pathways, through rapid spontaneous base flipping or constrained motion, converge to form a stable recognition complex (Figure 7B(iv)). Careful analysis of all kymographs (N~1600) generated for this study did not yield any definitive transition in either direction between the two diffusive modes of the protein. We thus speculate that both diffusive states are stabilized by binding energy contributed from protein-DNA interactions and the barrier to transitions are large such that these transitions are rare and rapid, therefore unlikely to be observed or recorded.
CONCLUSION
In summary, using single-molecule fluorescence microscopy, we have shown that Rad4-Rad23 performs both random walk and subdiffusion to facilitate damage recognition at different lesions. Evidence suggests that Rad4 β-hairpin domains 1 & 2 induced DNA bending, independent of β-hairpin domain 3 and thus most likely an early step and allows damage recognition for Fl-dT. Taken together our data support a dynamic multi-step damage recognition model utilizing different structural domains for distinct stages of damage detection.
EXPERIMENTAL PROCEDURES
Single-Molecule DNA Tightrope Assays
DNA tightrope assays were performed as previously described (Ghodke et al., 2014) at room temperature in binding buffer containing 5 mM BTP-HCl (pH 6.8), 75 mM NaCl (unless otherwise noted), 5% glycerol, 0.74 mM CHAPS, 0.5 mg/ml BSA and 5 mM DTT. Purified His-tagged Rad4-Rad23 was labeled with 655 nm streptavidin-coated quantum dots (Invitrogen) through biotinylated α-His antibody (Qiagen) and visualized with oblique angle illumination (see Supplemental Experimental Procedures). Images were collected at ~10 Hz and analyzed in ImageJ (NIH). Diffusion parameters and lifetime analysis were accomplished with custom Matlab scripts.
Atomic Force Microscopy
AFM imaging was conducted as previously described (Chen et al., 2015). Briefly, sample was diluted with AFM deposition buffer (25 mM HEPES pH 7.5, 25 mM NaOAc, and 10 mM MgOAc) before transferred to freshly cleaved mica. Mica surface was then rinsed with filtered H2O, dried in a stream of nitrogen gas, and imaged on a Multimode V microscope (Bruker Corp.). Binding positions and bend angles were extracted using ImageJ software (NIH).
UV Survival Assays
RAD4 deletion mutants were generated in yeast using the CRISPR-Cas9 system (Table S2). For UV survival assays, yeast cells were irradiated with 254 nm UV light at indicated doses before recovery in the dark and colony counting. FLAG-tagged Rad4 protein levels in each strain were assessed by lysing yeast cells and western blotting with α-FLAG antibody.
For full description of experimental data collection and analysis, DNA substrates, and details on in vivo experiments, refer to the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
The authors wish to thank Emily Beckwitt for critically reading and commenting on the manuscript; Dr. Brenda Diergaarde for helping with statistical analyses; Dr. Elise Fouquerel for helping with slot blot assays; and Drs. Guillermo Romero, Marcel Bruchez, and Patricia Opresko for providing helpful discussions. This work was made possible through funding from the National Institutes of Health 5R01ES019566 to B.V.H., 5R01ES024872 to K.A.B., 5R01ES002614 to J.J.W., and 2P30CA047904 to University of Pittsburgh Cancer Institute; and from the National Science Foundation MCB-1412692 to J.-H.M.
Footnotes
AUTHOR CONTRIBUTIONS
M.K. designed, performed, and analyzed all single-molecule tightrope experiments and slot blot assays. M.K., L.L., and K.I.D. designed, performed, and analyzed all AFM experiments. P.M., S.B., K.A.B., and J.J.W. designed and conducted yeast UV-survival experiments. P.M. and J.J.W. designed and conducted CPD repair rate experiments. X.C. and J-H.M. purified the proteins. S.C.W. provided imaging resources. N.M.K. provided expertise and feedback. M.K. drafted the paper, which was reviewed and edited by all authors. J-H.M. and B.V.H. directed all aspects of the project.
References
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KL, Roginskaya M, Zou Y, Altamirano A, Basu AK, Stone MP. Binding of the human nucleotide excision repair proteins XPA and XPC/HR23B to the 5R-thymine glycol lesion and structure of the cis-(5R,6S) thymine glycol epimer in the 5′-GTgG-3′ sequence: destabilization of two base pairs at the lesion site. Nucleic acids research. 2010;38:428–440. doi: 10.1093/nar/gkp844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch U, Trautlein D, Clement FC, Fei J, Leitenstorfer A, Ferrando-May E, Naegeli H. Two-stage dynamic DNA quality check by xeroderma pigmentosum group C protein. The EMBO journal. 2009;28:2387–2399. doi: 10.1038/emboj.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Velmurugu Y, Zheng G, Park B, Shim Y, Kim Y, Liu L, Van Houten B, He C, Ansari A, et al. Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nature communications. 2015;6:5849. doi: 10.1038/ncomms6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Molecular cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. The Journal of investigative dermatology. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AR, Kad NM, Nelson SR, Warshaw DM, Wallace SS. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic acids research. 2011;39:7487–7498. doi: 10.1093/nar/gkr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein IJ, Greene EC. Molecular traffic jams on DNA. Annual review of biophysics. 2013;42:241–263. doi: 10.1146/annurev-biophys-083012-130304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Friedberg EC, American Society for Microbiology . DNA repair and mutagenesis. Washington D.C.: ASM Press; 2006. p. xxix, 1118. [Google Scholar]
- Ghodke H, Wang H, Hsieh CL, Woldemeskel S, Watkins SC, Rapic-Otrin V, Van Houten B. Single-molecule analysis reveals human UV-damaged DNA-binding protein (UV-DDB) dimerizes on DNA via multiple kinetic intermediates. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E1862–1871. doi: 10.1073/pnas.1323856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chemical reviews. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546, 563–547. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, Greene EC. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Molecular cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S. Yeast Rad7-Rad16 complex, specific for the nucleotide excision repair of the nontranscribed DNA strand, is an ATP-dependent DNA damage sensor. The Journal of biological chemistry. 1997;272:21665–21668. doi: 10.1074/jbc.272.35.21665. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S. Affinity of yeast nucleotide excision repair factor 2, consisting of the Rad4 and Rad23 proteins, for ultraviolet damaged DNA. The Journal of biological chemistry. 1998;273:31541–31546. doi: 10.1074/jbc.273.47.31541. [DOI] [PubMed] [Google Scholar]
- Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic acids research. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nature reviews. Molecular cell biology. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hey T, Lipps G, Sugasawa K, Iwai S, Hanaoka F, Krauss G. The XPC-HR23B complex displays high affinity and specificity for damaged DNA in a true-equilibrium fluorescence assay. Biochemistry. 2002;41:6583–6587. doi: 10.1021/bi012202t. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Hofling F, Franosch T. Anomalous transport in the crowded world of biological cells. Rep Prog Phys. 2013;76:046602. doi: 10.1088/0034-4885/76/4/046602. [DOI] [PubMed] [Google Scholar]
- Hughes CD, Wang H, Ghodke H, Simons M, Towheed A, Peng Y, Van Houten B, Kad NM. Real-time single-molecule imaging reveals a direct interaction between UvrC and UvrB on DNA tightropes. Nucleic acids research. 2013;41:4901–4912. doi: 10.1093/nar/gkt177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaciuk M, Nowak E, Skowronek K, Tanska A, Nowotny M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nature structural & molecular biology. 2011;18:191–197. doi: 10.1038/nsmb.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicijevic A, Sugasawa K, Shimizu Y, Hanaoka F, Wijgers N, Djurica M, Hoeijmakers JH, Wyman C. DNA bending by the human damage recognition complex XPC-HR23B. DNA repair. 2003;2:325–336. doi: 10.1016/s1568-7864(02)00222-7. [DOI] [PubMed] [Google Scholar]
- Jansen LE, Verhage RA, Brouwer J. Preferential binding of yeast Rad4.Rad23 complex to damaged DNA. The Journal of biological chemistry. 1998;273:33111–33114. doi: 10.1074/jbc.273.50.33111. [DOI] [PubMed] [Google Scholar]
- Kad NM, Van Houten B. Dynamics of lesion processing by bacterial nucleotide excision repair proteins. Progress in molecular biology and translational science. 2012;110:1–24. doi: 10.1016/B978-0-12-387665-2.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Molecular cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Patel D, Choi BS. Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem Photobiol. 1995;62:44–50. doi: 10.1111/j.1751-1097.1995.tb05236.x. [DOI] [PubMed] [Google Scholar]
- Krasikova YS, Rechkunova NI, Maltseva EA, Pestryakov PE, Petruseva IO, Sugasawa K, Chen X, Min JH, Lavrik OI. Comparative analysis of interaction of human and yeast DNA damage recognition complexes with damaged DNA in nucleotide excision repair. The Journal of biological chemistry. 2013;288:10936–10947. doi: 10.1074/jbc.M112.444026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper J, Braun C, Elias A, Michels G, Sauer F, Schmitt DR, Poterszman A, Egly JM, Kisker C. In TFIIH, XPD helicase is exclusively devoted to DNA repair. PLoS biology. 2014;12:e1001954. doi: 10.1371/journal.pbio.1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumoto R, Masutani C, Sugasawa K, Iwai S, Araki M, Uchida A, Mizukoshi T, Hanaoka F. Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutation research. 2001;485:219–227. doi: 10.1016/s0921-8777(00)00082-3. [DOI] [PubMed] [Google Scholar]
- Legerski R, Peterson C. Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature. 1992;359:70–73. doi: 10.1038/359070a0. [DOI] [PubMed] [Google Scholar]
- Lettieri T, Kraehenbuehl R, Capiaghi C, Livingstone-Zatchej M, Thoma F. Functionally distinct nucleosome-free regions in yeast require Rad7 and Rad16 for nucleotide excision repair. DNA repair. 2008;7:734–743. doi: 10.1016/j.dnarep.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Li CL, Golebiowski FM, Onishi Y, Samara NL, Sugasawa K, Yang W. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Molecular cell. 2015;59:1025–1034. doi: 10.1016/j.molcel.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Countryman P, Buncher N, Kaur P, E L, Zhang Y, Gibson G, You C, Watkins SC, Piehler J, et al. TRF1 and TRF2 use different mechanisms to find telomeric DNA but share a novel mechanism to search for protein partners at telomeres. Nucleic acids research. 2014;42:2493–2504. doi: 10.1093/nar/gkt1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13:127–135. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Masutani C, Sugasawa K, Yanagisawa J, Sonoyama T, Ui M, Enomoto T, Takio K, Tanaka K, van der Spek PJ, Bootsma D, et al. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. The EMBO journal. 1994;13:1831–1843. doi: 10.1002/j.1460-2075.1994.tb06452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Mitchell DL, Haipek CA, Clarkson JM. (6-4)Photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane pyrimidine dimers. Mutation research. 1985;143:109–112. doi: 10.1016/s0165-7992(85)80018-x. [DOI] [PubMed] [Google Scholar]
- Mu H, Geacintov NE, Zhang Y, Broyde S. Recognition of Damaged DNA for Nucleotide Excision Repair: A Correlated Motion Mechanism with a Mismatched cis-syn Thymine Dimer Lesion. Biochemistry. 2015;54:5263–5267. doi: 10.1021/acs.biochem.5b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag R, Smerdon MJ. Altering the chromatin landscape for nucleotide excision repair. Mutation research. 2009;682:13–20. doi: 10.1016/j.mrrev.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Savir Y, Tlusty T. Conformational proofreading: the impact of conformational changes on the specificity of molecular recognition. PloS one. 2007;2:e468. doi: 10.1371/journal.pone.0000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harbor perspectives in biology. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophysical journal. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TJ, Tock MR, Egelhaaf SU, Poon WC, Dryden DT. DNA bending by M.EcoKI methyltransferase is coupled to nucleotide flipping. Nucleic acids research. 2005;33:3235–3244. doi: 10.1093/nar/gki618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Molecular cell. 2009;36:642–653. doi: 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Shimizu Y, Iwai S, Hanaoka F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA repair. 2002;1:95–107. doi: 10.1016/s1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- Tafvizi A, Mirny LA, van Oijen AM. Dancing on DNA: kinetic aspects of search processes on DNA. Chemphyschem: a European journal of chemical physics and physical chemistry. 2011;12:1481–1489. doi: 10.1002/cphc.201100112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmurugu Y, Chen X, Slogoff Sevilla P, Min JH, Ansari A. Twist-open mechanism of DNA damage recognition by the Rad4/XPC nucleotide excision repair complex. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E2296–2305. doi: 10.1073/pnas.1514666113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage R, Zeeman AM, de Groot N, Gleig F, Bang DD, van de Putte P, Brouwer J. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Molecular and cellular biology. 1994;14:6135–6142. doi: 10.1128/mcb.14.9.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harbor perspectives in biology. 2013;5:a012625. doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, et al. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2737–2746. doi: 10.1073/pnas.1110067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo JE, Khoo A, Fagbemi AF, Scharer OD. The efficiencies of damage recognition and excision correlate with duplex destabilization induced by acetylaminofluorene adducts in human nucleotide excision repair. Chemical research in toxicology. 2012;25:2462–2468. doi: 10.1021/tx3003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.