

Abstract
Primary glomerulocystic kidney disease is a special form of renal cystic disorder characterized by Bowman’s space dilatation in the absence of tubular cysts. ZEB2 is a SMAD-interacting transcription factor involved in Mowat-Wilson syndrome, a congenital disorder with an increased risk for kidney anomalies. Here we show that deletion of Zeb2 in mesenchyme-derived nephrons with either Pax2-cre or Six2-cre causes primary glomerulocystic kidney disease without tubular cysts in mice. Glomerulotubular junction analysis revealed many atubular glomeruli in the kidneys of Zeb2 knockout mice, which explains the presence of glomerular cysts in the absence of tubular dilatation. Gene expression analysis showed decreased expression of early proximal tubular markers in the kidneys of Zeb2 knockout mice preceding glomerular cyst formation, suggesting that defects in proximal tubule development during early nephrogenesis contribute to the formation of congenital atubular glomeruli. At the molecular level, Zeb2 deletion caused aberrant expression of Pkd1, Hnf1β, and Glis3, three genes causing glomerular cysts. Thus, Zeb2 regulates the morphogenesis of mesenchyme-derived nephrons and is required for proximal tubule development and glomerulotubular junction formation. Our findings also suggest that ZEB2 might be a novel disease gene in patients with primary glomerular cystic disease.
Keywords: kidney development, obstructive nephropathy, pediatric nephrology, proximal tubule, ADPKD
INTRODUCTION
Cystic kidney disease is a group of genetically heterogeneous disorders that are characterized mainly by renal tubular dilatation.1,2 Glomerulocystic kidney disease (GCKD) is a special form of cystic kidney disease that confines cystic dilatation to the Bowman’s space.3–5 GCKD is defined as two- to three-fold dilatation of Bowman’s space in more than 5% of identifiable glomeruli in the plane of a kidney section, and the glomerular cysts in primary GCKD are mainly localized to the subcapsular region of the kidney.4–6 Glomerular cysts in association with tubular cysts can be found in several syndromes including tuberous sclerosis complex, orofaciodigital syndrome 1, and Meckel-Gruber syndrome.3,7–9 Glomerular cysts can also be found in patients with mutations in the UMOD and HNF1β genes,10,11 and in patients of autosomal dominant polycystic kidney disease with PKD1 mutations, other ciliopathies such as nephronophthisis, multicystic dysplastic kidney or urinary tract obstruction.3 However, the genetic basis of primary glomerulocystic kidney disease without tubular dilatation remains largely unknown.
ZEB2 is a zinc finger E-box binding homeobox transcription factor that promotes epithelial-mesenchymal transition via a TGF-β/SMAD/ZEB/miR-200 signaling network.12–14 ZEB2 mutations in human cause Mowat-Wilson syndrome (MWS: OMIM 235730), a congenital disorder characterized by intellectual disability, craniofacial abnormalities, Hirschsprung’s disease, congenital heart defects, and an increased risk for congenital kidney anomalies.15 However, the pathological features of its kidney defects have not been examined and defined. 15,16 ZEB2 is also a known target for microRNA miR-200.17 Recently, it has been shown that upregulation of ZEB2 in Hnf1b knockout mice and downregulation of miR-200 in Dicer knockout mice are associated with glomerular and tubular cysts.18,19 However, no direct link between ZEB2 downregulation and renal cystic disease has been established and the pathological effect of ZEB2 downregulation in early nephrogenesis remains unknown.
Here we report that deletion of Zeb2 in early mouse nephrogenesis with either Pax2-cre or Six2-cre resulted in primary glomerulocystic disease without tubular dilatation. We found that loss of Zeb2 caused renal proximal tubule hypotrophy and reduced glomerulotubular junction integrity and maturation, which contributed to the formation of congenital atubular glomeruli leading to glomerulocystic disease. By gene expression analysis, we found that Zeb2 nephron specific knockout kidney had aberrant expression of Pkd1, Hnf1β, and Glis3, three genes associated with glomerular cysts. These results suggest that Zeb2 regulates the morphogenesis of metanephric mesenchyme derived early nephrons and is required for proximal tubule development and normal glomerulotubular junction formation.
RESULTS
Because aberrant expressions of Zeb2 and miR-200 have been reported in young mice with a cystic kidney phenotype and ZEB2 loss-of-function mutations are associated with an increased risk for renal anomalies in MWS patients,15,18,19 we hypothesized that ZEB2 plays an important role in early kidney development. To test this hypothesis, we analyzed a Zeb2 floxed conditional knockout (cKO) mouse line20 as Zeb2 null mice die at E9.5 before kidney development begins.21 We first crossed the Zeb2 floxed homozygotes (Zeb2flox/flox) with a Pax2-cre+ deleter strain22 that expresses the Cre recombinase in both the metanephric mesenchyme and ureteric bud (Supplemental Figure 1a). After genotyping 96 three-week old weanlings of Zeb2flox/+;Pax2-cre+ heterozygous matings, we did not find any Zeb2flox/flox;Pax2-cre+ homozygotes (Supplemental Figure 1b). We then analyzed newborn mice and E18.5 embryos from timed-pregnant females. Five dead newborn Zeb2flox/flox;Pax2-cre+ homozygotes and 21/55 (38%) E18.5 homozygous embryos were found (Supplemental Figure 1b), suggesting that Zeb2flox/flox;Pax2-cre+ homozygotes die at birth.
The E18.5 Zeb2flox/flox;Pax2-cre+ homozygous embryos did not display discernible gross structural defects of the kidney or ureter. However, histological examination of the kidneys from eight E18.5 Zeb2flox/flox;Pax2-cre+ homozygous embryos revealed that 100% of the homozygotes had renal cysts, while none of the seven littermate controls (0%) had a cystic phenotype (Table 1, Figure 1). The renal cysts first appeared at E16.5 as no cysts could be detected at E15.5 (Table 1, Figure 1). The renal cysts were apparently from a glomerular origin as 47% of the glomeruli were cystic in the E16.5 Zeb2 cKO embryos and 30% of the cysts had a visible glomerular tuft (Tables 1 and 2). This phenotype is sufficient for a diagnosis of glomerulocystic kidney disease (GCKD) according to the established criteria.4
Table 1.
Cystic phenotype observed in H&E stained kidney samples of Zeb2flox/flox;Pax2-cre+ and wild-type littermates between E16.5 and E18.5
| E16.5 | E17.5 | E18.5 | ||||
|---|---|---|---|---|---|---|
| Zeb2+/+ | Zeb2flox/flox; Pax2-cre+ | Zeb2+/+ | Zeb2flox/flox; Pax2-cre+ | Zeb2+/+ | Zeb2flox/flox; Pax2-cre+ | |
| Embryos with kidney cysts (more than 1 cyst) | 0/2 (0%) | 3/3 (100%) | 0/3 (0%) | 3/3 (100%) | 0/7 (0%) | 8/8 (100%) |
| Glomerular Cysts/Number of glomeruli | 1/48 2%, (n=4)00 |
26/55 47%, (n=5) |
0/40 0%, (n=3) |
18/59 30.5%, (n=4) |
1/363 0.2%, (n=13) |
61/391 15.6%, (n=15) |
n= number of kidneys analyzed
Figure 1. Deletion of Zeb2 with Pax2-cre leads to embryonic glomerulocystic kidney disease without tubular dilatation.
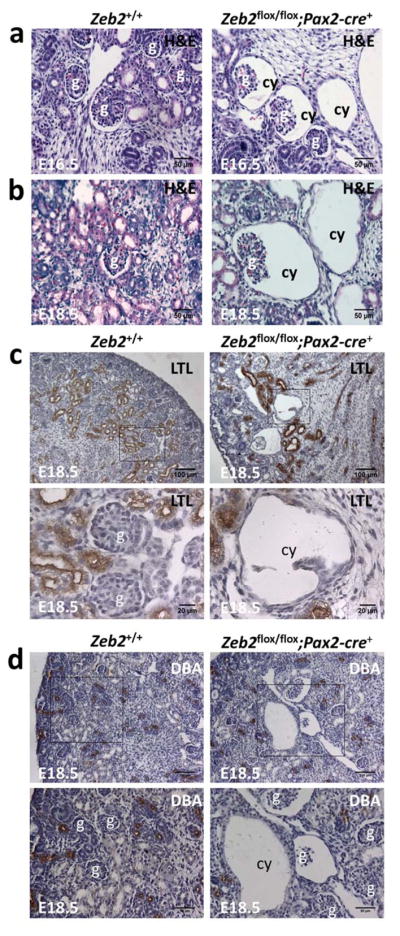
Congenital glomerular cysts were observed in the kidney of E16.5 (a) and E18.5 (b) Zeb2flox/flox;Pax2-cre+ homozygous embryos but not in the Zeb2+/+ littermate controls (H&E staining, 200x magnification). The cysts in the Zeb flox/flox;Pax2-cre+ kidneys at E18.5 are negative for the proximal tubules marker Lotus Tetragonolobus Lectin (LTL) (c), and the collecting duct and distal tubules marker Dolichos Biflorus Agglutinin (DBA) (d). Upper panels - 100x magnification; lower panels - enlarged images of boxed regions from upper panels (n≥3 for each group). Scale bars are shown. Abbreviations: g-glomerulus; cy-cyst.
Table 2.
Percentage of cysts with glomerular tufts on H&E stained kidney samples between E16.5 and E18.5.
| Cysts with Glomerular tufts/total cysts (n= number of kidneys analyzed) Zeb2flox/flox;Pax2-cre+ |
|
|---|---|
| E16.5 | 26/85 (30%, n=5) |
| E17.5 | 18/46 (39%, n=4) |
| E18.5 | 61/226 (27%, n=15) |
n= number of kidneys analyzed
To determine whether Zeb2 cKO embryonic kidneys also develop renal tubular cysts, we examined the kidney sections with both proximal tubule specific marker Lotus tetragonolobus lectin (LTL) and distal tubules and collecting duct specific marker Dolichos biflorus agglutinin (DBA). The cysts were negative for both LTL and DBA staining at E18.5 (Figure 1c and 1d), suggesting that the Zeb2flox/flox;Pax2-cre+ homozygous embryos develop primary glomerular cysts. To determine if glomerular cyst formation is due to a deletion of Zeb2 in the developing nephron, we examined ZEB2 expression during kidney development by analyzing a ZEB2-EGFP reporter mouse that has been reported previously.23 In addition to the stromal cells, ZEB2 was detected in a subset of cells in which it can be deleted by Pax2-cre in the developing nephron (Supplemental Figure 2), including the S-shaped bodies (Supplemental Figure 2e) and the glomeruli (Supplemental Figure 2f). By co-staining, we confirmed that ZEB2 is co-expressed in a subset of cells with PAX2 and JAG1, two markers of developing nephrons (Supplemental Figure 3).
Zeb2flox/flox;Pax2-cre+ homozygous mice died at birth (probably due to high expression of Pax2-cre in the nervous system where ZEB2 also plays an important role),22,23 precluding a longitudinal analysis of glomerulocystic kidney disease progression after birth. A recent study shows that SIX2 regulates ZEB2 expression through miR-200.24 SIX2 is also a nephron progenitor marker and is expressed in the metanephric mesenchyme that gives rise to all segments of the mature nephron including the parietal and visceral epithelial cells in the glomeruli and the proximal and distal tubular epithelial cells.25 To further delineate the role of ZEB2 in nephron development and to study the pathological effect of Zeb2 deletion in mesenchyme-derived nephrons as well as glomerular cystic phenotype in postnatal mature kidney, we crossed Zeb2flox/flox mice with Six2-cre mice. The Zeb2flox/flox;Six2-cre+ homozygous mice survived after birth and were weaned at a Mendelian distribution. Histological examination of the kidneys from nine Zeb2flox/flox;Six2-cre+ homozygous embryos revealed that, like the Zeb2flox/flox;Pax2-cre+ embryos, all homozygous embryos had glomerular cysts starting at E16.5 (Figure 2a). At postnatal day 12 (P12), the glomerular cysts in the Zeb2flox/flox;Six2-cre+ mice were mainly located in the subcapsular region of the kidney that is consistent with the primary glomerulocystic kidney disease diagnosis (Figure 2b).4 The cysts were also negative for both LTL and DBA staining (Figure 2c and 2d), confirming their glomerular origin. In comparison, none of the five wild-type littermate controls had a cystic phenotype at P12 (Figure 2b).
Figure 2. Mesenchyme-specific deletion of Zeb2 using Six2-cre also leads to glomerulocystic kidney disease without tubular dilatation.

(a) Dilated glomerular Bowman’s space (asterisk) was observed in the Zeb2flox/flox;Six2-cre+ embryonic kidneys at E16.5 but not in littermate controls (H&E staining, 200x magnification). (b) Glomerular cysts (asterisk) in the subcapsular region of the kidney cortex at P12 in the Zeb2flox/flox;Six2-cre+ kidneys but not in the littermate controls (H&E staining, 200x magnification). (c) IHC staining shows the cyst (cy) in a P12 Zeb2flox/flox;Six2-cre+ kidney is negative for the proximal tubules marker Lotus Tetragonolobus Lectin (LTL). Upper panels - 100x magnification; lower panels - high magnification of boxed regions in the upper panels (n≥3 for each group). (d) IHC staining shows the cysts (cy) in a P12 Zeb2flox/flox;Six2-cre+ kidney is negative for the collecting duct and distal tubules marker Dolichos Biflorus Agglutinin (DBA). Upper panels - 100x magnification; lower panels - high magnification of boxed regions in the upper panels (n≥3 for each group). Abbreviations: g-glomerulus; cy-cyst.
To determine the longitudinal effect of glomerulocystic kidney disease on mature kidney, we followed the Zeb2flox/flox;Six2-cre+ mice to adulthood. At 7 weeks of age, all Zeb2flox/flox;Six2-cre+ mice (n=5, 100%) developed macroscopic renal cysts that were visualized on the surface of the kidney (Figure 3a). Histological analysis revealed the presence of numerous large glomerular cysts in the area of the renal cortex and outer medulla (Figure 3b). Although no significant interstitial fibrosis was observed (Figure 4a), some remaining non-cystic glomeruli in adult Zeb2flox/flox;Six2-cre+ mice displayed glomerulosclerosis lesions (Figure 4b). Similar lesions were also observed in few non-cystic glomeruli in P8 Zeb2 cKO kidneys (Supplemental Figure 4). By 8 weeks of age, all Zeb2flox/flox;Six2-cre+ mice (n=5, 100%) developed albuminuria (Figure 4c and 4d) with significantly elevated serum blood urea nitrogen (BUN) levels (Figure 4e). Albuminuria was not directly caused by loss of ZEB2 in podocytes or reduced proximal tubule endocytosis as deletion of Zeb2 specifically in the podocyte using Nphs2-cre did not lead to proteinuria (data not shown), and the expressions of megalin and cubilin were upregulated in Zeb2 adult cKO kidneys (Supplemental Figure 5), which may increase endocytosis of albumin as previously reported.26 These data suggest that loss of Zeb2 in mesenchyme-derived nephrons alone is sufficient to cause glomerulocystic disease in mature kidneys, which leads to secondary glomerulosclerosis, albuminuria and renal failure in adult mice.
Figure 3. Mesenchyme-specific deletion of Zeb2 with Six2-cre causes glomerulocystic kidney disease in adult mice.

(a) Macroscopic images of normal kidneys from a 7 weeks old control mouse (left panel) and pale and cystic kidneys (marked by asterisk) from a Zeb2flox/flox;Six2-cre+ homozygous mouse (right panel); n=5 in each group. (b) Histological images of kidneys from 7 weeks old control Zeb2+/+ mice and Zeb2flox/flox;Six2-cre+ cKO mice show large cysts in the renal cortex and outer medulla region in the cKO mice (n=3 in each group). Upper panels - 25x magnification; lower panels - 100x magnification with scale bars at 100 μm; g-glomerulus; cy-cyst.
Figure 4. Mesenchyme-specific deletion of Zeb2 with Six2-cre causes glomerulosclerosis, albuminuria and renal failure in adult mice.

(a) Masson Trichrome staining (MTS) shows minimal fibrosis (blue) in the kidney of a 7 weeks old Zeb2flox/flox;Six2-cre+ mouse. Upper panels - 100x magnification; lower panels - high magnification of boxed regions in the upper panels (n=3 for each group). (b) Periodic acid Schiff (PAS) staining shows glomerulosclerosis (arrow) in non-cystic glomeruli of a 7 weeks old Zeb2flox/flox;Six2-cre+ mouse kidney. Upper panels - 100x magnification; lower panels - high magnification of boxed regions in the upper panels (n=3 for each group). (c) Representative SDS-page gel with Coomassie blue staining shows albuminuria (arrow) in two Zeb2flox/flox;Six2-cre+ 5 weeks old mice but not in two littermate controls (n=5 mice in each group); alb - albumin. (d) Increased albumin-to-creatinine ratio (ACR) in 5 weeks old Zeb2flox/flox;Six2-cre+ mice compared to Zeb2+/+ littermate controls (n=4 mice in each group, *p < 0.05) (e) Significantly elevated BUN in 5 weeks old Zeb2flox/flox;Six2-cre+ mice compared to Zeb2+/+ littermate controls (n=3 mice in each group, **p-value< 0.001). Data are represented as means +/− standard deviation.
One common cause of acquired glomerular cysts is the loss of glomerulotubular junction integrity leading to atubular glomeruli.27,28 Glomerulotubular integrity can be quantified by examining the connection of the proximal tubule to the Bowman’s capsule with the proximal tubule marker LTL.29 To determine whether the Zeb2 knockout mice have decreased glomerulotubular integrity, we examined glomeruli from five P12 Zeb2flox/flox;Six2-cre+ mice and five littermate controls by serial sections. We found that only 63/604 (10%) glomeruli had a visible LTL positive staining (i.e. glomerulotubular junction) in the Zeb2 cKO mice as compared to 115/324 (36%) in the wild-type littermate controls (Figure 5), a statistically significant decrease of glomerulotubular integrity, suggesting that Zeb2 deletion causes formation of atubular glomeruli.
Figure 5. Congenital atubular glomeruli in Zeb2 mesenchyme-specific knockout mice.

(a) LTL staining shows many glomerular Bowman’s capsule and proximal tubule connections (arrows) in P12 Zeb2+/+ wild-type control mice but very few connections in Zeb2flox/flox;Six2-cre+ mutant mice. Upper panels - 100x magnification; lower panels - 400x magnification; g -glomerulus. (n=5 in each group); Scale bars are shown. (b) Percentage of LTL-positive glomeruli out of total glomeruli counted in sagittal kidney sections from five Zeb2 cKO and five Zeb2+/+ littermate controls (n=604 glomeruli in Zeb2 cKO and n=324 glomeruli in wild-type were counted, *p < 10−3). (c) Representative serial sections of P12 Zeb2+/+ wild-type kidneys (upper panels) and Zeb2flox/flox;Six2-cre+ cKO kidneys (lower panels) show the detection of glomerulotubular junctions (arrows) in the wild-type glomeruli but absence in many Zeb2 cKO glomeruli (* indicating the same glomerulus on serial sections).
Acquired atubular glomeruli can be caused by atrophy of the proximal tubules in polycystic kidney disease.30 To determine if reduced glomerulotubular integrity in Zeb2 cKO is associated with renal proximal tubule atrophy (acquired defect) or hypotrophy (developmental defect), we analyzed the mRNA levels of markers for the proximal tubule (Hnf1a and Vil1), the podocyte (Nphs1 and Nphs2), and the collecting duct (Upk3a).31 We found that only the proximal tubule mRNA markers were significantly reduced in the Zeb2flox/flox;Six2-cre+ mice compared to the levels in the wild-type littermates at postnatal day 8 (P8) (Figure 6a). To determine if the reduction of proximal tubule mRNA at P8 is caused by an early developmental defect preceding the formation of glomerular cysts in Zeb2 cKO mice, we analyzed mRNA markers in E14.5 kidneys. Similar to P8, a significant decrease of the proximal tubular markers was detected in the E14.5 Zeb2flox/flox;Six2-cre+ kidneys as compared to their wild-type littermates (Figure 6b). Consistent with this finding, the expression of the proximal tubule brush border protein villin 1 (VIL1),31 was also downregulated in both E15.5 Zeb2flox/flox;Pax2-cre+ and E16.5 Zeb2flox/flox;Six2-cre+ mutant kidneys (Figure 6c and 6d). Finally, the mean kidney size of the E16.5 Zeb2 cKO embryos was smaller than that of their wild-type littermate controls (Figure 6e). Taken together, these data suggest that loss of Zeb2 in the mesenchyme-derived nephrons causes early proximal tubule developmental defects resulting in tubular hypotrophy, reduced glomerulotubular junction integrity, and congenital atubular glomeruli formation.
Figure 6. Decreased early proximal tubular marker expression in Zeb2 mesenchyme-specific knockout mice.

(a) At postnatal day 8 (P8), TaqMan assays show decreased mRNA expression levels of proximal tubular markers (Vil1 and Hnf1a) but not podocyte markers (Nphs1 and Nphs2) in Zeb2flox/flox;Six2-cre+ kidneys when normalized to a collecting duct marker Ppp1r3c. Collecting duct marker Upk3a was used as a control (n= 3 in each group, mean relative quantification adjusted to Ppp1r3c, *p < 0.05). (b) At E14.5, TaqMan assays show decreased mRNA expression levels of proximal tubular mRNA markers (Vil1 and Hnf1a) in the cKO kidneys compared to wild-type kidneys. There are no differences for the metanephric mesenchyme markers (Pax2 and Wt1) and the collecting duct markers (Ppp1r3c and Upk3a) in cKO and control kidneys (mean relative quantification adjusted to Gapdh, n=3 in each group, *p <0.05). (c) Immunofluorescent staining shows decreased expression of VIL1 (villin-1) protein in E15.5 Zeb2flox/flox;Pax2-cre+ cKO proximal tubules (arrowhead) as compared to the proximal tubules in wild-type littermates (arrows). (d) Decreased expression of VIL1 (villin-1) protein in E16.5 Zeb2flox/flox;Six2-cre+ cKO proximal tubules (arrowhead) as compared to the proximal tubules in wild-type littermates (arrows). (e) The Zeb2 cKO embryos have smaller kidney length at E16.5 as compared to wild-type controls (n=7 for cKO kidneys and n=6 for wild-type kidneys, *p <10−3).
Glomerular cysts are reported in several mouse models of renal cystic kidney disease, including Wwtr1, Glis3, Ofd1 and Pkhd1 knockout mice, the Hnf1β and Dicer cKO mice, and Pkd1 over expression transgenic mice.19,31–36 Interestingly, the Hnf1β cKO mice using Six2-cre develop glomerular cysts due to a drastic reduction in the levels of proximal tubular markers at E14.5, resembling the Zeb2 cKO phenotype.31 However, by immunostaining of JAG1, a marker for the renal vesicle and the S-shaped body,31 we did not observe differences between Zeb2 cKO and wild-type littermate controls (Supplemental Figure 6). To determine if loss of Zeb2 affects the expression of these six genes and the microRNA miR-200, we examined mRNA and microRNA levels in the kidney tissues of E14.5 and E18.5 Zeb2 cKO and wild-type controls. Although there was no significant difference in the expression levels of any of the six genes and miR-200 at E14.5 before glomerular cyst formation (Figure 7a), we detected a decreased expression of Glis3 and increased expression levels of Hnf1β and Pkd1 in E18.5 Zeb2 cKO compared to the wild-type littermate controls (Figure 7b). The expression level of Pkd1 mRNA was the most significantly upregulated in the Zeb2 cKO kidneys at postnatal day 8 (P8) (Figure 7c). Consistent with the mRNA levels, PKD1 coding protein polycystin-1 (PC1) was also found to be expressed at a higher level in the glomeruli of postnatal day 7 (P7) Zeb2flox/flox;Six2-cre+ mice but not at E16.5 and E17.5 when the glomerular cysts are initially observed (Figure 7d). Interestingly, the PC1 expression was upregulated in non-cystic glomeruli but not in the glomeruli with dilated Bowman’s space (Figure 7e). These data suggest that loss of Zeb2 in the kidney leads to upregulation of polycystin-1 expression in non-cystic glomeruli after the initial phase of glomerular cyst formation.
Figure 7. Abnormal expression of known glomerulocystic disease genes in Zeb2 conditional knockout kidneys.

(a) TaqMan assays show no differences of the mRNA levels for the 6 genes and miRNA miR200 between E14.5 Zeb2 cKO kidneys and wild-type controls (mean relative quantification adjusted to Gapdh, n≥3). (b) TaqMan assays show significant upregulation of mRNA levels of Pkd1 and Hnf1β and downregulation of Glis3 mRNA in E18.5 Zeb2 cKO kidneys compared to wild-type controls (mean relative quantification adjusted to Gapdh, n=5, *p <0.05) (c) Significant upregulation of Pkd1 mRNA at E18.5 and P8 but not E14.5 in Zeb2 cKO kidneys compared to wild-type controls (mean relative quantification adjusted to Gapdh, n=3, *p <0.05). (d) Immunofluorescence staining show increased levels of polycystin 1 (PC1) protein in the glomeruli (arrows) of Zeb2flox/flox;Six2-cre+ kidney at P7 (middle and lower panels), but not at E17.5 (upper panel). PC1 staining in the glomeruli was confirmed by co-expression of WT1, a glomerular podocyte marker (lower panel); Scale bars are shown. (e) Triple immunofluorescence staining with PC1, WT1 and DAPI in Zeb2 cKO kidney shows that increased level of polycystin 1 (PC1) protein was detected only in non-cystic glomeruli (glom 1) but not in an adjacent glomerulus (glom 2) with significantly enlarged Bowman’s space (cy). The podocytes (arrows) and parietal epithelial cells (arrowheads) are visible.
Renal cystogenesis is often associated with increased cell proliferation.37 To determine if abnormal cell proliferation also plays a role in the formation of glomerular cysts in Zeb2 cKO mice, we quantified the proliferation of parietal epithelial cells in the Bowman’s capsule of Zeb2 cKO kidneys and wild-type littermate controls using the cell proliferation marker phospho-Histone H3 (pHH3). No significant difference of cell proliferation was observed in Zeb2 cKO kidneys compared to wild-type littermate controls (Supplemental Figure 7), suggesting that cell proliferation does not play an important role in Zeb2 cKO glomerulocystic phenotype.
Apoptosis is part of normal kidney development during C-shaped body (CSB) and S-shaped body (SSB) formation.38,39 To determine if abnormal apoptosis in CSB and SSB may contribute to congenital atubular glomeruli formation in Zeb2 cKO mice, we performed TUNEL assays on E15.5 and E16.5 developing kidneys from five Zeb2 cKO and four littermate controls. Interestingly, 11/34 (32%) CSB/SSB were identified with at least one apoptotic cell in Zeb2 cKO kidneys, while 25/39 (64%) CSB/SSB were found in wild-type littermate controls (Supplemental Figure 8). These data suggest that loss of Zeb2 causes aberrant apoptosis in CSB and SSB during nephron development, which may contribute to proximal tubular hypotrophy and abnormal glomerulotubular junction maturation in Zeb2 cKO mice.
DISCUSSION
In this study, we found that ZEB2 is critical for the mesenchyme-derived nephron development. Conditional deletion of Zeb2 with Pax2-cre and Six2-cre, two Cre strains active in the metanephric mesenchyme derived nephron progenitor cells, resulted in the same glomerular cyst phenotype, indicating that ZEB2 regulates the morphogenesis of mesenchyme-derived nephrons and is required for normal nephron development. Decreased levels of proximal tubular markers at both mRNA and protein levels from E14.5 to E16.5 and reduced kidney size at E16.5 in the Zeb2 cKO mice suggest an early hypotrophy of the proximal tubule, leading to congenital atubular glomeruli formation, accumulation of glomerular filtrate and cystic expansion of the Bowman’s space. Although ZEB2 is also expressed in the stromal cells of the developing kidney, stromal cell-specific deletion of Zeb2 with Foxd1-cre did not result in a glomerular cystic phenotype (data not shown), which further supports the specific role of ZEB2 in early developing nephron. Consistent with our findings, haploinsufficiency of Zeb2 was also reported to be associated with delayed nephrogenesis in a transgenic rat model.40
Atubular glomeruli have been reported in polycystic kidney disease, obstructive uropathy, and many other glomerular or tubular diseases in humans and animal models.27,29,30,41 However, they are thought to be secondary to progressive injury to the proximal tubules inducing degenerative tubular cell changes and atrophy, eventually destroying proximal tubules at the glomerulotubular junctions.30,42 We found that loss of Zeb2 is associated with primary congenital atubular glomeruli due to developmental defects and hypotrophy of the proximal tubules and the glomerulotubular junction. Although the link between acquired atubular glomeruli and glomerular cyst formation was proposed two decades ago,28 our study now demonstrates that primary glomerulocystic disease can also be caused by congenital atubular glomeruli.
ZEB2 is also known as SIP1 (SMAD-interacting protein 1), which interacts with activated SMAD proteins and is part of the TGF-β/SMAD signaling pathway.43,44 SMADs are highly expressed in developing kidney.45 Elevated TGF-β/SMAD signaling has also been detected in experimental Pkd1 mouse models and human patients with polycystic kidney disease.46 We found abnormal expression of Glis3 gene in Zeb2 cKO kidneys at E18.5 compared to their wild type littermates. Transcription factor GLIS3 interacts with TAZ protein (encoded by Wwtr1 gene), which is also part of the TGF-β/SMAD pathway regulating SMAD shuttling between the cytoplasm and the nucleus.34,47 Interestingly, both Glis3 and Wwtr1 knockout mice develop glomerular cysts as Zeb2 cKO mice.34,36 In our study, we also found abnormal expression of Hnf1β in the Zeb2 cKO kidneys at E18.5 compared to the wild-type littermate controls. HNF1B mutations cause glomerulocystic disease in human and Hnf1β mesenchyme specific cKO mice also develops glomerular cysts.11,31 A recent study shows that the expressions of both Zeb2 and Pkd1 are upregulated in another Hnf1β renal specific knockout mouse model with polycystic kidney disease,18 suggesting that an optimal and balanced ZEB2 expression in the kidney is required to prevent renal cysts formation.
High levels of polycystin-1 are often detected in the kidney of ADPKD patients.48–52 Overexpression of polycystin-1 in mice also causes glomerular cysts.32,53 Hnf1β cKO and Dicer cKO mice develop renal cysts with increased levels of Pkd1 and decreased levels of miR-200.18,19 We found increased expression of Pkd1 mRNA and polycystin-1 in the glomeruli of Zeb2 cKO kidneys after the initial phase of glomerular cyst formation. Although miR-200 expression is repressed by ZEB254 and Zeb2 gene is a known target of miR-20055 in a double-negative feedback loop, we did not observe differential expression of miR-200 in Zeb2 cKO kidneys. Likewise, no differential expression of Zeb2 was observed in the Dicer cKO mice in which miR-200 is significantly downregulated.19 Therefore, Pkd1 upregulation in the non-cystic glomeruli of Zeb2 cKO mice is probably secondary to the glomerular stress (e.g. due to hyperfiltration of the non-cystic glomeruli) and not the result of a direct regulation of Pkd1 gene expression by the ZEB2/miR-200 signaling network. The effects of this increased expression of polycystin-1 in Zeb2 cKO mice are presently unknown and need further investigation.
In conclusion, by studying animal models of a disease gene causing a human syndrome associated with an increased risk of congenital kidney anomalies, we identified Zeb2 as a novel gene important in proximal tubule development and glomerulotubular junction formation. Loss of Zeb2 in mesenchyme-derived nephrons in mice results in reduced glomerulotubular integrity, congenital atubular glomeruli and primary glomerular cystic disease. Future studies are needed to elucidate the molecular pathway of ZEB2 signaling during kidney development and the cell type and mechanism of Pkd1 overexpression in non-cystic glomeruli, and to determine whether MWS patients also develop glomerular cysts and whether a subset of patients with glomerulocystic kidney disease carry ZEB2 mutations. As a transcription factor, ZEB2 may also provide a starting point for further identification of new genes important in glomerulotubular junction development and that, when mutated, may cause primary glomerulocystic kidney disease in patients.
MATERIALS AND METHODS
Animals
Zeb2 floxed conditional knockout mouse and ZEB2-EGFP reporter mouse were previously reported.20, 23 Pax2-cre+ mice were obtained from the MMRRC (#010569-UNC)22 and Six2-cre+ mice were purchased from the Jackson Lab (#009606).25 All animal studies were approved by the IACUC of Boston University.
Histology
The kidneys of mice at defined ages were dissected and fixed in 4% PFA and processed for paraffin embedding. Serial sections were cut and stained by H&E, Periodic acid–Schiff stain and Masson Trichrome stain using standard methods. Slides were viewed with an Olympus microscope and photographed using a DP72 digital camera.
Quantification of glomerular cysts and kidney length
The numbers of glomeruli with and without cysts were counted on an H&E-stained median sagittal kidney section from each animal. The glomerular cysts were quantified by counting the total number of glomeruli that are identified by glomerular tuft and by scoring them as cystic when a two- to threefold dilatation of the Bowman’s space was observed. Embryonic kidney length was measured using Olympus microscope and cellSens software.
Immunostaining
For immunohistochemistry (IHC), kidney sections were pretreated to quench endogenous peroxidase (3.0% hydrogen peroxide) and endogenous biotin (SP-2001, Vector Labs), and stained with biotinylated LTL (B-1325, Vector Labs) and DBA (B-1035, Vector Labs) following standard IHC protocol. For immunofluorescence staining, mouse kidneys were fixed in 4% PFA followed by incubation in 30% sucrose overnight at 4°C, embedded in OCT compound (Tissue-Tek), and cryosectioned at 10 μm. Frozen sections were permeabilized with 0.1% PBS- Triton X-100 for 10 min and blocked in 5% goat serum for 1 hour. Primary antibodies were incubated overnight at 4°C followed by secondary antibodies incubated at room temperature for 1 hour. Following primary antibodies were used: WT1 (sc-192, Santa Cruz), Polycystin-1 (sc-130554, Santa Cruz), Laminin (L9393, Sigma), GFP (GFP-1020, Aves), Villin-1 (2369, Cell Signaling), JAG1 (sc-8303, Santa Cruz), PAX2 (71-6000, Thermo Fisher), megalin (sc-16478, Santa Cruz), cubilin (sc-20609, Santa Cruz). Tissue sections were mounted in media containing DAPI and imaged by a Zeiss confocal microscope.
Urine and Plasma Analysis
Urine protein excretion was detected by SDS-PAGE followed by Coomassie blue staining and quantified using ImageJ. Urine creatinine was measured using a mouse creatinine assay kit (80350, Crystal Chem). Urine albumin/creatinine ratios were calculated. Serum BUN were measured using the Catalyst Dx Chemistry Analyzer by IDEXX.
Assessment of glomerulotubular integrity
The glomerulotubular integrity was assessed by analyzing the connection between the proximal tubules and the glomeruli on LTL-stained kidney sections as previously described,29, 30, 56 based on the knowledge that LTL-positive stained epithelial cells constitute part of the Bowman’s capsule with normal glomerulotubular junctions but are absent in atubular glomeruli. Briefly, consecutive serial sections of LTL-stained kidneys from Zeb2 cKO and wild-type controls (5 mice in each group) were used to make positive identification of atubular glomeruli. In order to quantify the glomerulotubular integrity, all glomeruli were counted on a single LTL-stained kidney section from each mouse and were divided into two categories on the basis of presence (positive) or absence (negative) of LTL staining in the Bowman’s capsule. Quantification of glomerulotubular integrity was presented as the percentage of LTL-positive glomeruli in total glomeruli counted in cKO mice and wild-type controls.29
Gene expression analysis
Total RNA was extracted from kidneys using miRNeasy Micro kit (217084, Qiagen). cDNA was synthesized using Verso cDNA Synthesis Kit (AB-1453, Life Technologies) and TaqMan MicroRNA RT kit (4366596, Life Technologies). Gene expression was analyzed using 7500FAST real-time PCR machine with TaqMan probes (Life Technologies). Relative gene expression data were analyzed by the delta-delta-Ct method and were normalized to the either Gapdh or Ppp1r3c.
Cell proliferation and apoptosis analyses
Proliferative cells were identified using an anti-pHH3 antibody (9701, Cell Signaling). To quantify cell proliferation, all the glomeruli were counted on a single kidney section from each of the 8 mice and were divided into two categories on the basis of presence (positive) or absence (negative) of pHH3 staining in the Bowman’s capsule. Quantification is presented as the percentage of pHH3-positive glomeruli out of total glomeruli counted. Apoptosis was analyzed as previously described using the TUNEL assay.57 Apoptotic cells were detected using Apoptag Peroxidase In Situ Apoptosis Detection Kit (S7100, Millipore). To quantify apoptosis in the CSB and SSB, we analyzed all the CSB/SSB in one kidney section from 9 different embryos, and counted them as either “positive” when at least one apoptotic cell was present or “negative” when no apoptotic cell was visualized. The immunofluorescent TUNEL staining was performed using the ApopTag Red In Situ Apoptosis Detection Kit (S7165, Millipore).
Statistical Analyses
Data are given as mean and standard deviation (SD). A minimum of three mice were used for each analysis, unless stated otherwise. Statistical analysis was performed by using the Student t-test or the chi-square test, and significance was determined at p < 0.05.
Supplementary Material
Supplemental Figure 1. Zeb2flox/flox;Pax2-cre+ conditional knockout mice die at birth.
(a) Breeding scheme for generating Zeb2 conditional knockout mice using the Pax2-cre+ allele. (b) Distributions of genotypes in weaned mice at 3 weeks from 20 litters (i) and at E18.5 from 10 litters (ii). Observed: the number of mice with each genotype obtained after breeding experiments; Expected: the number of mice with each genotype expected based on the parents genotypes and normal Mendelian distribution.
Supplemental Figure 2. ZEB2 expression in E16.5 developing mouse kidney.
(a–f) ZEB2 expression (red) in the developing kidney was detected using the ZEB2-EGFP reporter mouse at E16.5 with an anti-GFP antibody (red). The structure of the nephrons and collecting ducts were delineated with an anti-Laminin antibody (green). Magnification of 100x (a), 200x (b), and 400x (c, d) are indicated. (e) Enlarged boxed area from c shows ZEB2 positive cells (arrows) in an S-shaped body (ssb). (f) Enlarged boxed area from d shows ZEB2 positive cells (arrows) in the Bowman’s capsule of a glomerulus (g).
Supplemental Figure 3. ZEB2 and PAX2/JAG1 co-expression in E15.5 and E16.5 developing nephron from ZEB2-GFP transgenic mice.
(a) Co-localization of ZEB2 (green) and PAX2 (red) on an E15.5 Zeb2-GFP reporter mouse kidney shows co-expression of ZEB2-GFP and PAX2 in the same cell nuclei (arrows) of developing nephron. (b) ZEB2 (green) is expressed in the same cells (arrow) labeled by membrane protein JAG1 (red) in the S-shaped body of an E16.5 Zeb2-GFP reporter mouse kidney.
Supplemental Figure 4. Morphology of non-cystic glomeruli in Zeb2 cKO kidneys compared to wild-type littermate controls.
PAS staining of P8 kidneys from Zeb2flox/flox;Six2-cre+ cKO and Zeb2+/+ wild-type littermate controls show difference of the histomorphometry of cKO mutant non-cystic glomeruli (red arrows) compared with those of wild-type mice, including increased number of sclerotic glomeruli (blue arrow) and glomerular capillary dilation (yellow arrow) in the cKO mutant kidneys. Magnification: 200x in (a) and 400x in (b). Red asterisk marks glomerular cyst in cKO mutant kidney.
Supplemental Figure 5. Megalin and cubilin are upregulated in adult Zeb2 cKO renal proximal tubules.
Immunohistochemistry (IHC) staining with anti-megalin and anti-cubilin antibodies show similar levels of megalin and cubilin expression (arrows) in the proximal tubules in embryonic E18.5 kidneys of Zeb2 cKO and wild-type littermate controls. The expressions of megalin and cubilin are significantly upregulated in the proximal tubules of 7-week old adult Zeb2 cKO in comparison to the age-matched wild-type littermate controls, suggesting increased proximal tubule endocytosis of albumin in Zeb2 cKO. The albuminuria in Zeb2 cKO is probably coming through those nephrons that are patent and connected to the hyperfiltrative glomeruli.
Supplemental Figure 6. No major difference of JAG1 expression in E15.5 and E16.5 Zeb2 cKO kidney compared to littermate controls.
(a) No major difference of JAG1 expression patterns in developing nephron (arrow) of Zeb2flox/flox;Pax2-cre+ cKO and wild-type littermates at E15.5. (b) No major difference of JAG1 expression patterns in developing nephron (arrows) of Zeb2flox/flox;Six2-cre+ cKO and wild-type littermates at E16.5.
Supplemental Figure 7. No significant changes in cell proliferation in the Bowman’s capsule of Zeb2 knockout mice.
(a) Staining with an antibody against phospho-Histone H3 (pHH3) as a marker of cell proliferation in E17.5 Zeb2flox/flox;Pax2-cre+ embryonic kidneys (two kidneys in each group, 400x magnification). (b) Staining with an antibody against pHH3 as a marker of proliferation in P12 Zeb2flox/flox;Six2-cre+ kidneys (two kidneys in each group, 400x magnification). (c) No significant difference in cell proliferation between the wild-type and Zeb2 conditional knockout mice in the Bowman’s capsule (cKO mutant n=235 glomeruli analyzed, wild-type n=220 glomeruli analyzed from four kidneys in each group, ns - non significant).
Supplemental Figure 8. Decreased apoptosis in the C-shaped and S-shaped bodies in Zeb2 knockout mice.
(a) TUNEL (rhodamine fluorochrome) staining shows reduced positive apoptotic cells (arrows) in Zeb2 conditional knockout kidneys using both Pax2-cre+ and Six2-cre+ (n=5 mutant kidneys and n=4 wild-type kidneys, 400x magnification). (b) Immunofluorescence staining with TUNEL (red) and an antibody against Laminin (green) to delineate the C-shaped bodies and the S-Shaped bodies. (c) Reduced apoptosis in the C-shaped bodies and S-shaped bodies of the Zeb2 conditional knockouts (n=34 mutant C-shaped and S-shaped bodies and n=39 wild-type C-shaped and S-shaped bodies analyzed, *p-value < 10−2).
Acknowledgments
We thank Drs. Xueping Fan, Kenn Albrecht, Marc Lenburg, and Matthew Layne for helpful discussion, Dr. Richard Lu for helping with Zeb2 floxed mouse transfer, Kathleen Dashner for cryostat and microtome technical support. This work is supported by NIH grants R01DK078226 (WL) and R01HD060050 (RLM), a Cooperative Research Grant from Massachusetts Life Sciences Center (WL), and is also supported in part by Research Grant #1-FY12-426 from the March of Dimes Foundation (WL).
Footnotes
DISCLOSURE
All the authors declared no competing interests
References
- 1.Kurschat CE, Muller RU, Franke M, et al. An approach to cystic kidney diseases: the clinician’s view. Nature reviews. Nephrology. 2014;10:687–699. doi: 10.1038/nrneph.2014.173. [DOI] [PubMed] [Google Scholar]
- 2.Bonsib SM. The classification of renal cystic diseases and other congenital malformations of the kidney and urinary tract. Archives of pathology & laboratory medicine. 2010;134:554–568. doi: 10.5858/134.4.554. [DOI] [PubMed] [Google Scholar]
- 3.Bissler JJ, Siroky BJ, Yin H. Glomerulocystic kidney disease. Pediatr Nephrol. 2010;25:2049–2056. doi: 10.1007/s00467-009-1416-2. quiz 2056–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lennerz JK, Spence DC, Iskandar SS, et al. Glomerulocystic kidney: one hundred-year perspective. Archives of pathology & laboratory medicine. 2010;134:583–605. doi: 10.5858/134.4.583. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein J. Glomerulocystic kidney disease--nosological considerations. Pediatr Nephrol. 1993;7:464–470. doi: 10.1007/BF00857576. [DOI] [PubMed] [Google Scholar]
- 6.Gusmano R, Caridi G, Marini M, et al. Glomerulocystic kidney disease in a family. Nephrol Dial Transplant. 2002;17:813–818. doi: 10.1093/ndt/17.5.813. [DOI] [PubMed] [Google Scholar]
- 7.Feather SA, Winyard PJ, Dodd S, Woolf AS. Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: clinical, radiological and histopathological features of a new kindred. Nephrol Dial Transplant. 1997;12:1354–1361. doi: 10.1093/ndt/12.7.1354. [DOI] [PubMed] [Google Scholar]
- 8.Galliani CA, Gomez AM, Panniello G, Bisceglia M. Selected case from the Arkadi M. Rywlin International Pathology Slide Series: Asymmetric, segmental glomerulocystic kidney in an infant with tuberous sclerosis complex. Advances in anatomic pathology. 2015;22:135–143. doi: 10.1097/PAP.0000000000000055. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann C, Fliegauf M, Bruchle NO, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rampoldi L, Caridi G, Santon D, et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet. 2003;12:3369–3384. doi: 10.1093/hmg/ddg353. [DOI] [PubMed] [Google Scholar]
- 11.Bingham C, Bulman MP, Ellard S, et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219–224. doi: 10.1086/316945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong M, Jiang L, Zhou Y, et al. The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. 2012;302:F369–379. doi: 10.1152/ajprenal.00268.2011. [DOI] [PubMed] [Google Scholar]
- 13.Tang O, Chen XM, Shen S, et al. MiRNA-200b represses transforming growth factor-beta1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am J Physiol Renal Physiol. 2013;304:F1266–1273. doi: 10.1152/ajprenal.00302.2012. [DOI] [PubMed] [Google Scholar]
- 14.Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Molecular biology of the cell. 2011;22:1686–1698. doi: 10.1091/mbc.E11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet journal of rare diseases. 2007;2:42. doi: 10.1186/1750-1172-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada Y, Nomura N, Yamada K, et al. The spectrum of ZEB2 mutations causing the Mowat-Wilson syndrome in Japanese populations. Am J Med Genet A. 2014;164A:1899–1908. doi: 10.1002/ajmg.a.36551. [DOI] [PubMed] [Google Scholar]
- 17.Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature cell biology. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 18.Hajarnis SS, Patel V, Aboudehen K, et al. Transcription Factor HNF-1beta Regulates MicroRNA-200 Expression Through a Long Noncoding RNA. J Biol Chem. 2015 doi: 10.1074/jbc.M115.670646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel V, Hajarnis S, Williams D, et al. MicroRNAs regulate renal tubule maturation through modulation of Pkd1. J Am Soc Nephrol. 2012;23:1941–1948. doi: 10.1681/ASN.2012030321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higashi Y, Maruhashi M, Nelles L, et al. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis. 2002;32:82–84. doi: 10.1002/gene.10048. [DOI] [PubMed] [Google Scholar]
- 21.Van de Putte T, Maruhashi M, Francis A, et al. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am J Hum Genet. 2003;72:465–470. doi: 10.1086/346092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- 23.Nishizaki Y, Takagi T, Matsui F, Higashi Y. SIP1 expression patterns in brain investigated by generating a SIP1-EGFP reporter knock-in mouse. Genesis. 2014;52:56–67. doi: 10.1002/dvg.22726. [DOI] [PubMed] [Google Scholar]
- 24.Wang CA, Drasin D, Pham C, et al. Homeoprotein Six2 promotes breast cancer metastasis via transcriptional and epigenetic control of E-cadherin expression. Cancer Res. 2014;74:7357–7370. doi: 10.1158/0008-5472.CAN-14-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi A, Valerius MT, Mugford JW, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell stem cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terryn S, Tanaka K, Lengele JP, et al. Tubular proteinuria in patients with HNF1alpha mutations: HNF1alpha drives endocytosis in the proximal tubule. Kidney Int. 2016;89:1075–1089. doi: 10.1016/j.kint.2016.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol. 2008;19:197–206. doi: 10.1681/ASN.2007080862. [DOI] [PubMed] [Google Scholar]
- 28.Gibson IW, Downie TT, More IA, Lindop GB. Atubular glomeruli and glomerular cysts--a possible pathway for nephron loss in the human kidney? J Pathol. 1996;179:421–426. doi: 10.1002/(SICI)1096-9896(199608)179:4<421::AID-PATH616>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol. 2011;301:F110–117. doi: 10.1152/ajprenal.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galarreta CI, Grantham JJ, Forbes MS, et al. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am J Pathol. 2014;184:1957–1966. doi: 10.1016/j.ajpath.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massa F, Garbay S, Bouvier R, et al. Hepatocyte nuclear factor 1beta controls nephron tubular development. Development. 2013;140:886–896. doi: 10.1242/dev.086546. [DOI] [PubMed] [Google Scholar]
- 32.Pritchard L, Sloane-Stanley JA, Sharpe JA, et al. A human PKD1 transgene generates functional polycystin-1 in mice and is associated with a cystic phenotype. Hum Mol Genet. 2000;9:2617–2627. doi: 10.1093/hmg/9.18.2617. [DOI] [PubMed] [Google Scholar]
- 33.Williams SS, Cobo-Stark P, James LR, et al. Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2008;23:733–741. doi: 10.1007/s00467-007-0735-4. [DOI] [PubMed] [Google Scholar]
- 34.Kang HS, Beak JY, Kim YS, et al. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol. 2009;29:2556–2569. doi: 10.1128/MCB.01620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrante MI, Zullo A, Barra A, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112–117. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 36.Hossain Z, Ali SM, Ko HL, et al. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A. 2007;104:1631–1636. doi: 10.1073/pnas.0605266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paul BM, Vanden Heuvel GB. Kidney: polycystic kidney disease. Wiley interdisciplinary reviews. Developmental biology. 2014;3:465–487. doi: 10.1002/wdev.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho J. The regulation of apoptosis in kidney development: implications for nephron number and pattern? Frontiers in pediatrics. 2014;2:128. doi: 10.3389/fped.2014.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coles HS, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993;118:777–784. doi: 10.1242/dev.118.3.777. [DOI] [PubMed] [Google Scholar]
- 40.El-Kasti MM, Wells T, Carter DA. A novel long-range enhancer regulates postnatal expression of Zeb2: implications for Mowat-Wilson syndrome phenotypes. Hum Mol Genet. 2012;21:5429–5442. doi: 10.1093/hmg/dds389. [DOI] [PubMed] [Google Scholar]
- 41.Forbes MS, Thornhill BA, Galarreta CI, et al. Chronic unilateral ureteral obstruction in the neonatal mouse delays maturation of both kidneys and leads to late formation of atubular glomeruli. Am J Physiol Renal Physiol. 2013;305:F1736–1746. doi: 10.1152/ajprenal.00152.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanner GA, Tielker MA, Connors BA, et al. Atubular glomeruli in a rat model of polycystic kidney disease. Kidney Int. 2002;62:1947–1957. doi: 10.1046/j.1523-1755.2002.00689.x. [DOI] [PubMed] [Google Scholar]
- 43.Conidi A, Cazzola S, Beets K, et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFbeta/BMP signaling in vivo. Cytokine & growth factor reviews. 2011;22:287–300. doi: 10.1016/j.cytogfr.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 44.Hegarty SV, Sullivan AM, O’Keeffe GW. Zeb2: A multifunctional regulator of nervous system development. Progress in neurobiology. 2015;132:81–95. doi: 10.1016/j.pneurobio.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Vrljicak P, Myburgh D, Ryan AK, et al. Smad expression during kidney development. Am J Physiol Renal Physiol. 2004;286:F625–633. doi: 10.1152/ajprenal.00152.2003. [DOI] [PubMed] [Google Scholar]
- 46.Hassane S, Leonhard WN, van der Wal A, et al. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222:21–31. doi: 10.1002/path.2734. [DOI] [PubMed] [Google Scholar]
- 47.Varelas X, Sakuma R, Samavarchi-Tehrani P, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nature cell biology. 2008;10:837–848. doi: 10.1038/ncb1748. [DOI] [PubMed] [Google Scholar]
- 48.Peters DJ, Spruit L, Klingel R, et al. Adult, fetal, and polycystic kidney expression of polycystin, the polycystic kidney disease-1 gene product. Lab Invest. 1996;75:221–230. [PubMed] [Google Scholar]
- 49.Geng L, Segal Y, Peissel B, et al. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest. 1996;98:2674–2682. doi: 10.1172/JCI119090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palsson R, Sharma CP, Kim K, et al. Characterization and cell distribution of polycystin, the product of autosomal dominant polycystic kidney disease gene 1. Molecular medicine. 1996;2:702–711. [PMC free article] [PubMed] [Google Scholar]
- 51.Ward CJ, Turley H, Ong AC, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93:1524–1528. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song X, Di Giovanni V, He N, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18:2328–2343. doi: 10.1093/hmg/ddp165. [DOI] [PubMed] [Google Scholar]
- 53.Thivierge C, Kurbegovic A, Couillard M, et al. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol. 2006;26:1538–1548. doi: 10.1128/MCB.26.4.1538-1548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 55.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thornhill BA, Forbes MS, Marcinko ES, Chevalier RL. Glomerulotubular disconnection in neonatal mice after relief of partial ureteral obstruction. Kidney Int. 2007;72:1103–1112. doi: 10.1038/sj.ki.5002512. [DOI] [PubMed] [Google Scholar]
- 57.Omori S, Hida M, Ishikura K, et al. Expression of mitogen-activated protein kinase family in rat renal development. Kidney Int. 2000;58:27–37. doi: 10.1046/j.1523-1755.2000.00137.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Zeb2flox/flox;Pax2-cre+ conditional knockout mice die at birth.
(a) Breeding scheme for generating Zeb2 conditional knockout mice using the Pax2-cre+ allele. (b) Distributions of genotypes in weaned mice at 3 weeks from 20 litters (i) and at E18.5 from 10 litters (ii). Observed: the number of mice with each genotype obtained after breeding experiments; Expected: the number of mice with each genotype expected based on the parents genotypes and normal Mendelian distribution.
Supplemental Figure 2. ZEB2 expression in E16.5 developing mouse kidney.
(a–f) ZEB2 expression (red) in the developing kidney was detected using the ZEB2-EGFP reporter mouse at E16.5 with an anti-GFP antibody (red). The structure of the nephrons and collecting ducts were delineated with an anti-Laminin antibody (green). Magnification of 100x (a), 200x (b), and 400x (c, d) are indicated. (e) Enlarged boxed area from c shows ZEB2 positive cells (arrows) in an S-shaped body (ssb). (f) Enlarged boxed area from d shows ZEB2 positive cells (arrows) in the Bowman’s capsule of a glomerulus (g).
Supplemental Figure 3. ZEB2 and PAX2/JAG1 co-expression in E15.5 and E16.5 developing nephron from ZEB2-GFP transgenic mice.
(a) Co-localization of ZEB2 (green) and PAX2 (red) on an E15.5 Zeb2-GFP reporter mouse kidney shows co-expression of ZEB2-GFP and PAX2 in the same cell nuclei (arrows) of developing nephron. (b) ZEB2 (green) is expressed in the same cells (arrow) labeled by membrane protein JAG1 (red) in the S-shaped body of an E16.5 Zeb2-GFP reporter mouse kidney.
Supplemental Figure 4. Morphology of non-cystic glomeruli in Zeb2 cKO kidneys compared to wild-type littermate controls.
PAS staining of P8 kidneys from Zeb2flox/flox;Six2-cre+ cKO and Zeb2+/+ wild-type littermate controls show difference of the histomorphometry of cKO mutant non-cystic glomeruli (red arrows) compared with those of wild-type mice, including increased number of sclerotic glomeruli (blue arrow) and glomerular capillary dilation (yellow arrow) in the cKO mutant kidneys. Magnification: 200x in (a) and 400x in (b). Red asterisk marks glomerular cyst in cKO mutant kidney.
Supplemental Figure 5. Megalin and cubilin are upregulated in adult Zeb2 cKO renal proximal tubules.
Immunohistochemistry (IHC) staining with anti-megalin and anti-cubilin antibodies show similar levels of megalin and cubilin expression (arrows) in the proximal tubules in embryonic E18.5 kidneys of Zeb2 cKO and wild-type littermate controls. The expressions of megalin and cubilin are significantly upregulated in the proximal tubules of 7-week old adult Zeb2 cKO in comparison to the age-matched wild-type littermate controls, suggesting increased proximal tubule endocytosis of albumin in Zeb2 cKO. The albuminuria in Zeb2 cKO is probably coming through those nephrons that are patent and connected to the hyperfiltrative glomeruli.
Supplemental Figure 6. No major difference of JAG1 expression in E15.5 and E16.5 Zeb2 cKO kidney compared to littermate controls.
(a) No major difference of JAG1 expression patterns in developing nephron (arrow) of Zeb2flox/flox;Pax2-cre+ cKO and wild-type littermates at E15.5. (b) No major difference of JAG1 expression patterns in developing nephron (arrows) of Zeb2flox/flox;Six2-cre+ cKO and wild-type littermates at E16.5.
Supplemental Figure 7. No significant changes in cell proliferation in the Bowman’s capsule of Zeb2 knockout mice.
(a) Staining with an antibody against phospho-Histone H3 (pHH3) as a marker of cell proliferation in E17.5 Zeb2flox/flox;Pax2-cre+ embryonic kidneys (two kidneys in each group, 400x magnification). (b) Staining with an antibody against pHH3 as a marker of proliferation in P12 Zeb2flox/flox;Six2-cre+ kidneys (two kidneys in each group, 400x magnification). (c) No significant difference in cell proliferation between the wild-type and Zeb2 conditional knockout mice in the Bowman’s capsule (cKO mutant n=235 glomeruli analyzed, wild-type n=220 glomeruli analyzed from four kidneys in each group, ns - non significant).
Supplemental Figure 8. Decreased apoptosis in the C-shaped and S-shaped bodies in Zeb2 knockout mice.
(a) TUNEL (rhodamine fluorochrome) staining shows reduced positive apoptotic cells (arrows) in Zeb2 conditional knockout kidneys using both Pax2-cre+ and Six2-cre+ (n=5 mutant kidneys and n=4 wild-type kidneys, 400x magnification). (b) Immunofluorescence staining with TUNEL (red) and an antibody against Laminin (green) to delineate the C-shaped bodies and the S-Shaped bodies. (c) Reduced apoptosis in the C-shaped bodies and S-shaped bodies of the Zeb2 conditional knockouts (n=34 mutant C-shaped and S-shaped bodies and n=39 wild-type C-shaped and S-shaped bodies analyzed, *p-value < 10−2).