

SUMMARY
New genetic tools are needed to understand the functional interactions between HIV and human host factors in primary cells. We recently developed a method to edit the genome of primary CD4+ T cells by electroporation of CRISPR/Cas9 ribonucleoproteins (RNPs). Here, we adapted this methodology to a high-throughput platform for the efficient, arrayed editing of candidate host factors. CXCR4 or CCR5 knock-out cells generated with this method are resistant to HIV infection in a tropism-dependent manner, whereas knock-out of LEDGF or TNPO3 results in a tropism-independent reduction in infection. CRISPR/Cas9 RNPs can furthermore edit multiple genes simultaneously, enabling studies of interactions among multiple host and viral factors. Finally, in an arrayed screen of 45 genes associated with HIV integrase, we identified several candidate dependency/restriction factors, demonstrating the power of this approach as a discovery platform. This technology should accelerate target validation for pharmaceutical and cell-based therapies to cure HIV infection.
Graphical abstract
INTRODUCTION
Despite extraordinary progress in the development and distribution of antiretroviral drugs, Human Immunodeficiency Virus (HIV) remains a worldwide health threat, infecting millions of new people each year. Even with strict adherence to a therapeutic regimen, patients remain chronically infected with the virus and thus require lifelong treatment (Finzi et al., 1997; Siliciano et al., 2003; Wong et al., 1997). To date, a complete cure has been achieved in only a single person, the “Berlin patient.” In this case, the virus was eradicated by allogeneic, hematopoietic stem cell transplantation from a donor with a natural genetic variant in the CCR5 gene that prevented HIV entry into these cells (Allers et al., 2011; Hutter et al., 2009). This success has motivated ongoing efforts to engineer human immune cells that lack host factors required for HIV pathogenesis as a means to achieve a permanent cure (Baltimore, 1988; Deeks and McCune, 2010; Leibman and Riley, 2015).
Several clinical trials are currently underway using zinc-finger nucleases (ZFNs) to delete the HIV co-receptors CXCR4 and CCR5 to generate immune cells that are resistant to HIV infection in a manner similar to the Berlin patient (Didigu et al., 2014; Hutter et al., 2009; Tebas et al., 2014). These approaches generally rely on viral-based delivery of a ZFN-expression cassette to generate HIV resistant T cells or hematopoietic stem cells ex vivo (Maier et al., 2013; Perez et al., 2008; Wilen et al., 2011; Yi et al., 2014; Yuan et al., 2012). Autologous transplantation can then be used to repopulate a resistant T cell population while antiretroviral therapies and natural immune responses clear the remaining infection (Baltimore, 1988; Deeks and McCune, 2010; Didigu et al., 2014; DiGiusto et al., 2010; Holt et al., 2010; Tebas et al., 2014). While these represent potentially viable approaches, the use of viral delivery and the degree of off-target editing that may occur over the course of long-term ZFN expression raises concerns in bringing such a treatment to the clinic (Gabriel et al., 2011; Pattanayak et al., 2011; Thomas et al., 2003).
The advent of CRISPR/Cas9 genome editing has revolutionized our ability to surgically modify the genomes of human cells, but efficient delivery of Cas9 to primary T cells has been a major challenge (Doudna and Charpentier, 2014; Hsu et al., 2014; Mandal et al., 2014; Ran et al., 2013). Recently, we reported that we can overcome this challenge through electroporation of Cas9 ribonucleoproteins (RNPs) directly into primary human CD4+ T cells isolated from the peripheral blood (Schumann et al., 2015). This transient delivery of editing Cas9 RNPs enables high efficiency “knock-out” and “knock-in” genome editing and could provide a high-throughput method for therapeutic engineering of HIV-resistant human T cells. This approach would have several benefits over the traditional methodologies currently in trial as it does not rely on viral delivery, does not involve long-term expression off a nucleic acid cassette, and has low rates of off-target editing (Kim et al., 2014; Schumann et al., 2015). As Cas9 technology is further developed, the efficiency and off-target rate should improve, making these advantages even more stark (Doench et al., 2016; Fu et al., 2014; Kleinstiver et al., 2016; Slaymaker et al., 2016).
Beyond CXCR4 and CCR5, other human host factors can affect HIV pathogenesis at different stages of viral life cycle (Brass et al., 2008; Goff, 2007; Konig et al., 2008; Zhou et al., 2008). However functional studies of these factors have been limited by significant technical challenges in primary cell types and a subsequent reliance on RNA interference (RNAi) and immortalized cell line models (Pache et al., 2011). The limitations of these systems underscore the need for improved technology to knock-out specific gene sequences in primary human cells in a manner that is simple, scalable, reproducible, and efficient. Systematic validation of host genes that act as HIV dependency factors could unveil new targets for therapeutic intervention, either when targeted alone or in combination (Didigu et al., 2014; Voit et al., 2013).
Here, we report a high-throughput platform for the efficient editing of host factors that control HIV infection in primary human T cells. Arrayed delivery of Cas9 RNPs permits the rapid generation of isogenic T cells with ablated candidate factors for ex vivo interrogation and investigation. Using this platform, we disrupted the HIV co-receptors CXCR4 or CCR5 in multiple donors and reproducibly generated cells resistant to infection in a tropism-dependent manner. Targeting other dependency factors that act after viral entry, including LEDGF or TNPO3, resulted in blocks to infection independent of tropism. Targeting CXCR4 and CD4 simultaneously, we demonstrated that this platform also supports multiplex gene editing and can be used to generate double knock-out cells at no cost to the efficiency of the individual Cas9 RNPs. Finally, we used our platform to screen 146 unique Cas9 RNPs targeting 45 genes previously shown to interact with HIV integrase, identifying novel host dependency factors. These studies collectively demonstrate the utility of Cas9 RNP T cell editing as an efficient means to validate host-dependency factors in the context of viral infection and generate HIV-resistant primary human T cells for scientific and potentially therapeutic use.
RESULTS
A Platform for Editing HIV Host Factors in Primary Human T Cells
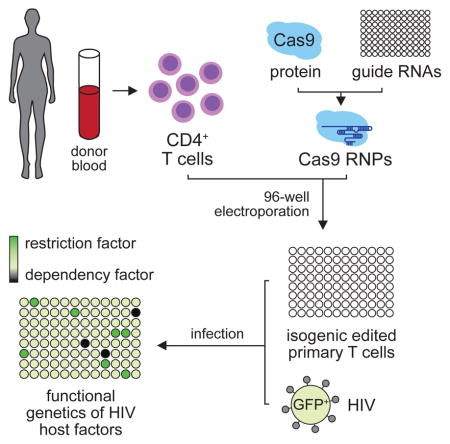
Based on our previous success editing primary human T cells via direct electroporation of Cas9 RNPs (Schumann et al., 2015), we established a high-throughput platform for gene editing that is fast, robust, efficient, and readily adaptable to a variety of downstream applications. Unique Cas9 RNPs were synthesized in 96-well format by complexing Cas9 recombinant protein with chemically synthesized CRISPR targeting RNAs (crRNAs) and trans-activating crRNA (tracrRNA) in vitro. These were subsequently delivered to primary human CD4+ T cells by 96-well plate electroporation, allowing for the rapid generation of hundreds of unique, genetically modified pools from a single blood donor (Figure 1). The protocols described here constitute significant improvements to our previous methodology in not only scalability, but also in enhanced efficiency, and improved availability, all reagents being commercially accessible. This platform therefore represents a valuable tool for the interrogation of genetic factors involved in a myriad of human immune cell processes including T cell signaling, differentiation, and infection by diverse pathogenic agents.
Figure 1.

Procedural overview of the multiplex platform for editing HIV host factors in primary human CD4+ T cells. CD4+ T cells are isolated from donor blood by negative selection and activated with plate-bound anti-CD3 and soluble anti-CD28. Cas9 RNPs are formed in vitro by incubating Cas9 protein with the appropriate crRNAs and tracrRNAs. These are electroporated into the activated T cells in a 96-well format to generate a polyclonal pool of homozygous edited (dark blue), heterozygous (light blue), and unedited (white) cells. These cells are activated again and expanded. Editing is verified at the DNA level by TIDE analysis or Sanger sequencing and at the protein level by immunostaining or immunoblot. These cells can then be infected with the appropriate viral strain(s) and infection monitored by flow cytometry either of an integrated GFP reporter or after staining for intracellular p24. See also Supplementary Table 1.
To test the efficacy of this platform for biological investigation, we targeted a series of HIV host factors for genetic modification. HIV infection has been thoroughly studied in a wide array of biological model systems, but primary cell data in a clean genetic system is difficult to obtain and remains a gold standard in host factor investigation. This is further complicated by variability not only observed between donors but between investigators depending on the methodology used for T cell stimulation, infection, and analysis. To account for this variability, we decided to test our platform across multiple loci, in multiple donors, with an array of different stimulation and infection protocols.
Briefly, CD4+ T cells were purified from human blood and activated with anti-CD3/CD28 stimulation. As described above, cells were electroporated with Cas9 RNPs and genome editing was validated at both the DNA and protein levels. The edited cell populations were then subjected to functional testing with various ex vivo HIV infection assays using both single-cycle reporter viruses as well as full molecular clones (Figure 1). Provided that this method is efficient and specific, deletion of bona fide host dependency factors is predicted to measurably reduce HIV infectivity compared to control electroporation treatments.
Cas9 RNP Ablation of HIV Co-Receptors Blocks Infection
HIV Envelope (Env) must bind to CD4 and subsequently one of two co-receptors, CXCR4 or CCR5, on the host cell to mediate membrane fusion and trigger successful entry (Alkhatib et al., 1996; Berson et al., 1996; Deng et al., 1996; Feng et al., 1996; Wu et al., 1996). The chemokine receptor CCR5 is predominantly used in vivo, but once an infection is established, viral variants that use the co-receptor CXCR4 commonly evolve (Connor et al., 1997; Moore et al., 2004). Several polymorphisms in the CCR5 gene have been linked with limited disease progression and even resistance to viral infection (Allers et al., 2011; Hutter et al., 2009; Liu et al., 1996; Samson et al., 1996). Targeted inhibition of CCR5 either genetically or chemically has proven to be a viable therapeutic route in vivo, and there is much interest in furthering and expanding such co-receptor based therapies (Allers et al., 2011; Didigu et al., 2014; Gulick et al., 2008; Holt et al., 2010; Hutter et al., 2009; Tebas et al., 2014). We used our platform to efficiently and specifically knock-out CXCR4 or CCR5, and measured the impact of this editing on co-receptor expression and HIV infectivity across multiple donors.
We previously demonstrated editing of CXCR4 in primary T cells (Schumann et al., 2015). Here we aimed to test the efficacy of this crRNA with our new platform across multiple donors and to measure the impact of these changes on HIV infection. We first confirmed that CXCR4 could be reproducibly and efficiently disrupted in multiple different human blood donors with the modified 96-well plate delivery format. The percent of CXCR4 positive cells decreased from roughly 90% in the unmodified and Cas9 protein only control populations to roughly 30–40% in the populations treated with CXCR4-targeting Cas9 RNPs in multiple independent donors (Figures 2A). Editing of the DNA was confirmed by Tracking of Indels by Decomposition (TIDE) analysis (Supplementary Table 1). Edited cells retained normal cell surface levels of CD4 and appropriately induced CD25 in response to stimulation with anti-CD2/CD3/CD28 beads (Figure 2B). The same holds true when we stimulate with soluble anti-CD3/CD28 or with PHA/IL-2 (data not shown). After stimulation, we challenged these cells with CXCR4-tropic HIV-1LAI, which includes a GFP marker in the place of nef, or an identical virus lacking native Env expression, but pseudotyped with pan-tropic VSV-G. Cells were co-stained for CXCR4 surface expression and infection rates quantified by flow cytometry. Unmodified cells and Cas9 control treated (without crRNA or tracrRNA) cells showed no difference in CXCR4 expression or infection with either virus (Figures 2B,C). CXCR4 knock-out cells, however, showed a substantial decrease in CXCR4+ cells and a concurrent decrease in infection with the CXCR4-tropic virus. Importantly, most of the cells in this population that did become infected still expressed high levels of CXCR4. On the other hand, CXCR4 knock-out cells showed no difference in infection with the VSV-G pseudotyped virus as compared to the controls (Figures 2B,C). This was consistently observed across all donors tested (Supplementary Figure 1). Therefore, these edited cells retained permissivity to HIV infection, but were rendered unsusceptible to specific viral strains in a tropism-dependent manner. These data demonstrate that treatment with Cas9 RNPs successfully ablated CXCR4 expression in human T cells across multiple donors and imposed a specific and targeted block to CXCR4-tropic HIV infection.
Figure 2.

Knock-out of HIV co-receptors CXCR4 and CCR5 confers resistance to infection in a tropism-dependent manner. (a) Primary T cells were isolated from three different donors and electroporated with either buffer alone, Cas9 protein alone, or Cas9 RNPs targeting CXCR4. Four days after electroporation, cells were stained with anti-CXCR4-APC in technical triplicate and analyzed by flow cytometry. (b) These cells were subsequently stained with anti-CD4-PE and anti-CXCR4-APC or anti-CD4-PE and anti-CD25-APC. Representative plots from one donor are shown above. (c) These cells were then infected with a CXCR4-tropic (LAI strain) GFP reporter virus or an identical VSV-G pseudotyped, Env-deficient virus in technical triplicate. After 36 hours, cells were co-stained with anti-CXCR4-APC and analyzed by flow cytometry. Representative plots from one donor are shown above. (d) Bar graphs depicting the average percent infected cells across technical triplicates +/− standard deviation, including both CXCR4− cells (red) and CXCR4+ cells (blue). Infections from one representative donor are shown with CXCR4-tropic virus on the left and VSV-G pseudotyped virus on the right. (e) Procedural schematic depicting the differences in stimulation/infection protocols for CXCR4 versus CCR5-tropic viruses. (f) Primary T cells were isolated from six different donors and electroporated with either buffer alone, Cas9 protein alone, or Cas9 RNPs targeting CCR5. Two days after electroporation, cells were stained with anti-CCR5-APC or for CD4-PE and CD25-APC and analyzed by flow cytometry. Representative plots from one donor are shown above. (g) These cells were then infected with a CCR5-tropic (JR-CSF strain) virus or a VSV-G pseudotyped reporter virus in technical triplicate. After 48 hours, cells were co-stained with p24-FITC and CCR5-APC before analysis by flow cytometry. The upper bar graph depicts the average percent CCR5+ cells (green), CCR5-tropic infected cells (blue), and VSV-G pseudotyped infected cells (red) across technical triplicates +/− standard deviation relative to the Cas9 control in one representative donor. The lower bar graph depicts the mean across six donors +/− standard error again relative to the Cas9 control. See also Supplementary Figures S1 and S2, and Supplementary Table S1.
While T cells uniformly express high levels of CXCR4, T cells in the peripheral blood generally express low to undetectable levels of CCR5 (Lee et al., 1999). By stimulating these cells with low levels of plate-bound anti-CD3 and soluble anti-CD28 antibodies, however, we were able to induce sufficient expression for infection and analysis (Figure 2D). We designed and tested three CCR5 targeting crRNAs and found that two of them successfully edited the CCR5 locus (Supplementary Table 1). While the percent CCR5+ cells achieved by our protocol varied slightly between donors, one of these crRNAs was able to routinely decrease this population by roughly 60–80% relative to controls (Figures 2E,F, Supplementary Figure 2). As with CXCR4 targeting, we saw no significant effects on CD4 expression or CD25 induction when targeting CCR5 (Figure 2F, G). Similarly, these cells showed resistance to HIV infection in a manner dependent on viral tropism. CCR5 targeted cells had lower rates of infection with the CCR5-tropic HIV-1 virus JR-CSF, whereas no significant difference was observed upon infection with a VSV-G pseudotyped, pan-tropic virus. This was observed across multiple independent donors (Supplementary Figure 2).
Altogether, these experiments demonstrate that this platform can be used to efficiently generate HIV-resistant edited primary cells by targeting specific host dependency factors. These cells respond normally to assorted stimuli and retain their permissivity to viral replication if a pathway of infection is present independent of the targeted host factor. More broadly, these data validate this platform as a means to study the effects of genetic perturbations in human T cells on HIV infection.
Validation of Candidate HIV Dependency Factors in Primary Human T Cells
We first demonstrated the utility of this platform to efficiently and reproducibly generate HIV-resistant primary T cells by targeted knock-out of CXCR4 and CCR5, host factors essential for virus entry. We next wanted to test the ability of this approach to interrogate host factors that act later in the viral lifecycle whose successful editing may also render cells resistant to early infection. Towards this end, we decided to target the chromatin-associated factor LEDGF/p75, a host co-factor shown to be important for HIV integration in several model systems (Ciuffi et al., 2005; Fadel et al., 2014; Llano et al., 2006a; Llano et al., 2004; Shun et al., 2007; Vandegraaff et al., 2006; Vandekerckhove et al., 2006). Knock-down experiments have been difficult to interpret due to residual functional activity of the gene (Llano et al., 2006b), but knock-out experiments in the mouse model and in cell lines have shown some effect (Fadel et al., 2014; Shun et al., 2007). While it is likely that there is some functional redundancy with other host factors, the use of LEDGF by the virus is thought to influence the integration site landscape and proviral latency (Gerard et al., 2015; Jordan et al., 2001; Llano et al., 2006a; Llano et al., 2004; Shun et al., 2007; Vandegraaff et al., 2006; Vandekerckhove et al., 2006). The inability to knockout LEDGF in primary T cells has greatly limited the study of this factor in ex vivo human systems.
To test the impact of LEDGF knock-out in primary human T cells, we first designed and tested an array of crRNAs with our multiplex delivery system (Figure 3A). Of the six LEDGF crRNAs we tested, two caused significant decreases in protein level by immunoblot and showed editing at the DNA level by TIDE analysis. (Figures 3A, Supplementary Table 1). These cell populations were infected with either a CXCR4-tropic HIV-1 GFP reporter virus or with a VSV-G pseudotyped virus as described earlier (Figure 2D). While CXCR4 knock-out specifically inhibited infection of the CXCR4-tropic virus, we saw similar inhibition of both viruses in the two populations where LEDGF had been depleted (Figure 3A). The level of inhibition observed is limited by not only the percent of cells that are effectively edited, but also by the length and MOI of the infection. To determine if we see a more pronounced effect using different infection conditions, we repeated these experiments using our most efficient crRNA with a replication competent HIV-1 GFP reporter virus in a spreading infection over 7 days (Figure 3B). Compared to controls, LEDGF targeted cells exhibited a 2 to 7-fold reduction in viral replication across multiple blood donors three days after infection (average 4-fold reduction, Figure 3C, Supplementary Figure 3). After 7 days of spread, these differences were even greater with LEDGF targeted cells exhibiting a 6 to 12-fold reduction in HIV replication (Figure 3C). Little change was observed in cellular viability over this time course as monitored by live cell gating (Supplementary Figure 3). These data collectively confirm a functional role for LEDGF/p75 in HIV infection in primary human CD4+ T cells. Furthermore, they demonstrate the utility of this platform for analyzing HIV host factors at later steps in infection, especially in analyzing those factors resistant to more traditional RNAi approaches.
Figure 3.

Knock-out of LEDGF and TNPO3 inhibit early events of HIV infection in primary T cells. (a) Primary T cells were isolated from two different donors and electroporated with the indicated Cas9 RNPs including 6 different RNPs targeting LEDGF. Four days after electroporation, cells were removed for immunoblotting and genomic DNA isolation. At the same time, cells were infected with a CXCR4-tropic (LAI strain) GFP reporter virus or an identical VSV-G pseudotyped, Env-deficient virus in technical triplicate. After 36 hours, percent infected cells (GFP+) were determined by flow cytometry. The bar graph depicts the average percent infected cells across technical triplicates +/− standard deviation for one representative donor. LEDGF protein levels were quantified and depicted immediately above the immunoblot. (b) Procedural schematic depicting the modified stimulation/infection protocol for analyzing the LEDGF knock-out cells over the course of a spreading infection. (c) Primary T cells were isolated from four different donors and electroporated with buffer alone, Cas9 alone, or the most effective Cas9 RNP targeting LEDGF (crRNA #5, see above). Cells were infected as indicated in (b) and percent infection determined by flow cytometry. Results for two donors are depicted at days 3 and 7 in the first two graphs. The final graph depicts the average percent infection observed in the Cas9 control and LEDGF KO cells across all four donors at day 3 +/− standard deviation. (d) Primary T cells were isolated from two different donors and electroporated with the indicated Cas9 RNPs. Four days after electroporation, cells were removed for immunoblotting. At the same time, cells were infected with a CXCR4-tropic (LAI strain) GFP reporter virus or an identical VSV-G pseudotyped, Env-deficient virus in technical triplicate. After 36 hours, percent infected cells (GFP+) were determined by flow cytometry. The bar graph depicts the average percent infected cells across technical triplicates +/− standard deviation for one representative donor. See also Figure S3 and Table S1.
We next tested ablation of three other candidate host dependency factors that act at three other stages of HIV replication to further generalize the applicability of the arrayed targeting method: TNPO3 (required for efficient cellular trafficking by proper localization of CPSF6) (Brass et al., 2008; Christ et al., 2008; De Iaco et al., 2013), NUP153 (required for nuclear import of the pre-integration complex) (Konig et al., 2008; Matreyek and Engelman, 2011), and CDK9 (required for viral gene expression) (Mancebo et al., 1997; Zhu et al., 1997). For each gene, we designed three crRNAs and tested them for knock-out generation in multiple donors by TIDE analysis at the DNA level and immunoblot at the protein level (Supplementary Table 1). Two of the three crRNAs for NUP153 and CDK9 reproducibly led to decreased cell viability and cell death, consistent with essential functions for these genes in cellular health (Albert et al., 2014; Ball and Ullman, 2005; Duheron et al., 2014; Price, 2000). The Cas9 RNPs that did not cause cell death showed low levels of editing at the DNA level, but no depletion of the respective protein (data not shown). We interpret these data to mean that these two genes are essential for cellular viability and they were not assayed further. In contrast, cells survived when the nuclear transporter TNPO3 was disrupted; two crRNAs were identified that generated insertions and deletions at the TNPO3 locus with roughly 40–70% efficiency depending on the donor (Supplementary Table 1).
To directly compare the efficacy of CXCR4, TNPO3, and LEDGF editing, we used the most efficient crRNA for each gene to generate isogenic knock-out populations in multiple, independent donors. Knock-out of each factor was specific and did not alter the protein level of the other host factors (Figure 3D). Upon infection with a CXCR4-tropic GFP reporter virus (as in Figure 2D), CXCR4 knockout nearly ablated infection while TNPO3 and LEDGF knock-out inhibited infection with roughly 60% efficacy. Pseudotyping the virus with VSV-G envelope was sufficient to completely overcome the barrier CXCR4 depletion poses to infection, but knock-out of TNPO3 and LEDGF still inhibited infection by the same amount relative to controls (Figure 3D). Thus, while some locus and donor variability in editing efficiency was observed, this Cas9 RNP platform serves as a robust method for disrupting key host factors at multiple stages of infection.
Cas9 RNP Multiplexing allows for the Generation of Double Knock-out Cells
A potential strength of using in vitro synthesized Cas9 RNPs is the ease with which double knockouts could be generated. Like siRNA methodologies, simply mixing the Cas9 RNPs prior to electroporation would be expected to yield a mixed population of cells, some of which are doubly modified. To test the feasibility of this approach, we attempted to knock-out both CXCR4 and CD4 by co-electroporation of their respective Cas9 RNPs. For CD4, we designed three crRNAs which yielded different efficiencies when electroporated the respective RNPs singly (Figure 4C). Immunostaining for both CXCR4 and CD4 demonstrated specific depletion of each cell surface marker only when the Cas9 RNP targeting that gene was included and furthermore demonstrated the clear accumulation of a double negative population in the multiplexed samples (representative flow plots, Figure 4A). Comparing the efficiency of CXCR4 knock-out alone or in multiplex with the CD4 RNPs showed no loss of knock-out efficiency in both donors tested (Figure 4B). Similarly, all CD4 RNPs demonstrated equivalent efficiencies with or without inclusion of the CXCR4 RNP (data not shown).
Figure 4.

CRISPR RNP multiplexing allows for the generation of double knock-out primary T cells. (a) Primary T cells were isolated from two different donors and electroporated with the indicated Cas9 RNPs. CXCR4 and one of three different CD4 crRNA were mixed at a 1:1 ratio prior to incubation with the Cas9 protein. Cells were stained with anti-CD4-PE and anti-CXCR4−APC 48 hours after electroporation. Flow plots from one representative donor are depicted. (b) Percent CXCR4− cells as measured by flow cytometry in two donors electroporated with either Cas9 protein alone, the CXCR4 RNP alone, or the CXCR4 RNP mixed 1:1 with three different CD4 RNPs. (c) Percent CXCR4− or CD4− cells after electroporation with each RNP singly are recorded for each donor under single KO efficiency. The observed percent of double knock-out cells are then reported alongside the percent editing predicted given an independent probability of editing at each locus. (d) Primary T cells were isolated from three different donors and electroporated with the indicated Cas9 RNPs. CXCR4 and LEDGF crRNPs were mixed at a 1:1 ratio prior to electroporation in the double targeted population. Four days after electroporation, cells were removed for immunoblotting as well as immunostaining against CXCR4. Average percent CXCR4+ cells in each population across technical triplicate +/− standard deviation are depicted in the bar graph above the associated immunoblot for LEDGF. (e) Cells were infected with a CXCR4-tropic (LAI strain) GFP reporter or an identical VSV-G pseudotyped, Env-deficient virus in technical triplicate. After 36 hours, cells were co-stained for CXCR4-APC and percent infected cells (GFP+) determined by flow cytometry. The bar graph depicts the average percent infected cells across technical triplicates +/− standard deviation for one representative donor. p-values were calculated by pairwise Student’s t-test. See also Supplementary Figure S4 and Supplementary Table S1.
Several methods for delivering nucleic acid or protein to cells result in preferential delivery of multiple reagents to the same cell at once such that a single cell that receives one packaged molecule tends to receive all others in the mixture (Ma et al., 2007). Cas9 RNPs, however, then must direct to the chromosomal DNA, successfully cleave their target site and introduce a functional mutation to result in a phenotypic readout, a process that may or may not be related to cleavage success at independent loci. Based on the efficiency of each CXCR4 and CD4 RNP when delivered alone, and given that we see no loss in efficiency during multiplexing, we calculated the predicted percent of double knock-out cells when delivering the Cas9 RNPs at a 1:1 ratio assuming editing at each locus was independent of the other (Figure 4C). We found that this nearly perfectly reflected the observed percent double knock-out cells when staining for CD4 and CXCR4 (Figure 4C). The generation of double knock-out cells by Cas9 RNP multiplexing therefore is dependent on the efficiency of each individual RNP and the probability of editing at each locus appears to be independent.
Given the ability of this platform to generate double knock-out cells, we decided to multiplex two of our most efficient Cas9 RNPs, against CXCR4 and LEDGF, to see if we could generate cells doubly resistant to HIV infection. As observed with CD4 and CXCR4, we saw no loss of efficiency for either the CXCR4 or LEDGF RNPs when delivered simultaneously as opposed to individually (Figure 4D). These cell populations were infected with a CXCR4-tropic HIV-1 GFP reporter virus or with a VSV-G pseudotyped virus as previously described and normalized to the Cas9 only control (Figure 2E). As observed before (Figure 3F), CXCR4 disruption caused cells to become almost completely resistant to CXCR4-tropic HIV infection, but had no impact on the infectivity of the VSV-G pseudotyped virus (Figure 4B,C). On the other hand, LEDGF knock-out resulted in a modest decrease in the infectivity of both viruses regardless of their envelope in agreement with prior results (Figures 3D, 4B,C). The cells treated with both RNPs demonstrated a dual phenotype, restricting the CXCR4-tropic virus as effectively as the CXCR4 knock-out cells and restricting the VSV-G pseudotyped virus as effectively as the LEDGF knockout cells (Figures 4B,C). These viral replication phenotypes are consistent with our protein level analyses and indicate the efficient generation of double knock-out primary T cells. This was repeated in multiple, independent donors with nearly identical results (Supplementary Figure 4). Taken together, these data show that our multiplex editing platform can successfully generate double knock-out populations. Not only could this be useful for the analysis of functional redundancy and epistatic relationships among genes, such combinatorial editing could be an effective tool for the generation of multiply HIV-resistant cells for curative transplantation therapy (Didigu et al., 2014; Voit et al., 2013).
An Arrayed Platform for Primary T Cell Genetics using CRISPR Cas9 RNPs
One major benefit to this approach is the ability to perform high efficiency gene editing in primary T cells in an arrayed format for screening and high-throughput phenotypic analysis. Unlike pooled CRISPR libraries that rely on selection and sequencing for the identification of impactful genes, this system allows for the generation of hundreds of physically distinct cellular pools for analysis and a myriad of downstream applications. This is especially ideal for high-throughput screens that may require the interrogation of hundreds of genes with specific and specialized readouts not suitable for pooled analysis.
As proof-of-principle, we designed a screen of 45 genes described in the published literature that either directly or indirectly affect the function of HIV integrase. These included 21 genes supported by at least two publications in the HIV-1, human interaction database as well as 24 genes identified by HIV-1, human protein-protein interaction mapping by affinity-purification mass spectrometry (AP-MS) (Ako-Adjei et al., 2015; Jager et al., 2012). Three crRNAs were designed per gene alongside multiple non-targeting controls and several previously analyzed crRNAs, including those targeting CXCR4 and LEDGF as positive controls and CDK9 as a toxicity control. In total, 146 crRNAs were designed and synthesized in 96-well plate arrayed format (Supplementary Tables 1 & 2). These crRNAs were incubated with tracrRNA and Cas9 protein to form Cas9 RNPs, which were subsequently electroporated into activated primary T cells from two donors (Figure 5A). These cells were reactivated and replica plated into five 96-well plates. 48 hours later, one plate was lysed for protein, one was lysed for genomic DNA, and three were infected with HIV-1NL4–3 with IRES:GFP immediately following the Nef reading frame. Every two days for six days, half of the cultures were removed and fixed for analysis by automated flow cytometry and the cultures supplemented with an equal volume of fresh media (full data set provided in Supplementary Table 2).
Figure 5.

A primary cell knock-out screen of genes predicted to influence HIV integrase confirms the role of several proteins in HIV replication. (a) CD4+ T cells were isolated from the blood of two donors by negative selection and activated with plate-bound anti-CD3 and soluble anti-CD28. 146 independent Cas9 RNPs were formed in vitro by incubating Cas9 protein with the appropriate crRNAs and tracrRNA. These were electroporated into the activated T cells in 96-well format to generate polyclonal pools of edited cells. These cells were activated again and replica plated. Cells were infected in triplicate with replication-competent HIV-1 (NL4-3 strain) with IRES:GFP cloned behind Nef. Every 48 hours for 6 days, half of the culture was removed to monitor infection by flow cytometry while an equal volume of fresh media was replaced. (b) Percent HIV infected cells (GFP positive) in each polyclonal knock-out pool are plotted as the average across technical triplicates +/− standard deviation as measured at day 4 post-infection in each donor. crRNAs are ordered left to right by average infectivity. The median value +/− 1.5 standard deviations across all wells are annotated by the dotted lines. (c) Percent HIV infected cells plotted over 6 days in primary T cells nucleofected with the indicated CRISPR Cas9 RNPs. Six non-targeting controls included in the plates are annotated in gray. The spreading infection results are displayed for all RNPs that scored above (blue) or below (red) 1.5 standard deviations from the median infectivity across the plate at any given time point in the series. See also Supplementary Figure S5, and Supplementary Tables S1 and S2.
At each timepoint, we observed a roughly normal distribution of infection rates tightly clustered around the median value (Figure 5B). Cells treated with non-targeting controls distributed evenly around the median while the cells treated with CXCR4 RNPs consistently resulted in the lowest rate of infection. Any RNP that scored more or less than 1.5 standard deviations from the median at any given timepoint was considered significantly impactful and the full spreading infection profile for these hits was plotted alongside the non-targeting controls (Figure 5C). Of the RNPs with significant effects, including the positive control RNPs targeting validated host dependency factors, more resulted in decreased rates of infection (1.5 standard deviations less than the median; red line), but some resulted in increased rates of infection (1.5 standard deviations than the median; blue line).
For both donors, the strongest blocks to infection were dictated by knock-out of the positive controls: CXCR4, CDK9, and LEDGF (Figure 5C). CDK9 likely exerted its effect based on both toxicity of the editing event as well as specific blocks to the HIV replication cycle. No other RNP elicited cellular toxicity as severe as those targeting CDK9 as determined by flow cytometry measuring cellular density (data not shown). GEMIN2 knock-out also consistently resulted in blocks to infection in both donors, as predicted from studies in cell lines (Hamamoto et al., 2006; Nishitsuji et al., 2009). KPNA1, KPNA5, KAT2A also significantly decreased replication in at least one donor, confirming previously published studies (Armon-Omer et al., 2004; Gallay et al., 1997; Levin et al., 2010; Terreni et al., 2010). FAM96B and UNC45A resulted in significant blocks to infection as well, but no role for these proteins in HIV infection has been published beyond an interaction with HIV integrase (Jager et al., 2012).
In contrast to the above factors, SMARCB1 (INI1/SNF5) and XRCC6 knock-out both resulted in strong increases in infection in both donors (Figure 5C). SMARCB1 is known to inhibit HIV replication in cell lines, possibly through the stabilization of pre-intregration complexes in non-reactive conformations (Das et al., 2009; Maroun et al., 2006; Pyeon et al., 2015). XRCC6 (Ku70) and other components of the NHEJ pathway, however, have an unclear role in the HIV lifecycle, though some results suggest a role in post-integration expression (Baekelandt et al., 2000; Jeanson et al., 2002; Li et al., 2001; Manic et al., 2013). These results suggest a possible role for Ku70 as a restriction factor in primary T cells and highlights the importance of this approach to directly test the impact of genetic alterations in relevant primary models. Additionally, PLOD1, SUCLA2, HDGFRP2, and EEF1A1 each scored significantly as a potential restriction factor in at least one of the two donors.
Importantly, we cannot rule out the role of any gene that failed to score as significant in this assay without validation of the crRNAs used at the DNA and/or protein level. Conversely, correlation between crRNA effectiveness at the protein level and the functional impact on infection can provide secondary validation in screens of untested crRNAs. For example, XRCC6 crRNA 2 resulted in the most efficient knock-out by immunoblot, crRNA 3 was of intermediate efficiency, and crRNA 1 was ineffective (Supplementary Figure 5 and Figure 5C). This correlates well with the spreading infection data where we see crRNA 2 as effective in both donors, crRNA 3 as effective in one donor, and crRNA 1 failing to score significantly. Similar parallels are observed with UNC45A, where crRNAs 1 and 3 are the only two to effectively result in gene knock-out by immunoblot and are the only two to result in significant inhibition of HIV infection (Supplementary Figure 5 and Figure 5C). TIDE analysis of editing for each gene observed to alter HIV infectivity further validated these findings. In every case, only those crRNAs that caused a phenotypic change in infection were successful at inducing DNA edits at the corresponding genetic loci (Supplementary Table 1).
Overall, these data demonstrate the strength of this platform for the high-throughput screening of genes directly in primary cells. This approach has the potential to unveil a number of new host factors playing essential roles in HIV infection and pathogenesis. Given the ease of use, effectiveness of editing, and universal availability of the reagents, we hope this platform can be broadly adapted for the study of a broad variety of T cell processes.
DISCUSSION
In this study, we describe an efficient, high-throughput method for systematically editing genes in primary human lymphocytes to facilitate the study of viral host factors, T cell genetic interactions, and therapeutic approaches for the cure of HIV. This arrayed Cas9 RNP platform is notable for its efficiency, adaptability, and scalability. Each reaction consists only of the primary T cells, recombinant Cas9 protein, electroporation buffer, tracrRNA, and crRNA. We found targeting to be robust across different donors and different loci, with one to two of every three crRNAs tested achieving significant editing and detectable decreases in protein expression. The T cells must be activated, and generally require 24 to 48 hours for recovery after electroporation, but are otherwise amenable for use in a wide variety of downstream applications. We found minimal adverse effects on cell replication, surface marker expression, response to stimulation, or infection broadly. These experiments were performed with 300,000 cells per reaction in 96-well plate format, but the cell number could be even further reduced to scale up target discovery efforts. Coupled to the chemical synthesis of crRNAs in 96-well plates, this platform allows for the screening of hundreds of genes in isogenic sets of primary cells from a single donor. The method should be readily adaptable to diverse primary human cell types that are targets of a variety of pathogenic organisms.
In this study, we used this Cas9 RNP platform to knock-out genes that are required for HIV infection in T cells. Targeting of the HIV co-receptors CXCR4 and CCR5 elicits strong protective phenotypes to infection that strongly correlate with the effectiveness of the RNP in a tropism dependent manner. CCR5 has already been developed as a target for treatment with the antiretroviral drug Marviroc, and cell-based therapies with CCR5-deficient cells are under development (Didigu et al., 2014; Gulick et al., 2008; Tebas et al., 2014). However, the natural variant in CCR5 that confers resistance to HIV may increase susceptibility to West Nile Virus, suggesting that CCR5 may have pleotropic affects on T cell function and infection with diverse pathogens (Glass et al., 2006). Similarly, CXCR4 is essential in hematopoietic stem cells, but appears largely dispensable in T cells, suggesting multiple roles in a cell type dependent manner (Berson et al., 1996; Feng et al., 1996; Zou et al., 1998). The ability to readily and reproducibly generate knock-out cells ex vivo provides a system for testing pleiotropic effects of these modifications, an essential consideration for the development of next generation therapeutics.
This approach was also successful in targeting genes required at later stages of initial infection. Disruptions of LEDGF and TNPO3 were well tolerated in primary T cells and both resulted in clear defects in HIV replication. The ability to measure the impact of LEDGF on infection demonstrates the advantage of this technique over RNAi approaches. Even treatment with ubiquitously penetrating RNAi can leave cells with persisent low levels of target gene expression, which has complicated functional validation of factors required at only low concentrations, such as LEDGF (Llano et al., 2006a; Llano et al., 2006b). In contrast, Cas9 RNPs can generate true genetic knock-outs in a substantial subset of targeted cells. While still a heterogenous pool, a large fraction of Cas9 RNP treated cells are completely devoid of the target protein, allowing for functional interrogation in a clean genetic system. Furthermore, unlike siRNA electroporation, transient treatment with Cas9 RNPs generates permanent genetic modifications in the T cells, allowing for the examination of later time points in infection where more robust effects may be observed.
While LEDGF and TNPO3 knock-outs resulted in clear replication defects upon single round infection, neither inhibited infection fully, even after adjusting for observed editing efficiency. This strongly suggests alternate routes to infection may exist independent of these factors or that some level of functional redundancy with other unidentified host factors is occurring in primary T cells (Levin et al., 2010; Vandegraaff et al., 2006; Vandekerckhove et al., 2006). The ease in generating double knock-out populations using this system make it an attractive platform for analyzing these redundant factors and perhaps for systematically determining epistatic relationships among host dependency genes to better identify gene pathways (Roguev et al., 2013). The fact that editing at different loci appears to occur independently of each other, however, mandates the validiation of highly efficient RNPs to ensure high efficiency of double editing. Such multiplex editing could generate cells that are resistant to HIV infection through multiple independent mechanisms, analogous to current combinatorial antiretroviral therapies (Didigu et al., 2014; Voit et al., 2013).
In contrast to LEDGF and TNPO3, NUP153 and CDK9 appeared essential for T cell survival ex vivo. While not unexpected, these results rule out therapeutic ablation of these factors. While genes that impact cell viability or growth are obviously excluded from this possibility, ablation of any gene would need to be studied more thoroughly in a mouse model prior to consideration for any cell-based therapy. As mentioned above, while CXCR4 and CCR5 deletion may not cause an obvious deficiency in vitro, deletions of these genes have been linked to immune defects in vivo. One potential way around this problem is to focus on identifying and engineering specific, naturally occuring polymorphisms that are known to be well tolerated and potentially protective against infection. We have previously demonstrated that Cas9 RNPs can generate knock-in edits in primary T cells in addition to the knock-out disruptions that were the focus of the current studies (Schumann et al., 2015). While the efficiency of knock-in edits is lower than knock-outs, as the technology improves we envision this platform could be just as readily employed to study the impact of point mutants and haplotypes on infection.
Beyond using this CRISPR/Cas9 RNP approach to explore the roles of known host factors, genetic relationships between genes, and functional redundancy, the ease of scalability in performing these manipulations in primary human T cells opens up the possibility of discovery based screening. As a proof-of-principle, we screened 45 genes with known or suspected roles in HIV integrase function as curated from principally cell line based data in the literature. Beyond confirming the expected phenotype of several host factors in primary cells, we also uncovered novel roles for genes with no previously published replication phenotype, including XRCC6 and UNC45A. While much more follow-up work remains to be done with these factors, it demonstrates the power of the platform to achieve high quality genetic perturbation in medium to high throughput scale.
This platform has multiple applications well beyond the identification of genes involved in HIV replication. The capacity to perform high content screens in human T cells allows for the genetic dissection of pathways essential for immune cell effector function in a variety of healthy and diseased states. One major concern in applying genome editing to primary human cells has been that CRISPR/Cas9 generates a heterogeneous mixture of cells with various phenotypes as a result of imprecise repair mechanisms at the targeted site (Doudna and Charpentier, 2014; Ran et al., 2013). While this platform routinely can target genes with 70-90% efficiency, not every cell is edited in the final population. Moving forward, we will need improved ways of purifying edited cells for both research and therapeutic purposes. Nevertheless, this study demonstrates that we can already link specific, edited genotypes to complex phenotypes, including HIV infection. The efficiency of gene disruption is high enough that we are able to observe quantitative changes in HIV infection with multiple in vitro assays. Heterogeneously edited cells may actually prove beneficial for experiments performed under selective conditions, identifying genetic variants that protect against infection, but leave normal gene function unperturbed. Efforts to understand these and other questions are ongoing and promise to shed more light on the potential of CRISPR/Cas9 technology for therapeutic purposes.
Multiplex Cas9 RNP mediated editing of primary CD4+ T cells is a powerful method for the study of T cell processes and for the identification/analysis of next-generation cell and drug-based therapies. It allows for interrogation of microbial dependencies on human host genes directly in the relevant primary cells, identifying pathways that could potentially be exploited for therapeutic development. Specifically coupling Cas9 RNP editing with cell-based therapies could provide a direct path from target discovery and validation to the engineering of therapeutic cells for the cure of HIV infection and other disease states.
EXPERIMENTAL PROCEDURES
Detailed experimental procedures and associated references are available in the supplementary data.
Cas9 RNP mediated Editing of Primary Human T cells
In brief, primary CD4+ T cells were isolated from human whole blood by Ficoll gradient centrifugation and negative selection via the Easysep Human CD4+ T-cell enrichment kit (Stemcell Technologies). T cells were stimulated with bound anti-CD3 and soluble anti-CD28 in the presence of IL-2 for 48 hours prior to electroporation. Electroporation was performed using the Amaxa P3 Primary Cell 96-well Nucleofector kit and 4D-Nucleofecter (Lonza). Recombinant S. pyogenes Cas9 protein used in this study expresses a C-terminal HA tag and two nuclear localization signal (NLS) peptides that facilitate transport across the nuclear membrane. The protein was expressed and purified as described (Anders and Jinek, 2014) and obtained from the QB3 Macrolab, University of California, Berkeley. Purified Cas9 protein was stored in 20 mM HEPES at pH 7.5 plus 150mM potassium chloride, 10% glycerol, and 1mM tris(2-carboxyethyl)phosphine (TCEP) at −80C. crRNA for each gene were designed using the online tool developed by the Zhang lab at the Massachusetts Institute of Technology (http://crispr.mit.edu/)(Hsu et al., 2013). Each crRNA and the associated tracrRNA were chemically synthesized (Dharmacon) and suspended in 10mM Tris-HCl pH 7.4 to generate 80μM RNA stocks.
Cas9 RNPs were prepared fresh for each experiment. crRNA and tracrRNA were first mixed 1:1 and incubated 30 minutes at 37C to generate 40μM crRNA:tracrRNA duplexes. An equal volume of 40μM Cas9-NLS was slowly added to the crRNA:tracrRNA and incubated for 15 minutes at 37C to generate 20μM Cas9 RNPs. For each reaction, roughly 300,000 stimulated T cells were pelleted and re-suspended in 20μL P3 buffer (Lonza). 3μl of 20μM Cas9 RNP mix was added directly to these cells and the entire volume transferred to the 96-well reaction cuvette. For double editing reactions, 3μL of each Cas9 RNP was added to 20μL cells. Cells were electroporated using program EH-115 on the Amaxa 4D-Nucleofector (Lonza). 80μL pre-warmed, complete RPMI was added to each well and the cells were allowed to recover for 30 minutes at 37C. Cells were then re-stimulated on plates coated overnight with 10μg/ml anti-CD3 (UCHT1, Tonbo Biosciences) and 10μg/ml anti-CD28 (CD28.2, Tonbo Biosciences) for 24 hours prior to infection.
Supplementary Material
Acknowledgments
We thank all members of Marson, Krogan, and Simon labs for suggestions and technical assistance. This research was supported by the UCSF MPHD T32 Training Grant (J.F.H.), a fellowship of the Deutsche Forschungsgemeinschaft (SCHU 3020/2-1, K.S.), a UCSF Sandler Fellowship (A.M.), a gift from Jake Aronov (A.M.), NIH/NIDA Avenir New Innovator Award (DP2DA042423, A.M.), NIH/NIAID funding for HIV studies (R01 AI064001, V.S.), NIH/NIGMS funding for the HIV Accessory & Regulatory Complexes (HARC) Center (P50 GM082250, A.M. and N.J.K.), NIH funding for the FluOMICs cooperative agreement (U19 AI106754, J.F.H. and N.J.K.), NIH/NIAID funding for the HIV Immune Networks Team (P01 AI090935, N.J.K. & V.S.), and NIH funding for the UCSF-Gladstone Institute of Virology & Immunology Center for AIDS Research (CFAR, P30 AI027763). Special thanks to Ethan Brookes, Matthew Hall, and Olivier Cantada at Lonza Bioscience for their support with the Nucleofection transfection technology. We also thank Anja Smith and Darrick Chow at Dharmacon for support and assistance with crRNA and tracrRNA synthesis. A patent has been filed on the use of Cas9 RNPs to edit the genome of human primary cells (A.M., J.A.D. and K.S.). A.M. serves as an advisor to Juno Therapeutics and the Marson lab has sponsored research agreements with Juno Therapeutics and Epinomics. J.D. is a co-founder of Editas Medicine, Intellia Therapeutics, and Caribou Biosciences and serves as a scientific advisor to Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, and Driver.
Footnotes
AUTHOR CONTRIBUTIONS
J.F.H, K.S., N.J.K and A.M conceived the project. Primary cell isolation, crRNA design, and knock-out cell generation were conducted by K.S. Target identification, HIV infection, and subsequent analyses were performed by J.F.H. and M.J.M. TIDE analysis was performed by J.W. LEDGF spreading infections were performed by L.M. The figures were designed and assembled by J.F.H. Text was written by J.F.H. and A.M. with input from K.S., L.M., J.D., N.J.K., and V.S. M.J.M., J.F.H, K.S., and A.M. edited the manuscript for publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ako-Adjei D, Fu W, Wallin C, Katz KS, Song G, Darji D, Brister JR, Ptak RG, Pruitt KD. HIV-1, human interaction database: current status and new features. Nucleic acids research. 2015;43:D566–570. doi: 10.1093/nar/gku1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert TK, Rigault C, Eickhoff J, Baumgart K, Antrecht C, Klebl B, Mittler G, Meisterernst M. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. British journal of pharmacology. 2014;171:55–68. doi: 10.1111/bph.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, Schneider T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- Anders C, Jinek M. In vitro enzymology of Cas9. Methods in enzymology. 2014;546:1–20. doi: 10.1016/B978-0-12-801185-0.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armon-Omer A, Graessmann A, Loyter A. A synthetic peptide bearing the HIV-1 integrase 161–173 amino acid residues mediates active nuclear import and binding to importin alpha: characterization of a functional nuclear localization signal. Journal of molecular biology. 2004;336:1117–1128. doi: 10.1016/j.jmb.2003.11.057. [DOI] [PubMed] [Google Scholar]
- Baekelandt V, Claeys A, Cherepanov P, De Clercq E, De Strooper B, Nuttin B, Debyser Z. DNA-Dependent protein kinase is not required for efficient lentivirus integration. Journal of virology. 2000;74:11278–11285. doi: 10.1128/jvi.74.23.11278-11285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball JR, Ullman KS. Versatility at the nuclear pore complex: lessons learned from the nucleoporin Nup153. Chromosoma. 2005;114:319–330. doi: 10.1007/s00412-005-0019-3. [DOI] [PubMed] [Google Scholar]
- Baltimore D. Gene therapy. Intracellular immunization. Nature. 1988;335:395–396. doi: 10.1038/335395a0. [DOI] [PubMed] [Google Scholar]
- Berson JF, Long D, Doranz BJ, Rucker J, Jirik FR, Doms RW. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strains. Journal of virology. 1996;70:6288–6295. doi: 10.1128/jvi.70.9.6288-6295.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, et al. Transportin-SR2 imports HIV into the nucleus. Current biology : CB. 2008;18:1192–1202. doi: 10.1016/j.cub.2008.07.079. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nature medicine. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. The Journal of experimental medicine. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Cano J, Kalpana GV. Multimerization and DNA binding properties of INI1/hSNF5 and its functional significance. The Journal of biological chemistry. 2009;284:19903–19914. doi: 10.1074/jbc.M808141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology. 2013;10:20. doi: 10.1186/1742-4690-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, McCune JM. Can HIV be cured with stem cell therapy? Nature biotechnology. 2010;28:807–810. doi: 10.1038/nbt0810-807. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Didigu CA, Wilen CB, Wang J, Duong J, Secreto AJ, Danet-Desnoyers GA, Riley JL, Gregory PD, June CH, Holmes MC, et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123:61–69. doi: 10.1182/blood-2013-08-521229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, Mi S, Yam P, Stinson S, Kalos M, et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Science translational medicine. 2010;2:36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Duheron V, Chatel G, Sauder U, Oliveri V, Fahrenkrog B. Structural characterization of altered nucleoporin Nup153 expression in human cells by thin-section electron microscopy. Nucleus. 2014;5:601–612. doi: 10.4161/19491034.2014.990853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel HJ, Morrison JH, Saenz DT, Fuchs JR, Kvaratskhelia M, Ekker SC, Poeschla EM. TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. Journal of virology. 2014;88:9704–9717. doi: 10.1128/JVI.01397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Fu BX, Hansen LL, Artiles KL, Nonet ML, Fire AZ. Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic acids research. 2014;42:13778–13787. doi: 10.1093/nar/gku1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature biotechnology. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Gallay P, Hope T, Chin D, Trono D. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Natl Acad Sci U S A. 1997;94:9825–9830. doi: 10.1073/pnas.94.18.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard A, Segeral E, Naughtin M, Abdouni A, Charmeteau B, Cheynier R, Rain JC, Emiliani S. The integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for postintegration silencing of HIV-1 gene expression in latently infected cells. Cell Host Microbe. 2015;17:107–117. doi: 10.1016/j.chom.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. CCR5 deficiency increases risk of symptomatic West Nile virus infection. The Journal of experimental medicine. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SP. Host factors exploited by retroviruses. Nat Rev Microbiol. 2007;5:253–263. doi: 10.1038/nrmicro1541. [DOI] [PubMed] [Google Scholar]
- Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. The New England journal of medicine. 2008;359:1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto S, Nishitsuji H, Amagasa T, Kannagi M, Masuda T. Identification of a novel human immunodeficiency virus type 1 integrase interactor, Gemin2, that facilitates efficient viral cDNA synthesis in vivo. Journal of virology. 2006;80:5670–5677. doi: 10.1128/JVI.02471-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nature biotechnology. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. The New England journal of medicine. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanson L, Subra F, Vaganay S, Hervy M, Marangoni E, Bourhis J, Mouscadet JF. Effect of Ku80 depletion on the preintegrative steps of HIV-1 replication in human cells. Virology. 2002;300:100–108. doi: 10.1006/viro.2002.1515. [DOI] [PubMed] [Google Scholar]
- Jordan A, Defechereux P, Verdin E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. The EMBO journal. 2001;20:1726–1738. doi: 10.1093/emboj/20.7.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome research. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1999;96:5215–5220. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibman RS, Riley JL. Engineering T Cells to Functionally Cure HIV-1 Infection. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:1149–1159. doi: 10.1038/mt.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Hayouka Z, Friedler A, Loyter A. Transportin 3 and importin alpha are required for effective nuclear import of HIV-1 integrase in virus-infected cells. Nucleus. 2010;1:422–431. doi: 10.4161/nucl.1.5.12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. The EMBO journal. 2001;20:3272–3281. doi: 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science. 2006a;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. Journal of virology. 2004;78:9524–9537. doi: 10.1128/JVI.78.17.9524-9537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano M, Vanegas M, Hutchins N, Thompson D, Delgado S, Poeschla EM. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. Journal of molecular biology. 2006b;360:760–773. doi: 10.1016/j.jmb.2006.04.073. [DOI] [PubMed] [Google Scholar]
- Ma ZL, Werner M, Korber C, Joshi I, Hamad M, Wahle P, Hollmann M. Quantitative analysis of cotransfection efficiencies in studies of ionotropic glutamate receptor complexes. Journal of neuroscience research. 2007;85:99–115. doi: 10.1002/jnr.21096. [DOI] [PubMed] [Google Scholar]
- Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, Zheng Z, Cotte J, Carpenito C, Wood T, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Human gene therapy. 2013;24:245–258. doi: 10.1089/hum.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes & development. 1997;11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, et al. Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells using CRISPR/Cas9. Cell stem cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manic G, Maurin-Marlin A, Laurent F, Vitale I, Thierry S, Delelis O, Dessen P, Vincendeau M, Leib-Mosch C, Hazan U, et al. Impact of the Ku complex on HIV-1 expression and latency. PloS one. 2013;8:e69691. doi: 10.1371/journal.pone.0069691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun M, Delelis O, Coadou G, Bader T, Segeral E, Mbemba G, Petit C, Sonigo P, Rain JC, Mouscadet JF, et al. Inhibition of early steps of HIV-1 replication by SNF5/Ini1. The Journal of biological chemistry. 2006;281:22736–22743. doi: 10.1074/jbc.M604849200. [DOI] [PubMed] [Google Scholar]
- Matreyek KA, Engelman A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. Journal of virology. 2011;85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Kitchen SG, Pugach P, Zack JA. The CCR5 and CXCR4 coreceptors--central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS research and human retroviruses. 2004;20:111–126. doi: 10.1089/088922204322749567. [DOI] [PubMed] [Google Scholar]
- Nishitsuji H, Hayashi T, Takahashi T, Miyano M, Kannagi M, Masuda T. Augmentation of reverse transcription by integrase through an interaction with host factor, SIP1/Gemin2 Is critical for HIV-1 infection. PloS one. 2009;4:e7825. doi: 10.1371/journal.pone.0007825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pache L, Konig R, Chanda SK. Identifying HIV-1 host cell factors by genome-scale RNAi screening. Methods. 2011;53:3–12. doi: 10.1016/j.ymeth.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nature methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nature biotechnology. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Molecular and cellular biology. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyeon D, Price L, Park IW. Comparative molecular genetic analysis of simian and human HIV-1 integrase interactor INI1/SMARCB1/SNF5. Archives of virology. 2015;160:3085–3091. doi: 10.1007/s00705-015-2585-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Talbot D, Negri GL, Shales M, Cagney G, Bandyopadhyay S, Panning B, Krogan NJ. Quantitative genetic-interaction mapping in mammalian cells. Nature methods. 2013;10:432–437. doi: 10.1038/nmeth.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes & development. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature medicine. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. The New England journal of medicine. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terreni M, Valentini P, Liverani V, Gutierrez MI, Di Primio C, Di Fenza A, Tozzini V, Allouch A, Albanese A, Giacca M, et al. GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology. 2010;7:18. doi: 10.1186/1742-4690-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nature reviews Genetics. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- Vandegraaff N, Devroe E, Turlure F, Silver PA, Engelman A. Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology. 2006;346:415–426. doi: 10.1016/j.virol.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove L, Christ F, Van Maele B, De Rijck J, Gijsbers R, Van den Haute C, Witvrouw M, Debyser Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. Journal of virology. 2006;80:1886–1896. doi: 10.1128/JVI.80.4.1886-1896.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit RA, McMahon MA, Sawyer SL, Porteus MH. Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:786–795. doi: 10.1038/mt.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ, Sherrill-Mix SA, Patro SC, Secreto AJ, Jordan AP, et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS pathogens. 2011;7:e1002020. doi: 10.1371/journal.ppat.1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- Yi G, Choi JG, Bharaj P, Abraham S, Dang Y, Kafri T, Alozie O, Manjunath MN, Shankar P. CCR5 Gene Editing of Resting CD4(+) T Cells by Transient ZFN Expression From HIV Envelope Pseudotyped Nonintegrating Lentivirus Confers HIV-1 Resistance in Humanized Mice. Molecular therapy Nucleic acids. 2014;3:e198. doi: 10.1038/mtna.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL, Gregory PD, Holmes MC, Torbett BE. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20:849–859. doi: 10.1038/mt.2011.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes & development. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.