

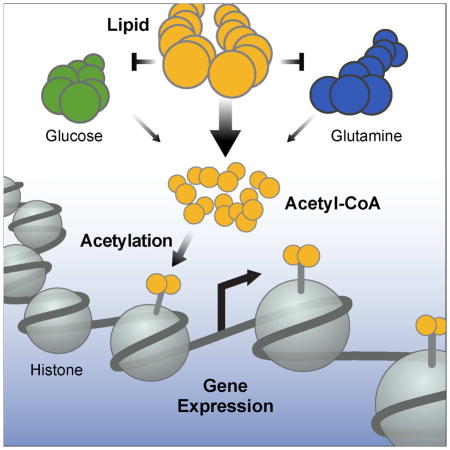
SUMMARY
Cells integrate nutrient sensing and metabolism to coordinate proper cellular responses to a particular nutrient source. For example, glucose drives a gene expression program characterized by activating genes involved in its metabolism, in part, by increasing glucose-derived histone acetylation. Here, we find that lipid-derived acetyl-CoA is a major source of carbon for histone acetylation. Using 13C-carbon tracing combined with acetyl-proteomics, we show that up to 90% of acetylation on certain histone lysines can be derived from fatty acid carbon, even in the presence of excess glucose. By repressing both glucose and glutamine metabolism, fatty acid oxidation reprograms cellular metabolism leading to increased lipid-derived acetyl-CoA. Gene expression profiling of octanoate-treated hepatocytes shows a pattern of upregulated lipid metabolic genes, demonstrating a specific transcriptional response to lipid. These studies expand the landscape of nutrient sensing and uncover how lipids and metabolism are integrated by epigenetic events that control gene expression.
Graphical Abstract
INTRODUCTION
Cells sense nutrient availability and integrate signals into proper metabolic and cellular responses by several mechanisms. For example, nutrients influence cellular processes by providing substrates for post-translational modification of proteins, which control enzymatic activity or affect the epigenome to alter gene expression patterns (Gut and Verdin, 2013). In this way, nutrients and their metabolites can directly influence a wide range of cellular processes. Proper integration of these metabolites and signals is crucial for health, as dysregulation of protein and histone modifications are associated with several disease states, such as neurodegenerative diseases and cancer (Kaelin and McKnight, 2013).
The amino acid methionine is required for synthesis of S-adenosylmethionine, which is the universal donor of methyl groups for histone or DNA methylation. Levels of methionine and methionine uptake dictate levels of histone methylation in vitro, as well as in animals and humans (Dann et al., 2015; Mentch et al., 2015). Similarly, acetyl-CoA is the substrate for histone acetylation. The acetyl moiety from acetyl-CoA is added to lysines on histone proteins by histone/lysine acetyl-transferase (HAT/KAT) proteins. In general, increases in histone acetylation are associated with chromatin relaxation, allowing access for transcription factors and activation of gene expression.
In yeast, the major source of acetyl-CoA for histone acetylation is acetate, which produces acetyl-CoA via acetyl-CoA synthetase enzymes Acs1p and Acs2p (Takahashi et al., 2006). In higher organisms, glucose has been described as the carbon source of acetyl-CoA for histone acetylation. Glucose-derived carbon produces citrate in mitochondria, which crosses into the nucleus where the nuclear isoform of the enzyme ATP-citrate lyase (ACLY) cleaves citrate to produce acetyl-CoA for histone acetylation (Wellen et al., 2009). The addition of glucose and serum to cells after serum starvation leads to increased histone acetylation with a corresponding induction of genes involved in glucose metabolism in vitro (Wellen et al., 2009). These studies demonstrate that cells have sophisticated epigenetic mechanisms to sense and integrate nutrient signals for activation of coordinated gene expression programs.
While the relationship between glucose and histone acetylation is well-known, we questioned whether histone acetylation was unique to glucose-derived carbon and a glucose metabolic gene expression program. Recently, studies have shown relationships between histone acetylation and metabolites other than glucose (Donohoe et al., 2012; Gao et al., 2016), suggesting that multiple sources of carbon might integrate into the epigenome. Because lipids can be a major source of cellular acetyl-CoA, we set out to test whether lipid carbon contributes to histone acetylation.
RESULTS
Lipids induce histone hyperacetylation
To test whether lipids could influence histone acetylation, we used an eight-carbon, saturated fatty acid called octanoate, which is a readily oxidized lipid known to increase ketone body production (McGarry and Foster, 1971). Long-chain fatty acids (LCFAs) (e.g. palmitate) have multiple cellular fates, including re-esterification, desaturation, elongation or oxidation (Currie et al., 2013). The oxidation of LCFAs is controlled primarily via the enzyme carnitine palmitolyltransferase 1 (CPT1), which is required for the transport of LCFAs across the mitochondrial membrane. In contrast, medium-chain fatty acids (6–12 carbons), such as octanoate, uniquely drive fatty acid oxidation, by diffusing across mitochondrial membranes and bypassing CPT1, thereby leading to rapid oxidation to acetyl-CoA (McGarry and Foster, 1974). Using octanoate to drive lipid oxidation in immortalized hepatocytes [alpha mouse liver 12 (AML12)], we found a striking, dose-dependent increase in histone acetylation, represented by the robust and well-characterized histone 3 lysine 9 (H3K9) acetylation mark (Fig. 1A).
Figure 1. Octanoate causes histone hyperacetylation.

(A) Western blot of histone acetylation in AML12 cells treated for 24 hours with octanoate or PBS added to complete media. Data is presented as fold change relative to control, error bars are standard deviations of triplicate samples. Data is representative of experiments repeated at least 5 times. (B) Western blot of acetylated histone H3K9 in cell lysates from AML12 cells. Cells were serum starved overnight and then treated for 24 hours with base DMEM media (deplete of glucose, glutamine, or pyruvate; containing 10% FBS) alone or in combination with glucose (25 mM), glutamine (4 mM) or octanoate (2 mM). Graph shows the fold change of mean signal intensity of duplicate experiments relative to vehicle control +/− standard error mean (SEM) with representative western blot shown. (C) Quantitative proteomic analysis of protein acetylation in AML12 cells treated for 24 hours with vehicle or 2mM octanoate added to complete media, performed in triplicate. The graph shows log2 fold-change of acetylated peptides in octanoate vs. PBS treated samples with an adjusted p-value (Padjusted) ≤ 0.05 (5% FDR). (D) Pie chart representing the top 50 most hyperacetylated peptides upon octanoate treatment with Padjusted ≤ 0.05. (E) Illustration summarizing the top 10 most acetylated histone peptides upon octanoate treatment on that make up the core nucleosome are labeled in red. (F) Western blot of histone H3K9 acetylation in AML12 cells treated with various fatty acids (2 mM) for 6 hours; the graph displays the mean fold-change compared to control of the signal intensity from 3 independent experiments; +/− SEM with representative western blot shown. (G) Western blot of histone acetylation in multiple cell lines treated for 24 hours with 2 mM octanoate. See also Fig. S1 and Table S1.
We then compared histone acetylation after octanoate treatment to the other major cellular fuel sources in the cell, namely glucose and glutamine. Cells were serum starved overnight, followed by treatment with various nutrients in the presence of serum for 24 hours, as was performed previously (Wellen et al., 2009). Cells were treated with base Dulbecco’s Modified Eagle Medium (DMEM, a medium containing no glucose, glutamine, or pyruvate; with 10% Fetal Bovine Serum [FBS] added) or with the addition of 25 mM glucose, 4 mM glutamine, 2 mM octanoate, or a combination of glucose and glutamine (complete media) with octanoate. Following treatment, histone acetylation was measured by western blotting for acetylated H3K9. Treatment with glucose alone, but not glutamine alone, led to a modest increase in histone acetylation (~20% over glucose-free media, Fig. 1B). Remarkably, the addition of octanoate, either alone or in the presence of glucose and glutamine, led to a striking increase in histone acetylation (Fig. 1B). Interestingly, simultaneously starving the cells of glucose and glutamine increased histone acetylation over nutrient-rich media (Fig. 1B), which also may be due to oxidation of lipids. These results show that lipids can induce histone acetylation, and suggest that lipid-induced histone acetylation may be a dominant mechanism over glucose-induced histone acetylation in the setting of complete nutrients.
To determine the breadth of changes in protein acetylation upon octanoate treatment, as well as the specificity for different histone lysines, we performed an unbiased, quantitative tandem mass tag (TMT)-based mass spectrometry acetyl-proteomic analysis, as has been performed previously (Grimsrud et al., 2012). In whole cell lysates of AML12 cells cultured in complete media with octanoate, we identified 573 acetylated peptides, among which histones were the most hyperacetylated class of proteins (Figs. 1C–1D and Table S1, ProteomeXchange identifier PXD005040). Additionally, we found the top 10 most acetylated histone peptides were on lysines on histone H2A, H3 and H4 (Fig. 1E), demonstrating a broad scope of histone hyperacetylation upon octanoate treatment.
Next, we tested whether histone acetylation was specific to octanoate or if other readily oxidized medium-chain fatty acids could also induce histone hyperacetylation. Like octanoate (C8, caprylic acid), other medium-chain fatty acids hexanoate (C6, caproic acid), decanoate (C10, capric acid) and dodecanoate (C12, lauric acid) induced significant increases in histone acetylation when added to complete media (Fig. 1F). Remarkably, these fatty acids lead to a rapid induction of histone hyperacetylation, where all lipids increased histone acetylation after only 6 hours (Fig. 1F). Additionally, when palmitate (C16) oxidation is activated, we see increased histone acetylation at physiological lipid doses (Fig. S1A). Thus, these data show that a range of fatty acids induce histone hyperacetylation, suggesting that increasing lipid oxidation generally increases histone acetylation.
HDAC inhibition cannot explain histone hyperacetylation by lipids
The ketone body β-hydroxybutyrate (βOHB) is an end-product of fatty acid metabolism and is a known histone deacetylase (HDAC) inhibitor (Shimazu et al., 2013). Thus, we considered whether ketones, a well-known fate octanoate oxidation (McGarry and Foster, 1974), could be inhibiting HDACs and increasing histone acetylation. We found that treating AML12 cells with exogenous βOHB at concentrations mimicking endogenous ketone levels produced from octanoate (Fig. S1B), or up to 30-fold higher, showed no change in histone acetylation (Fig. S1C). These data suggest that endogenous ketone inhibition of HDACs cannot explain the increases in histone acetylation. Further, we found an induction of histone acetylation in octanoate-treated cells across a panel of non-ketogenic cell lines, including a hepatocyte cell line derived from hepatocellular carcinoma (HepG2), human embryonic kidney cells (HEK 293T), as well as two breast cancer cell lines (MCF7 and MDA-MB-231), demonstrating octanoate increases histone acetylation in many different cell types in the absence of ketone production (Fig. 1G and S1D). Together, these data support the idea that histone acetylation is not due to production of βOHB by octanoate and subsequent HDAC inhibition.
Given that the short-chain fatty acid butyrate is a well-known HDAC inhibitor (Candido et al., 1978), we considered whether the medium-chain fatty acid octanoate could be directly inhibiting HDACs. We measured enzyme activity for a range of recombinant HDACs (HDAC1, 2, 3, 6 and 8) when incubated with octanoate, and then compared these to the values achieved by the HDAC inhibitors butyrate, trichostatin A (TSA), and suberanilohydroxamic acid (SAHA) (Fig. S1E). Relative to these well-known inhibitors, we found that octanoate was a poor HDAC inhibitor, consistent with previous results (Chang et al., 2013); therefore, HDAC inhibition is unlikely to account for octanoate-induced histone acetylation.
Lipid-derived carbon directly acetylates histones
We then sought to definitively determine whether lipid-derived carbon was acetylating histones by tracing [U-13C]octanoate-derived carbon onto acetyl-lysine carbon on histones. AML12 cells were treated with vehicle control or [U-13C]octanoate added to complete media for 24 hours, followed by mass spectrometry analyses of purified histones to measure 13C-enrichment on acetylated lysines, similar to a previous approach (Evertts et al., 2013). In cells treated with 13C-octanoate, we found that numerous acetylated histone peptides displayed striking mass shifts in MS1 spectra compared to vehicle control, consistent with the incorporation of octanoate-derived carbon directly onto histones (Fig. 2A–D). Representative MS1 spectra from peptides containing the doubly-acetylated H3K9,K14 histone peptide (Figs. 2A and 2C) or the triply-acetylated H4K8,K12,K16 histone peptide (Figs. 2B and 2D) showed remarkable incorporation of 13C-acetyl from 13C-octanoate (Table S2). To quantify the extent of enrichment of 13C carbon on these peptides, we corrected the MS1 mass isotopomer distributions for natural 13C-isotope abundance and found that 85–90% of the acetyl carbon on these histone peptides was derived from octanoate (Fig. 2E–2F), demonstrating that the overwhelming majority of histone acetylation on these peptides is from octanoate. Thus, these data show that lipid-derived carbon is a bona fide source of carbon for histone acetylation and can modify the epigenome.
Figure 2. Lipid carbon directly acetylates histones.

Representative MS1 spectra from AML12 cells treated with vehicle (A) or [U-13C]octanoate (C) added to complete media, highlighting the isotope distribution of histone H3 peptides acetylated on lysines 9 and 14 (z=2). The same is shown for precursor ions (z=3) detecting histone H4 acetylation on lysines 8, 12 and 16 in vehicle (B) and [U-13C]octanoate (D) treated samples. Mass shifts to the right indicate presence of [U-13C]octanoate-derived carbon at those specific histone lysines. Relative enrichments of acetylated sites for histone H3 (E) and histone H4 (F) corrected for natural abundance. Single 13C-acetyl indicates one lysine in a given peptide was found to be modified with a 13C2-acetyl group; double or triple acetyl means that two or three lysines of a given peptide are acetylated with [U-13C]octanoate-derived carbon. Red lower-case lysines indicate acetylated lysines (regardless of 13C enrichment). See also Figure S2 and Table S2.
Octanoate is preferentially oxidized and generates acetyl-CoA
We then considered the mechanism by which medium-chain fatty acids might produce carbon for histone acetylation. To measure how lipids influence the overall cellular metabolic state, we first performed steady-state metabolite profiling using targeted mass spectrometry of acyl-carnitines, amino acids, and organic acids (Figs. 3A and S2). Octanoyl-carnitine and hexanoyl-carnitine were markedly elevated in octanoate-treated cells, consistent with octanoate uptake and oxidation (Fig. S2C). Furthermore, we observed specific increases in the organic acids pyruvate, citrate, fumarate, and malate (Fig. 3A), but no differences in amino acids (Fig. S2A), suggesting that the addition of octanoate to complete culture medium was uniquely affecting the tricarboxylic acid (TCA) cycle.
Figure 3. Octanoate reprograms cellular metabolism.

(A) Organic acid levels measured by tandem mass spectrometry of AML12 cells treated in quadruplicate for 24 hours with 2 mM octanoate compared to vehicle added to complete media. Data is fold-change over control, +/− SEM. 14C-labeled pyruvate (B) or glutamine (C) oxidation in AML12 cells treated with or without octanoate in complete media for 24 hours prior to capture and measurement of 14CO2. Mean normalized data shown from 3 independent experiments +/− SEM. *p<0.05, **p<0.01 (Students t-test) (D) Volcano plot of protein phosphorylation changes measured by TMT-based quantitative mass spectrometry of whole cell AML12 lysates treated for 24 hours with octanoate or vehicle added to complete media; fold-change on a log2 scale vs. p-value on a negative log10 scale. Phoshopeptides showing statically significant (Padjusted <0.1) increases or decreases in abundance in response to octanoate treatment are colored red or blue, respectively. Phosphopeptides identifying Pdha1 serine 232 phosphorylation residues are highlighted (E) 13C-labeling of TCA cycle intermediates in AML12 cells treated with [U-13C]glucose or [U-13C]glutamine in the presence or absence of unlabeled 2 mM octanoate compared to [U-13C]octanoate labeling in the presence of unlabeled glucose and glutamine. Mass isotopomer distribution (MID) of citrate from [U-13C]glucose (F) or [U-13C]glutamine (G) in the presence or absence of unlabeled 2 mM octanoate. (H) Citrate MID from [U-13C]octanoate in the presence of unlabeled glucose and glutamine. (I) Acetate levels in media collected from AML12 cells with [U-13C] labeled substrates for 24 hours. (J) Acetyl-CoA labeling pattern from AML12 cells treated with [U-13C] labeled substrates; all data collected after 24 hours. All experiments were done in complete media.
To test this hypothesis, we interrogated TCA cycle metabolism (Fig. 3B–C). Increased pyruvate levels suggested that pyruvate metabolism was altered in the presence of octanoate. Therefore, we measured the oxidation of [2-14C]pyruvate to 14CO2 in octanoate-treated AML12 cells and found reduced pyruvate oxidation (~60% of control, Fig. 3B), consistent with a glucose-sparing effect known as the “Randle cycle” (Randle et al., 1963). Fatty acid oxidation inhibits glucose oxidation in part by inhibiting pyruvate dehydrogenase (PDH) (Hue and Taegtmeyer, 2009). Acetyl-CoA, reduced nicotinamide adenine dinucleotide (NADH), and ATP generated by fatty acid oxidation activate pyruvate dehydrogenase kinases and/or inhibit PDH phosphatases, leading to increased phosphorylation and inhibition of PDH (Sugden and Holness, 2003). Accordingly, we found serine 232 of PDH to have the greatest increase in phosphorylation in octanoate-treated AML12 cells compared to vehicle out of 267 phosphopeptides measured with significantly (Padjusted ≤ 0.05) increased phosphorylation (Fig. 3D and Table S1).
Reduced pyruvate metabolism, via genetic ablation of PDH or the mitochondrial pyruvate carrier, can cause compensatory increases in glutamine oxidation to supply anaplerotic substrates for the TCA cycle (Rajagopalan et al., 2015; Yang et al., 2014). Therefore, we predicted glutamine oxidation might be increased in the presence of octanoate. However, when we measured [U-14C]glutamine oxidation by 14CO2 capture, we found instead that glutamine oxidation was also reduced upon octanoate treatment (~55% of control, Fig. 3C), demonstrating that in addition to the known glucose-sparing effect in the setting of fatty acid oxidation, octanoate induces a glutamine-sparing effect.
To further characterize how altered metabolism could lead to lipid-derived histone acetylation, we determined the relative contribution of three major fuel sources in our system (glucose, glutamine and octanoate) to global metabolite pools using uniformly labeled 13C-stable isotope tracers in AML12 cells treated with octanoate. We found that under normal media conditions, glucose and glutamine are the major fuel sources contributing to the TCA cycle, approaching 80% of 13C-labeling for most TCA cycle intermediates (Fig. 3E). However, adding octanoate to complete culture medium diminished the relative contributions of glucose and glutamine to TCA cycle intermediates, which were replaced by octanoate-derived carbons (Fig. 3E).
Further testing TCA cycle function, we traced carbon into the TCA cycle by examining the isotope labeling pattern of citrate—a central TCA cycle metabolite that can be produced from glucose, glutamine and fatty acids. From [U-13C]glucose, we found the predominantly labeled isotopomer of citrate was M+2, indicating pyruvate passed through PDH to form acetyl-CoA and condensed with unlabeled four-carbon oxaloacetate to form citrate (Fig. 3F). However, when cells were given octanoate in the presence of [U-13C]glucose, M+2 citrate decreased by more than 60% compared to control samples (Fig. 3F). Furthermore, M+4 and M+5 isotopomers, which represent further turns of glucose carbon in the TCA cycle, were almost undetectable upon octanoate treatment (Fig. 3F), consistent with PDH inhibition, and reduced contribution of glucose to the TCA cycle.
Consistent with lower glutamine oxidation (Fig. 3C), we observed a decrease in the M+4 citrate isotopomer from [U-13C]glutamine in the presence of octanoate (Fig. 3G). Additionally, we observed increased unlabeled citrate (M+0), indicating reduced glutamine anaplerosis into the TCA cycle (Fig. 3G). Importantly, we found that reductive glutamine metabolism, which produces M+5 citrate, was also reduced in the setting of octanoate, showing an overall inhibition of glutamine flux into the TCA cycle.
When we monitored the fate of octanoate carbon in complete media, nearly 90% of the total citrate pool contained at least one carbon labeled from [U-13C]octanoate (Fig. 3H). Importantly, octanoate treatment resulted in labeled citrate isotopomers M+4–M+6, which requires octanoate-derived carbons to traverse multiple turns of the TCA cycle. Together, these isotope tracing data show that octanoate becomes the predominant fuel source in the cell over both glucose and glutamine.
Because citrate was heavily labeled by octanoate carbon, we predicted the source of this carbon was from mitochondrial acetyl-CoA generated by octanoate oxidation. However, octanoate is also oxidized to acetyl-CoA in peroxisomes, generating 2–4 equivalents of acetate, a source of acetyl-CoA for histone acetylation (Kasumov et al., 2005). We found octanoate-treated cells excreted nearly 100 μM acetate after 24 hours (Fig. 3I), supporting the idea that peroxisomal octanoate oxidation generates a significant amount of acetyl-CoA that is hydrolyzed to acetate for excretion. Finally, we directly tested the contribution of octanoate to the total cellular acetyl-CoA pool and found that 75% of the acetyl-CoA pool was octanoate-derived after 24 hours (Fig. 3J). Together, these studies show that octanoate oxidation leads to metabolic reprogramming whereby lipid carbon is preferentially oxidized over other fuel sources resulting in octanoate-derived carbon labeling a significant fraction of the acetyl-CoA pool (Fig. 3K); in this way, lipid-derived carbon becomes the dominant source of acetyl carbon for histone acetylation.
Octanoate activates a specific gene expression program
To identify the physiological role of lipid-induced histone acetylation, we measured global changes in gene expression. Indeed, histone acetylation is a key regulator of gene transcription by “opening” chromatin to allow transcriptional regulators to bind to those regions of DNA and drive gene expression. To identify the genes and pathways that were responding to octanoate treatment, we performed a gene microarray analysis of cells treated with increasing amounts of octanoate (1 mM, 2 mM, 5 mM) compared to control (Fig. 4A, GEO Accession: GSE87156). Gene set enrichment analyses revealed that ‘fatty acid metabolic process’ was the most highly upregulated process (Fig. 4B), demonstrating a specific gene expression response to lipid exposure. We identified several lipid metabolism and lipid handling genes upregulated in the gene microarray and confirmed these by quantitative polymerase chain reaction (qPCR) (Table S3 and Fig. 4C).
Figure 4. Octanoate causes a specific lipid metabolism gene expression signature.

Microarray gene expression analysis of AML12 cells treated with vehicle or with 1 mM, 2 mM or 5 mM octanoate added to complete media for 24 hours. (A) Heat map of all genes identified with upregulated genes in red and downregulated genes in blue for the given doses of octanoate. (B) Gene set enrichment analysis (GSEA) of microarray data comparing vehicle to 2 mM octanoate. Ranked by p-value on a −log10 scale with upregulated pathways in red and downregulated in blue. (C) qPCR analysis of lipid metabolic genes and genes found to be upregulated with octanoate treatment in microarray dataset. Cells were treated with vehicle or 2 mM octanoate for 24 hours. (D) qPCR analysis of genes associated with glucose-induced histone acetylation in AML12 cells treated with 2 mM octanoate. (E) qPCR data of AML12 cells treated with vehicle, 2 mM octanoate or 20 nM TSA alone or in combination for 24 hours in complete media. qPCR gene expression data are representative of repeated experiments each performed in triplicate +/− SD. All experiments were done in complete media. See also Figure S3 and Table S3.
Interestingly, pathways involving retinoid X receptor (RXR) activation, including liver X receptor (LXR)/RXR and farnesoid X receptor (FXR)/RXR activation, were identified by pathway analysis (Ingenuity) as activated upon octanoate treatment (Fig. S3). Among the genes that were most highly expressed in response to octanoate were retinoid metabolism genes, including transthyretin (Ttr) and retinal dehydrogenase (Aldh1a1), which are responsible for transporting retinol into cells and converting retinal to retinoic acid, respectively (Fig. 4C). Retinoic acid activates the nuclear hormone receptors RAR (retinoic acid receptor) and RXR, activation of which are known to increase the expression of genes involved in lipid and retinoid metabolism (Bonet et al., 2012). The action of retinoic acid is potentiated by HDAC inhibition and subsequent histone acetylation (Minucci et al., 1997). Thus, our findings suggest that activation of RXR or its partners could contribute lipid-specific gene expression from octanoate, which may be potentiated by octanoate-induced histone acetylation.
Importantly, octanoate did not cause an induction of glucose-metabolizing genes (Fig. 4D), which have previously been shown to be induced via glucose-derived histone acetylation (Wellen et al., 2009). These data further support the idea that cells specifically respond to specific nutrients by inducing specific metabolic genes. Additionally, we measured expression of several octanoate-induced genes after increasing histone acetylation via HDAC inhibition. While histone acetylation induced by TSA leads to a unique gene expression signature (Van Lint et al., 1996), it did not lead to the same gene expression pattern as octanoate (Fig. 4E), demonstrating that histone acetylation alone does not lead to generic gene expression signatures. Together, these data show that signals emanating from lipids drive histone acetylation and a specific lipid metabolic gene expression pattern, which is different than glucose-driven gene expression patterns or generic histone acetylation gene expression patterns.
DISCUSSION
In this study, we describe a mechanism by which fatty acids are sensed and integrated into the epigenome. Using stable isotope tracing, we found lipid-derived carbons are deposited onto histone lysines, demonstrating that lipids are a bona fide source of carbon for histone acetylation. This occurs via a coordinated mechanism where lipid oxidation reprograms metabolism, leading to the production of lipid-derived acetyl-CoA. We show histone acetylation is co-incident with activation of a lipid-specific gene expression program, which is different than the program activated by glucose or TSA-induced histone acetylation. Together, these finding identify a system where metabolism, epigenetics, and gene expression coordinate the appropriate cellular response to a nutrient signal.
Emerging evidence shows that metabolism and histone acetylation are inextricably linked. Glucose is a conical example of how nutrient-derived carbon integrates into the epigenome by histone acetylation (Wellen et al., 2009). More recently, de novo fatty acid synthesis and histone acetylation have been shown to use the same pool of acetyl-CoA, where knockdown of acetyl-CoA carboxylase (ACC1, a crucial enzyme in fatty acid synthesis), leads to histone hyperacetylation (Galdieri and Vancura, 2012). Our data further define the relationship between fatty acid metabolism and histone acetylation. Acetyl-CoA derived from lipid oxidation is the predominant contributor to the global acetyl-CoA pool, which is reflected in increased acetate, citrate, and histone acetylation.
Interestingly, when we tested the possibility that canonical acetate or citrate handling enzymes might be mediating lipid-induced histone acetylation, we found no role. We knocked down Acyl-CoA synthetase short-chain family member 2 (Acss2) or Acly, which have been shown to be involved in regulation of acetate or glucose-induced histone acetylation, respectively, and found no reduction in histone acetylation with octanoate (Fig. S4A). In fact, we found that ACLY expression was decreased with increasing doses of octanoate while ACSS2 expression increased in AML12 cells (Fig. S4B–C). ACLY is a crucial enzyme in fatty acid synthesis; because fatty acid synthesis and fatty acid oxidation are reciprocal processes, decreased ACLY expression would be expected during lipid oxidation. These data suggest against a role for ACLY and ACSS2 in lipid-induced histone acetylation in this system; however, it does not exclude the possibility that they might play a role in lipid-induced histone acetylation in other settings.
The absence of a known metabolic or enzymatic intermediate step mediating histone acetylation in our study suggests that other processes could be involved. For example, various acetyl-CoA producing enzymes could influence the acetyl-CoA pool therefore histone acetylation. Acetyl-carnitine formed in mitochondria can be a source of acetyl groups for histone acetylation via transportation out of mitochondria by carnitine-acylcarnitine translocase (CAT) and converted back to acetyl-CoA in the nucleus via nuclear carnitine acetyltransferase (CrAT) (Madiraju et al., 2009). This model could be important for transporting fatty acid derived acetyl-CoA from the mitochondria to produce nuclear acetyl-CoA for histone acetylation (Taylor et al., 2014). An alternate possibility is that the compartmentalization of acetyl-CoA is not as rigid as is thought. The levels of labeling of octanoate carbon into acetyl-CoA were remarkably consistent with labeling in histone acetylation, as well as key metabolites synthesized from acetyl-CoA (i.e. citrate or acetate formation), regardless of compartment. While more work is required to identify which enzymes and metabolic intermediates lie between lipids and acetyl-CoA, and how their transported for histone acetylation, our findings show that the source of carbon for histone acetylation is dictated by the predominant contributor to the global acetyl-CoA pool.
Overall, our data support a model where multiple sources of acetyl-CoA can lead to histone acetylation, which then promotes gene expression. However, if multiple sources of acetyl-CoA can increase histone acetylation, then the specificity of gene expression is likely encoded by nutrient-specific transcription factors or transcriptional co-regulators. Consistent with this model, we identified RAR/RXR activation by octanoate, which could mediate the lipid metabolism-specific gene expression program observed. Future studies will determine how metabolic and epigenetic processes work together to ensure the appropriate cellular response.
Dysregulated epigenetic modifications are known to play a role in several disease states (Portela and Esteller, 2010). Targeting epigenetics and metabolism are aggressively being pursued as independent strategies to treat a variety of diseases (Arrowsmith et al., 2012). Our data suggest that metabolism and epigenetic regulation are inextricably linked and that efficacious strategies may emerge from better understanding the connections between metabolism and epigenetic regulation.
EXPERIMENTAL PROCEDURES
Cell culture
AML12 mouse hepatocytes, 293T cells and HepG2 cells were purchased from ATCC. MDA-MB-231 and MCF7 cells were gifts from Donald McDonnell’s laboratory. All cell lines were cultured in DMEM High Glucose media (Sigma Cat #5796) without sodium pyruvate and were supplemented with 10% FBS (Gibco). Cells were treated with the medium-chain fatty acids sodium hexanoate (C6), sodium octanoate (C8) and sodium decanoate (C10) (Sigma) dissolved in phosphate buffered saline (PBS) and sodium dodecanoate (C12) dissolved in 10% ethanol in PBS. PBS was used as the vehicle control in all experiments. In experiments containing C12 (Fig. 1F), all samples also contained final concentration of 0.1% ethanol added to the medium. Cells were counted using a Beckman Coulter Countess cell counter using trypan blue exclusion and seeded at the indicated densities. All experiments were done in complete DMEM with 10% FBS (referred to as complete media) with fatty acids or vehicle supplemented, unless otherwise noted.
qPCR
Cells were treated with octanoate for 24 hours washed with PBS, lysed with Bio-Rad Aurum RNA lysis buffer and frozen at −80°C. RNA was isolated using the Bio-Rad Aurum RNA mini kit as per protocol (Cat #7326820). cDNA was synthesized using the Bio-Rad iScript cDNA synthesis kit. cDNA was diluted 1:10 with nuclease-free water prior to addition to quantitative real-time PCR reaction (qPCR). qPCR reaction mix contained 2.5 μl 1:10 cDNA, 0.5 μM of 10μM forward and reverse primer mix, 4 μL iTAQ Universal SYBR green Supermix (BioRad) and 1 μL RNase/DNase free water. qPCR was run using a Viia 7 Real-Time PCR system (Applied Biosystems).
Western blotting
Cells were plated and allowed to adhere overnight, and then treated the next day for the indicated times. Cells were collected prior to confluency, washed with PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer including protease and/or phosphatase inhibitors. Lysis buffer contained either tablet protease and phosphatase inhibitors (Roche) or Halt protease and phosphatase inhibitor cocktail (ThermoFisher Scientific #78440). Cells were blocked in 5% milk diluted in a mixture of tris-buffered saline and Polysorbate 20 (TBST; containing 0.1% tween) for at least 1 hour at room temperature or overnight at 4°C with rocking, and then washed with TBST. Membranes were incubated in primary antibody either overnight at 4°C or at room temperature for 1–2 hours with rocking. Primary antibodies were all purchased from Cell Signaling Technologies and used at a concentration of 1:1000. Blots were washed with TBST and then incubated for 1 hour with rocking in Licor secondary antibodies used at a concentration of 1:10,000 in TBST. Western blots were developed using a Licor Odyssey CLx. Blots were quantified using the Licor software. Histone blots were performed with the acetyl-histone antibody blot and total histone antibody probed on duplicate blots run concurrently.
Statistical analysis
All statistics performed were two-way Student’s t-tests with significance being defined as p ≤ 0.05. Statistics for the proteomics and microarray data are outlined in the supplemental experimental procedures.
Supplementary Material
Acknowledgments
We would like to thank Brett Peterson for thoughtful input throughout the project, Chris Newgard for critical review of the manuscript, Maciek R. Antoniewicz’s laboratory (University of Delaware) for assistance in developing the acetate assay, Karen Abramson and Simon Gregory for assistance with the gene microarray study, David Corcoran for GSEA and computational support, and the entire Hirschey laboratory for feedback. We would also like to acknowledge funding support from the American Heart Association 12SDG8840004 (to MDH), the National Institutes of Health (NIH) and the National Institute of Aging (NIA) grant R01AA022146 (to MDH), the Duke Pepper Older Americans Independence Center (OAIC) Program in Aging Research supported by the NIA (P30AG028716-01), and the Duke Cancer Institute (P30CA014236). EM is supported by an NIH training grant to the Duke University Molecular Cancer Biology Training Program (5T32-CA059365). CAO thanks the Carlsberg Foundation (2011_01_0169, 2013_01_0333, and CF15-0115), the Novo Nordisk Foundation (NNF15OC0017334), and the Lundbeck Foundation for the Group Leader Fellowship. The funding sources had no role in the conduct or presentation of this research.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, EM, PAG, and MDH.; Investigation, EM, SBC, DBF, BK, ORI, PAG; Writing - Original Draft, EM and MDH; Writing - Review & Editing, all authors; Supervision, CAO and MDH; Project Administration, MDH; Funding Acquisition, MDH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Bonet ML, Ribot J, Palou A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochimica et biophysica acta. 2012;1821:177–189. doi: 10.1016/j.bbalip.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RS. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology. 2013;69:105–114. doi: 10.1016/j.neuropharm.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell metabolism. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann SG, Ryskin M, Barsotti AM, Golas J, Shi C, Miranda M, Hosselet C, Lemon L, Lucas J, Karnoub M, et al. Reciprocal regulation of amino acid import and epigenetic state through Lat1 and EZH2. The EMBO journal. 2015;34:1773–1785. doi: 10.15252/embj.201488166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Molecular cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, Coller HA, Garcia BA. Quantitative dynamics of the link between cellular metabolism and histone acetylation. The Journal of biological chemistry. 2013;288:12142–12151. doi: 10.1074/jbc.M112.428318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdieri L, Vancura A. Acetyl-CoA carboxylase regulates global histone acetylation. The Journal of biological chemistry. 2012;287:23865–23876. doi: 10.1074/jbc.M112.380519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nature communications. 2016;7:11960. doi: 10.1038/ncomms11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, et al. Cell metabolism. 2012. A Quantitative Map of the Liver Mitochondrial Phosphoproteome Reveals Posttranslational Control of Ketogenesis; pp. 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. American journal of physiology Endocrinology and metabolism. 2009;297:E578–591. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumov T, Adams JE, Bian F, David F, Thomas KR, Jobbins KA, Minkler PE, Hoppel CL, Brunengraber H. Probing peroxisomal beta-oxidation and the labelling of acetyl-CoA proxies with [1-(13C)]octanoate and [3-(13C)]octanoate in the perfused rat liver. The Biochemical journal. 2005;389:397–401. doi: 10.1042/BJ20050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics. 2009;4:399–403. doi: 10.4161/epi.4.6.9767. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Foster DW. The regulation of ketogenesis from octanoic acid. The role of the tricarboxylic acid cycle and fatty acid synthesis. The Journal of biological chemistry. 1971;246:1149–1159. [PubMed] [Google Scholar]
- McGarry JD, Foster DW. The metabolism of (minus)-octanoylcarnitine in perfused livers from fed and fasted rats. Evidence for a possible regulatory role of carnitine acyltransferase in the control of ketogenesis. The Journal of biological chemistry. 1974;249:7984–7990. [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell metabolism. 2015;22:861–873. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S, Horn V, Bhattacharyya N, Russanova V, Ogryzko VV, Gabriele L, Howard BH, Ozato K. A histone deacetylase inhibitor potentiates retinoid receptor action in embryonal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11295–11300. doi: 10.1073/pnas.94.21.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nature biotechnology. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Rajagopalan KN, Egnatchik RA, Calvaruso MA, Wasti AT, Padanad MS, Boroughs LK, Ko B, Hensley CT, Acar M, Hu Z, et al. Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer & metabolism. 2015;3:7. doi: 10.1186/s40170-015-0134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. American journal of physiology Endocrinology and metabolism. 2003;284:E855–862. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Molecular cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Taylor EM, Jones AD, Henagan TM. A Review of Mitochondrial-derived Fatty Acids in Epigenetic Regulation of Obesity and Type 2 Diabetes. J Nutrit Health Food Sci. 2014;2:1–4. doi: 10.15226/jnhfs.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.