

Abstract
Human dentition development is a long and complex process which involves a series of reciprocal and sequential interactions between the embryonic stomodeal epithelium and the underlying neural crest–derived mesenchyme. Despite environment disturbances, tooth development is predominantly genetically controlled. To date, more than 200 genes have been identified in tooth development. These genes implied in various signaling pathways such as the bone morphogenetic protein, fibroblast growth factor, sonic hedgehog homolog, ectodysplasin A, wingless-type MMTV integration site family (Wnt), and transform growth factor pathways. Mutations in any of these strictly balanced signaling cascades may cause arrested odontogenesis and/or other dental defects. This article aims to review current knowledge about the genetic mechanisms responsible for selective nonsyndromic tooth agenesis in humans and to present a detailed summary of syndromes with hypodontia as regular features and their causative genes.
Keywords: tooth agenesis, hypodontia, oligodontia, genes
Introduction
Tooth agenesis affects approximately 2 to 9% of the population,1 making it one of the most prevalent craniofacial anomalies in humans. Population studies have revealed that up to 20% of the normal population may lose at least one-third molar, and primary dentition is rarely affected than permanent dentition.1 2 The most frequently absent teeth are third molars (10–25%), followed by lower second premolars and upper lateral incisors.3 Agenesis of the first and second molars is very rare. When more than six teeth are missing (excluding third molars), the condition is referred to as severe hypodontia or oligodontia. An extreme case of hypodontia is anodontia, denoting complete loss of all teeth.2 Tooth agenesis can either occur as an isolated condition (nonsyndromic form) or can be occur as part of a genetic syndrome (syndromic form), reflecting the genetically and phenotypically heterogeneity of the condition. Nonsyndromic tooth agenesis is classified as a sporadic or familial form, inherited in an autosomal dominant, autosomal recessive, or X-linked mode. It is often associated with some other oral defects, such as tooth size and/or shape defects,2 4 cleft lip (CL) and/or cleft palate (CLP),5 enamel hypoplasia,4 and altered craniofacial growth.5
Although tooth agenesis is occasionally caused by environmental factors, such as infections, trauma, chemotherapy, or radiotherapy, the majority of cases are due to genetic factors.6 7 8 9 10 Molecular studies of odontogenesis, using the mouse tooth as a model system, have shown that tooth development is under strict genetic control (Table 1). During early tooth development, multiple signaling molecules are expressed in the dental lamina epithelium and induced the dental mesenchyme.7 8 Important signaling pathways are involved in organogenesis including, but are not limited to the transcription factors, signaling molecules and its receptors, extracellular matrix molecules, and growth factors.9 Abnormal gene function (loss-of-function, gain-of-function) may disrupt specific signaling networks and cause a wide variety of selective tooth agenesis patterns or other oral malformations.10 Some genes responsible for syndromic tooth agenesis, including ectodermal dysplasias (EDs), Rieger and Witkop syndrome (Table 2), as well as those causing inherited oligodontia as an independent trait, have been identified.
Table 1. Genes involved in tooth development of mouse models.
| Gene | Mouse model | OMIM | Type of molecule | Dental phenotypes | Mechanisms |
|---|---|---|---|---|---|
| ACTBA | ActßA −/− | 102630 | Signal molecule | Arrested at bud stage | Failure of signaling |
| ACTRIIA | ActRIIA −/− | 604221 | Receptor | Mandibular incisors agenesis | Failure of signaling |
| ACTRIIA, ACTRIIB | Double mutant | Receptor | Molar and mandibular incisors agenesis | Failure of signaling | |
| BMPR1A | Bmpr1a −/− | 601299 | Receptor | Arrested at early bud stage | Impaired BMP signaling pathway |
| DKK1 | Overexpression | 605189 | Extracellular matrix | Arrested before or at placode stage | Decreased Wnt signaling |
| DLX1/DLX2 | Double ko | Transcription factor | Arrested before or at placode stage | Failure of signaling | |
| EDA | Tabby mutant | 300451 | Signal molecule | Partial agenesis, abnormal size, shape | Abnormal epithelial and enamel knot signaling |
| Overexpression | Supernumerary teeth | Overactivation of EDA signaling | |||
| EDAR | Downless mutant | 604095 | Receptor | Partial agenesis, abnormal size, shape | Abnormal epithelial and enamel knot signaling |
| EDARADD | Crinkled mutant | 606603 | Intracellular protein | Partial agenesis, abnormal size, shape | Abnormal epithelial and enamel knot signaling |
| EVC | Evc −/− | 604831 | Intracellular protein | Agenesis of maxillary incisors | Abnormal hedgehog signaling |
| FGF8 | Conditional ko | 600483 | Signal molecule | Molar agenesis | Failure of signal transmission |
| FGFR2 | Fgfr2IIIb −/− | 176943 | Receptor | Arrested at bud stage | Failure of signal transmission |
| FST | Fst −/− | 136470 | Extracellular protein | Reduced mandibular incisors | Failure of signal transmission |
| GAS1 | Gas1 −/− | 139185 | Extracellular protein | Fused maxillary incisors | Abnormal sonic hedgehog signaling |
| GLI2 | Gli2 −/− | 165230 | Transcription factor | Fused maxillary incisors | Abnormal sonic hedgehog signaling |
| GLI2/GLI3 | Double mutant | Transcription factor | Arrested before or at placode stage | Abnormal sonic hedgehog signaling | |
| IKKA | Ikka −/− | 600664 | Intracellular protein | Flat cusp, incisors evaginated | Abnormal epithelial and enamel knot signaling |
| IKKR | Ikkr −/− | 300248 | Intracellular protein | Male lethal, female agenesis, or conical teeth | Abnormal epithelial and enamel knot signaling |
| IRF6 | Irf6 +/− | 607199 | Transcription factor | Hypodontia, cleft palate | Affected Jag2-Notch1 signaling |
| LEF1 | Lef1 −/− | 153245 | Transcription factor | Arrested at late bud stage | Abnormal epithelial and enamel knot signaling |
| MSX1 | Msx1 −/− | 142983 | Transcription factor | Arrested at late bud stage | Abnormal BMP signaling |
| MSX1/MSX2 | Double mutant | Transcription factor | Arrested before or at placode stage | Abnormal BMP signaling | |
| P63 | p63 −/− | 603273 | Transcription factor | Arrested before or at placode stage | Abnormal epithelial signaling |
| PAX9 | Pax9 −/− | 167416 | Transcription factor | Arrested at bud stage | Failure of signaling |
| PITX2 | Pitx2 −/− | 601542 | Transcription factor | Arrested before or at placode stage | Failure of signaling |
| PRX1/PRX2 | Double mutant | 167420 | Transcription factor | Arrested before or at placode stage | Abnormal mandibular patterning |
| RUNX2 | Runx2 −/− | 600211 | Transcription factor | Arrested at bud stage | Failure of signaling |
| SHH | Shh −/− | 600725 | Signal molecule | Delayed and fused teeth | Failure of signaling |
| SMAD2 | Smad2 +/− | 601366 | Signal transducer | Incisor, mandibular molar agenesis | Failure of signal transduction |
| SMO | Smo −/− | 601500 | Signal transducer | Molars fused and delayed eruption | Failure of Shh signal transduction |
Abbreviations: BMP, bone morphogenetic protein; EDA, ectodysplasin A; ko, knockout.
Table 2. Human syndromic tooth agenesis and known genes.
| Gene | Type of molecule | Syndrome | OMIM | Dental/oral phenotypes | Mechanisms |
|---|---|---|---|---|---|
| ADAMTS2 | Procollagen I N-proteinase | Ehlers–Danlos hypermobility type VII | 225410 | Hypodontia, microdontia, dentin dysplasia, severe gingival hyperplasia, open bite, TMJ mobility restriction | Unknown |
| ANTXR1 | Tumor-specific endothelial marker | GAPO syndrome | 230740 | Pseusoanodontia, growth retardation, alopecia | Defect in extracellular matrix homeostasis |
| BCOR | Transcription regulator | Oculo-facio-cardio-dental syndrome | 113650 | Male lethal, no tooth development, female fused teeth | Abnormal transcription regulator |
| COL1A1/2 | Extracellular matrix | Osteogenesis imperfect type I | 166200 | Hypodontia | Abnormal extracellular matrix |
| CREBBP | Transcriptional coactivator | Rubinstein–Taybi syndrome | 180849 | Hypodontia, hyperdontia, natal teeth, micrognathia | Inactivate transcription of cAMP responsive genes |
| COL3A1 | Collagen | Ehlers–Danlos hypermobility type | 130020 | Hypodontia of permanent mandibular incisors, dentin dysplasia | Abnormal collagen I fibrillogenesis |
| CXORF5 | Intracellular protein | Orofacial digital syndrome type I | 311200 | Male lethal, female hypodontia | Cilia formation failure |
| DTDST | Sulfate transporter | Diastrophic dysplasia | Hypodontia | Impaired proteoglycan synthesis | |
| EDA | Signal molecule | Hypohidrotic ectodermal dysplasia | 305100 | Oligodontia/anodontia, peg or misshaped teeth | Abnormal epithelial and enamel knot signaling |
| EDAR | Receptor | Hypohidrotic ectodermal dysplasia | 129490 | Oligodontia/anodontia, peg or misshaped teeth | Abnormal epithelial and enamel knot signaling |
| EDARADD | Intracellular protein | Hypohidrotic ectodermal dysplasia | 224900 | Oligodontia/anodontia, peg or misshaped teeth | Abnormal epithelial and enamel knot signaling |
| EVC | Intracellular protein | Ellis–van Creveld syndrome | 225500 | Agenesis of incisors, conical teeth, taurodontism | Abnormal hedgehog signaling |
| EVC2 | Intracellular protein | Ellis–van Creveld syndrome; Weyers syndrome | 225500 | Agenesis of incisors, conical teeth, taurodontism | Abnormal hedgehog signaling |
| EYA1 | Phosphatase | Branchio-oro-renal syndrome | 113650 | Branchial fistulas or cysts, hearing loss | Disrupt phosphorylation-dependent transcription factor modulation |
| FGF10 | FGFs | Lacrimoauriculodentodigital syndrome | 149730 | Enamel dysplasia, microdontia, hypodontia | Failure of FGF signaling |
| FGFR2 | FGFs receptor | Apert syndrome | 101200 | Hypodontia | Abnormal epithelial and enamel knot signaling |
| FGFR3 | FGFs receptor | Crouzonodermoskeletal syndrome | 612247 | Hypodontia, delayed eruption, cementomas | Unknown |
| FLNB | Filamin B | Larsen syndrome | 150250 | Delayed eruption, flattened midface, cleft palate | Affect segmentation and endochondral ossification |
| FOXC1 | Transcription factors | Axenfeld–Rieger syndrome type 3 | 602482 | Oligodontia, microdontia, and short roots | Impaired Notch signaling |
| GJA1 | Gap junction protein | Oculodentodigital dysplasia | 164200 | Small teeth | Abnormal gap junctions |
| GRHL2 | Transcription factor | Autosomal recessive ectodermal dysplasia | 616029 | Hypodontia, enamel hypoplasia | Unknown |
| HOXB1 | Transcription factor | Moebius syndrome | 157900 | Facial paresis, missing teeth, ectrodactyly | Disrupted DN-protein binding, altered transcriptional activity |
| IKBKG | Intracellular protein | Hypohidrotic ectodermal dysplasia with immune deficiency | 300291 | Agenesis or conical teeth | Abnormal epithelial and enamel knot signaling |
| IKKγ | Intracellular protein | Incontinentia pigmenti | 308300 | Agenesis or conical teeth in females | Abnormal epithelial and enamel knot signaling |
| IRF6 | Transcription factor | Van der Woude syndrome | 211370 | Hypodontia | Failure of signaling |
| JAG1 | Notch receptor ligand | Alagille syndrome | 118450 | Oligodontia, coronal craniosynostosis | Impaired dominant-negative inhibition of Notch signaling |
| KMT2D | Lysine (K)-specific methyltransferase | Kabuki syndrome | 147920 | Missing incisors, cleft lip/palate, bifid tongue, and uvula | Disrupted histone methylation |
| KDM6A | Lysine (K)-specific demethylase | 300867 | Impaired histone demethylation | ||
| MKKS | Chaperonin | Laurence–Moon–Bardet–Biedl syndrome | 245800 | Hypodontia, small teeth, short roots | Unknown |
| NSD1 | Androgen receptor | Sotos syndrome | 117550 | Premolar missing, enamel hypoplasia | Unknown |
| P63 | Transcription factor | P63 syndromes | 129900 | Hypodontia | Failure of epithelial signaling |
| PHGDH | Phosphoglycerate dehydrogenase | Neu–Laxova syndrome | 256520 | Hypodontia | Unknown |
| PITX2 | Transcription factor | Rieger syndrome type I | 180500 | Oligodontia | Failure of epithelial signaling |
| PORCN | Transmembrane protein | Focal dermal hypoplasia | 305600 | Lip papillomas, hypoplastic teeth | Abnormal Wnt signaling |
| PVRL1 | Adhesion protein | Cleft palate–ectodermal dysplasia syndrome | 225060 | Oligodontia | Perturbed cell adhesion |
| RECQL4 | DNA helicase | Rothmund–Thomson syndrome | 268400 | Hypodontia | Unknown |
| RSK2 | Ribosomal kinase | Coffin–Lowry syndrome | 303600 | Thick lips, high palate, hypodontia, microdontia, delayed eruption, premature tooth loss | Abnormal phosphorylation and activation of MAPK signaling |
| SHH | Signal molecule | Holoprosencephaly | 142945 | Fused teeth and delayed eruption | Failure of signaling |
| TBX3 | Transcription factor | Ulnar mammary syndrome | 181450 | Hypodontia | Failure of epithelial signaling |
| TFAP2B | Transcription factor | Char syndrome | 169100 | Agenesis of premolar or molars | Reduced transcription |
| TCOF1 | Intracellular protein | Treacher–Collins syndrome | 154500 | Hypodontia | Abnormal nuclear trafficking |
| WNT10A | Signal molecule | Odonto-onycho-dermal dysplasia | 257980 | Oligodontia, abnormal shape and size | Failure of signaling |
| UBR1 | Recognin | Johanson–Blizzard syndrome | 243800 | Oligodontia, nasal alae aplasia, hearing loss | Unknown |
Abbreviations: cAMP, cyclic adenosine monophosphate; FGF, fibroblast growth factor; GAPO, growth retardation, alopecia, pseudo-anodontia, optic atrophy; TMJ, temporomandibular joint.
Genes Involved in Nonsyndromic Tooth Agenesis
Muscle Segment Homeobox, Homolog 1 (MSX1)
Muscle segment homeobox, homolog 1 (MSX1) belongs to a family of transcription factor that contains a highly conserved DNA-binding homeodomain.11 12 The principle phenotypes in homozygous MSX1-deficient mice were complete secondary CLP, failure of incisor development, and bud-stage arrest of molar development,13 suggesting that MSX1 is involved in multiple epithelial–mesenchymal interactions during mouse embryogenesis and appears to be critical during early tooth germ (bud and cap) developmental stages.12 13
MSX1 was the first causative gene identified in human nonsyndromic tooth agenesis.14 To date, more than 28 MSX1 heterozygous mutations have been reported to cause various types of selective tooth agenesis, suggesting that the mutant phenotype was due to haploinsufficiency, rather than a dominant-negative mechanism. Among these mutations, the majority are missense or nonsense mutations. The phenotype caused by a premature stop codon was slightly more severe than that caused by the substitution of one amino acid. This might be attributed to the increased susceptibility of degradation and lack of stability of the truncated protein messenger RNA (mRNA) (nonsense-mediated decay) compared with the missense mutated protein. The most distinguishing feature of MSX1-associated tooth agenesis is the frequent absence of second premolars and third molars (75%). Only one case of autosomal recessive oligodontia is known for a MSX1 missense mutation.15 In addition to isolated tooth agenesis, mutations of MSX1 cause other phenotypes. Some affected individuals also had CL or CLP, extending the MSX1 mutations phenotypes and supporting relationship between MSX1 and nonsyndromic CL and CLP.14 A MSX1 Ser202Stop mutation was reported to be associated with the Witkop syndrome, which includes tooth agenesis and nail dystrophy.12 Most MSX1 mutations (70%) causing isolated tooth agenesis clustered in the homeodomain, whereas all mutations associated with CL and/or CLP clustered outside the homeodomain. Also, abnormalities of the short arm of chromosome 4 including deletion of the entire MSX1 gene can cause Wolf–Hirschhorn syndrome, which exhibited oligodontia as part of the phenotype.16 These indicate that mutations in different domains of MSX1 have different effects during craniofacial development. The exact mechanisms through which variant forms of MSX1 mutations lead to variable phenotypes remain unknown. It is speculated that interactions of mutant MSX1 with other regulatory proteins such as DLX2 and DLX5, LHX2, PAX3, and PAX9 may have been disturbed.
Paired Box Gene 9 (PAX9)
PAX9 is a transcription factor characterized by a paired domain, an octapeptide motif, and a C-terminal transcriptional regulatory domain. PAX genes all share a unique paired domain which is an evolutionarily conserved 128-amino-acid region binding with DNA in a sequence-specific manner. This paired domain consists of two independent subdomains that structurally resemble a helix-turn-helix motif.17 PAX9 is a key regulator in mouse embryogenesis. It is widely expressed in the neural crest–derived mesenchyme that involved in craniofacial and tooth development, and is required for expression of BMP4, MSX1, and LEF1.18 PAX9 null mice die shortly after birth. In addition, all teeth fail to develop beyond the bud stage. Secondary CLP and other skeletal abnormalities in the head and limbs also have been observed.18 Another PAX9-neo mouse model which produced decreased levels of PAX9 mRNA and showed oligodontia. Homozygous PAX9 neo/neo mice exhibited hypoplastic or missing lower incisors and third molars. When combined with a PAX9 null allele, the PAX9 neo/null mutant mice developed severe oligodontia.19 The missing molars were arrested consistently at an earlier stage, suggesting that a reduction of PAX9 dosage may be essential in tooth development. PAX9 neo/neo and neo/null mice also showed defects in enamel formation of the continuously growing incisors, whereas molars exhibited increased attrition and reparative dentin formation.20
So far, more than 20 PAX9 mutations have been detected, most of them located in the paired domain. These mutations, ranging from missense to premature terminations or even deletion of the whole gene, are all heterozygous, indicating that a loss-of-function mechanism contributed to haploinsufficiency of PAX9.21 Unlike the phenotypes caused by MSX1 mutations, PAX9 mutations preferentially lead to second molars absence,20 21 and sometimes in combination with other types of tooth agenesis, most commonly second premolars and mandibular incisors.3 10 Moreover, it has been reported that missense mutations in the paired domain clustered near the N-terminal resulted in the molar and canine agenesis phenotype. The precise mechanism about PAX9 mutations is still unknown, mutant PAX9 may impair DNA binding and by modifying downstream bone morphogenetic protein (BMP)4 promoter transcriptional activities.20
MSX1 and PAX9 Interactions
Multiple lines of evidence have demonstrated that MSX1 and PAX9 are dosage-sensitive genes and can interact both at the gene and protein levels.22 PAX9 interacts synergistically with MSX1 to activate both MSX1 and the downstream BMP4 promoter in vitro.23 BMP4 is a key MSX1-dependent mesenchymal odontogenic signal for driving tooth morphogenesis through the bud-to-cap transition and enamel knot induction at the late cap stage.24 Mutations in either PAX9 or MSX1 can lead to defective interactions that disrupt normal BMP4 expression and function which is essential for the tooth morphogenesis. A recent in vivo study showed that PAX9(+/ − ) and MSX1(+/ − ) double mutants exhibited consistently absent lower incisors and an incompletely penetrant CL phenotype and transgenic BMP4 expression can partly rescue this phenotype.24 25 These data suggest that MSX1 and PAX9 function together throughout tooth and craniofacial development and affect multiple signaling pathways that influence incisor size and symmetry. Also, a combined reduction of PAX9 and MSX1 gene dosages may increase the risk for orofacial clefting and oligodontia.25 Recently, it was revealed that a zinc finger transcription factor odd-skipped-related 2 acts downstream of PAX9 and patterns the mesenchymal odontogenic field through protein–protein interactions with MSX1 and PAX9 during early tooth development.26
Axis Inhibitor 2 (AXIN2)
Axis inhibitor 2 (AXIN2) is an important negative regulator of catenin Wnt signaling pathway. Mutations in genes of the Wnt signaling pathway that prevent the degradation of β-catenin can lead to tumorigenesis. Somatic AXIN2 mutations have been described in a variety of human cancers, including skin, gastrointestinal, liver, and ovarian cancer.27 In mouse embryos, Axin2 is expressed intensively in dental papilla mesenchyme and enamel knots during odontogenesis, suggesting the involvement of Axin2 in regulating tooth formation.27 28 Reports of AXIN2 mutations in isolated tooth agenesis are rare in comparison with investigations of other causative genes. The first germline AXIN2 mutation was identified in a large Finnish family presenting isolated oligodontia and a variable colorectal phenotype with full penetrance. In this family, the oligodontia phenotype was entirely penetrant in mutation carriers. No ectodermal findings were described. Almost all family members with oligodontia had colorectal neoplasms, ranging from polyposis to colorectal malignancy with no polyps.27 29 Another de novo germline mutation was also detected in a 13-year-old boy with oligodontia. Of note, this mutation was identical to a frameshift mutation described in a colorectal cancer tissue. In another three-generation family segregating autosomal dominant oligodontia variably associated with sparse hair and eyebrows, colon or gastric polyps, early-onset colorectal, and/or breast cancer, a novel heterozygous nonsense mutation was identified in AXIN2.30 Subsequently, AXIN2 mutations were reported in patients with syndromic or nonsyndromic tooth agenesis and oral clefts.3 27 A significant association between AXIN2 variants and cases with at least one missing incisor were also identified.3 When compared with the pattern of agenesis of the posterior dentition seen in MSX1 and PAX9 affected families, individuals with loss-of-function mutation in AXIN2 display a mixed pattern of dental agenesis (Figs. 1 and 2).31
Fig. 1.
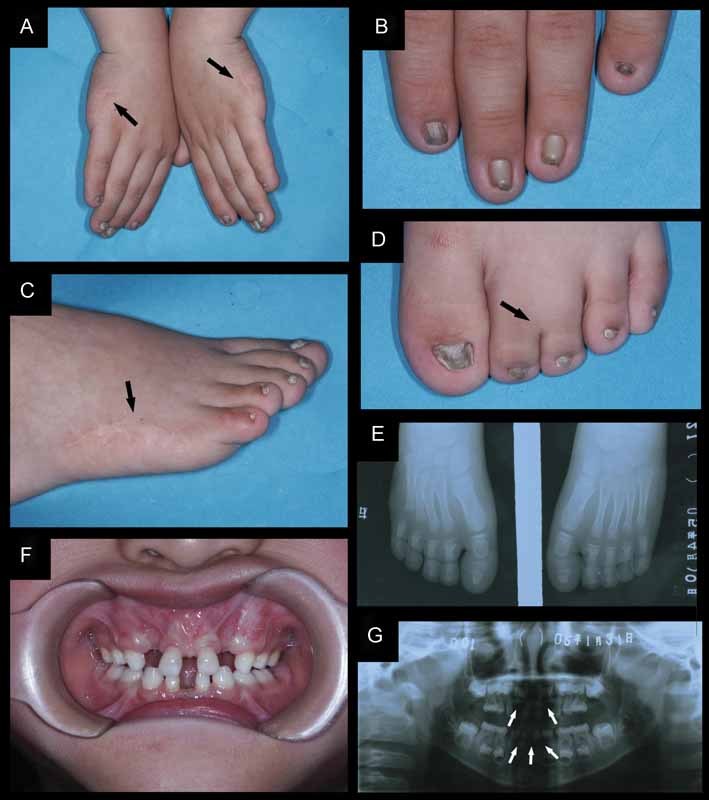
Clinical photographs and radiographs of a patient affecting with Weyers acrofacial dysostosis. (A and C) Bilateral postaxial polydactyly type A was removed at birth by surgical operations on both hands and feet, only scars (black arrows) were seen after amputation. (B and D) Apparent dysplastic fingernails and toenails. (D and E) Partial syndactyly at the second and third toes on both feet (black arrows). X-ray shows that the phalanges were not affected. (F) Oral examination of the upper lip reveals multiple oral frenula, diastemas, teeth which were abnormal in size and shape, delayed teeth eruption. (G) Panoramic radiographs showed the absence of permanent incisors (white arrows).
Fig. 2.
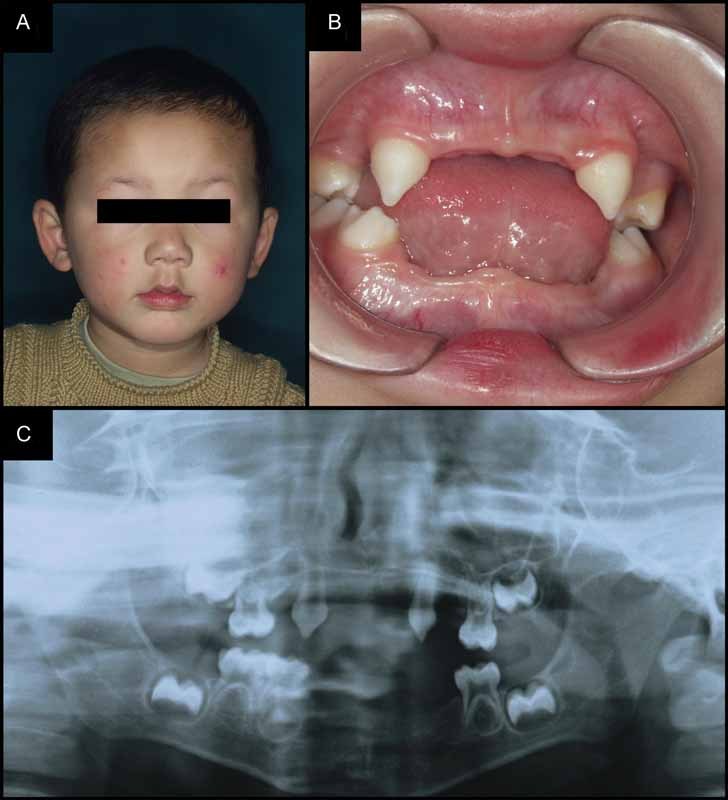
Characterization of a patient with X-linked recessive isolated hypodontia. (A and B) The patient is a 13-year-old boy, who had fine scalp and body hair, nails, and no complaints about tolerance to heat. His facial features appeared normal, but dental examination revealed multiple missing and misshapen deciduous teeth. (C) A panoramic radiograph depicts the typical pattern of congenitally missing teeth and tooth buds.
Ectodysplasin A (EDA)
X-linked hypohidrotic ED (HED) (XLHED), the most frequent form of HED, is characterized by abnormalities of teeth, hair, and eccrine sweat glands.32 Affected males usually present all of the typical phenotypes, including frontal bossing, sparse hair, and dry skin. Teeth are often missing or misshapen. In heterozygous female carriers, however, the severity of the disorder varies widely, even within families. The gene responsible for XLHED was ectodysplasin A (EDA). EDA is a type II transmembrane protein with a small N-terminal intracellular domain, a larger extracellular domain containing a (Gly-X-Y)19 collagen-like repeat, and a C-terminal tumor necrosis factor (TNF) homology domain.32 EDA, being a member of the TNF ligand superfamily, has extensive alternative splicing that leads to various isoforms. The most common EDA splice isoforms (EDA-A1 and EDA-A2) promote transcription by binding to distinct receptors.32 That is, EDA-A1 binds to the EDA receptor (EDAR), which interacts with the EDAR-associated death domain (EDARADD) and activates the downstream nuclear factor-κB (NF-κB) kinase signaling pathways.
More than 100 mutations have been reported in EDA, most of which are localized within exons 1, 3, and 5 and are mainly point mutations, clustering in the four functional domains of EDA. Intragenic insertions or deletions, large deletions including entire exon loss, and complete gene deletion have also been reported. Recently, mutations of EDA have been reported in some X-linked nonsyndromic hypodontia families.33 Unlike patients previously described with XLHED, affected patients from both families did not have any clinical features except oligodontia, and almost all female carriers were normal or had a milder phenotype, suggesting that EDA mutations can lead to a unique form of isolated X-linked recessive hypodontia.34 35
Wingless-Type MMTV Integration Site Family, Member 10A (WNT10A)
Odonto-onycho-dermal dysplasia (OODD) is a rare autosomal recessive syndrome mainly characterized by dry hair, oligodontia, smooth tongue, onychodysplasia, keratoderma, and hyperhidrosis. Schöpf–Schulz–Passarge syndrome (SPSS) is distinguished by the presence of multiple eyelid cysts, histologically corresponding to apocrine hidrocystomas. A homozygous nonsense mutation in WNT10A was found in all patients from three consanguineous families affecting OODD36 37 38 and an allelic disease SPSS. This demonstrated that ED can be caused by WNT10A which altered the Wnt signaling pathway. WNT10A is strongly expressed in embryonic limb, skin, hair follicle, and dental epithelium at the dental lamina and bud stages.39 During cap stage, its expression in the enamel knot is marked.
Recently, a large proportion (> 50%) of patients with isolated oligodontia and mild manifestations of ED were reported with biallelic or monoallelic WNT10A mutations, In an American family with variable hypodontia involving the lateral incisors and premolar teeth missing, two missense mutations F228I and D217N in WNT10A were identified, for which affected members were heterozygous or compound heterozygous. None of the family members had other ED manifestations.40 Recently, WNT10A mutations were found in patients with nonsyndromic tooth agenesis.41 The most frequent mutation, F228I, represented 62% of the WNT10A mutations in these nonsyndromic hypodontia patients. Individuals with biallelic mutations had a relatively severe phenotype than those with monoallelic mutations. Upper and lower premolars were the most affected missing teeth. Probands with biallelic mutations had a higher frequency of absent maxillary and mandibular molars and mandibular central incisors. In six Thai patients with isolated hypodontia of the maxillary permanent canines, three heterozygous mutations were identified in WNT10A. One of the affected individuals also had pegged maxillary permanent lateral incisors with dens invaginatus. Two mothers of the patients carried the mutation and had pegged maxillary permanent lateral incisors,41 indicating that WNT10A is a major gene in the etiology of isolated hypodontia.
Latent Transforming Growth Factor β-Binding Protein 3 (LTBP3)
Latent transforming growth factor β-binding protein 3 (LTBP3) was first identified through its similarity to human fibrillin-1 epidermal growth factor-like repeats. It is an extracellular matrix protein and binds to the latent form of the multipotent cytokine transform growth factor (TGF)-β. Several genes in the TGF-β signaling pathway have been implicated in skeletal developmental disorders, emphasizing the important role of this pathway in bone growth. Through genome-wide linkage study and sequencing, a homozygous nonsense mutation was identified in LTBP3 causing an autosomal recessive form of familial severe oligodontia, short stature, as well as apparent increased bone density in the spine and skull base.42 43 These findings suggested key roles for LTBP3-mediated transcription in development of the human tooth and axial skeleton. However, LTBP3 null mice had no obvious dental abnormalities comparable to the human phenotypes. The adult LTBP3 null mice were more than 50% smaller than sex-matched wild-type littermates. By day 12, null mice developed craniofacial malformations. At 3 months, there was a pronounced rounding cranial vault, shortened upper jaw, underbite, and kyphosis. The mutant mice also developed osteosclerosis of the long bones, thoracic kyphosis, distorted ribcage along with cranial base synchondroses. Between 6 and 9 months of age, mutant mice also developed osteosclerosis and osteoarthritis. These subsequent phenotypic changes were consistent with those described for mice that have perturbed TGF-β signaling.44 The mechanism responsible for the synostosis in LTBP3 null mice is not clear. LTBP3 may have a structural function within the synchondroses, which in the absence of LTBP3 ossify prematurely, or LTBP3 may play an important role in regulating chondrocyte differentiation by controlling the TGF-β availability in the synchondrosis, and decreased TGF-β level may regulate parathyroid hormone-related protein expression either downstream of Indian hedgehog (Ihh) or independently of Ihh signaling.45
SPARC-Related Modular Calcium-Binding Protein 2 (SMOC2)
Secreted protein acidic and rich in cysteine (SPARC)-related modular calcium-binding protein 2 (SMOC2) is an early dental developmental gene in human beings. It belongs to a family of BM-40 matricellular proteins which is known as SPARC, and regulates interactions between cells and the extracellular matrix. SMOC2 expression was found in the oral ectoderm and the outer dental epithelium at E14.5 in mouse and in mesenchymal papilla facing the epithelial loops of molars and the only lingual loop of incisors.46 Recently, by genome-wide scan and Sanger sequencing, a homozygous splicing mutation was detected in a highly consanguineous Turkish family which manifested extreme microdontia, oligodontia, and dental dysplasia type I.46 The unaffected parents and siblings were heterozygous for the mutation. A second nonsense SMOC2 mutation was reported in a consanguineous Pakistan family with autosomal recessive oligodontia and microdontia.47 Although the function of SMOC2 in mammalian development has yet to be fully investigated, there are some evidences that it may affect the receptor-mediated signaling of many growth factors. Knockdown of SMOC2 in zebrafish showed that the first two bilateral teeth were smaller than those of the controls at day 5, the size and presence of the teeth were probably dependent on the level of SMOC2 depletion, and affected pharyngeal teeth development which had abnormalities resemble of the human phenotype. Expression of three major odontogenesis genes altered: DLX2, BMP2, and PITX2,47 suggesting that SMOC2 has an evolutionarily conserved role in tooth and oropharyngeal development.
Other Genes
EDAR and EDARADD are two genes that can cause autosomal dominant as well as autosomal recessive HED and nonsyndromic tooth agenesis.48 49 50 Keratin 17 (KRT17) is expressed in the sebaceous gland, nail bed, hair shaft, and epidermal appendages. Mutations in KRT17 have been identified in patients with pachyonychia congenita type 2 and oligodontia.51 FGFR1, transmembrane receptor of fibroblast growth factors (FGFs), one of the genes that is also involved in human craniosynostosis, has been associated with families of nonsyndromic hypodontia, in particular, premolar agenesis.52 FGFR1 is broadly expressed in the facial primordia and plays important roles in advancing skeletogenesis by regulating osteoblast and chondroblast differentiation.52
Syndromic Tooth Agenesis and Genes
More than 200 CL/CLP syndromes exhibit varying levels of hypodontia as part of their phenotypes. The prevalence of hypodontia increases with cleft severity, and upper lateral incisor is the most frequently affected tooth in the cleft area both in primary and in permanent dentitions. A higher incidence of dental agenesis outside the cleft region, more in particular involving the permanent maxillary dentition has also been reported. At present, several syndromic CLP-specific gene mutations have been identified (Table 2); however, the role of these genes in the development of dental agenesis remains unclear.
Van der Woude Syndrome
Van der Woude syndrome (VWS) is one of the most common human autosomal dominant disorders associated with CL/CLP (∼1% of syndromic CL/CLP cases), characterized by lower lip pits, CL/CLP, and hypodontia (in 70% of cases). Mutations in interferon regulatory factor 6 (IRF6) have been identified in more than 100 unrelated families with VWS. IRF6 encodes a transcription factor that is highly expressed in a variety of embryonic craniofacial tissues, including the medial edges of the fusing palatal processes, tooth buds, hair follicles, and skin, and it has been suggested that IRF6 mediates interactions between members of the TGF-β superfamily of signaling peptides.53
P63 Syndromes
P63 gene mutations have been found in several oral clefting syndromes, which are namely P63 syndromes. Ectrodactyly–ED–clefting (EEC) syndrome is a rare autosomal dominantly inherited condition characterized by split hands and feet, ED, and CLP. Additional EEC features include lacrimal duct anomalies, urogenital defects, conductive hearing loss, chronic respiratory infections, ventricular cardiomyopathy, and developmental delay. Ankyloblepharon ectodermal defects CL/CLP syndrome, limb mammary syndrome, acro-dermato-ungual-lacrimal-tooth syndrome, and Rapp–Hodgkin syndrome are EEC-like conditions and allelic to the EEC syndrome. Several different mutations of P63 gene have been revealed in these families.54
CLP–Ectodermal Dysplasia Syndrome
A homozygous loss-of-function mutation in the PVRL1 gene has been linked to CLPED-1, an autosomal recessive CLP–ED syndrome. Patients with CLPED-1 have scanty eyebrows, sparse and dry hair, syndactyly of the fingers and toes, CL/CLP, nail dysplasia, hypodontia of the upper central incisors, and abnormal size and shape of tooth crowns.55
Ectodermal Dysplasia Syndromes
The term ED is used to designate a heterogenous group of disorders characterized by a constellation of findings involving a primary defect of the skin, teeth, and appendageal structures including hair, nail, exocrine, and sebaceous glands. The different types of ED are caused by the mutation or deletion of certain genes located on different chromosomes.
HED is the most common ED, usually inherited as an X-linked recessive trait, or inherited in autosomal dominant or recessive forms. X-linked recessive HED is caused by mutations in EDA gene. The protein product, EDA, is a novel member of the TNF family and functions as a signaling molecule during epithelial morphogenesis.48 Autosomal dominant and recessive forms of HED are caused by mutations in EDAR or EDARADD coding for a TNF receptor.
Incontinentia pigmenti (IP) is a rare multisystem disorder classified as an ED condition with additional ocular and central nervous anomalies. IP is an X-linked dominant disorder due to mutations in NEMO gene. Mostly, females are affected as males with the mutation usually do not survive through gestation. More than 90% of patients with IP have associations of oligodontia, microdontia, and supernumerary cusps in the posterior permanent teeth, delayed tooth eruption, and/or taurodontia. A distinct form of HED with immunodeficiency segregates as an X-linked recessive trait, which is allelic to IP. NEMO, the protein in focus, is required for activation of NF-κB, a transcription factor transducing TNF signaling.
Witkop syndrome is another autosomal dominant ED characterized by nail dysplasia, severe hypodontia, and conical teeth. Mutations in MSX1 gene have been identified in several unrelated families with Witkop syndrome.10
Other Syndromes
Tooth agenesis not only appears in some common syndromes but also in several syndromes with rather low prevalence, such as Rieger syndrome,56 Ellis–van Creveld syndrome,57 and Sotos syndrome (Table 2).
Conclusion
In mammals, tooth development is governed by a sequential and reciprocal signaling process between two adjacent tissues: the primitive epithelium lining the stomodeum and mesenchymal cells arising from cranial neural crest cells.58 Therefore, genes implicated in epithelial–mesenchymal interactions during different stages of odontogenesis serve as good candidates for tooth agenesis. A variety of dental anomalies, either morphologically, numerically, or structurally may be due to abnormal function of these specific proteins. Recent years, several gene mutations have been detected to be responsible for the tooth agenesis; however, known mutations explain only a restricted number of cases of agenesis, little is known about the genetic defects underlying this complex condition. Oral clefts and syndromic forms of tooth agenesis may be the best models for isolated tooth agenesis. New techniques such as next-generation sequencing are already available for the early diagnosis of mutations that imply a risk of developing genetic diseases. These would bring new possibilities of early diagnosis and treatment.
References
- 1.Peters H, Balling R. Teeth. Where and how to make them. Trends Genet. 1999;15(2):59–65. doi: 10.1016/s0168-9525(98)01662-x. [DOI] [PubMed] [Google Scholar]
- 2.Line S RP. Molecular morphogenetic fields in the development of human dentition. J Theor Biol. 2001;211(1):67–75. doi: 10.1006/jtbi.2001.2333. [DOI] [PubMed] [Google Scholar]
- 3.Matalova E, Fleischmannova J, Sharpe P T, Tucker A S. Tooth agenesis: from molecular genetics to molecular dentistry. J Dent Res. 2008;87(7):617–623. doi: 10.1177/154405910808700715. [DOI] [PubMed] [Google Scholar]
- 4.Rølling S, Poulsen S. Agenesis of permanent teeth in 8138 Danish schoolchildren: prevalence and intra-oral distribution according to gender. Int J Paediatr Dent. 2009;19(3):172–175. doi: 10.1111/j.1365-263X.2008.00958.x. [DOI] [PubMed] [Google Scholar]
- 5.Vieira A R. Oral clefts and syndromic forms of tooth agenesis as models for genetics of isolated tooth agenesis. J Dent Res. 2003;82(3):162–165. doi: 10.1177/154405910308200303. [DOI] [PubMed] [Google Scholar]
- 6.Maas R, Bei M. The genetic control of early tooth development. Crit Rev Oral Biol Med. 1997;8(1):4–39. doi: 10.1177/10454411970080010101. [DOI] [PubMed] [Google Scholar]
- 7.Vastardis H. The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop. 2000;117(6):650–656. [PubMed] [Google Scholar]
- 8.Thesleff I. The genetic basis of tooth development and dental defects. Am J Med Genet A. 2006;140(23):2530–2535. doi: 10.1002/ajmg.a.31360. [DOI] [PubMed] [Google Scholar]
- 9.De Coster P J, Marks L A, Martens L C, Huysseune A. Dental agenesis: genetic and clinical perspectives. J Oral Pathol Med. 2009;38(1):1–17. doi: 10.1111/j.1600-0714.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 10.Nieminen P. Genetic basis of tooth agenesis. J Exp Zoolog B Mol Dev Evol. 2009;312B(4):320–342. doi: 10.1002/jez.b.21277. [DOI] [PubMed] [Google Scholar]
- 11.Thomas B L, Sharpe P T. Patterning of the murine dentition by homeobox genes. Eur J Oral Sci. 1998;106 01:48–54. doi: 10.1111/j.1600-0722.1998.tb02153.x. [DOI] [PubMed] [Google Scholar]
- 12.Lidral A C, Reising B C. The role of MSX1 in human tooth agenesis. J Dent Res. 2002;81(4):274–278. doi: 10.1177/154405910208100410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6(4):348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 14.van den Boogaard M J, Dorland M, Beemer F A, van Amstel H K. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24(4):342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- 15.Chishti M S, Muhammad D, Haider M, Ahmad W. A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J Hum Genet. 2006;51(10):872–878. doi: 10.1007/s10038-006-0037-x. [DOI] [PubMed] [Google Scholar]
- 16.Vastardis H, Karimbux N, Guthua S W, Seidman J G, Seidman C E. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13(4):417–421. doi: 10.1038/ng0896-417. [DOI] [PubMed] [Google Scholar]
- 17.Stockton D W, Das P, Goldenberg M, D'Souza R N, Patel P I. Mutation of PAX9 is associated with oligodontia. Nat Genet. 2000;24(1):18–19. doi: 10.1038/71634. [DOI] [PubMed] [Google Scholar]
- 18.Peters H, Neubüser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12(17):2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kist R, Watson M, Wang X. et al. Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum Mol Genet. 2005;14(23):3605–3617. doi: 10.1093/hmg/ddi388. [DOI] [PubMed] [Google Scholar]
- 20.Kapadia H, Frazier-Bowers S, Ogawa T, D'Souza R N. Molecular characterization of a novel PAX9 missense mutation causing posterior tooth agenesis. Eur J Hum Genet. 2006;14(4):403–409. doi: 10.1038/sj.ejhg.5201574. [DOI] [PubMed] [Google Scholar]
- 21.Suda N, Ogawa T, Kojima T, Saito C, Moriyama K. Non-syndromic oligodontia with a novel mutation of PAX9. J Dent Res. 2011;90(3):382–386. doi: 10.1177/0022034510390042. [DOI] [PubMed] [Google Scholar]
- 22.Mostowska A, Kobielak A, Trzeciak W H. Molecular basis of non-syndromic tooth agenesis: mutations of MSX1 and PAX9 reflect their role in patterning human dentition. Eur J Oral Sci. 2003;111(5):365–370. doi: 10.1034/j.1600-0722.2003.00069.x. [DOI] [PubMed] [Google Scholar]
- 23.Ogawa T, Kapadia H, Feng J Q, Raghow R, Peters H, D'Souza R N. Functional consequences of interactions between Pax9 and Msx1 genes in normal and abnormal tooth development. J Biol Chem. 2006;281(27):18363–18369. doi: 10.1074/jbc.M601543200. [DOI] [PubMed] [Google Scholar]
- 24.Nakatomi M, Wang X P, Key D. et al. Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev Biol. 2010;340(2):438–449. doi: 10.1016/j.ydbio.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 25.Paixão-Côrtes V R, Braga T, Salzano F M, Mundstock K, Mundstock C A, Bortolini M C. PAX9 and MSX1 transcription factor genes in non-syndromic dental agenesis. Arch Oral Biol. 2011;56(4):337–344. doi: 10.1016/j.archoralbio.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J, Gao Y, Zhang Z. et al. Osr2 acts downstream of Pax9 and interacts with both Msx1 and Pax9 to pattern the tooth developmental field. Dev Biol. 2011;353(2):344–353. doi: 10.1016/j.ydbio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Dong X, Mai M. et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26(2):146–147. doi: 10.1038/79859. [DOI] [PubMed] [Google Scholar]
- 28.Lammi L, Arte S, Somer M. et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callahan N, Modesto A, Meira R, Seymen F, Patir A, Vieira A R. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch Oral Biol. 2009;54(1):45–49. doi: 10.1016/j.archoralbio.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marvin M L, Mazzoni S M, Herron C M, Edwards S, Gruber S B, Petty E M. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A. 2011;155A(4):898–902. doi: 10.1002/ajmg.a.33927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong S, Liu H, Bai B. et al. Novel missense mutations in the AXIN2 gene associated with non-syndromic oligodontia. Arch Oral Biol. 2014;59(3):349–353. doi: 10.1016/j.archoralbio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Kere J, Srivastava A K, Montonen O. et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13(4):409–416. doi: 10.1038/ng0895-409. [DOI] [PubMed] [Google Scholar]
- 33.Fan H, Ye X, Shi L. et al. Mutations in the EDA gene are responsible for X-linked hypohidrotic ectodermal dysplasia and hypodontia in Chinese kindreds. Eur J Oral Sci. 2008;116(5):412–417. doi: 10.1111/j.1600-0722.2008.00555.x. [DOI] [PubMed] [Google Scholar]
- 34.van der Hout A H, Oudesluijs G G, Venema A. et al. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur J Hum Genet. 2008;16(6):673–679. doi: 10.1038/sj.ejhg.5202012. [DOI] [PubMed] [Google Scholar]
- 35.Song S, Han D, Qu H. et al. EDA gene mutations underlie non-syndromic oligodontia. J Dent Res. 2009;88(2):126–131. doi: 10.1177/0022034508328627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adaimy L, Chouery E, Megarbane H. et al. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet. 2007;81(4):821–828. doi: 10.1086/520064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bohring A, Stamm T, Spaich C. et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet. 2009;85(1):97–105. doi: 10.1016/j.ajhg.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kantaputra P, Sripathomsawat W. WNT10A and isolated hypodontia. Am J Med Genet A. 2011;155A(5):1119–1122. doi: 10.1002/ajmg.a.33840. [DOI] [PubMed] [Google Scholar]
- 39.van den Boogaard M J, Créton M, Bronkhorst Y. et al. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 2012;49(5):327–331. doi: 10.1136/jmedgenet-2012-100750. [DOI] [PubMed] [Google Scholar]
- 40.Plaisancié J, Bailleul-Forestier I, Gaston V. et al. Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am J Med Genet A. 2013;161A(4):671–678. doi: 10.1002/ajmg.a.35747. [DOI] [PubMed] [Google Scholar]
- 41.Arzoo P S, Klar J, Bergendal B, Norderyd J, Dahl N. WNT10A mutations account for ¼ of population-based isolated oligodontia and show phenotypic correlations. Am J Med Genet A. 2014;164A(2):353–359. doi: 10.1002/ajmg.a.36243. [DOI] [PubMed] [Google Scholar]
- 42.Dabovic B, Chen Y, Colarossi C. et al. Bone abnormalities in latent TGF-[beta] binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-[beta] bioavailability. J Cell Biol. 2002;156(2):227–232. doi: 10.1083/jcb.200111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dabovic B, Chen Y, Colarossi C, Zambuto L, Obata H, Rifkin D B. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J Endocrinol. 2002;175(1):129–141. doi: 10.1677/joe.0.1750129. [DOI] [PubMed] [Google Scholar]
- 44.Dabovic B, Levasseur R, Zambuto L, Chen Y, Karsenty G, Rifkin D B. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone. 2005;37(1):25–31. doi: 10.1016/j.bone.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 45.Noor A, Windpassinger C, Vitcu I. et al. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am J Hum Genet. 2009;84(4):519–523. doi: 10.1016/j.ajhg.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bloch-Zupan A, Jamet X, Etard C. et al. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in SMOC2, causing major dental developmental defects. Am J Hum Genet. 2011;89(6):773–781. doi: 10.1016/j.ajhg.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alfawaz S, Fong F, Plagnol V, Wong F S, Fearne J, Kelsell D P. Recessive oligodontia linked to a homozygous loss-of-function mutation in the SMOC2 gene. Arch Oral Biol. 2013;58(5):462–466. doi: 10.1016/j.archoralbio.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 48.Bergendal B, Klar J, Stecksén-Blicks C, Norderyd J, Dahl N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A. 2011;155A(7):1616–1622. doi: 10.1002/ajmg.a.34045. [DOI] [PubMed] [Google Scholar]
- 49.Cluzeau C, Hadj-Rabia S, Jambou M. et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat. 2011;32(1):70–72. doi: 10.1002/humu.21384. [DOI] [PubMed] [Google Scholar]
- 50.Vieira A R, Meira R, Modesto A, Murray J C. MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J Dent Res. 2004;83(9):723–727. doi: 10.1177/154405910408300913. [DOI] [PubMed] [Google Scholar]
- 51.Ruf S, Klimas D, Hönemann M, Jabir S. Genetic background of nonsyndromic oligodontia: a systematic review and meta-analysis. J Orofac Orthop. 2013;74(4):295–308. doi: 10.1007/s00056-013-0138-z. [DOI] [PubMed] [Google Scholar]
- 52.Chhabra N, Goswami M, Chhabra A. Genetic basis of dental agenesis—molecular genetics patterning clinical dentistry. Med Oral Patol Oral Cir Bucal. 2014;19(2):e112–e119. doi: 10.4317/medoral.19158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye X Q, Jin H X, Shi L S. et al. Identification of novel mutations of IRF6 gene in Chinese families with Van der Woude syndrome. Int J Mol Med. 2005;16(5):851–856. [PubMed] [Google Scholar]
- 54.Yin W, Ye X, Shi L. et al. TP63 gene mutations in Chinese P63 syndrome patients. J Dent Res. 2010;89(8):813–817. doi: 10.1177/0022034510366804. [DOI] [PubMed] [Google Scholar]
- 55.Sözen M A, Suzuki K, Tolarova M M, Bustos T, Fernández Iglesias J E, Spritz R A. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat Genet. 2001;29(2):141–142. doi: 10.1038/ng740. [DOI] [PubMed] [Google Scholar]
- 56.Semina E V, Reiter R, Leysens N J. et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14(4):392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 57.Ye X, Song G, Fan M. et al. A novel heterozygous deletion in the EVC2 gene causes Weyers acrofacial dysostosis. Hum Genet. 2006;119(1–2):199–205. doi: 10.1007/s00439-005-0129-2. [DOI] [PubMed] [Google Scholar]
- 58.Jernvall J, Thesleff I. Tooth shape formation and tooth renewal: evolving with the same signals. Development. 2012;139(19):3487–3497. doi: 10.1242/dev.085084. [DOI] [PubMed] [Google Scholar]