

Abstract
Microtia is a genetic condition affecting the external ears and presents clinically along a wide spectrum: minimally affected ears are small with minor shape abnormalities; extremely affected ears lack all identifiable structures, with the most extreme being absence of the entire external ear. Multiple genetic causes have been linked to microtia in both animal models and humans, which are improving our understanding of the condition and may lead to the identification of a unified cause for the condition. Microtia is also a prominent feature of several genetic syndromes, the study of which has provided further insight into the possible causes and genetic mechanisms of the condition. This article reviews our current understanding of microtia including epidemiological characteristics, classification systems, environmental and genetic causative factors leading to microtia. Despite our increased understanding of the genetics of microtia, we do not have a means of preventing the condition and still rely on complex staged, surgical correction.
Keywords: microtia, genetic causes, gene, anotia, embryology
Introduction
Microtia encompasses a spectrum of congenital anomalies of the auricle that range in severity from mild partial structural abnormalities to complete absence of the ear (anotia).1 Currently, there is no consensus on the terminology that should be used to describe and classify this condition. Some authors prefer to use the term “microtia”2 3 4 5 while others use “microtia–anotia” or “microtia/anotia.”6 7 8 9 10 The term “microtia” includes anotia (complete absence of the ear) as the most severe end of the microtia spectrum for the purpose of this review.
The condition presents with aesthetic concerns for the child and parents and can lead to severe psychological sequelae secondary to the condition itself and the complexity of surgical treatments to correct associated anomalies as well as the ear deformity. Because microtia is associated with absence of the external ear canal, the condition may present with hearing problems, and if the clinical presentation is bilateral, may be associated with significant language delays.
Embryology and Anatomy
The external ear is composed of several important landmarks that may be affected to varying degrees in microtia and are depicted in Fig. 1. The external ear begins to develop during the 5th week of gestation from the first and second pharyngeal arches on the ventral surface of the embryo.5 11 Mesenchymal cells of mesodermal and cranial neural crest origin are the primary cell type in the pharyngeal arches. Reciprocal signaling between neural crest cells (NCC) and the craniofacial ectoderm plays an important role in driving facial development, including that of the external ear.12 13 The historically accepted embryological pattern for ear development has been that the pharyngeal arches give rise to six hillocks that form the major anatomic structures of the ear. Hillocks one to three arise from the first arch and form the tragus, helical root, and helix. The second arch gives rise to hillocks four to six which comprise the antihelix, concha, and antitragus (Fig. 2). During the 7th week of gestation, the auricular hillocks differentiate, enlarge, and eventually fuse. This morphogenesis determines the final auricular shape, size, and position. The ears gradually move from their initial position low on the neck to their final more cranial position.11
Fig. 1.
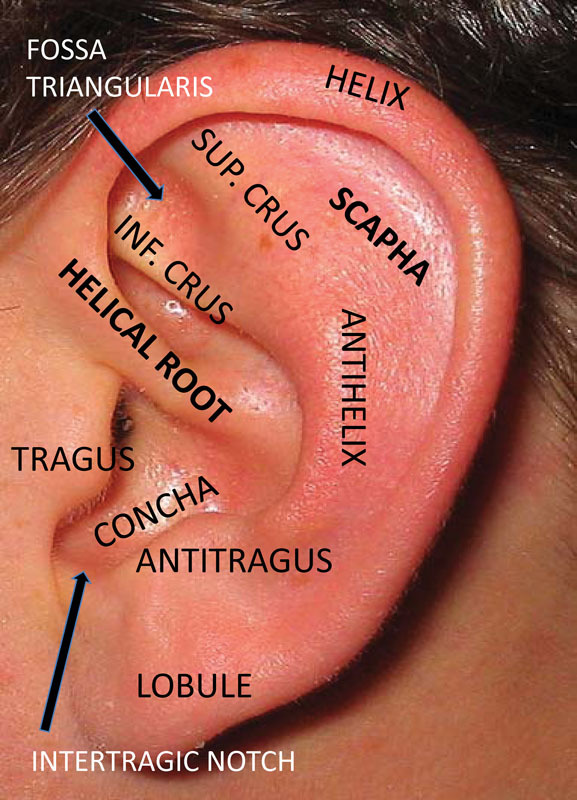
Normal anatomy of the external ear.
Fig. 2.
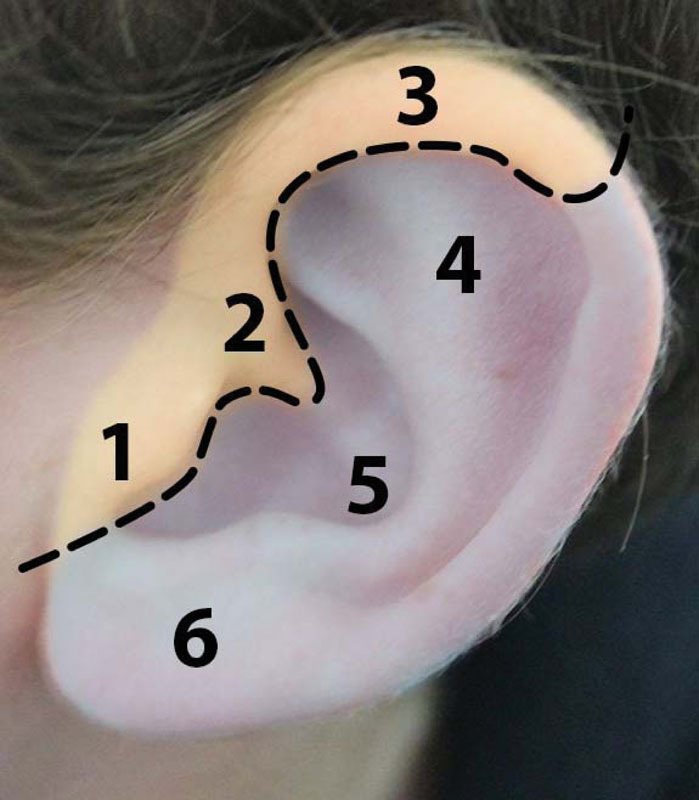
Yellow shading indicates embryological origin from branchial arch 1: 1, tragus; 2, helical root; 3, helix. Light purple indicates a branchial arch 2 origin: 4, antihelix; 5, antitragus; 6, lobule.
Recent reports, however, raise doubts as to the actual origin of the major anatomic subunits of the ear including which tissues actually give rise to the external auditory canal (EAC). Minoux et al found that the mouse pinna derives from the HOXA2-expressing neural crest–derived mesenchyme of the second pharyngeal arch, and not from a composite of first and second arch mesenchyme as previously proposed using genetic fate mapping.14 These authors also demonstrated that the mouse EAC is entirely lined by HOXA2-negative first arch mesenchyme and does not develop at the first pharyngeal cleft, as previously assumed. Given these new findings, a reorganization of our understanding of the embryonic origin of the anatomical structures of the ear is underway.
Incidence
Population-based studies on the incidence of microtia in Italy, France, Sweden, Finland, and the United States range between 0.83 and 4.34 per 10,000 births.3 7 8 9 10 13 Studies conducted using nonpopulation-based data reported higher rates for Ecuadorians,2 15 Chileans, and among Native Americans in the United States.16 17 18 Among U.S. Native Americans, the overall incidence is between 1/4,000 and 1/6,500 live births. This is even higher in the Navajo population. In a study of 15,890 Navajo Indians, 1:935 were found to have microtia, specifically clustered to one quarter of the Western reservation.18 Differing rates between studies may be due to under- or overreporting in hospital records because there is no standard definition or classification system used for what constitutes microtia—not at the extreme end (anotia), which is easy to identify clinically, but at the less extreme end, where the auricle is small with more limited structural abnormalities.
Classification Systems
Many classification systems have been proposed for ear abnormalities on the microtia spectrum, the first of which was proposed by Hermann Marx in 1926. In 1978, Tanzer attempted to classify the anomalies according to their surgical correction.19 Finally, Weerda modified and combined these classifications and added embryologic development as part of the metric for organization.13 20 Hunter et al published a standardized terminology article in the American Journal of Genetics in an attempt to improve reporting on microtia and chose the Weerda classification system as the basis for their proposed classification system (Table 1; Fig. 3).
Table 1. Summary of Weerda and Hunter et al classifications for microtia.
| Weerda classification | Hunter et al classification |
|---|---|
| First degree dysplasia | Microtia, first degree |
| Most structures of a normal auricle are recognizable (minor deformities): (A) macrotia, (B) protruding ears, (C) cryptoptia, (D) absence of upper helix, (E) Small deformities, (F) colobomata, (G) lobule deformities, (H) cup ear deformities | Presence of all the normal ear components and the median longitudinal length more than 2 SD below the mean |
| Second degree dysplasia | Microtia, second degree |
| Some structures of a normal auricle are recognizable: (A) cup ear deformity type III and (B) miniear | Median longitudinal length of the ear more than 2 SD below the mean in the presence of some, but not all, parts of the normal ear |
| Third degree dysplasia | Microtia, third degree |
| None of the structures of a normal auricle are recognizable: (A) unilateral, (B) bilateral, and (C) anotia (peanut ears are included in this group) | Presence of some auricular structures, but none of these structures conforms to recognized ear components |
| Anotia | |
| Complete absence of the ear |
Abbreviation: SD, standard deviation.
Fig. 3.

Degrees of dysplasia associated with microtia: Anotia (A), with variations of third degree dysplasia (B and C); second degree dysplasia (D–F), and first degree dysplasia, including cup ear (G) and cryptotia (H).
Despite the difficulties associated with accurate classification of the range of presentations in microtia, it is imperative to adopt a clear classification system to assist with data collection, practitioner communication, and our understanding of the genetic links to variations in presentation of microtia.
Risk Factors for Developing Microtia
Vascular Disruption
Though vascular disruption as an explanation for the development of microtia is losing ground, there continue to be adherents of this theory.21 Localized ischemia and tissue necrosis may develop with disruption in the vascular supply leading to malformation of the auricular tissues. This may arise by several mechanisms including underdevelopment of the arterial system to specific tissues, occlusion of the vessels secondary to compression or hemorrhage, or by means of vasoconstriction secondary to a pharmacological or genetic cause.
Poswillo conducted the key study suggesting vascular disruption as a cause for craniofacial deformities in monkeys and mice exposed to thalidomide and triazine, respectively. They showed that these exposures led to ipsilateral hematomas at the junction of the pharyngeal and hyoid arteries with associated unilateral ear and mandibular defects.22 23 Further supporting this concept, a transgenic mouse line carrying a nonexpressed transgene was noted to develop a phenotype similar to hemifacial microsomia.24 25 These authors reported rupture of the vasculature of the second pharyngeal arch with histologically confirmed hemorrhage and subsequent phagocytosis. However, a causative gene has not been identified.
Several authors refute the vascular disruption theory. A reassessment of Poswillo's work by Johnston and Bronsky led them to conclude that the hematomas occurred too late in relation to drug delivery, at a time after which the underdevelopment of tissues was already present.26 Furthermore, they argue, the vascular disruption of a single vessel in the head and neck region could not adequately explain cases of bilateral microtia and the occurrence of microtia with other noncraniofacial malformations affecting the hands, kidneys, and heart.
Environmental Factors
Rick factors for developing microtia include maternal illness and anemia during pregnancy, diabetes, and maternal race7 8; high maternal or paternal age2 7 9; and multiple births.8 9 A Japanese study analyzing 592 patients with microtia found 28% of the patients' mothers had a cold, imminent spontaneous abortion, or anemia during pregnancy.27 A study conducted in Latin America found acute maternal illness during the first trimester to be a major risk factor in the development of microtia in that patient population.28 Mothers with chronic type I diabetes are at significantly higher risk for having a child with microtia. In patients with microtia, low birth weight is more common than in healthy children.4 6 29
There is strong evidence for association between microtia and exposure to the teratogens retinoic acid (RA), thalidomide,1 and mycophenolate mofetil.30 31 In mothers exposed to isotretinoin, 83% of pregnancies result in infants with serious birth defects including microtia.32 Exposure to high altitude,2 16 33 and being of Hispanic, Asian, or Native American ethnicity2 8 9 10 15 34 are also established risk factors.
Genetic Factors
There are multiple studies involving animal models suggesting that particular genetic pathways cause microtia (Table 2). In an excellent review on the genetics of microtia, Luquetti et al34 propose that a genetic cause for microtia is suggested by five observations: (1) higher concordance in monozygotic twins (38.5%) than in dizygotic twins (4.5%),35 (2) estimates of familial cases ranging from 3 to 34%,2 6 27 36 (3) reports of familial cases with autosomal recessive or dominant modes of inheritance with variable expression and incomplete penetrance,37 38 39 40 41 42 43 44 45 46 47 48 49 (4) more than 18 distinct microtia-associated syndromes for which single-gene defects or chromosomal aberrations have been reported, and (5) mouse models demonstrating that mutations in specific genes HOXA2, SIX and eyes absent (EYA), TBX1, IRF6, and CHUK result in microtia.
Table 2. Genes identified in animal studies with microtia as a major feature.
| Animal model | Gene(s) identified |
|---|---|
| Mouse | HOXA1, HOXA2, HOXB1, SIX1–4, TBX1, IRF6, CHUK, EYA, SALL1, Prx1, Prx 2, TCOF1, GSC, HMX1, BMP5, FGFR1, FGF8, FGF10, Wnt5a |
| Chick | HMX1, SIX1 |
| Frog | SIX1 |
| Rat | HMX1 |
| Cow | HMX1 |
Note: The underlined genes are also present in human studies.
Genetic Studies in Animals
Murine models have elucidated the mechanisms of aberrations in craniofacial development, including microtia. Auricular defects in these mice range from mild deformities to complete anotia (similar to the spectrum in humans). The Hox genes are a large group of homeobox genes, which express critical transcription factors in embryonic development and are strongly linked to microtia. NCC in the second branchial arch express HOXA2 over a prolonged period of time.4 50 HOXA2 knockout mice present with microtia.51 52 53 In addition, inactivation of HOXA1 in a mouse model results in hypoplastic external ears, and a combination of HOXA1 and HOXB1 knockout mice present with complete anotia.54
Members of the SIX homeobox gene family (SIX1–6) have been implicated in external ear development.55 SIX functions appear conserved across evolutionary development evidenced by the fact that knockout of SIX1 in frogs, chicks, and mice, all result in craniofacial abnormalities.56 57 58 SIX1 and SIX4 exert their effect via PAX3 gene expression, which controls the early steps of myogenic cell delamination and migration from the somite.59 Heterogeneous SIX1 to SIX4 mice present with microtia as well as severe rib anomalies.
EYA forms a complex with SIX (EYA–SIX) to regulate the development of several tissues and organs. Natural target genes of the EYA–SIX complex include SIX2 and (sal-like-1) SALL1. Studies on EYA1 expression have shown a major role in pinna development, apparently related to cartilage formation, while knockout mice for EYA1 present with anotia.
TBX1 is a member of the T-box gene family of transcription factors. Mutations in TBX1 result in failure of middle and outer ear development in a mouse model. Inactivation of TBX1 in the pharyngeal arch endoderm causes similar defects in outer ear formation indicating a primary role for this gene in pharyngeal arch morphogenesis.60
Mice that are homozygous null for IRF6 lack external ears in addition to exhibiting abnormal skin, limbs, and short snouts and jaws. A similar phenotype was observed in mice deficient for CHUK (also known as IKK-α). Abnormalities in the IRF6 and CHUK mice are secondary to defects in epidermal differentiation and cell proliferation.61 62 In the double mutant mice model for Prx1/Prx2 homeobox genes, defects in the external, middle, and inner ear, a lack of tympanic rings, cleft mandible, and polydactyly are noted.63 Treacher Collins–Franceschetti 1 (TCOF1) encodes a protein named treacle. Expression studies of TCOF1 in the mouse embryo support a role for treacle in the development of the craniofacial complex. Creation of a knockout mouse model for TCOF1 results in heterozygous mice with severe craniofacial malformations.4 64 Human goosecoid (GSC) is a homeodomain transcription factor and a downstream target of endothelin. Mice with a homozygous disruption of GSC have multiple developmental defects including the ear.65
HMX1 is a homeodomain transcription factor found in the developing mouse and chick nervous system and eye.66 HMX1 expression appears in the branchial arches in the mouse model.67 Mutations in the HMX1 locus have also been identified in rats and cows, leading to what appear to be isolated ear malformations. These isolated auricular malformations have been attributed to disruption of a conserved noncoding element downstream.
Signaling Pathways
Growth factors involved in signaling pathways of outer ear development include the bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs), RA, and wingless/INT (Wnts). Dysregulation of these signaling pathways can lead to malformations of the auricle. BMPs, especially BMP5, have been considered as candidate genes for microtia in humans; however, studies in mice have shown that BMP5 is more related to ear growth than the actual formation of the external ear.13 FGFR1–3 play various roles in pinna development.68 69 FGF8 and -10 mutant mice present with small outer ears.68 Mice homozygous for a hypomorphic FGFR1 allele present with very small ears and abnormal EACs.70 Retinoid embryopathy results in apoptosis of NCC before migration into the pharyngeal arches has been observed and may interfere with cell survival once present in the pharyngeal arches.71 Members of the Wnt family have been implicated in NCC formation and development. It has been shown that Wnt5a is expressed in the mesenchyme of the developing outer ear and Wnt5a knockout mice present with small ears.72
Human Studies in Microtia
A wide range of patients with microtia (15–60%) present with additional abnormalities.2 4 8 10 13 15 Among 5 million live and still births, 818 cases were identified as having at least one major associated congenital anomaly; these findings form the basis for defining specific syndromes. The most frequent simultaneous dysmorphic features associated with microtia are cleft palate (12.8%), cleft lip and palate (11.5%), anophthalmia/microphthalmia (11.5%), facial asymmetry (10.6%), macrostomia (6.4%), preaxial polydactyly (2.2%), holoprosencephaly (2.2%), and epibulbar dermoids (1.7%).34
The most common syndromes associated with microtia are oculoauriculovertebral spectrum (OAVS), Goldenhar syndrome (GS)/hemifacial microsomia/craniofacial microsomia (CFM), Treacher Collins, Nager, DiGeorge, or 22qdeletion syndrome, Townes–Brock syndrome (TBS), and branchio-oto-renal (BOR) syndrome.4 13
Oculoauriculovertebral Spectrum
OAVS is the most extensively studied syndrome associated with microtia. The reader is referred to the article recently published by Beleza-Meireles et al for a comprehensive review of all available genetic literature on OAVS.73 OAVS is a complex heterogeneous disorder involving the first and second branchial arch derivatives. The OAVS is broad, with anomalies including facial asymmetry resulting from maxillary and/or mandibular hypoplasia; preauricular or facial tags; ear malformations such as microtia, anotia, or aural atresia; and hearing loss. In a Turkish population with GS, microtia was present in 52% of patients.4 74
Forty percent of patients with OAVS show a strong allelic expression of BAPX1, a gene that belongs to the NK-2 family of transcription factors and plays an essential role in craniofacial development.75 BAPX1 anomalies are present in patient fibroblasts, suggesting that epigenetic dysregulation of BAPX1 plays an important role in this syndrome.76
Goldenhar Syndrome/Hemifacial Microsomia/Craniofacial Microsomia
CFM is a congenital condition characterized by asymmetric hypoplasia of the craniofacial structures, most commonly including the mandible and ear.77 Other clinical features for diagnosis include cranial nerve palsies, epibulbar dermoids, maxillary hypoplasia, soft tissue deficiency, orbital asymmetry, and extracranial malformations.78 79 Heterozygous mutations in the EFTUD2 gene have been identified in a subgroup of patients with mandibulofacial dysostosis with microcephaly that overlaps with CFM.
A suggestive linkage to a region on chromosome 14q32 was found by genome-wide linkage analysis in two families with features of CFM.80 The most interesting candidate gene in the linked region was GSC. No disease-causing mutation in the coding region of the gene analyzed by Southern blotting could be identified in these two families or in 120 sporadic cases of CFM.78
Treacher Collins Syndrome
Treacher Collins syndrome is an autosomal dominant disorder that presents phenotypically with hypoplastic facial bones, microtia, micrognathia, cleft palate, and hearing abnormalities.64 81 Mutations in the TCOF1 gene have been identified as the cause of Treacher Collins syndrome in up to 78% of patients.82 83 84 85 TCOF1 encodes a protein called treacle, which plays an active role in the early embryonic development in structures that become bones and other tissues of the face.
Nager Syndrome
Nager syndrome presents with micrognathia, external ear defects, EAC stenosis, bilateral conductive hearing loss, cleft palate, down-slanting palpebral fissures, a high nasal bridge, hypoplastic or absent thumbs, and variable lower limb and toe defects.86 87 88 Most cases of Nager syndrome are sporadic, although both autosomal recessive and dominant familial cases have been reported.89 90 DNA sequencing of eight patients with Nager syndrome for a possible mutation in either of the PRX1 and PRX2 genes was performed, but no pathogenic variant was found.91
DiGeorge Syndrome
DiGeorge syndrome is one of the most common presentations of microdeletion associated with 22.q11.2 deletion syndrome. In most cases, the deletion eliminates 3 mbp of DNA encoding for approximately 30 genes.92 Specifically, the human TBX1 gene, which is required for ear development and is expressed in multiple tissues during embryogenesis, is deleted.93 Features of DiGeorge syndrome include ear defects, hearing impairment, craniofacial abnormalities, thymus and parathyroid gland hypoplasia, and heart malformations.94 The ears are typically low-set, small and with abnormal folding of the pinna.
Townes–Brock Syndrome
TBS is a rare autosomal dominant syndrome with a combination of anal, renal, limb, and ear anomalies. TBS is caused by mutations in the SALL1 gene on chromosome 16q.4 TBS and GS have a significant number of overlapping features, including first and second arch defects and preaxial defects of the upper limbs. The phenotypic similarities between TBS and GS suggest that they may have a common genetic etiology.95
Branchio-oto-renal Syndrome
Branchio-otic syndrome (BOS) is an autosomal dominant developmental disorder characterized by branchial cleft cysts, auricular or EAC abnormalities, preauricular pits, and hearing loss. BOR syndrome is diagnosed when BOS is accompanied by additional malformations of the kidney or urinary tract.96 97 98 Mutations in SIX1 and EYA1 have been shown to cause BOS, while mutations in SIX5 and EYA1 can cause BOR syndrome. Both are associated with microtia, among several other craniofacial defects.99 100 101 102 103
Four genetic loci have been mapped for BOS/BOR4: BOR1,100 BOR2,104 BOS2,101 and BOS3.102 Except for BOS2, the corresponding genes have been identified. EYA1 was the first gene identified for BOR syndrome at the BOR1 locus100 and is found in approximately 40% of cases.98 The gene SIX5 has been cloned for the BOR2 locus and missense mutations were identified in SIX5 in 5.2% (5/95) of the patients with BOR.104 SIX1 is the responsible gene at the BOS3 locus.97
Genetic Studies in Humans
Microtia has been reported in individuals with trisomies 21 and 22, as well as with mosaicism of trisomies 13 and 18105 106; and in deletions of 4p, 5p; 18p, 18q; and 22q11.2.13 Chromosomal translocations involving the 6p24 region have also been associated with bilateral microtia and orofacial clefting.107 Microtia, usually anotia, occurs in approximately two-thirds of all patients who have a terminal deletion of 18q. The extent and nature of the chromosome 18 deletions has been studied by array comparative genomic hybridization and a critical region of 5 Mb was deleted in all patients with anotia on 18q22.3–18q23, making this a candidate chromosomal region for anotia.108
A missense mutation in exon 3 of GSC was found by sequence analysis in 2/121 patients with isolated microtia. In the same study, screening of the BMP5 locus revealed a missense mutation in four patients. None of these mutations were detected in control subjects, suggesting a potential causative role in the development of microtia (Table 3).109
Table 3. Genes identified in human syndromes with microtia as a major feature.
| Human syndrome | Gene(s) identified |
|---|---|
| Oculoauriculovertebral spectrum | HMX1 |
| Treacher Collins | TCOF1, POL1RC, POL1RD |
| Craniofacial Microsomia | GSC |
| Branchio-oto | SIX1, EYA1 |
| Branchio-oto-renal | SIX5, EYA1 |
| Townes–Brock | SALL1 |
| Nager | SF3B4 |
| Mandibulofacial dysostosis with microcephaly | EFTUD2 |
| Auriculocondylar | PLCB4, GNAI3 |
| CHARGE | CHD7 |
| Lacrimo-auriculo-dental-digital | FGFR2, FGFR3, FGF10 |
| Kabuki | MLL2, KDM6A |
| Fraser | FRAS1, FREM2, GRIP1 |
Note: The underlined genes are also present in animal studies.
The methylation status of the EYA1 gene promoter was analyzed in 64 individuals with microtia and 36 healthy controls. The methylation levels at this locus were significantly lower in individuals with microtia than in controls; based on this information, these authors suggest that hypomethylation may be related to the pathogenesis of microtia.110
A coding variant of the HMX1 gene underlies a recessive disorder referred to as oculoauricular syndrome which is characterized by malformations of the pinna accompanied by variable eye defects.111 112 The same HMX1 noncoding element, when present in different species (mouse, rats, cows, and humans), yields similar phenotypes, providing some of the strongest evidence to date for noncoding, regulatory elements playing an important role in these more “isolated” disease presentations.113 This finding also highlights that any noncoding mutations, in HMX1 or any other gene associated with syndromic microtia, could be sufficient to cause isolated microtia phenotypes in humans.
Current Genetic Hypotheses for Microtia
The most likely underlying cause for the development of microtia is a disturbance of NCC, although the exact mechanism(s) remain unknown. However, given the clinical heterogeneity of microtia, it is possible that different pathogenic processes affecting the NCC lead to the different grades of microtia (Fig. 3).13 In addition, defects in NCC function have been associated with numerous craniofacial syndromes.114 In Treacher Collins syndrome, TCOF1 mutations result in haploinsufficiency of the protein treacle, as discussed previously, leading to insufficient ribosome genesis, diminished cell proliferation, and increased neuroepithelial apoptosis. This process results in depletion of NCC precursors leading to a reduced number of cells migrating into the first and second pharyngeal arches which results in the most severe Treacher Collins phenotype, including severe, bilateral microtia.115
Apoptosis of NCC before migration into the pharyngeal arches has been observed in retinoid embryopathy and may interfere with cell survival once present in the pharyngeal arches. The endothelin signaling pathway, which regulates Hox gene expression, is also affected by retinoid exposure and may affect the positional identity of NCC within the pharyngeal arches. In diabetes, hyperglycemia causes downregulation of PAX3, which encodes a transcription factor critical for early NCC survival and migration.71 Finally, the effects of thalidomide may include downregulation of FGF8116 and BMP signaling,116 117 though direct antiangiogenic effects and oxidative stress are also postulated as independent mechanisms,118 119 that disrupt NCC.
Future Directions
The final common pathway in both environmental and genetic causes resulting in the development of microtia appears to involve disruption of the number, migration, and final position of NCC within the pharyngeal arches. Though many genetic causes for microtia have been identified, with the development of animal models that predictably result in either isolated microtia or a constellation of abnormalities that includes microtia, we still do not understand the abnormal processes well enough to control them. Future work must focus on treatment strategies for microtia in animal models that may eventually be translatable to humans.
References
- 1.Carey J C, Park A H, Muntz H R. Oxford, New York: Oxford University Press; 2006. External ear; pp. 329–338. [Google Scholar]
- 2.Castilla E E, Orioli I M. Prevalence rates of microtia in South America. Int J Epidemiol. 1986;15(3):364–368. doi: 10.1093/ije/15.3.364. [DOI] [PubMed] [Google Scholar]
- 3.Suutarla S, Rautio J, Ritvanen A, Ala-Mello S, Jero J, Klockars T. Microtia in Finland: comparison of characteristics in different populations. Int J Pediatr Otorhinolaryngol. 2007;71(8):1211–1217. doi: 10.1016/j.ijporl.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 4.Alasti F, Van Camp G. Genetics of microtia and associated syndromes. J Med Genet. 2009;46(6):361–369. doi: 10.1136/jmg.2008.062158. [DOI] [PubMed] [Google Scholar]
- 5.Hunter A, Frias J L, Gillessen-Kaesbach G, Hughes H, Jones K L, Wilson L. Elements of morphology: standard terminology for the ear. Am J Med Genet A. 2009;149A(1):40–60. doi: 10.1002/ajmg.a.32599. [DOI] [PubMed] [Google Scholar]
- 6.Mastroiacovo P, Corchia C, Botto L D, Lanni R, Zampino G, Fusco D. Epidemiology and genetics of microtia-anotia: a registry based study on over one million births. J Med Genet. 1995;32(6):453–457. doi: 10.1136/jmg.32.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris J, Källén B, Robert E. The epidemiology of anotia and microtia. J Med Genet. 1996;33(10):809–813. doi: 10.1136/jmg.33.10.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw G M, Carmichael S L, Kaidarova Z, Harris J A. Epidemiologic characteristics of anotia and microtia in California, 1989-1997. Birth Defects Res A Clin Mol Teratol. 2004;70(7):472–475. doi: 10.1002/bdra.20042. [DOI] [PubMed] [Google Scholar]
- 9.Forrester M B, Merz R D. Descriptive epidemiology of anotia and microtia, Hawaii, 1986-2002. Congenit Anom (Kyoto) 2005;45(4):119–124. doi: 10.1111/j.1741-4520.2005.00080.x. [DOI] [PubMed] [Google Scholar]
- 10.Canfield M A, Langlois P H, Nguyen L M, Scheuerle A E. Epidemiologic features and clinical subgroups of anotia/microtia in Texas. Birth Defects Res A Clin Mol Teratol. 2009;85(11):905–913. doi: 10.1002/bdra.20626. [DOI] [PubMed] [Google Scholar]
- 11.Schoenwolf G C, Bleyl S B, Brauer P R, Francis-West P H. New York: Churchill Livingstone; 2015. Development of the ears. In: Larsen's Human Embryology. 5th ed; pp. 473–487. [Google Scholar]
- 12.Noden D M, Trainor P A. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207(5):575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luquetti D V, Heike C L, Hing A V, Cunningham M L, Cox T C. Microtia: epidemiology and genetics. Am J Med Genet A. 2012;158A(1):124–139. doi: 10.1002/ajmg.a.34352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minoux M, Kratochwil C F, Ducret S. et al. Mouse Hoxa2 mutations provide a model for microtia and auricle duplication. Development. 2013;140(21):4386–4397. doi: 10.1242/dev.098046. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Andrade F, Lopez-Pulles R, Espín V H, Paz-y-Miño C. High altitude and microtia in Ecuadorian patients. J Neonatal Perinatal Med. 2010;3(2):109–116. [Google Scholar]
- 16.Jaffe B F. The incidence of ear diseases in the Navajo Indians. Laryngoscope. 1969;79(12):2126–2134. doi: 10.1288/00005537-196912000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Aase J M Tegtmeier R E Microtia in New Mexico: evidence for multifactorial causation Birth Defects Orig Artic Ser 197713(3A):113–116. [PubMed] [Google Scholar]
- 18.Nelson S M, Berry R I. Ear disease and hearing loss among Navajo children—a mass survey. Laryngoscope. 1984;94(3):316–323. doi: 10.1288/00005537-198403000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Tanzer R C. The constricted (cup and lop) ear. Plast Reconstr Surg. 1975;55(4):406–415. [PubMed] [Google Scholar]
- 20.Weerda H. Classification of congenital deformities of the auricle. Facial Plast Surg. 1988;5(5):385–388. doi: 10.1055/s-2008-1064778. [DOI] [PubMed] [Google Scholar]
- 21.Sadler T W, Rasmussen S A. Examining the evidence for vascular pathogenesis of selected birth defects. Am J Med Genet A. 2010;152A(10):2426–2436. doi: 10.1002/ajmg.a.33636. [DOI] [PubMed] [Google Scholar]
- 22.Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973;35(3):302–328. doi: 10.1016/0030-4220(73)90070-4. [DOI] [PubMed] [Google Scholar]
- 23.Poswillo D. Hemorrhage in development of the face. Birth Defects Orig Artic Ser. 1975;11(7):61–81. [PubMed] [Google Scholar]
- 24.Otani H, Tanaka O, Naora H. et al. Microtia as an autosomal dominant mutation in a transgenic mouse line: a possible animal model of branchial arch anomalies. Anat Anz. 1991;172(1):1–9. [PubMed] [Google Scholar]
- 25.Naora H, Kimura M, Otani H. et al. Transgenic mouse model of hemifacial microsomia: cloning and characterization of insertional mutation region on chromosome 10. Genomics. 1994;23(3):515–519. doi: 10.1006/geno.1994.1537. [DOI] [PubMed] [Google Scholar]
- 26.Johnston M C, Bronsky P T. Prenatal craniofacial development: new insights on normal and abnormal mechanisms. Crit Rev Oral Biol Med. 1995;6(4):368–422. doi: 10.1177/10454411950060040601. [DOI] [PubMed] [Google Scholar]
- 27.Okajima H, Takeichi Y, Umeda K, Baba S. Clinical analysis of 592 patients with microtia. Acta Otolaryngol Suppl. 1996;525:18–24. [PubMed] [Google Scholar]
- 28.Lopez-Camelo J S, Orioli I M. Heterogeneous rates for birth defects in Latin America: hints on causality. Genet Epidemiol. 1996;13(5):469–481. doi: 10.1002/(SICI)1098-2272(1996)13:5<469::AID-GEPI3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 29.Ewart-Toland A, Yankowitz J, Winder A. et al. Oculoauriculovertebral abnormalities in children of diabetic mothers. Am J Med Genet. 2000;90(4):303–309. [PubMed] [Google Scholar]
- 30.Ang G S, Simpson S A, Reddy A R. Mycophenolate mofetil embryopathy may be dose and timing dependent. Am J Med Genet A. 2008;146A(15):1963–1966. doi: 10.1002/ajmg.a.32420. [DOI] [PubMed] [Google Scholar]
- 31.Anderka M T, Lin A E, Abuelo D N, Mitchell A A, Rasmussen S A. Reviewing the evidence for mycophenolate mofetil as a new teratogen: case report and review of the literature. Am J Med Genet A. 2009;149A(6):1241–1248. doi: 10.1002/ajmg.a.32685. [DOI] [PubMed] [Google Scholar]
- 32.Stern R S Rosa F Baum C Isotretinoin and pregnancy J Am Acad Dermatol 198410(5 Pt 1):851–854. [DOI] [PubMed] [Google Scholar]
- 33.Castilla E E, Lopez-Camelo J S, Campaña H. Altitude as a risk factor for congenital anomalies. Am J Med Genet. 1999;86(1):9–14. doi: 10.1002/(sici)1096-8628(19990903)86:1<9::aid-ajmg3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 34.Luquetti D V, Cox T C, Lopez-Camelo J, Dutra MdaG, Cunningham M L, Castilla E E. Preferential associated anomalies in 818 cases of microtia in South America. Am J Med Genet A. 2013;161A(5):1051–1057. doi: 10.1002/ajmg.a.35888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Artunduaga M A, Quintanilla-Dieck MdeL, Greenway S. et al. A classic twin study of external ear malformations, including microtia. N Engl J Med. 2009;361(12):1216–1218. doi: 10.1056/NEJMc0902556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Llano-Rivas I, González-del Angel A, del Castillo V, Reyes R, Carnevale A. Microtia: a clinical and genetic study at the National Institute of Pediatrics in Mexico City. Arch Med Res. 1999;30(2):120–124. doi: 10.1016/s0188-0128(98)00023-2. [DOI] [PubMed] [Google Scholar]
- 37.Ellwood L C, Winter S T, Dar H. Familial microtia with meatal atresia in two sibships. J Med Genet. 1968;5(4):289–291. doi: 10.1136/jmg.5.4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konigsmark B W, Nager G T, Haskins H L. Recessive microtia, meatal atresia, and hearing loss. Report of a sibship. Arch Otolaryngol. 1972;96(2):105–109. doi: 10.1001/archotol.1972.00770090179002. [DOI] [PubMed] [Google Scholar]
- 39.Balci S. Familial microtia with meatal atresia in father and son. Turk J Pediatr. 1974;16(3):140–143. [PubMed] [Google Scholar]
- 40.Guizar-Vázquez J, Arredondo-Vega F, Rostenberg I, Manzano C, Armendares S. Microtia and meatal atresia in mother and son. Clin Genet. 1978;14(2):80–82. doi: 10.1111/j.1399-0004.1978.tb02110.x. [DOI] [PubMed] [Google Scholar]
- 41.Zankl M, Zang K D. Inheritance of microtia and aural atresia in a family with five affected members. Clin Genet. 1979;16(5):331–334. doi: 10.1111/j.1399-0004.1979.tb01011.x. [DOI] [PubMed] [Google Scholar]
- 42.Schmid M, Schröder M, Langenbeck U. Familial microtia, meatal atresia, and conductive deafness in three siblings. Am J Med Genet. 1985;22(2):327–332. doi: 10.1002/ajmg.1320220216. [DOI] [PubMed] [Google Scholar]
- 43.Strisciuglio P, Ballabio A, Parenti G. Microtia with meatal atresia and conductive deafness: mild and severe manifestations within the same sibship. J Med Genet. 1986;23(5):459–460. doi: 10.1136/jmg.23.5.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orstavik K H, Medbø S, Mair I W. Right-sided microtia and conductive hearing loss with variable expressivity in three generations. Clin Genet. 1990;38(2):117–120. doi: 10.1111/j.1399-0004.1990.tb03558.x. [DOI] [PubMed] [Google Scholar]
- 45.Gupta A, Patton M A. Familial microtia with meatal atresia and conductive deafness in five generations. Am J Med Genet. 1995;59(2):238–241. doi: 10.1002/ajmg.1320590223. [DOI] [PubMed] [Google Scholar]
- 46.Balci S, Boduroğlu K, Kaya S. Familial microtia in four generations with variable expressivity and incomplete penetrance in association with type I syndactyly. Turk J Pediatr. 2001;43(4):362–365. [PubMed] [Google Scholar]
- 47.Klockars T, Suutarla S, Kentala E, Ala-Mello S, Rautio J. Inheritance of microtia in the Finnish population. Int J Pediatr Otorhinolaryngol. 2007;71(11):1783–1788. doi: 10.1016/j.ijporl.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 48.Alasti F, Sadeghi A, Sanati M H. et al. A mutation in HOXA2 is responsible for autosomal-recessive microtia in an Iranian family. Am J Hum Genet. 2008;82(4):982–991. doi: 10.1016/j.ajhg.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chafai Elalaoui S, Cherkaoui Jaouad I, Rifai L, Sefiani A. Autosomal dominant microtia. Eur J Med Genet. 2010;53(2):100–103. doi: 10.1016/j.ejmg.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 50.Couly G, Grapin-Botton A, Coltey P, Ruhin B, Le Douarin N M. Determination of the identity of the derivatives of the cephalic neural crest: incompatibility between Hox gene expression and lower jaw development. Development. 1998;125(17):3445–3459. doi: 10.1242/dev.125.17.3445. [DOI] [PubMed] [Google Scholar]
- 51.Gendron-Maguire M, Mallo M, Zhang M, Gridley T. Hoxa-2 mutant mice exhibit homeotic transformation of skeletal elements derived from cranial neural crest. Cell. 1993;75(7):1317–1331. doi: 10.1016/0092-8674(93)90619-2. [DOI] [PubMed] [Google Scholar]
- 52.Rijli F M, Mark M, Lakkaraju S, Dierich A, Dollé P, Chambon P. A homeotic transformation is generated in the rostral branchial region of the head by disruption of Hoxa-2, which acts as a selector gene. Cell. 1993;75(7):1333–1349. doi: 10.1016/0092-8674(93)90620-6. [DOI] [PubMed] [Google Scholar]
- 53.O'Gorman S. Second branchial arch lineages of the middle ear of wild-type and Hoxa2 mutant mice. Dev Dyn. 2005;234(1):124–131. doi: 10.1002/dvdy.20402. [DOI] [PubMed] [Google Scholar]
- 54.Gavalas A, Studer M, Lumsden A, Rijli F M, Krumlauf R, Chambon P. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development. 1998;125(6):1123–1136. doi: 10.1242/dev.125.6.1123. [DOI] [PubMed] [Google Scholar]
- 55.Kawakami K, Sato S, Ozaki H, Ikeda K. Six family genes—structure and function as transcription factors and their roles in development. BioEssays. 2000;22(7):616–626. doi: 10.1002/1521-1878(200007)22:7<616::AID-BIES4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 56.Laclef C, Souil E, Demignon J, Maire P. Thymus, kidney and craniofacial abnormalities in Six 1 deficient mice. Mech Dev. 2003;120(6):669–679. doi: 10.1016/s0925-4773(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 57.Brugmann S A, Pandur P D, Kenyon K L, Pignoni F, Moody S A. Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development. 2004;131(23):5871–5881. doi: 10.1242/dev.01516. [DOI] [PubMed] [Google Scholar]
- 58.Christophorou N A, Bailey A P, Hanson S, Streit A. Activation of Six1 target genes is required for sensory placode formation. Dev Biol. 2009;336(2):327–336. doi: 10.1016/j.ydbio.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 59.Grifone R, Demignon J, Houbron C. et al. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development. 2005;132(9):2235–2249. doi: 10.1242/dev.01773. [DOI] [PubMed] [Google Scholar]
- 60.Arnold J S, Braunstein E M, Ohyama T. et al. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum Mol Genet. 2006;15(10):1629–1639. doi: 10.1093/hmg/ddl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Y, Baud V, Delhase M. et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284(5412):316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 62.Ingraham C R, Kinoshita A, Kondo S. et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38(11):1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ten Berge D, Brouwer A, Korving J, Martin J F, Meijlink F. Prx1 and Prx2 in skeletogenesis: roles in the craniofacial region, inner ear and limbs. Development. 1998;125(19):3831–3842. doi: 10.1242/dev.125.19.3831. [DOI] [PubMed] [Google Scholar]
- 64.Dixon M J Treacher Collins syndrome Hum Mol Genet 19965(Spec No):1391–1396. [DOI] [PubMed] [Google Scholar]
- 65.Yamada G, Mansouri A, Torres M. et al. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development. 1995;121(9):2917–2922. doi: 10.1242/dev.121.9.2917. [DOI] [PubMed] [Google Scholar]
- 66.Wang W, Lufkin T. Hmx homeobox gene function in inner ear and nervous system cell-type specification and development. Exp Cell Res. 2005;306(2):373–379. doi: 10.1016/j.yexcr.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 67.Yoshiura K, Leysens N J, Reiter R S, Murray J C. Cloning, characterization, and mapping of the mouse homeobox gene Hmx1. Genomics. 1998;50(1):61–68. doi: 10.1006/geno.1998.5284. [DOI] [PubMed] [Google Scholar]
- 68.Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers E N. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129(19):4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- 69.Wright T J, Mansour S L. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130(15):3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- 70.Partanen J, Schwartz L, Rossant J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 1998;12(15):2332–2344. doi: 10.1101/gad.12.15.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zabihi S, Loeken M R. Understanding diabetic teratogenesis: where are we now and where are we going? Birth Defects Res A Clin Mol Teratol. 2010;88(10):779–790. doi: 10.1002/bdra.20704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaguchi T P, Bradley A, McMahon A P, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126(6):1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 73.Beleza-Meireles A, Clayton-Smith J, Saraiva J M, Tassabehji M. Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet. 2014;51(10):635–645. doi: 10.1136/jmedgenet-2014-102476. [DOI] [PubMed] [Google Scholar]
- 74.Engiz O, Balci S, Unsal M, Ozer S, Oguz K K, Aktas D. 31 cases with oculoauriculovertebral dysplasia (Goldenhar syndrome): clinical, neuroradiologic, audiologic and cytogenetic findings. Genet Couns. 2007;18(3):277–288. [PubMed] [Google Scholar]
- 75.Tribioli C, Lufkin T. Molecular cloning, chromosomal mapping and developmental expression of BAPX1, a novel human homeobox-containing gene homologous to Drosophila bagpipe. Gene. 1997;203(2):225–233. doi: 10.1016/s0378-1119(97)00520-9. [DOI] [PubMed] [Google Scholar]
- 76.Fischer S, Lüdecke H J, Wieczorek D, Böhringer S, Gillessen-Kaesbach G, Horsthemke B. Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum Mol Genet. 2006;15(4):581–587. doi: 10.1093/hmg/ddi474. [DOI] [PubMed] [Google Scholar]
- 77.Heike C L, Hing A V, Aspinall C A. et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet. 2013;163C(4):271–282. doi: 10.1002/ajmg.c.31373. [DOI] [PubMed] [Google Scholar]
- 78.Cohen M M Jr, Rollnick B R, Kaye C I. Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J. 1989;26(4):276–286. [PubMed] [Google Scholar]
- 79.Gorlin R J, Cohen M M, Jr, Hennekam R CM. New York: Oxford University Press; 2001. Syndromes of the Head and Neck; p. 1344. [Google Scholar]
- 80.Kelberman D, Tyson J, Chandler D C. et al. Hemifacial microsomia: progress in understanding the genetic basis of a complex malformation syndrome. Hum Genet. 2001;109(6):638–645. doi: 10.1007/s00439-001-0626-x. [DOI] [PubMed] [Google Scholar]
- 81.Marres H A. Hearing loss in the Treacher Collins syndrome. Adv Otorhinolaryngol. 2002;61:209–215. doi: 10.1159/000066811. [DOI] [PubMed] [Google Scholar]
- 82.Su P H, Chen J Y, Chen S J, Yu J S. Treacher Collins syndrome with a de Novo 5-bp deletion in the TCOF1 gene. J Formos Med Assoc. 2006;105(6):518–521. doi: 10.1016/S0929-6646(09)60194-7. [DOI] [PubMed] [Google Scholar]
- 83.Hao J, Liu Z, Kong W, Wang J. [Treacher Collins syndrome: case report and literature review] Lin Chuang Er Bi Yan Hou Ke Za Zhi. 2006;20(13):582–584. [PubMed] [Google Scholar]
- 84.Edwards S J, Gladwin A J, Dixon M J. The mutational spectrum in Treacher Collins syndrome reveals a predominance of mutations that create a premature-termination codon. Am J Hum Genet. 1997;60(3):515–524. [PMC free article] [PubMed] [Google Scholar]
- 85.Teber O A, Gillessen-Kaesbach G, Fischer S. et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004;12(11):879–890. doi: 10.1038/sj.ejhg.5201260. [DOI] [PubMed] [Google Scholar]
- 86.Opitz J M, Mollica F, Sorge G, Milana G, Cimino G, Caltabiano M. Acrofacial dysostoses: review and report of a previously undescribed condition: the autosomal or X-linked dominant Catania form of acrofacial dysostosis. Am J Med Genet. 1993;47(5):660–678. doi: 10.1002/ajmg.1320470517. [DOI] [PubMed] [Google Scholar]
- 87.Buchanan E PMD, Xue A SMD, Hollier L HJMD Jr. Craniofacial syndromes. Plast Reconstr Surg. 2014;134(1):128e–153e. doi: 10.1097/PRS.0000000000000308. [DOI] [PubMed] [Google Scholar]
- 88.Forrest C RMD, Hopper R A. Craniofacial syndromes and surgery. Plast Reconstr Surg. 2013;131(1):86e–109e. doi: 10.1097/PRS.0b013e318272c12b. [DOI] [PubMed] [Google Scholar]
- 89.Chemke J, Mogilner B M, Ben-Itzhak I, Zurkowski L, Ophir D. Autosomal recessive inheritance of Nager acrofacial dysostosis. J Med Genet. 1988;25(4):230–232. doi: 10.1136/jmg.25.4.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McDonald M T, Gorski J L. Nager acrofacial dysostosis. J Med Genet. 1993;30(9):779–782. doi: 10.1136/jmg.30.9.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Norris R A, Scott K K, Moore C S. et al. Human PRRX1 and PRRX2 genes: cloning, expression, genomic localization, and exclusion as disease genes for Nager syndrome. Mamm Genome. 2000;11(11):1000–1005. doi: 10.1007/s003350010193. [DOI] [PubMed] [Google Scholar]
- 92.McDermid H E, Morrow B E. Genomic disorders on 22q11. Am J Hum Genet. 2002;70(5):1077–1088. doi: 10.1086/340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lindsay E A, Vitelli F, Su H. et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 94.Emanuel B S, McDonald-McGinn D, Saitta S C, Zackai E H. The 22q11.2 deletion syndrome. Adv Pediatr. 2001;48:39–73. [PubMed] [Google Scholar]
- 95.Kosaki R, Fujimaru R, Samejima H. et al. Wide phenotypic variations within a family with SALL1 mutations: Isolated external ear abnormalities to Goldenhar syndrome. Am J Med Genet A. 2007;143A(10):1087–1090. doi: 10.1002/ajmg.a.31700. [DOI] [PubMed] [Google Scholar]
- 96.Dixon J, Jones N C, Sandell L L. et al. Tcof1/treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103(36):13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruf R G, Xu P X, Silvius D. et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101(21):8090–8095. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matsunaga T, Okada M, Usami S, Okuyama T. Phenotypic consequences in a Japanese family having branchio-oto-renal syndrome with a novel frameshift mutation in the gene EYA1. Acta Otolaryngol. 2007;127(1):98–104. doi: 10.1080/00016480500527185. [DOI] [PubMed] [Google Scholar]
- 99.Hone S W, Smith R J. Genetics of hearing impairment. Semin Neonatol. 2001;6(6):531–541. doi: 10.1053/siny.2001.0094. [DOI] [PubMed] [Google Scholar]
- 100.Abdelhak S, Kalatzis V, Heilig R. et al. Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum Mol Genet. 1997;6(13):2247–2255. doi: 10.1093/hmg/6.13.2247. [DOI] [PubMed] [Google Scholar]
- 101.Kumar S Deffenbacher K Cremers C W Van Camp G Kimberling W J Branchio-oto-renal syndrome: identification of novel mutations, molecular characterization, mutation distribution, and prospects for genetic testing Genet Test 1997. –1998; 14243–251. [DOI] [PubMed] [Google Scholar]
- 102.Rodríguez Soriano J. Branchio-oto-renal syndrome. J Nephrol. 2003;16(4):603–605. [PubMed] [Google Scholar]
- 103.Ruf R G, Berkman J, Wolf M T. et al. A gene locus for branchio-otic syndrome maps to chromosome 14q21.3-q24.3. J Med Genet. 2003;40(7):515–519. doi: 10.1136/jmg.40.7.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoskins B E, Cramer C H, Silvius D. et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet. 2007;80(4):800–804. doi: 10.1086/513322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Giannatou E, Leze H, Katana A. et al. Unilateral microtia in an infant with trisomy 18 mosaicism. Genet Couns. 2009;20(2):181–187. [PubMed] [Google Scholar]
- 106.Griffith C B, Vance G H, Weaver D D. Phenotypic variability in trisomy 13 mosaicism: two new patients and literature review. Am J Med Genet A. 2009;149A(6):1346–1358. doi: 10.1002/ajmg.a.32883. [DOI] [PubMed] [Google Scholar]
- 107.Davies A F, Imaizumi K, Mirza G. et al. Further evidence for the involvement of human chromosome 6p24 in the aetiology of orofacial clefting. J Med Genet. 1998;35(10):857–861. doi: 10.1136/jmg.35.10.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Veltman J A, Jonkers Y, Nuijten I. et al. Definition of a critical region on chromosome 18 for congenital aural atresia by arrayCGH. Am J Hum Genet. 2003;72(6):1578–1584. doi: 10.1086/375695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Q, Zhang J, Yin W. Pedigree and genetic study of a bilateral congenital microtia family. Plast Reconstr Surg. 2010;125(3):979–987. doi: 10.1097/PRS.0b013e3181ccdbba. [DOI] [PubMed] [Google Scholar]
- 110.Lin L, Pan B, Jiang H Y. et al. Study of methylation of promoter of EYA1 gene in microtia[in Chinese] Zhonghua Zheng Xing Wai Ke Za Zhi. 2009;25(6):436–439. [PubMed] [Google Scholar]
- 111.Schorderet D F, Nichini O, Boisset G. et al. Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am J Hum Genet. 2008;82(5):1178–1184. doi: 10.1016/j.ajhg.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vaclavik V, Schorderet D F, Borruat F X, Munier F L. Retinal dystrophy in the oculo-auricular syndrome due to HMX1 mutation. Ophthalmic Genet. 2011;32(2):114–117. doi: 10.3109/13816810.2011.562955. [DOI] [PubMed] [Google Scholar]
- 113.Turner E E, Cox T C. Genetic evidence for conserved non-coding element function across species-the ears have it. Front Phys. 2014;5:7. doi: 10.3389/fphys.2014.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Passos-Bueno M R, Ornelas C C, Fanganiello R D. Syndromes of the first and second pharyngeal arches: a review. Am J Med Genet A. 2009;149A(8):1853–1859. doi: 10.1002/ajmg.a.32950. [DOI] [PubMed] [Google Scholar]
- 115.Trainor P A. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet A. 2010;152A(12):2984–2994. doi: 10.1002/ajmg.a.33454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hansen J M, Gong S G, Philbert M, Harris C. Misregulation of gene expression in the redox-sensitive NF-kappab-dependent limb outgrowth pathway by thalidomide. Dev Dyn. 2002;225(2):186–194. doi: 10.1002/dvdy.10150. [DOI] [PubMed] [Google Scholar]
- 117.Knobloch J, Shaughnessy J D Jr, Rüther U. Thalidomide induces limb deformities by perturbing the Bmp/Dkk1/Wnt signaling pathway. FASEB J. 2007;21(7):1410–1421. doi: 10.1096/fj.06-7603com. [DOI] [PubMed] [Google Scholar]
- 118.Ito T, Ando H, Suzuki T. et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 119.Parman T, Wiley M J, Wells P G. Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat Med. 1999;5(5):582–585. doi: 10.1038/8466. [DOI] [PubMed] [Google Scholar]