

Abstract
Objective
ATP binding cassette transporter A1 (ABCA1) is the principal protein responsible for cellular cholesterol efflux. Abundance and functionality of ABCA1 is regulated both transcriptionally and post-translationally, with endocytosis of ABCA1 being an important element of post-translational regulation. Functional ABCA1 resides on the plasma membrane, but can be internalized and either degraded or recycled back to the plasma membrane. The interaction between the degradative and recycling pathways determines the abundance of ABCA1 and may contribute to the efflux of intracellular cholesterol.
Approach and Results
Here, we show that the principal pathway responsible for the internalization of ABCA1 leading to its degradation in macrophages is ARF6-dependent endocytic pathway. This pathway was predominant in regulation of ABCA1 abundance and efflux of plasma membrane cholesterol. Conversely, the efflux of intracellular cholesterol was predominantly controlled by ARF6-indepednent pathways, and inhibition of ARF6 shifted ABCA1 into recycling endosomes enhancing efflux of intracellular cholesterol.
Conclusions
We conclude that ARF6-dependent pathway is the predominant route responsible for the ABCA1 internalization and degradation, while ARF6-independent endocytic pathways may contribute to ABCA1 recycling and efflux of intracellular cholesterol.
Keywords: cholesterol, ABCA1, endocytosis, cholesterol efflux, ARF6, clathrin, Cdc42, dynamin-2
Subject codes: Lipids and Cholesterol, Cell Biology/Structural Biology
Introduction
ATP binding cassette transporter A-I (ABCA1) is a key element of cellular cholesterol trafficking machinery. ABCA1 is responsible for the specific efflux of cellular cholesterol to extracellular acceptors, principally, apolipoprotein A-I (apoA-I) 1, and for the trafficking of cholesterol inside the cells 2. Cholesterol efflux is a critical element of reverse cholesterol transport pathway (RCT) and plays a key role in biogenesis of high density lipoprotein (HDL) 3. Both RCT and HDL are important for maintaining cellular cholesterol homeostasis; impairment of cholesterol homeostasis plays a prominent role in pathogenesis of many diseases, from atherosclerosis 4 and diabetes 5 to Alzheimer’s disease 6 and cancer 7.
ABCA1 abundance and activity are regulated on multiple levels. Transcription of ABCA1 is principally regulated by the Liver X Receptors (LXR), and LXR agonists are potent activators of cholesterol efflux 8. Post-transcriptionally, ABCA1 is regulated by several microRNAs, with miR-33 being a best investigated example 9. There are also several post-translational regulatory pathways involving calpain-dependent degradation of ABCA1 10 and ubiquitination followed by either proteasomal 11 or lysosomal 12 degradation. Activity and stability of ABCA1 are also regulated by phosphorylation of ABCA1 13 and formation of complexes with ABCA12 14 and LXRβ 15. Furthermore, ABCA1 is regulated by endocytic recycling between plasma membrane and intracellular compartments, a poorly investigated pathway.
Several studies demonstrated that internalization of resident plasma membrane ABCA1 is followed by either degradation or recycling 12, 16, 17. Endocytosis of ABCA1 was found to be an important determinant of ABCA1 abundance and functionality and is regulated by many factors, such as apoA-I 16, infectious agents (e.g. HIV and prions) 18, 19 or LXR agonists 12. It has been suggested that apart from regulating ABCA1 abundance on the cell surface, recycling of ABCA1 may be functionally involved in trafficking lipids between intracellular compartments and plasma membrane for the efflux or during formation of HDL; evidence was reported both in favour 2, 20, 21 and against 22, 23 this possibility. Earlier studies extensively documented endocytosis of HDL 24; however, these reports preceded the discovery of ABCA1, and the role of the latter in HDL endocytosis and retroendocytosis could only be assumed. Recently, recycling and intracellular trafficking of ABCA1 were implicated in trafficking of lipoproteins across the endothelium 25. While it seems certain that regulated endocytosis of ABCA1, alone or complexed with apoA-I or HDL, does occur, the involvement of the specific endocytic pathway(s) is yet to be characterized.
Endocytosis and endocytic recycling of proteins residing at the plasma membrane are facilitated by a complex network of distinctive, but partially overlapping and redundant pathways (for review see 26–28). The principal pathways that can be implicated in endocytosis and/or endocytic recycling of ABCA1 (i.e. carrying a cargo similar to ABCA1) include clathrin-dependent pathway, caveolae-dependent pathway and two clathrin-independent pathways, Cdc42-dependent and ARF6-dependent pathways. Azuma et al. reported that endocytosis of ABCA1 occurs via clathrin-dependent pathway and part of internalized ABCA1 is recycled back to the cell surface via Rab4-mediated pathway 20. To our knowledge this is the only study which investigated a specific pathway of ABCA1 endocytosis. In our study, we investigated several endocytic pathways and established the key role of ARF6 in regulation of ABCA1 endocytosis and cholesterol efflux under various metabolic conditions.
Materials and Methods
Materials and Methods are available as online-only Data Supplement. Schematic representation of the experimental design is shown in Supplemental Fig. I.
Results
ARF6 and dynamin-2 regulate endocytosis of ABCA1
To investigate the contribution of various pathways to endocytosis of ABCA1 we employed siRNA silencing in mouse macrophage cells RAW 264.7 to reduce the abundance of four proteins playing key roles in different endocytic pathways: clathrin, dynamin-2, Cdc42 and Arf6. Transfection of cells with the corresponding siRNA resulted in reduction of the abundance of each of the proteins by 50–70% (Supplemental Fig. IIA); higher levels of silencing affected cell viability. There was no off-target inhibition, i.e. silencing of one protein did not affect the abundance of any of the three other proteins (data not shown).
Silencing of clathrin and Cdc42 did not affect the abundance of total Abca1, while silencing of dynamin-2 and Arf6 increased the abundance of Abca1 in the cells by approximately 20% (Fig. 1A). When the abundance of Abca1 on the cell surface was measured, silencing of clathrin and Cdc42 again had no effect, while silencing of dynamin-2 and Arf6 increased the abundance of the cell-surface Abca1 by 70% (Fig. 1B). Given that silencing was partial for all four factors, the observed difference in the effects indicates the predominant role of dynamin-2 and Arf6 in regulation of Abca1 abundance. To further elucidate the mechanism responsible for the effects of silencing of dynamin-2 and Arf6 on the Abca1 abundance, we measured internalization and degradation of Abca1. Internalization was assessed by biotinylation of cell-surface Abca1 and measuring the abundance of the intracellular biotinylated Abca1 after 30 min incubation. Silencing of dynamin-2 and Arf6 reduced the amount of internalized Abca1 by 40% (Fig. 1C). Degradation of Abca1was assessed in pulse-chase experiments after labelling Abca1 with 35S; silencing of dynamin-2 and Arf6 reduced the rate of Abca1 degradation by approximately 40%, although due to high variability the difference was not statistically significant (Fig. 1D). Finally, we evaluated the functional consequences of the inhibition of different endocytic pathways by measuring cholesterol efflux from cells to apolipoprotein A-I (apoA-I), a process exclusively controlled by Abca1. Consistent with the effects on Abca1 abundance, silencing of Arf6 and dynamin-2 increased cholesterol efflux to apoA-I by 50%, while silencing of clathrin and Cdc42 had no effect (Fig. 1E). At the same time, a non-specific efflux to methyl-β-cyclodextrin (MBC, 200 μg/ml) was only slightly increased (<10%) with silencing of all four genes (Fig. 1 F). Thus, the abundance of functional Abca1 on the cell surface is controlled by endocytic pathways involving dynamin-2 and Arf6, with clathrin-dependent and Cdc42-dependent pathways not playing a substantial role.
Figure 1. The effect of silencing of individual endocytic pathways on ABCA1 abundance and functionality.

A – The abundance of total Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). B – The abundance of cell-surface Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). C – The abundance of internalized Abca1 in cells with silenced dynamin-2 (Dy) or Arf6 (Ar). Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). D – Rates of degradation of Abca1 in a pulse-chase experiment after silencing dynamin-2 (Dy) or Arf6 (Ar). Means±SEM of triplicate determinations are shown. E- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. ***p<0.001. F- Cholesterol efflux to methyl-β-cyclodextrin (MBC, final concentration 200 μg/ml) from cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. *p<0.05; ***p<0.001 G – The abundance of total Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). *p<0.05 (versus mock transfected cells with the same treatment). H – The abundance of cell-surface Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). *p<0.05 (versus mock transfected cells with the same treatment). I – The abundance of internalized Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). J- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells transiently transfected with Arf6 or mock plasmid. Means ± SEM of quadruplicate determinations are shown.
The experiments with Arf6 silencing were complemented by experiments with overexpression of Arf6. Heterologous Arf6-GFP was transiently expressed in RAW 264.7 cells (Supplemental Fig. IIB). The abundances of total (Fig. 1G), cell-surface (Fig. 1H) and internalized (Fig. 1I) Abca1were reduced in cells overexpressing Arf6. Cholesterol efflux, however, was unaffected by Arf6 overexpression (Fig. 1J). Thus, the effect of Arf6 overexpression on Abca1 abundance and localization were opposite to those of Arf6 silencing, however, excessive Arf6 did not affect cholesterol efflux.
Finally, to ensure that the effects of Arf6 silencing were specific, the experiments were repeated with an alternative siRNA directed to a different site of Arf6 mRNA (see Materials and Methods). Both original and alternative siRNA caused similar reductions of Arf6 abundance and similar increases of total and cell-surface Abca1 (Supplemental Fig. IIC, D). ARF6-dependent endocytic pathway is considered to be dynamin-independent 26. There is, however, mounting evidence that dynamin-2 is an essential component of all endocytic pathways, including ARF6-dependent pathway. Furthermore, recent report demonstrated that dynamin-2 is a potent activator of ARF6 29. To investigate if inhibition of dynamin-2 and Arf6 regulates two independent endocytic pathways or different steps of the same pathway, we compared the effects of silencing of Arf6 or dynamin-2 with the effects of co-silencing of the two proteins together. Co-silencing of Arf6 and dynamin-2 resulted in silencing of both proteins by 60–70% (Supplemental Fig. IIIA), and led to an increase in the abundance of total Abca1 similar to that when each protein was silenced individually (Supplemental Fig. IIIA). There was no additive effect of co-silencing of Arf6 and dynamin-2 on the abundance of cell-surface Abca1; on the contrary, co-silencing of Arf6 and dynamin-2 seems to reduce Abca1 abundance on the cell surface (Supplemental Fig. IIIB). Furthermore, silencing of Arf6, dynamin-2, or co-silencing of the both proteins resulted in similar increases of cholesterol efflux to apoA-I (Supplemental Fig. IIIC). Thus, it is likely that ARF6 and dynamin-2 target two steps of the same pathway.
To confirm that silencing of ARF6 and dynamin-2 targets a posttranslational regulatory step, we repeated the experiments in the presence of protein synthesis inhibitor, cycloheximide. Silencing of Arf6 or dynamin-2 similarly increased the abundance of total Abca1 in the presence or absence of cycloheximide (Supplemental Fig. IIID), confirming that ARF6 and dynamin-2 affect post-translational steps of regulation of ABCA1 abundance.
Endocytosis of ABCA1 in cholesterol-loaded cells
Cholesterol regulates many aspects of cellular metabolism, and excessive cholesterol plays a role in pathogenesis of many diseases, most notably, atherosclerosis. Excessive cholesterol is often a consequence of impairment of cellular cholesterol metabolism, but at the same time it regulates many pathways responsible for cholesterol homeostasis 12. To evaluate how excessive cholesterol affects ABCA1 endocytosis, we investigated relevant endocytic pathways in cholesterol-loaded cells. Loading of cells by incubating them with methyl-β-cyclodextrin/cholesterol complex resulted in almost tripling of cell total cholesterol content (Fig. 2A). We then used methyl-β-cyclodextrin/BODIPY-cholesterol complex to determine which cellular cholesterol pool is enriched by this method of cholesterol-loading. Time-course of the loading is shown in Supplemental Fig. IVA demonstrating that although initially plasma membrane pool gets enriched with exogenous cholesterol, within 30 min excessive cholesterol equilibrated among all cholesterol pools. Abundance of total and cell-surface Abca1 and cholesterol efflux were elevated in cells loaded with cholesterol (Supplemental Fig. IVB–D).
Figure 2. The effect of silencing of individual endocytic pathways on ABCA1 abundance and functionality in cells loaded with cholesterol.

A – Cholesterol content of cells loaded with cholesterol. B – The abundance of total Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). C – The abundance of cell-surface Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). D – The abundance of internalized Abca1 in cells with silenced dynamin-2 (Dy) or Arf6 (Ar). Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). E – Rates of degradation of Abca1 in a pulse-chase experiment after silencing Arf6 (Ar) or dynamin-2 (Dy). Means±SEM of triplicate determinations are shown. F- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. *p<0.05, ***p<0.001. G – The abundance of total Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). H – The abundance of cell-surface Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). I – The abundance of internalized Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). J- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells transiently transfected with Arf6 or mock plasmid. Means ± SEM of quadruplicate determinations are shown.
Similar to the cells not loaded with cholesterol, silencing of clathrin and Cdc42 had no effect on Abca1 abundance, while silencing of dynamin-2 and Arf6 increased the abundance of Abca1, although the magnitude of the effect was reduced compared to cells with unmanipulated cholesterol content (Fig. 2B). When cell-surface Abca1 was analysed, silencing of dynamin-2, caused statistically significant increase in the abundance of cell-surface Abca1 in cholesterol-loaded cells, while silencing of Arf6 caused only marginal increase of Abca1 (Fig. 2C). Unexpectedly, both the amount of internalized Abca1 (Fig. 2D) and the rate of Abca1 degradation (Fig. 2E) were increased after silencing of dynamin-2, with silencing of Arf6 having no effect on the amount of internalized Abca1 or the rate of its degradation. The rate of cholesterol efflux from cholesterol-loaded cells was elevated after silencing of dynamin-2 and Arf6, but also after silencing of Cdc42 (Fig. 2F).
The experiments with Arf6 silencing were complemented by the experiments with overexpression of Arf6. The abundance of total (Fig. 2G), cell-surface (Fig. 2H) and internalized (Fig. 3I) Abca1 as well as cholesterol efflux (Fig. 2J), were unaffected by overexpression Arf6 in cholesterol-loaded cells.
Figure 3. The effect of silencing of individual endocytic pathways on ABCA1 abundance and functionality in cholesterol-depleted cells.

A – Cholesterol content of cholesterol-depleted cells. B – The abundance of total Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). *p<0.05. C – The abundance of cell-surface Abca1 in cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). **p<0.01. D – The abundance of internalized Abca1 in cells with silenced dynamin-2 (Dy) or Arf6 (Ar). Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). *p<0.05. E – Rates of degradation of Abca1 in a pulse-chase experiment after silencing Arf6 (Ar) or dynamin-2 (Dy). Means±SEM of triplicate determinations are shown. F- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells with silenced clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. **p<0.01, ***p<0.001. G – The abundance of total Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of total Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). *p<0.05 (versus mock transfected cells with the same treatment). H – The abundance of cell-surface Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of cell-surface Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). ***p<0.001 (versus mock transfected cells with the same treatment); I – The abundance of internalized Abca1 in cells transiently transfected with Arf6 (+Ar) or mock (M) plasmid. Left – Western blot of internalized Abca1; right – densitometric quantitation of bands from three independent experiments (Means±SEM). J- Cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells transiently transfected with Arf6 or mock plasmid. Means ± SEM of quadruplicate determinations are shown. **p<0.01 (versus mock transfected cells with the same treatment).
Thus, it appears that the contribution of the ARF6-dependent endocytosis to regulation of ABCA1 abundance is reduced in cholesterol-loaded cells, whereas dynamin-2 continues to play a role. There was a further inconsistency in the effects of silencing of dynamin-2 on intracellular distribution of Abca1 with increased abundance of cell-surface Abca1 coinciding with increased internalization and degradation of Abca1. The most plausible explanation for these inconsistencies is that silencing of dynamin-2 affects not only ARF6-dependent endocytosis leading to degradation, but also other, ARF6-independent, pathways leading to recycling of ABCA1.
Endocytosis of ABCA1 in cholesterol-depleted cells
To further investigate how ABCA1 endocytosis is regulated by cell cholesterol content, we studied endocytic pathways in cells depleted of cholesterol. Treatment of cells with methyl-β-cyclodextrin resulted in 70% reduction of cellular total cholesterol content (Fig. 3A). It was previously demonstrated that treatment of cells with MBD selectively depletes plasma membrane cholesterol 30, a finding also confirmed in this study by dramatic reduction of the abundance of lipid rafts after treatment with MBD (Supplemental Fig. IVE). Abundance of total and cell-surface Abca1 and cholesterol efflux were reduced in cholesterol depleted cells (Supplemental Fig. IVB–D). Under these conditions we again found that silencing of clathrin, dynamin-2 and Cdc42 had no effect on total Abca1 abundance, while silencing of Arf6 increased it (Fig. 3B). Cell-surface Abca1 was also increased after silencing of Arf6 and dynamin-2, it was increased also after silencing of Cdc42 (Fig. 3C). The abundance of internalized ABCA1 was increased after silencing of Arf6 and dynamin-2 (Fig. 3D). Conversely, silencing of Arf6 and dynamin-2 had a tendency to reduce Abca1 degradation (Fig. 3E). Unexpectedly, however, despite increased abundance of total and cell-surface Abca1, silencing of dynamin-2, Arf6 and Cdc42 significantly reduced cholesterol efflux (Fig. 3F).
The experiments with Arf6 silencing were complemented by experiments with overexpression of Arf6. The effects of the overexpression of Arf6 on Abca1 abundance were opposite to those of Arf6 silencing: the abundance of total (Fig. 3G), cell-surface (Fig. 3H) and internalized (Fig. 4I) Abca1was reduced. Cholesterol efflux was also reduced in cholesterol-depleted, Arf6 overexpressing cells (Fig. 3J).
Figure 4. The effect of silencing of individual endocytic pathways on the efflux of intracellular cholesterol.

A – Incorporation of [3H]acetate into cholesterol in cells with unmanipulated cholesterol content, cholesterol loaded and cholesterol-depleted cells. Means ± SEM of quadruplicate determinations are shown. B – Intracellular cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cells with unmanipulated cholesterol content after silencing of clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. C – Intracellular cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cholesterol-loaded cells after silencing of clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. D – Intracellular cholesterol efflux to apoA-I (final concentration 30 μg/ml) from cholesterol-depleted cells after silencing of clathrin (Cl), dynamin-2 (Dy), Arf6 (Ar) or Cdc42 (Cd). Means ± SEM of quadruplicate determinations are shown. *p<0.05; **p<0.01, ***p<0.001 (versus mock-transfected cells); #p<0.05 (versus clathrin, dynamin-2 and Cdc42 silencing).
Thus, the effects of silencing of endocytic pathways on Abca1 abundance in cholesterol-depleted cells were qualitatively similar to those in cells with unmanipulated cholesterol content. However, a paradoxical reduction of cholesterol efflux after silencing of Arf6, dynamin-2 and Cdc42 suggested that in cholesterol-depleted cells involvement of endocytic pathways in cholesterol regulation may go beyond regulation of ABCA1 abundance.
Efflux of intracellular cholesterol
Inconsistencies in the effects of inhibition of endocytic pathways on ABCA1 abundance and cholesterol efflux suggested that the involvement of these pathways may not be limited to regulation of ABCA1 abundance. In addition to regulating the abundance and localization of ABCA1, endocytosis may also be involved in the efflux of intracellular cholesterol. That may be achieved through retroendocytosis, when ABCA1-apoA-I complex cycles between intracellular compartments and plasma membrane contributing to cholesterol trafficking inside the cell. Recently, it was demonstrated that ABCA1 is essential for transfer of plasma membrane cholesterol to the endoplasmic reticulum and that clathrin-independent endocytic pathways are involved 2. We investigated the effect of silencing of selected endocytic pathways on the efflux of the newly synthesized, intracellular cholesterol. In these experiments, labelling of newly synthesized cholesterol by incorporation of [3H]acetate was combined with the efflux incubation, and a proportion of newly synthesized [3H]cholesterol released to apoA-I was assessed. Loading of cells with cholesterol had no effect on the rate of cholesterol biosynthesis (Fig. 4A), however, cholesterol depletion resulted in significant elevation of the rate of cholesterol biosynthesis (Fig. 4A). In cells with unmanipulated cholesterol content, silencing of any of the endocytic pathways (clathrin-, dynamin-2-, Arf6-, and Cdc42-dependent) reduced the efflux of intracellular cholesterol by up to 80% (Fig. 4B). Interestingly, while there was no significant difference between the effects of silencing of clathrin, dynamin-2 and Cdc42, silencing of Arf6 was less effective in reducing intracellular cholesterol efflux. When cells were loaded with cholesterol, silencing of any of the pathways had no effect on the efflux of intracellular cholesterol (Fig. 4C), arguing against the contribution of endocytic pathways to the efflux of intracellular cholesterol in cholesterol-loaded cells. In cholesterol-depleted cells, however, while silencing of clathrin, dynamin and Cdc42 reduced the efflux of intracellular cholesterol, silencing of Arf6 caused 15-fold increase of intracellular cholesterol efflux (Fig. 4D). It appears that under the condition of unmanipulated cholesterol content, but especially in cholesterol-depleted cells, silencing of Arf6 prevented trafficking of Abca1 into lysosomes, reducing its degradation and instead redirecting it back to the cell surface, at the same time facilitating the efflux of intracellular cholesterol.
Intracellular itinerary of ABCA1
Collectively, our findings suggest that ARF6 plays a key role in endocytosis leading to degradation of ABCA1 while other endocytic pathways may be involved in ABCA1 recycling. Therefore, we investigated intracellular itinerary of ABCA1 hypothesizing that inhibition of ARF6 would redirect ABCA1 from a degradative to a recycling pathway. We used confocal microscopy to assess co-localization of Abca1 with lysosome marker Lamp1 and recycling endosome marker Rab11 27. Under condition of unmanipulated cholesterol content silencing of dynamin-2 and Arf6 almost halved co-localization of Abca1 with lysosome marker Lamp1 (Fig. 5A, C) at the same time increasing by 50% co-localization with Rab11 (Fig. 5B, D). In cholesterol-loaded cells, there was no statistically significant impact of the dynamin-2 or Arf6 silencing on co-localization of Abca1 with Lamp1 (Fig. 5E, G) or Rab11 (Fig. 5F, H). In cholesterol-depleted cells, silencing of Arf6, but not of dynamin-2, reduced co-localization of Abca1 with Lamp1 by 25% (Fig. 5I, K) with no statistically significant effect on co-localization of Abca1 and Rab11 (Fig. 5J, L). These findings were consistent with the outcomes of pulse-chase experiments (Figs. 1D, 2E, 3E) suggesting that silencing of ARF6 reduces endocytosis leading to degradation of ABCA1 under unmanipulated and cholesterol-depleted conditions.
Figure 5. The effect of silencing of Arf6 endocytic pathway on intracellular localization of Abca1.

A – Confocal microscopy analysis of co-localization of Abca1 and Lamp1 in cells with unmanipulated cholesterol content after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. B – Confocal microscopy analysis of co-localization of Abca1 and Rab11 in cells with unmanipulated cholesterol content after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. C -Quantitation of co-localization of Abca1 and Lamp1 (Means±SEM for 50–70 cells for each bar). ***p<0.001. D -Quantitation of co-localization of Abca1 and Rab11 (Means±SEM for 50–70 cells for each bar). *p<0.05. E – Confocal microscopy analysis of co-localization of Abca1 and Lamp1 in cholesterol-loaded cells after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. F – Confocal microscopy analysis of co-localization of Abca1 and Rab11 in cholesterol-loaded cells after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. G -Quantitation of co-localization of Abca1 and Lamp1 (Means±SEM for 50–70 cells for each bar). G -Quantitation of co-localization of Abca1 and Lamp1 (Means±SEM for 50–70 cells for each bar). H -Quantitation of co-localization of Abca1 and Rab11 (Means±SEM for 50–70 cells for each bar). I – Confocal microscopy analysis of co-localization of Abca1 and Lamp1 in cholesterol-depleted cells after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. J – Confocal microscopy analysis of co-localization of Abca1 and Rab11 in cholesterol-depleted cells after silencing of dynamin-2 (Dy) or Arf6 (Ar). Bar – 10 μm. K -Quantitation of co-localization of Abca1 and Lamp1 (Means±SEM for 50–70 cells for each bar). ***p<0.001. L -quantitation of co-localization of Abca1 and Rab11 (Means±SEM for 50–70 cells for each bar).
We also investigated the effect of Arf6 overexpression on degradation of Abca1 by assessing co-localization of Abca1 with Lamp1 (Supplemental Fig. V - note that colour of Abca1 was digitally changed to green for better visualization of co-localization). Consistent with the measurements of Abca1 abundance, Abca1/Lamp1 co-localization was increased in cells with unmanipulated cholesterol content overexpressing Arf6 (Supplemental Fig. VA, D). In cholesterol-loaded cells Abca1/Lamp1 co-localization was reduced relative to cells with unmanipulated cholesterol content and there was no effect of Arf6 overexpression on Abca1/Lamp1 co-localization (Supplemental Fig. VB, D). In cholesterol-depleted cells Abca1/Lamp1 co-localization was increased and there was a reduction in Abca1/Lamp1 co-localization in cells overexpressing Arf6 (Supplemental Fig. VC, D). It appears that increased abundance of Arf6 regulates Abca1 degradation in cells with unmanipulated cholesterol content and in cholesterol-depleted cells, but plays limited role in cholesterol-loaded cells.
Combined, our findings are consistent with the hypothesis that inhibition of Arf6-dependent degradative endocytic pathway may re-direct Abca1 toward a recycling endocytic pathway, at least in cells with unmanipulated cholesterol content.
ARF6 regulates endocytosis of ABCA1 in bone marrow derived macrophages
Finally, to confirm the main findings of this study in primary cells, we tested the effects of Arf6 silencing in bone marrow derived macrophages (BMDM). Abca1 abundance in these cells was significantly higher compared to RAW 264.7 cells; therefore, the experiments with BMDM were done without stimulation of Abca1 expression with an LXR agonist. Transfection of BMDM with siRNAAr resulted in approximately 50% reduction in Arf6 abundance in cells with unmanipulated cholesterol content, as well as in cholesterol-loaded and cholesterol-depleted cells (Supplemental Fig. VIA). Silencing of Arf6 caused approximately a 20% increase of Abca1/αNa/K-ATPase ratio under all three conditions (Supplemental Fig. VIA). Silencing of Arf6 also caused significant increase in the abundance of cell-surface Abca1, especially in cholesterol-depleted cells (Supplemental Fig. VIB). Consistent with changes in the abundance of cell-surface Abca1, cholesterol efflux was significantly stimulated by Arf6 silencing (Supplemental Fig. VIC). An inconsistency in the effects of Arf6 silencing on cholesterol efflux from cholesterol-depleted BMDM and RAW 264.7 cells is most likely the result of much higher abundance of total and cell-surface Abca1 in BMDM.
Discussion
Endocytosis of the receptors on the surface of the cells, including lipoprotein receptors, is a well-established phenomenon 27. Such endocytosis serves, on the one hand, to deliver receptor ligands or cargo inside the cells, and on the other hand, to regulate receptor abundance and exposure on the cell surface. Numerous evidence suggest that ABCA1, a key transporter in cholesterol efflux pathway, is also a subject of endocytosis 16, 21, 31, 32, but the mechanisms and physiological role of endocytosis of ABCA1 are unclear.
One suggested role of ABCA1 endocytosis is regulation of ABCA1 abundance and its exposure at the cell surface. According to this hypothesis, one may expect that inhibition of a pathway responsible for ABCA1 endocytosis and degradation would increase ABCA1 abundance and exposure at the cell surface as well as cholesterol efflux. Indeed, in this study we demonstrated that silencing of dynamin-2 and ARF6 in cells with unmanipulated cholesterol content increased the abundance of total and cell-surface ABCA1, reduced internalization and degradation of ABCA1 and increased cholesterol efflux. Silencing of the key elements of the two other pathways, clathrin-dependent pathway and CDC42-dependent pathway, had no effect on ABCA1 abundance, localization and function. Caveolin-dependent endocytic pathway was not directly investigated in this study. However, treatment of cells with MBC, which reduced the abundance of lipid rafts inhibiting caveolin-dependent pathway, resulted in a decreased, rather than an increased, abundance of ABCA1, suggesting that caveolin-dependent endocytosis is unlikely to contribute significantly to the regulation of ABCA1 abundance, at least in macrophages. Our findings are consistent with a suggestion that endocytic pathway involving dynamin-2 and ARF6 plays an important role in determining the rate of ABCA1 endocytosis with subsequent lysosomal degradation in cells with unmanipulated cholesterol content.
The involvement of the ARF6-dependent endocytosis in the regulation of ABCA1 abundance, localization and function became equivocal when cell cholesterol content was altered. In cholesterol-loaded cells silencing of ARF6 had only a limited effect on ABCA1 abundance, localization and degradation, presumably reflecting limited role of ABCA1 degradation in cells overloaded with cholesterol. Several pathways of ABCA1 degradation were described, some of them only become active in response to loading of cells with cholesterol 11, 12, and we speculate that cholesterol loading switches ABCA1 degradation away from ARF6-dependent endocytosis. The effects of silencing of endocytic pathways on ABCA1abundance in cholesterol-depleted cells were generally similar to those in cells with unmanipulated cholesterol content. It was therefore surprising that cholesterol efflux, although overall reduced by cholesterol depletion, was further reduced by silencing of clathrin-independent endocytic pathways and also after overexpression of ARF6, conflicting with the effects of these treatments on the abundance of total and cell-surface ABCA1. It is possible that clathrin-independent endocytic pathways are involved in delivery of cholesterol from plasma membrane to intracellular compartments, as was recently suggested by Yamauchi et al 2 reducing availability of cholesterol for the efflux. However, if this were the case, inhibition of clathrin-independent endocytosis (CIE) would result in relative enrichment of plasma membrane with cholesterol and increased cholesterol efflux, which was not observed in our experiments. We therefore suggest that the effects of modulating endocytic pathways on cholesterol efflux in cholesterol-depleted cells may affect the efflux not only from the depleted pool of plasma membrane cholesterol, but also from the pool of intracellular cholesterol.
Another suggested role of ABCA1 endocytosis was its participation in intracellular cholesterol trafficking. Two mechanisms were previously proposed. First, it was suggested that after binding to apoA-I, ABCA1-apoA-I complex may be endocytosed, bind intracellular lipids and recycle back to the cell surface releasing apoA-I along with associated lipids into extracellular space 31, 33–35. Second, it was suggested that ABCA1 in the absence of extracellular acceptors may facilitate transfer of cholesterol from the plasma membrane to endoplasmic reticulum through CIE and that ABCA1 abundance affects the efficiency of certain CIE pathways 2, which may in turn affect the egress of cholesterol from intracellular compartments. Involvement of ABCA1 in trafficking of intracellular cholesterol to the plasma membrane is supported by the recent finding that newly synthesized sterols are a preferential substrate for ABCA1-dependent efflux and that ABCA1 and newly synthesized sterols selectively accumulate in the same plasma membrane domain, rafts 36. When we investigated the involvement of endocytic pathways in efflux of specifically intracellular cholesterol, we found that it was significantly reduced by inhibition of both clathrin-dependent and clathrin-indepenedent pathways in cells with unmanipulated cholesterol content. However, inhibition of ARF6 was the least efficient; moreover, localization of the intracellular ABCA1 shifted from the lysosomes to the recycling endosomes after silencing of ARF6. In cholesterol-depleted cells, where cholesterol efflux was low, silencing of ARF6 resulted in a dramatic elevation of the rate of intracellular cholesterol efflux. Collectively, these findings suggest that inhibition of ARF6 endocitic pathway may redirect ABCA1 into alternative endocytic pathways involved in ABCA1 recycling and trafficking of intracellular cholesterol.
Overall, based on our findings, we suggest the following model of involvement of different endocytic pathways in regulation of ABCA1 abundance and function (Fig. 6). We propose that ARF6-dependent pathway is responsible for endocytosis of ABCA1, directing it into late endosomes with subsequent degradation. This pathway regulates the abundance of ABCA1 on plasma membrane and consequently the efflux of plasma membrane cholesterol. Other clathrin-dependent and clathrin-independent pathways may be responsible for the recycling of ABCA1; they are not involved in regulation of ABCA1 abundance, but may be involved in the efflux of intracellular cholesterol. In cholesterol-loaded cells, the contribution of the endocytic pathways to the regulation of the ABCA1 abundance and function is reduced, leaving ABCA1 on the cell surface to maximize cholesterol efflux. In cholesterol-depleted cells, a significant proportion of the effluxed cholesterol comes from intracellular compartments and cholesterol efflux is predominantly affected by the rate of ABCA1 recycling rather than by its abundance on plasma membrane. Endocytic pathways may compete with each other, and inhibition of ARF6-dependent degradative pathway may redirect ABCA1 into recycling pathways leading to increased intracellular cholesterol efflux at the expense of plasma membrane cholesterol.
Figure 6. Schematic representation of the proposed involvement of endocytic pathways in the regulation of ABCA1 endocytosis.
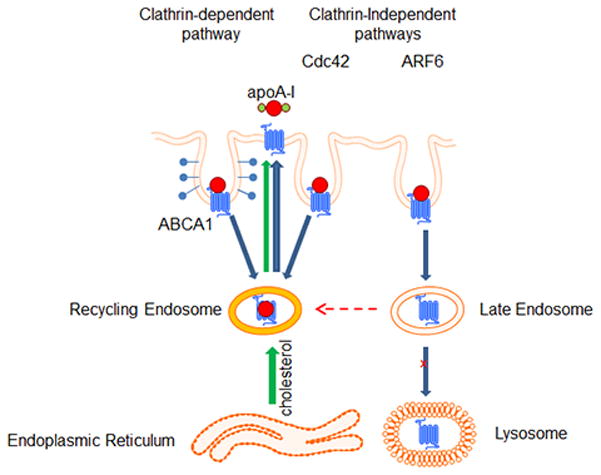
ARF6-dependent pathway is responsible for endocytosis of ABCA1 directing it into late endosomes followed by degradation of ABCA1. This pathway is responsible for regulation of the abundance of ABCA1 on cell surface. Other clathrin-dependent and clathrin-indepedndent pathways may be responsible for the recycling of ABCA1; they are not involved in regulation of ABCA1 abundance, but may be involved in the efflux of intracellular cholesterol. Endocytic pathways compete with each other and inhibition of ARF6 (degradation) pathway may re-direct ABCA1 into recycling pathways leading to increased activity of intracellular cholesterol efflux.
In conclusion, our findings demonstrate the critical role of ARF6 in regulation of ABCA1 abundance and function. However, further studies are required to test the proposed model of endocytic pathway interactions.
Supplementary Material
Highlights.
ARF6-dedpendent endocytosis is the predominant pathway of ABCA1 degradation
ARF6-independent pathways control intracellular cholesterol efflux
ABCA1 endocytosis may shift between degradation and recycling
Acknowledgments
Photomicrographs were collected with equipment provided by Monash micro imaging facility.
Sources of Funding
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) (GNT1036352) and in part by the Victorian Government’s OIS Program.
Abbreviations
- ABCA1
ATP binding cassette transporter A1
- apoA-I
apolipoprotein A-I
- BMDM
bone marrow derived macrophages
- CIE
clathrin-independent endocytosis
- HDL
high density lipoprotein
- LXR
Liver X Receptor
- MBC
methyl-β-cyclodextrin
- RCT
reverse cholesterol transport
Footnotes
Disclosures
None
References
- 1.Oram JF. HDL Apolipoproteins and ABCA1: Partners in the Removal of Excess Cellular Cholesterol. Arterioscler Thromb Vasc Biol. 2003;23:720–727. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- 2.Yamauchi Y, Iwamoto N, Rogers MA, Abe-Dohmae S, Fujimoto T, Chang CCY, Ishigami M, Kishimoto T, Kobayashi T, Ueda K, Furukawa K, Chang T-Y, Yokoyama S. Deficiency in the Lipid Exporter ABCA1 Impairs Retrograde Sterol Movement and Disrupts Sterol Sensing at the Endoplasmic Reticulum. J Biol Chem. 2015;290:23464–23477. doi: 10.1074/jbc.M115.662668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attie AD, Kastelein JP, Hayden MR. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res. 2001;42:1717–1726. [PubMed] [Google Scholar]
- 4.Singaraja RR, Brunham LR, Visscher H, Kastelein JJP, Hayden MR. Efflux and Atherosclerosis: The Clinical and Biochemical Impact of Variations in the ABCA1 Gene. Arterioscler Thromb Vasc Biol. 2003;23:1322–1332. doi: 10.1161/01.ATV.0000078520.89539.77. [DOI] [PubMed] [Google Scholar]
- 5.von Eckardstein A, Sibler RA. Possible contributions of lipoproteins and cholesterol to the pathogenesis of diabetes mellitus type 2. Curr Opin Lipidol. 2011;22:26–32. doi: 10.1097/MOL.0b013e3283412279. [DOI] [PubMed] [Google Scholar]
- 6.Koldamova R, Fitz NF, Lefterov I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim Biophys Acta. 2010;1801:824–830. doi: 10.1016/j.bbalip.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith B, Land H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012;2:580–590. doi: 10.1016/j.celrep.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DJ, Cooke AJ, Edwards AS. Non-steroidal LXR agonists; an emerging therapeutic strategy for the treatment of atherosclerosis. Recent Pat Cardiovasc Drug Discov. 2006;1:21–46. doi: 10.2174/157489006775244245. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Hernando C, Moore KJ. MicroRNA Modulation of Cholesterol Homeostasis. Arterioscler Thromb Vasc Biol. 2011;31:2378–2382. doi: 10.1161/ATVBAHA.111.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang N, Tall AR. Regulation and Mechanisms of ATP-Binding Cassette Transporter A1-Mediated Cellular Cholesterol Efflux. Arterioscler Thromb Vasc Biol. 2003;23:1178–1184. doi: 10.1161/01.ATV.0000075912.83860.26. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh V, Kim M-J, Gelissen IC, Brown AJ, Sandoval C, Hallab JC, Kockx M, Traini M, Jessup W, Kritharides L. Cellular Cholesterol Regulates Ubiquitination and Degradation of the Cholesterol Export Proteins ABCA1 and ABCG1. J Biol Chem. 2014;289:7524–7536. doi: 10.1074/jbc.M113.515890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizuno T, Hayashi H, Kusuhara H. Cellular Cholesterol Accumulation Facilitates Ubiquitination and Lysosomal Degradation of Cell Surface–Resident ABCA1. Arterioscler Thromb Vasc Biol. 2015;35:1347–1356. doi: 10.1161/ATVBAHA.114.305182. [DOI] [PubMed] [Google Scholar]
- 13.Roosbeek S, Peelman F, Verhee A, Labeur C, Caster H, Lensink MF, Cirulli C, Grooten J, Cochet C, Vandekerckhove J, Amoresano A, Chimini G, Tavernier J, Rosseneu M. Phosphorylation by Protein Kinase CK2 Modulates the Activity of the ATP Binding Cassette A1 Transporter. J Biol Chem. 2004;279:37779–37788. doi: 10.1074/jbc.M401821200. [DOI] [PubMed] [Google Scholar]
- 14.Fu Y, Mukhamedova N, Ip S, D’Souza W, Henley Katya J, DiTommaso T, Kesani R, Ditiatkovski M, Jones L, Lane Rachael M, Jennings G, Smyth Ian M, Kile Benjamin T, Sviridov D. ABCA12 Regulates ABCA1-Dependent Cholesterol Efflux from Macrophages and the Development of Atherosclerosis. Cell Metabolism. 2013;18:225–238. doi: 10.1016/j.cmet.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Hozoji-Inada M, Munehira Y, Nagao K, Kioka N, Ueda K. Liver X Receptor beta (LXRbeta) Interacts Directly with ATP-binding Cassette A1 (ABCA1) to Promote High Density Lipoprotein Formation during Acute Cholesterol Accumulation. J Biol Chem. 2011;286:20117–20124. doi: 10.1074/jbc.M111.235846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu R, Arakawa R, Ito-Osumi C, Iwamoto N, Yokoyama S. ApoA-I Facilitates ABCA1 Recycle/Accumulation to Cell Surface by Inhibiting Its Intracellular Degradation and Increases HDL Generation. Arterioscler Thromb Vasc Biol. 2008;28:1820–1824. doi: 10.1161/ATVBAHA.108.169482. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein AI involves endocytosis and resecretion in a calcium-dependent pathway [see comments] Proc Natl Acad Sci U S A. 1999;96:11358–11363. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui HL, Guo B, Scicluna B, Coleman BM, Lawson VA, Ellett L, Meikle PJ, Bukrinsky M, Mukhamedova N, Sviridov D, Hill AF. Prion Infection Impairs Cholesterol Metabolism in Neuronal Cells. J Biol Chem. 2014;289:789–802. doi: 10.1074/jbc.M113.535807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui HL, Grant A, Mukhamedova N, Pushkarsky T, Jennelle L, Dubrovsky L, Gaus K, Fitzgerald ML, Sviridov D, Bukrinsky M. HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J Lipid Res. 2012;53:696–708. doi: 10.1194/jlr.M023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azuma Y, Takada M, Shin HW, Kioka N, Nakayama K, Ueda K. Retroendocytosis pathway of ABCA1/apoA-I contributes to HDL formation. Genes Cells. 2009;14:191–204. doi: 10.1111/j.1365-2443.2008.01261.x. [DOI] [PubMed] [Google Scholar]
- 21.Hassan HH, Bailey D, Lee D-YD, Iatan I, Hafiane A, Ruel I, Krimbou L, Genest J. Quantitative Analysis of ABCA1-dependent Compartmentalization and Trafficking of Apolipoprotein A-I: IMPLICATIONS FOR DETERMINING CELLULAR KINETICS OF NASCENT HIGH DENSITY LIPOPROTEIN BIOGENESIS. J Biol Chem. 2008;283:11164–11175. doi: 10.1074/jbc.M707720200. [DOI] [PubMed] [Google Scholar]
- 22.Denis M, Landry YD, Zha X. ATP-binding Cassette A1-mediated Lipidation of Apolipoprotein A-I Occurs at the Plasma Membrane and Not in the Endocytic Compartments. J Biol Chem. 2008;283:16178–16186. doi: 10.1074/jbc.M709597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faulkner LE, Panagotopulos SE, Johnson JD, Woollett LA, Hui DY, Witting SR, Maiorano JN, Davidson WS. An analysis of the role of a retroendocytosis pathway in ABCA1-mediated cholesterol efflux from macrophages. J Lipid Res. 2008;49:1322–1332. doi: 10.1194/jlr.M800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kambouris AM, Roach PD, Calvert GD, Nestel PJ. Retroendocytosis of high density lipoproteins by the human hepatoma cell line, HepG2. Arteriosclerosis. 1990;10:582–590. doi: 10.1161/01.atv.10.4.582. [DOI] [PubMed] [Google Scholar]
- 25.Cavelier C, Rohrer L, von Eckardstein A. ATP-Binding Cassette Transporter A1 Modulates Apolipoprotein A-I Transcytosis Through Aortic Endothelial Cells. Circ Res. 2006;99:1060–1066. doi: 10.1161/01.RES.0000250567.17569.b3. [DOI] [PubMed] [Google Scholar]
- 26.Doherty GJ, McMahon HT. Mechanisms of Endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 27.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada R, Yamauchi Y, Hongu T, Funakoshi Y, Ohbayashi N, Hasegawa H, Kanaho Y. Activation of the Small G Protein Arf6 by Dynamin2 through Guanine Nucleotide Exchange Factors in Endocytosis. Sci Rep. 2015;5:14919. doi: 10.1038/srep14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu SM, Cogny A, Kockx M, Dean RT, Gaus K, Jessup W, Kritharides L. Cyclodextrins differentially mobilize free and esterified cholesterol from primary human foam cell macrophages. J Lipid Res. 2003;44:1156–1166. doi: 10.1194/jlr.M200464-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein AI. J Lipid Res. 2004;45:635–644. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Yokoyama S, Arakawa R, Wu C-a, Iwamoto N, Lu R, Tsujita M, Abe-Dohmae S. Calpain-mediated ABCA1 degradation: Post-translational regulation of ABCA1 for HDL biogenesis. Biochim Biophys Acta. 2012;1821:547–551. doi: 10.1016/j.bbalip.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Yamauchi Y, Chang CCY, Hayashi M, Abe-Dohmae S, Reid PC, Chang T-Y, Yokoyama S. Intracellular cholesterol mobilization involved in the ABCA1/apolipoprotein-mediated assembly of high density lipoprotein in fibroblasts. J Lipid Res. 2004;45:1943–1951. doi: 10.1194/jlr.M400264-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Neufeld EB, Stonik JA, Demosky SJ, Jr, Knapper CL, Combs CA, Cooney A, Comly M, Dwyer N, Blanchette-Mackie J, Remaley AT, Santamarina-Fojo S, Brewer HB., Jr The ABCA1 Transporter Modulates Late Endocytic Trafficking: INSIGHTS FROM THE CORRECTION OF THE GENETIC DEFECT IN TANGIER DISEASE. J Biol Chem. 2004;279:15571–15578. doi: 10.1074/jbc.M314160200. [DOI] [PubMed] [Google Scholar]
- 35.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB., Jr Cellular Localization and Trafficking of the Human ABCA1 Transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 36.Yamauchi Y, Yokoyama S, Chang T-Y. ABCA1-dependent sterol release: sterol molecule specificity and potential membrane domain for HDL biogenesis. J Lipid Res. 2016;57:77–88. doi: 10.1194/jlr.M063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.