

Summary
Androgen receptor (AR) signaling is a key driver of prostate cancer (PC). While androgen-deprivation therapy is transiently effective in advanced disease, tumors often progress to a lethal castration-resistant state (CRPC). We show that recurrent PC-driver mutations in SPOP stabilize the TRIM24 protein, which promotes proliferation under low androgen conditions. TRIM24 augments AR signaling, and AR and TRIM24 co-activated genes are significantly up-regulated in CRPC. Expression of TRIM24 protein increases from primary PC to CRPC, and both TRIM24 protein levels and the AR/TRIM24 gene signature predict disease-recurrence. Analyses in CRPC cells reveal that the TRIM24 bromodomain and the AR-interacting motif are essential to support proliferation. These data provide a rationale for therapeutic TRIM24 targeting in SPOP-mutant and CRPC patients.
Graphical abstract
Introduction
Prostate cancer (PC) remains one of the most common causes of male cancer deaths worldwide (Jemal et al., 2011). Many patients with organ-confined tumors at initial diagnosis relapse following radical prostatectomy or local radiotherapy and develop recurrent disease. On a molecular level, the steroid hormone androgen activates the androgen receptor (AR), which in turn functions as a nuclear receptor transcription factor and executes specific tumorigenic gene expression programs (Matsumoto et al., 2013; Wang et al., 2009). Therefore, advanced cancer therapy includes androgen-deprivation approaches through inhibition of androgen synthesis and the administration of competitive AR antagonists (Niraula et al., 2012). Unfortunately, most patients develop resistance to treatment and subsequently progress to castration-resistant disease (CRPC) that in most cases continues to rely on AR signaling (Heinlein and Chang, 2004; Scher and Sawyers, 2005). As CRPC is often fatal, there is a significant need for improved treatment options. How AR regulates CRPC growth is incompletely characterized, but has been reported to involve mechanisms that enable transactivation of AR under low androgen levels. Proposed processes include intratumoral production of androgens (Montgomery et al., 2008), genetic changes of the AR gene (Taplin et al., 1995; Visakorpi et al., 1995), the emergence of ligand-independent AR splice variants (Guo et al., 2009; Hu et al., 2009; Sun et al., 2010), cross talk between AR and other signaling pathways (Lamont and Tindall, 2011), and the altered action of transcriptional co-regulators (Agoulnik et al., 2006; Gregory et al., 2001; Linja et al., 2004; Taylor et al., 2010; Xu et al., 2012).
Genome sequencing studies have revealed recurrent founder mutations in the substrate-binding cleft of the cullin-RING ubiquitin ligase adaptor SPOP (speckle-type POZ protein) in approximately 10% of primary PC (Barbieri et al., 2012; Blattner et al., 2014; Kandoth et al., 2013). SPOP proteins harboring PC-specific mutations have been reported as being defective in mediating ubiquitylation and proteasomal degradation of AR and its co-activator NCOA3 and thus promote AR signaling (An et al., 2014; Geng et al., 2013; Geng et al., 2014). In agreement with this, enhanced AR signaling has been identified as a cardinal feature of SPOP mutant tumors (TCGA, 2015). Using an unbiased proteomic approach in prostate epithelial cells, we identified TRIM24 (tripartite motif-containing protein 24, also known as TIF1α) as another potential effector protein downstream of SPOP mutations (Theurillat et al., 2014). Moreover, TRIM24 showed reduced ubiquitylation that was accompanied by increased protein levels in the presence of SPOP mutations (Theurillat et al., 2014).
TRIM24 has been implicated in driving different tumor types through its ability to interfere with tumor suppressive and oncogenic pathways (Hatakeyama, 2011; Herquel et al., 2011). Its N-terminal tripartite motifs include a RING domain, which is involved in ubiquitylation and degradation of p53 (Allton et al., 2009). Moreover, a C-terminal tandem PHD finger-bromodomain confers TRIM24 with the ability to recognize unmodified histone H3K4 through the former domain, as well as H3K23-acetyl through the latter domain (Tsai et al., 2010). This chromatin interacting module has been implicated in general transcriptional co-regulation, as well as the activation of estrogen-responsive genes in breast cancer and the PIK3CA gene in glioma (Herquel et al., 2011; Tsai et al., 2010; Zhang et al., 2015). Through its conserved single LxxLL motif TRIM24 interacts with the AF2 domain of several nuclear receptors, including AR (Le Douarin et al., 1996; Thenot et al., 1997; vom Baur et al., 1996). In line with these observations, TRIM24 was shown to enhance AR-mediated gene activation in reporter assays (Kikuchi et al., 2009). Whether these different functions of TRIM24 influence PC progression is not known.
In this study we sought to investigate the role of TRIM24 in PC and to elucidate its suitability as a therapeutic target by combining a molecular characterization in PC cell lines with analyses in PC patients. We hypothesized that TRIM24 may be important for PC progression by functioning as an oncogenic transcriptional activator that cooperates with AR-dependent gene expression in CRPC settings, potentially opening new therapeutic avenues for treatment.
Results
TRIM24 mediates SPOP-mutant PC cell proliferation in low androgen
To investigate the impact of PC-associated SPOP mutants on TRIM24 deregulation and on androgen-mediated cell proliferation, we tested the effect of different SPOP mutations in the androgen-dependent LNCaP cell line. We observed that levels of TRIM24 protein were augmented upon expression of a number of different PC-associated SPOP mutations (Figure 1A), which is in agreement with our earlier results in prostate epithelial cells (Theurillat et al., 2014). Importantly, TRIM24 mRNA levels that were assessed in a parallel experiment did not follow the protein expression changes (Figure 1B), suggesting that these SPOP mutations regulate TRIM24 at the protein level in LNCaP cells. To examine how SPOP affects TRIM24 protein stability in LNCaP cells, we made use of a HA-tagged form of TRIM24 and found that this protein decayed more rapidly in the presence of wild-type (WT) SPOP than with the SPOP-F133L mutant (Figure S1A). In agreement with TRIM24 being an SPOP target, we found that SPOP-WT expression and treatment with the proteasome inhibitor MG132 increased TRIM24 ubiquitylation in 293T cells (Figure S1B), and SPOP knockdown augmented TRIM24 protein levels in LNCaP cells (Figure S1C). These results are consistent with our earlier findings in prostate epithelial cells (Theurillat et al., 2014) and support a model in which SPOP mutants impair ubiquitylation and degradation of TRIM24 through the proteasome.
Figure 1. Mutant SPOP stabilizes TRIM24 promoting PC cell growth under low androgen.
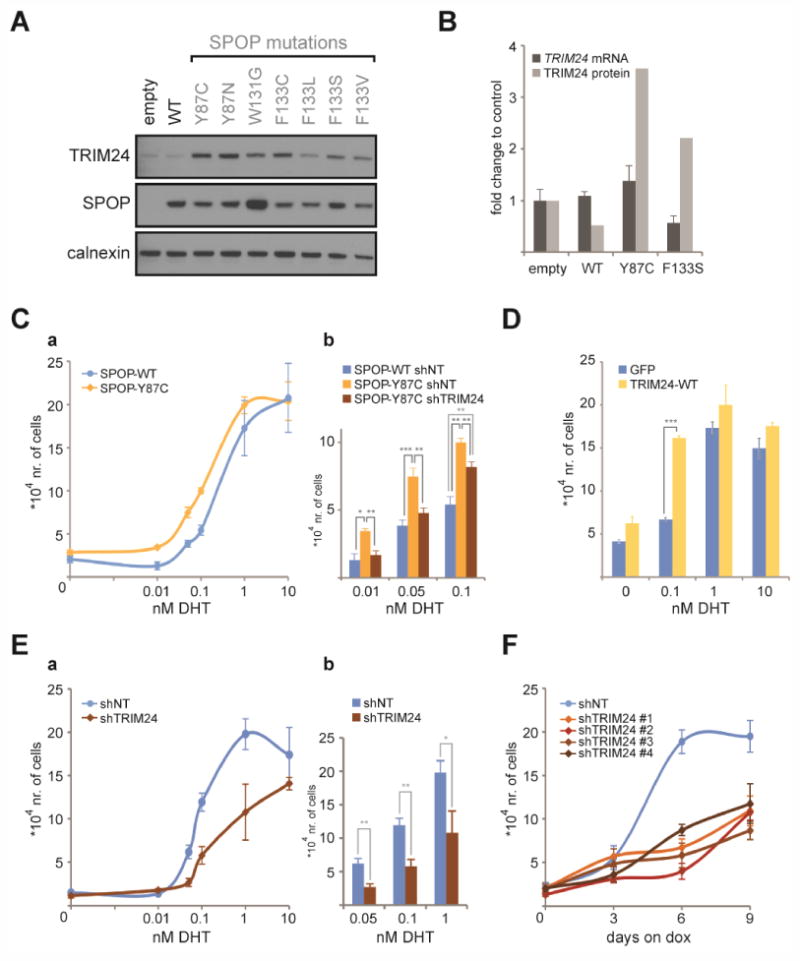
(A) TRIM24 and SPOP protein levels were detected by Western blot in LNCaP cells stably expressing wild-type (WT) or the indicated mutant SPOP constructs (in grey). Calnexin was used as a loading control. (B) TRIM24 protein levels from the indicated SPOP lines were compared to TRIM24 mRNA as assessed by quantitative PCR. (C) LNCaP cells stably expressing SPOP-WT or SPOP-Y87C were infected with lentiviral doxycycline (dox)-inducible shRNA targeting TRIM24 or a non-targeting (NT) control. Cells were treated with dox while being grown in indicated DHT concentrations and counted by hemacytometer (n=3) after 6 days. (a) Cell numbers were plotted against a log10 scale of the DHT concentration. (b) Cell numbers under low DHT levels are additionally highlighted in a bar plot. (D) LNCaP cells harboring dox-inducible TRIM24 or GFP cDNA were treated with dox for 6 days, while being grown in the presence of the indicated DHT concentration. Cell numbers were assessed as in (C). (E) LNCaP cells stably expressing dox-inducible shRNA targeting TRIM24 or a NT control were treated with dox for 6 days while being grown in the presence of the indicated concentration of DHT. Cell numbers were counted as in (C). (a) Cell numbers were plotted against a log10 scale of the DHT concentration. (b) Cell numbers under low DHT levels are additionally highlighted in a bar plot. (F) LNCaP-abl cells stably expressing 4 different dox-inducible shRNAs targeting TRIM24 or a NT control were treated with dox at day 0 and grown for 9 days. Cell numbers were counted after the indicated days and plotted. TRIM24 protein levels were assessed at the end of each experiment and are shown in Figures S1E, S1I, S1J and S1K. Data are represented as mean +/- SEM. Statistical analysis was performed using a two-tailed student's t-test assuming unequal variance *p<0.05, ** p<0.01, *** p<0.005.
(See also Figure S1).
Next we measured androgen-dependent LNCaP growth, in the presence of SPOP mutants, across a range of androgen concentrations. We found that when only limiting amounts of 5α-dihydrotestosterone (DHT) were available, SPOP mutant-expressing LNCaP cells all displayed a significant growth advantage compared to cells expressing SPOP-WT (Figures 1Ca and S1D). This growth advantage seen in SPOP-Y87C expressing cells cultured under low DHT concentrations was abrogated when TRIM24 expression was silenced by a specific shRNA (Figures 1Cb and S1E). The SPOP-W131G mutant showed comparable results (Figures S1F and S1G). At a higher DHT concentration, silencing of TRIM24 partially decreased the SPOP-Y87C mediated growth advantage (Figure 1Cb), suggesting that additional proteins may contribute to the SPOP mutant phenotype. This is also reflected in the DHT-induced values of proliferation, expressed as the half maximal effective concentration (EC50). The EC50 for the SPOP-Y87C mutant cells is 3-fold lower than the EC50 of SPOP-WT cells, while knock-down of TRIM24 in the mutant cells does not fully revert back to the WT EC50 level (Figure S1H). In summary, these findings reveal that the stabilization of TRIM24 protein by SPOP mutations is necessary to promote optimal PC proliferation under low androgen conditions.
TRIM24 promotes PC growth and sensitizes cells to low androgen availability
We then asked whether TRIM24 is sufficient to promote PC growth under low androgen availability. Indeed, over-expression of doxycycline (dox)-inducible TRIM24 (Figure S1I) was sufficient to mediate increased proliferation of LNCaP cells at 0.1nM DHT, when compared to GFP expressing control cells (Figure 1D). Conversely, when we decreased endogenous TRIM24 levels with shRNA (Figure S1J), we saw a reduction in androgen-dependent LNCaP proliferation (Figure 1Ea). Again, this phenotype was most prominent under low DHT concentrations (Figure 1Eb). Therefore, we tested the effect of TRIM24 depletion on the growth of the LNCaP-derived CRPC line LNCaP-abl (abl), which is continuously cultured in the absence of androgens. We found that reduction of TRIM24 with 4 different shRNA-based hairpins (Figure S1K) resulted in impaired proliferation rates for each of them (Figure 1F). More specifically, TRIM24-depleted abl cells exhibited a decreased capacity of G1/S cell cycle transition, without an increase in apoptosis (Figures S1L-S1O). In addition, TRIM24 knock-down triggered similar phenotypes in the androgen-independent lines CWR-22Rv1 and LNCaP95 (Figures S1P and S1Q), further supporting the conclusion that TRIM24 is essential for PC proliferation in castration-resistant settings.
TRIM24 binds to promoters and activates genes involved in cell proliferation and AR signaling in PC cells
To understand why hormone-starved PC cells were more dependent on TRIM24 than hormone-stimulated cells, we set out to identify TRIM24-dependent gene expression programs by overlapping TRIM24 cistromes with TRIM24-regulated genes. Direct genomic binding of TRIM24 was determined by performing ChIP-seq in abl as well as in LNCaP cells cultured in the absence or presence of hormone stimulation (Figure 2A). TRIM24 was found to be most abundant near promoters in all cistromes (Figures 2B and 2C). The number of TRIM24 sites went from 9744 and 7105 in hormone-starved and hormone-stimulated LNCaP cells, respectively, to 20621 sites in abl cells, with most LNCaP-specific TRIM24 sites also present in abl cells (Figure 2A). In addition, most of the over-represented transcription factor binding motifs associated with the TRIM24 cistromes are shared in both LNCaP and abl cells (Table S1). We also assessed the overlap of the LNCaP-specific TRIM24 ChIP-seq with published H3K27-acetyl data sets (Hazelett et al., 2014), which is a histone mark to which the TRIM24 bromodomain binds (Tsai et al., 2010). We identified a significant overlap between the respective cistromes both in the absence or presence of hormone-stimulation (Figures 2D and 2E, p<0.001, random permutation), suggesting that the TRIM24 bromodomain recognizes acetylated chromatin irrespective of cellular androgen levels.
Figure 2. TRIM24 binds to promoters and activates genes involved in cell proliferation and AR signaling in PC cells.
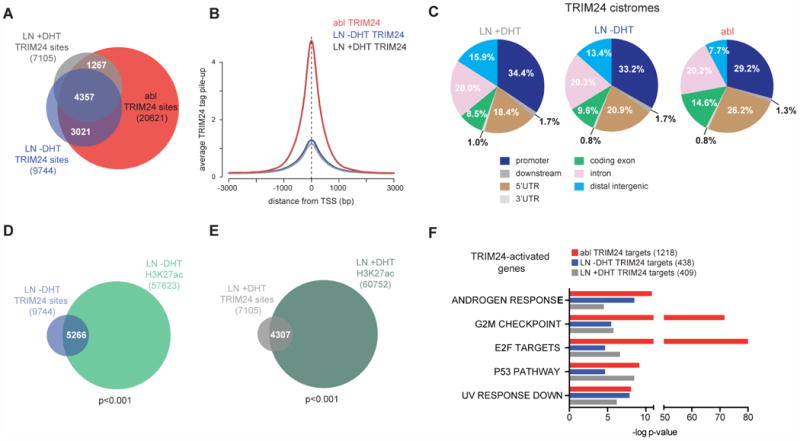
(A) Venn diagram overlapping the TRIM24 cistromes determined in hormone-starved (-DHT) and hormone-stimulated (+DHT) LNCaP (LN) cells, in addition to the hormone-refractory line LNCaP-abl (abl). The total number of genomic binding sites for each cistrome is indicated in parenthesis. (B) TRIM24 ChIP-seq signals from (A) were expressed relative to the location of TSS in the genome. (C) Cis-regulatory Element Annotation System (CEAS) (Ji et al., 2006) was used to characterize the genome-wide binding of the TRIM24-specific cistromes. Assessed binding distribution included regions up to 3kb upstream of TSS (promoter), up to 3kb downstream of TSS (downstream), 5′ and 3′ UTRs, coding exons, introns and distal intergenic regions. (D, E) Venn diagrams overlapping TRIM24 cistromes with published Histone H3 K27-acetylation (H3K27ac) cistromes (Hazelett et al., 2014) reveal (D) a 5266 site overlap in LN -DHT (p<0.001, random permutation), and (E) a 4307 site overlap in LN +DHT (p<0.001, random permutation). The total number of genomic binding sites for each cistrome is indicated in parenthesis. (F) Direct TRIM24-activated gene targets were determined by overlapping TRIM24 cistromes with TRIM24-dependent gene expression profiles in LN cells with our without DHT-stimulation and in abl cells. Direct targets were defined by TRIM24 promoter binding (3kb around TSS) and significant siRNA-mediated down-regulation (LIMMA, p<0.05, fold-change>1.3). Gene set enrichment analysis was used to compare TRIM24 targets with curated HALLMARK gene sets (Molecular Signatures Database). The top 5 common pathways together with their significance values (-log p-value) are depicted.
(See also Figure S2 and Tables S1-S3).
We identified TRIM24-dependent gene expression by microarray analysis in the LNCaP and abl cells. siRNA-mediated silencing of TRIM24 induced more genes to be down- rather than up-regulated (SAM, fold-change>2), consistent with TRIM24 acting as a transcriptional activator in this setting (Figure S2A). Moreover, 107 and 117 genes showed decreased levels after TRIM24 knockdown in LNCaP cells cultured without or with DHT stimulation, respectively (Figure S2A). In abl cells 278 genes were down-regulated upon TRIM24 depletion (Figure S2A). Many TRIM24-activated genes were involved in cell cycle processes (Figure S2B) and were up-regulated in abl cells compared to LNCaP cells (Figures S2C and S2D). Therefore, TRIM24 more prominently activates cell cycle genes in abl cells, thus providing an explanation for the increased dependency of these CRPC cells on intact TRIM24 levels for proliferation.
The correlation of the TRIM24 cistromes with TRIM24-regulated genes revealed a significant association between TRIM24 binding and transcriptional activation in both LNCaP and abl cells (Figure S2E). We further validated a set of TRIM24-activated gene targets by ChIP-quantitative PCR (qPCR) (Figures S2F and S2G) and Western blotting (Figure S2H). We then determined direct TRIM24-activated targets by identifying genes with promoter-bound TRIM24 (3kb around TSS) that were also significantly down-regulated by TRIM24 depletion (LIMMA, p<0.05, fold-change>1.3). To enable pathway analyses on the direct TRIM24-activated gene targets, we enlarged the target gene lists using a less stringent fold-change cutoff. In this analysis, 438 and 409 TRIM24 target genes were identified in LNCaP cells cultured under hormone-starvation (-DHT) and -stimulation (+DHT), respectively (Figure 2F and Table S2). In addition, we also found 1218 genes that were positively regulated by TRIM24 binding by applying the same criteria to the abl cell data sets (Figure 2F and Table S2).
We then used gene set enrichment analyses to compare the identified TRIM24-activated gene targets with the curated HALLMARK gene sets present in the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005). When we determined the top 10 most enriched biological processes, 5 of them were common among all the different TRIM24-activated target sets (Figure 2F and Table S3M). We then focused on identifying differences in the prevalence of genes present in the common pathways that would help explain our phenotypic findings in the different PC cell lines. In line with TRIM24 driving abl cell proliferation (Figure 1F), we found that cell cycle-related pathways, such as the G2M checkpoint and E2F targets were more prominently regulated in these CRPC cells compared to LNCaP cells (Figure 2F). Androgen responsive genes were more frequently activated by TRIM24 in LNCaP and abl cells under hormone-starved conditions, when compared to hormone-stimulated LNCaP cells (Figure 2F). This result is consistent with TRIM24 mediating PC cell growth under low DHT levels (Figures 1D and 1E) and suggests that TRIM24 co-activates AR-regulated genes more prominently under limiting hormone conditions and is thus more essential for PC proliferation in the castration-resistant setting.
To assess if TRIM24 and AR work together to co-activate target genes, we explored the interaction between AR and TRIM24. First, we found a significant overlap between the AR and the TRIM24 cistromes (Figures S3A and S3B) with a DHT-dependent increase of TRIM24 binding at the shared sites in LNCaP cells (Figure S3C). Second, we could confirm a weak interaction between the endogenous proteins when performing AR-specific immunoprecipitation on nuclear extracts (Figure S3D). Finally, by using peptide arrays (Figure S3E), we also validated the reported interaction between the TRIM24 LxxLL motif and the AR ligand binding domain (Cavailles et al., 1995; Kikuchi et al., 2009; Le Douarin et al., 1996; Thenot et al., 1997). Taken together, these results imply that AR and TRIM24 directly cooperate to activate genes in PC though we cannot exclude participation by another LxxLL-binding factor in the complex. Moreover, the overlap between AR and TRIM24 genomic binding is more pronounced in abl cells (2419 sites) when compared to LNCaP cells (526 sites) (Figures S3A and S3B), consistent with an increased interaction between the two factors in CRPC.
AR and TRIM24 directly activate genes that are up-regulated in CRPC and predict recurrence in primary tumors
To assess which genes are co-activated by AR and TRIM24 in CRPC cells, we derived an AR/TRIM24-regulated signature consisting of 21 genes using cistrome and gene expression data from abl cells. More specifically, genes within the signature were defined as those in a 100kb interval around overlapping AR and TRIM24 sites, which were also down-regulated by siRNA targeting AR and TRIM24 (LIMMA, p<0.05, fold-change>1.5). We could detect concomitant recruitment of AR and TRIM24 on the same allele by ChIP-reChIP for the AURKB enhancer and the PBK promoter (Figures S3F and S3G). We then used this signature to stratify publicly available gene expression data from PC patients (Taylor et al., 2010) and identified two prominent clusters (Figure 3A). When we calculated the average expression of all AR/TRIM24 targets for the two clusters, we found that one cluster showed a significantly higher average gene expression when compared to the other cluster (Figure 3B). Thus, we named the clusters “low” (grey) and “high” (yellow). The “high” cluster was significantly enriched in metastatic samples compared to the “low” cluster (Figure 3A, fisher exact test, p<0.0001), revealing that AR/TRIM24 co-activated targets are up-regulated in metastasis. We then assessed the probability of recurrence-free survival of primary PC patients stratified according to low or high AR/TRIM24 target gene activation. We found that patients with AR/TRIM24 co-activated genes in the “high” cluster showed a greater chance of recurrence, indicating a predictive value of our gene signature (Figure 3C).
Figure 3. AR and TRIM24 co-activated genes are up-regulated in CRPC and predict disease recurrence in primary tumors.
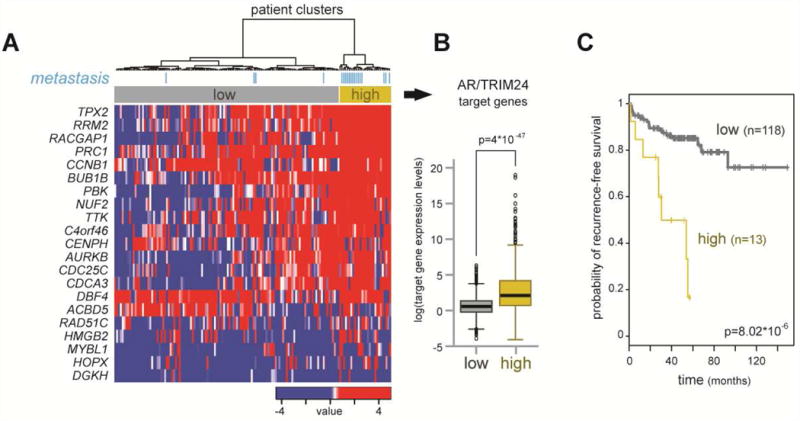
(A) AR/TRIM24 co-activated targets in CRPC were defined as the 21 genes bound by overlapping AR and TRIM24 sites, within 100kb of their TSS and down-regulated by siRNA-mediated knock-down of both factors in abl cells (LIMMA, p<0.05, fold-change>1.5). Hierarchical clustering of patient gene expression data (MSKCC/Taylor set) (Taylor et al., 2010) with the 21-gene signature resulted in two main clusters indicated in grey and yellow, respectively. The clusters were named based on the average expression of the 21-gene signature, which was lower in the grey cluster (low) compared to the yellow cluster (high). The metastatic samples in the data set are marked in light blue. (B) Average expression levels of the 21 AR/TRIM24 gene targets are depicted for both clusters. Data are represented as mean +/- SEM. Statistical analysis was performed using a two-tailed student's t-test assuming unequal variance p<0.001. (C) Kaplan-Meier curve for PSA-recurrence-free survival. Patients were stratified as high and low according to the 21-gene expression signature in primary tumors (p-value, log rank test). p<0.001.
(See also Figure S3).
TRIM24 protein expression is associated with tumor aggressiveness
Given the fact that a combined AR/TRIM24 gene expression signature in primary tumors was predictive for recurrent disease and was increased in metastatic PC, we tested if TRIM24 protein levels were, on their own, predictive in human tumor tissues by performing immunohistochemistry (IHC) analyses with a validated antibody (Figures S4A and S4B). We analyzed TRIM24 protein expression on PC tissue microarrays (TMAs) by IHC using a three-tiered scoring system as published previously (Theurillat et al., 2014). The level of TRIM24 expression gradually increased from samples of benign prostatic hyperplasia (BPH) to primary PC tumors (PPC), and was most abundant in samples of CRPC (Figures 4A and 4B, p<0.001, Chi2). Importantly, TRIM24 protein expression was also associated with increased risk of tumor recurrence after surgery in PPC as measured by PSA levels (Figure 4C, p<0.001), and thus had predictive power. In line with these findings, increased TRIM24 protein levels in primary tumors significantly correlated with features of tumor cell proliferation, such as higher Gleason score, tumor size (pT), and Ki-67 positive nuclei (Figures S4C and S4D). In contrast, metastatic spread to local lymph nodes (pN) did not significantly correlate (Figure S4C).
Figure 4. TRIM24 protein levels increase during PC progression and predict disease recurrence in primary tumors.
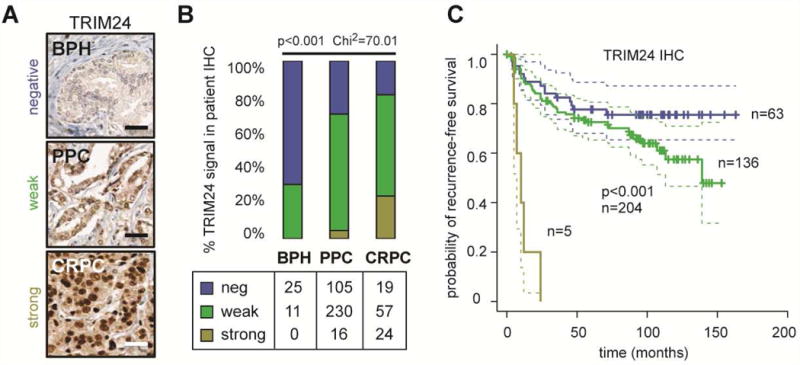
(A) TRIM24 immunohistochemistry (IHC) analysis on tumor tissue microarrays (TMA) of benign prostate hyperplasia (BPH), primary PC tumors (PPC), and advanced castration-resistant disease (CRPC) using a three-tiered scoring system as previously described (Theurillat et al., 2014). Bar represents 50 mm. (B) Quantification and percentage of negative, weak and strong TRIM24 nuclear staining of patients with BPH, PPC, and CRPC. (C) Kaplan-Meier curves with pointwise 95% confidence bands of PSA-based recurrence-free survival. Patients were stratified as negative, weak or strong TRIM24 nuclear staining (p-value, log rank test, p<0.001). Patients with primary PC undergoing radical prostatectomy and reaching the PSA nadir (<0.1ng/mL) postoperatively were used for analysis.
(See also Figure S4 and Table S4)
To test whether other AR signaling components predict disease recurrence, we analyzed AR and NCOA3, a known AR co-activator that we previously assessed, on these TMAs (Theurillat et al., 2014). AR (Figure S4E) and NCOA3 (Figure S4F), both measured by a three-tiered scoring system, correlated significantly with TRIM24 expression (Figures S4E and S4F, p<0.001 each), but failed to predict disease recurrence after surgery (Figures S4G and S4H). We interrogated whether TRIM24 protein drives the cell line-derived AR/TRIM24-dependent gene signature (Figure 3A) in human tissue samples. For this, we compared the expression level of the 21-gene signature with TRIM24 protein levels in an independent cohort of PPC patients (Figure S4I). We detected a positive correlation between the expression of the gene signature (Table S4) and elevated TRIM24 protein levels, consistent with both measurements harboring predictive power. Given the evidence that TRIM24 levels can be regulated through protein stabilization in PC (Figures 1A, 1B and S1A), we compared TRIM24 mRNA expression measured by qPCR, with protein levels measured by IHC, in a panel of primary and CRPC samples with negative, weak or strong TRIM24 nuclear staining. There was no significant correlation between TRIM24 mRNA and protein expression (Figure S4J, R=0.13, n.s., Spearman rank), consistent with the concept that TRIM24 protein levels are post-translationally regulated in PC patients.
Finally, we compared the power of TRIM24 in predicting recurrent disease to previously analyzed clinicopathological parameters and SPOP mutations. In univariate Cox regression analysis, TRIM24 levels, higher Gleason score, increased pT, positive pN, positive resection margins and increased PSA were significantly associated with tumor recurrence (Figure 5A). As expected neither AR levels (Figure S4E) nor NCOA3 levels (Figure S4F) were predictive (Figures 5A, S4G and S4H). Positive SPOP mutation status failed to have prognostic value (Figure 5A), indicating that TRIM24 levels and SPOP mutations are not strictly related to each other in patient samples. In fact, we previously reported that TRIM24 protein levels are increased in both SPOP-WT and mutant tumors (Theurillat et al., 2014), suggesting that additional mechanisms contribute to elevating TRIM24 protein in PC patients. In a multivariate Cox regression analysis, the three-tiered scoring system for TRIM24 failed to show independent prognostic value (not shown), whereas a two-tiered (negative/weak (negative), strong (positive)) scoring system did have independent predictive power (Figure 5B). Taken together, all these analyses provide support for TRIM24 promoting tumor aggressiveness and progression in PC patients.
Figure 5. Univariate and multivariate Cox regression analysis on a primary PC cohort.
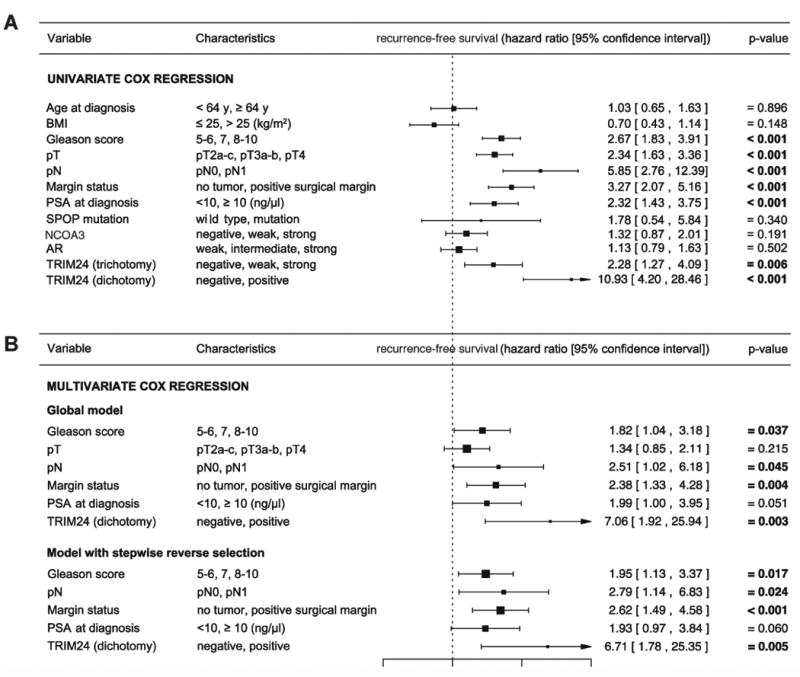
(A, B) Univariate (A) and multivariate (B) Cox regression analyses of the association between patients' characteristics at surgery and the probability of recurrence-free survival based on PSA levels. Patients' characteristics included age, BMI, Gleason score, tumor size (pT), metastatic spread to local lymph nodes (pN), margin status at resection, PSA at diagnosis, SPOP mutation status, NCOA3, AR and TRIM24 staining. TRIM24 trichotomy and dichotomy are defined as follows: TRIM24 trichotomy includes three-tiers corresponding to negative, weak or strong TRIM24 expression. TRIM24 dichotomy includes two-tiers where negative includes negative or weak TRIM24 expression and positive includes strong expression only. Time to PSA recurrence (cut-off 0.1ng/mL) was selected as clinical end point. The hazard ratios were estimated with a univariate (A) or multivariate (B) Cox proportional hazards model. The dashed vertical line was drawn at the no effect point (hazard ratio of 1.0). Horizontal lines represent a 95% confidence interval. The mid-point of the box represents the mean effect estimate and the area of the box represents the weight for each subgroup. Significant p-values (p<0.05) are marked in bold. Limit for reverse selection procedure was p=0.1.
TRIM24 accelerates intraprostatic tumor cell proliferation in vivo
We assessed the effects of TRIM24 over-expression on the intraprostatic tumor cell growth in mice using LNCaP cells that constitutively over-expressed TRIM24 or GFP (Figures 6A-6C). We found that TRIM24 over-expressing tumors proliferated more quickly than their GFP-expressing counterparts (Figure 6D). This difference was significant at days 22 and 29 after intraprostatic cell injection (Figure 6E). Since this difference was attenuated at later time points, we went on to surgically castrate the mice with established tumors to assess the effects of TRIM24 over-expression in a low androgen setting. 6 weeks after the castration, we found that 3 of the 10 (30%) TRIM24-expressing tumors continued growing, whereas only 1 of the 10 (10%) control tumors grew (Figures 6F and 6G). This suggests that TRIM24 over-expression in vivo increases the chance of castration resistance emerging during PC progression.
Figure 6. TRIM24 over-expression accelerates intraprostatic tumor cell growth in mice.
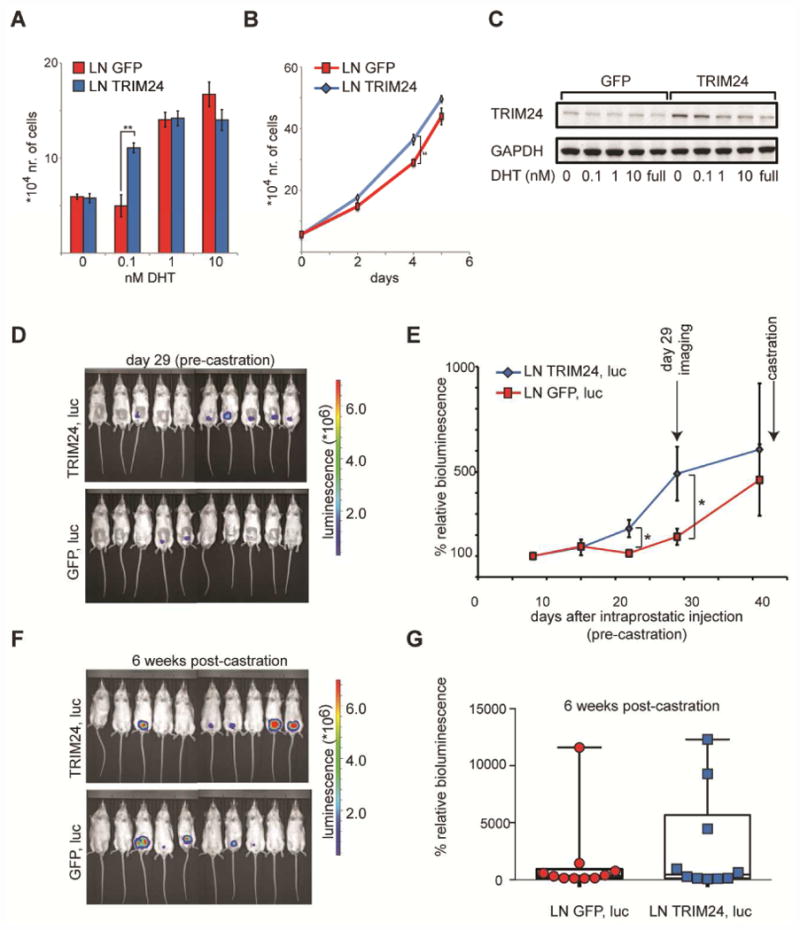
(A-C) LNCaP (LN) cells harboring constitutive expression of GFP or TRIM24 were grown (A) in the presence of the indicated concentration of DHT for 6 days or (B) in full media for 5 days and counted by hemacytometer (n=3). (C) TRIM24 protein levels were measured by Western blotting. Modified cells were either cultured in the presence of the indicated concentration of DHT or in full media (full). GAPDH served as a loading control. (D-G) Intraprostatic injection of modified LNCaP lines additionally expressing luciferase (luc) in mice. (D) Raw images from the bioluminescence detection 29 days after tumor cell injection. (E) Luminescence values were plotted as an average of % of the first measurement (% relative bioluminescence) for each mouse in each respective group. The measurements were done in intact male mice at day 8, 15, 22, 29 and 41 after tumor cell injection. (F, G) Mice were surgically castrated after the 41 day-measurement and (F) luminescence was detected again after 6 weeks and (G) the % relative bioluminescence values were expressed in a box plot. The two groups consisted of 10 mice each. The animal weight between the two groups was assessed and did not significantly change. Statistical analysis was performed using a two-tailed student's t-test assuming unequal variance * p<0.05, ** p<0.01. Data are represented as mean +/- SEM.
The TRIM24 bromodomain and AR-interacting LxxLL motif are required to support the proliferation of CRPC cells
To determine if TRIM24 over-expression was sufficient to increase the proliferation rate of CRPC cells, we over-expressed TRIM24-WT in abl cells. We observed increased growth, consistent with higher TRIM24 levels enhancing CRPC cell proliferation (Figures S5A and S5B). To assess which functional domains of TRIM24 are necessary to stimulate CRPC cell proliferation, we further conducted complementation experiments in abl cells. For this, we engineered abl cells expressing dox-inducible shRNA targeting TRIM24. As expected, addition of dox to deplete TRIM24 reduced proliferation (Figure 7A). Importantly, this phenotype could be rescued through the re-expression of shRNA-resistant TRIM24 cDNA encoding the WT form (Figures 7A and 7B). Proliferation rates were not restored by the re-expression of TRIM24 point mutants with a disrupted LxxLL motif (L763A, L764A) or a disrupted bromodomain (F979A, N980A) (Figures 7A and 7B). We found comparable results when we assessed the proliferation of TRIM24 over-expressing LNCaP cells (Figures S5C and S5D). In this setting, the growth advantage observed with TRIM24-WT over-expression under low DHT conditions (Figure S5C) is lost when either the LxxLL-motif mutant or the bromodomain mutant of TRIM24 are expressed at a comparable level (Figures S5C and S5D). Consistent with enhanced AR signaling, the DHT-induced value of proliferation EC50 decreased in the presence of TRIM24-WT but not in the presence of TRIM24 mutant over-expression in LNCaP cells (Figure S5E). When we monitored AR binding to the AURKB enhancer, a target site shared with TRIM24 in abl cells (Figure S3G), we found that TRIM24 depletion significantly reduced AR recruitment (Figure 7C). This phenotype was rescued when TRIM24-WT but not its bromodomain mutant was expressed in the complemented abl lines (Figure 7D). AR recruitment in the presence of TRIM24 L763A, L764A was more variable, suggesting that the two proteins may interact outside of the LxxLL domain (Figure 7D). Results from additional proliferation experiments reveal that combining enzalutamide treatment with TRIM24 inhibition has a larger effect on CRPC cell growth than either condition alone (Figures S5F and S5G). Taken together, these results support the notion that interactions of TRIM24 with both AR and acetylated chromatin are essential to drive CRPC cell proliferation and thus could be targeted by therapeutic interventions.
Figure 7. The TRIM24 bromodomain and AR-interacting LxxLL motif are required for proliferation of CRPC cells.
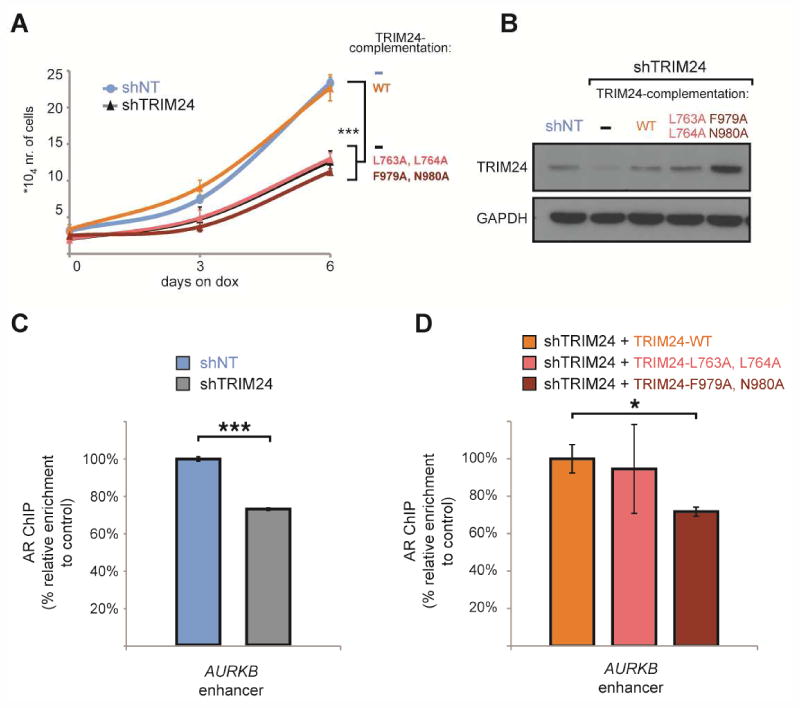
(A) Proliferation curves of abl cells stably expressing doxycycline (dox)-inducible shRNA targeting TRIM24 or a non-targeting (NT) control are shown. The shTRIM24 cell line was complemented with TRIM24 cDNA-rescue constructs resistant to shTRIM24, including TRIM24-WT (WT), TRIM24-L763A, L764A (L763A, L764A) and TRIM24-F979A, N980A (F979A, N980A). Cultures were treated with dox at day 0 and were grown and counted at 0, 3 and 6 days and values were plotted. Statistical analysis was performed using a two-tailed student's t-test assuming unequal variance ***p<0.005. (B) TRIM24 protein levels of the cell lines complemented with the indicated constructs were measured after 6 days of dox-treatment. (C, D) AR-specific ChIP followed by qPCR was performed at the AURKB enhancer, which is co-occupied by AR and TRIM24 (n=2). The different cell lines assayed are described in (A) and include (C) abl cells expressing shTRIM24 or shNT, and (D) abl shTRIM24 cell lines complemented with TRIM24-WT, TRIM24-L763A, L764A and TRIM24-F979A, N980A. All cell lines were treated with dox for 6 days to induce shRNA expression. The relative enrichment of the AR ChIP compared to the total input amount was set at 100% for the respective control condition. Statistical analysis was performed using a one-tailed student's t-test assuming equal variance * p<0.05, *** p<0.005. Data are represented as mean +/- SEM.
(See also Figure S5).
Discussion
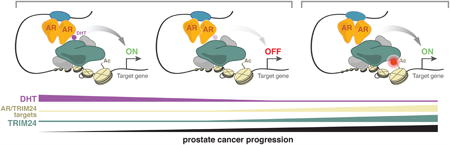
Here we establish a role for TRIM24 as a driver of SPOP mutant and advanced, castration-resistant PC. In both settings, TRIM24 promotes cancer cell proliferation under low levels of androgen. We show that TRIM24 acts as a transcriptional activator of genes involved in cell cycle progression by collaborating with AR signaling. Our molecular findings are supported by an assessment in PC patients. In these studies we reveal that nuclear levels of TRIM24 increase during PC progression. Moreover, in primary tumors we find that patients with high nuclear TRIM24 levels or high expression of AR/TRIM24 co-activated genes have a higher risk of disease recurrence. Mutational analyses revealed that the interaction of TRIM24 with both AR and acetylated chromatin is required for promoting CRPC cell proliferation. Therefore, we propose that these domains of TRIM24 may become essential for activating and recruiting AR in CRPC and could be potential drug targets (Figure 8).
Figure 8. Model of the oncogenic role of TRIM24 in CRPC.
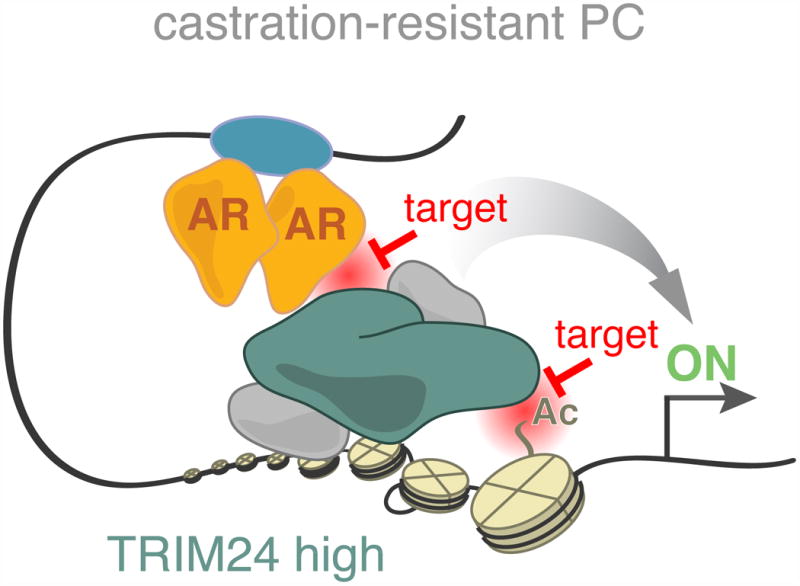
The functional role of TRIM24 is augmented in CRPC and depends on its interactions with AR (LxxLL motif) as well as acetylated histones (bromodomain) suggesting that. both TRIM24 functions could be targeted therapeutically
Our results reveal that part of the increase of TRIM24 protein observed during PC progression is due to post-translational dysregulation. More specifically, we show that expression of PC-specific SPOP mutations in androgen-dependent LNCaP cells stabilizes TRIM24 protein, which in turn enhances the proliferative androgen response. These results are in agreement with SPOP mutant tumors showing high levels of AR signaling in PC patients (TCGA, 2015). In addition, we also detect a positive correlation between SPOP mutant-activated and TRIM24-activated genes in abl cells, when we compare our TRIM24-dependent gene expression with a published SPOP mutant signature (Geng et al., 2014) in the same cell line (GSEA, NES: 1.24, p<0.001). Although all these findings imply a functional link between SPOP mutations and TRIM24 in mediating proliferation under low androgen, there is not a one-to-one correspondence between the two and increased TRIM24 protein levels in CRPC are likely regulated by additional mechanisms. This is also reflected by the result that elevated TRIM24 protein expression is observed in approximately 25% of cases we examined by IHC, whereas SPOP mutations in CRPC patients have only been reported at 5% and 8%, respectively and are not enriched compared to primary tumors (Grasso et al., 2012; Robinson et al., 2015).
Interestingly, the oncogenic role of TRIM24 in tumorigenesis is not restricted to AR signaling, as TRIM24 over-expression is detected in a wide variety of tumors (Chambon et al., 2011; Cui et al., 2013; Li et al., 2012; Liu et al., 2014; Miao et al., 2015; Tsai et al., 2010; Xue et al., 2015). Moreover, we detect TRIM24 binding to and activation of the PIK3CA gene, which has also been observed in glioma (Zhang et al., 2015). In addition, we identified EGFR as an AR-independent transcriptional target gene of TRIM24. Both PIK3CA and EGFR synergize to engage oncogenic PI3K/AKT signaling, which is a key driver of many different cancer types, including PC (Guo et al., 2006; Taylor et al., 2010). TRIM24 is also able to ubiquitylate and degrade phosphorylated p53 through its RING domain (Allton et al., 2009; Jain et al., 2014). Genetic alterations in AR, PTEN, and TP53 are enriched in CRPC, suggesting that factors that feed into these oncogenic pathways, such as TRIM24, may drive PC progression from primary disease to CRPC (Baca et al., 2013; Barbieri et al., 2012; Grasso et al., 2012; Robinson et al., 2015).
Our results show that TRIM24 drives the proliferation of CRPC cells. This observation aligns well with the concept that AR co-activators, such as TRIM24, drive CRPC by sustaining AR signaling under low hormone availability (Agoulnik et al., 2006; Gregory et al., 2001; Linja et al., 2004; Taylor et al., 2010; Xu et al., 2012). Moreover, we make use of the LNCaP-derived CRPC cell line abl (Culig et al., 1999), which serves as a model to address how AR-dependent transcription is activated to drive CRPC proliferation under hormone-starvation conditions (Eder et al., 2000; Wang et al., 2009). We previously showed that AR is recruited to novel genomic sites in abl cells, where it executes a distinct transcriptional program implicated in cell proliferation (Wang et al., 2009). Here, we provide evidence that this “AR reprogramming” may depend on TRIM24, which ultimately results in a marked dependency of the CRPC cells on TRIM24. In support of this idea, we find a significant increase in AR and TRIM24 co-activated genes in abl compared to LNCaP cells. This finding is also mirrored in PC patients where the levels of these AR/TRIM24-stimulated genes increase with disease progression.
Our study suggests that CRPC displays a marked dependency on TRIM24 to drive cellular proliferation. At the molecular level, both the AR co-activator (LxxLL motif) and the acetylated chromatin reader function (bromodomain) of TRIM24 are required for CRPC cell proliferation. These functions of TRIM24 may become essential for efficient AR transactivation in CRPC when low androgen levels mediate only suboptimal activation. We further propose that TRIM24 binding to acetylated histones through its bromodomain anchors AR to the genome under hormone-starvation. Interestingly, a new class of epigenetic drugs, the so-called bromodomain inhibitors, is being developed for cancer therapy (Filippakopoulos and Knapp, 2014). These small molecules target the interaction between acetyl-lysine containing chromatin and interacting bromodomains (Filippakopoulos and Knapp, 2014). Recently, tool compounds, consisting of acetyl-lysine mimetic benzimidazolones, were shown to act as dual TRIM24 and BRPF bromodomain inhibitors (Bennett et al., 2015; Palmer et al., 2015), suggesting that in the clinic specific targeting of TRIM24 chromatin binding may be achievable in the future.
In summary, we report an oncogenic role for TRIM24 as a transcriptional activator and mediator of hormone-refractory PC cell growth in SPOP-mutant and CRPC. We hypothesize that our new insights will provide the rationale for targeting TRIM24 in these settings.
Experimental Procedures
Gene expression and microarray analysis
Total RNA was isolated either from LNCaP or abl cells transfected with siRNA targeting either control or TRIM24 and subjected to reverse transcription, labeling, and hybridization to human U133 plus 2.0 expression arrays (Affymetrix).
Chromatin Immunoprecipitation (ChIP), ChIP-reChIP, ChIP-seq
ChIP and ChIP-reChIP were performed as described (Jehle et al., 2014; Xu et al., 2012). For ChIP-seq, sequencing libraries were generated according to manufacturer's instructions (ThruPLEX-FD Prep Kit, Rubicon Genomics) and then sequenced on the Illumina Hiseq-2000 platform.
Animal model experiments
The orthotopic intraprostatic injection of modified LNCaP cells was performed as previously published (Elsadek et al., 2011) and was carried out at the DFCI Lurie Family Imaging Center. The animal experiments were carried out under the Lurie Center IACUC protocol and were in accordance with the IACUC standards for the welfare of animals.
Recurrence-free Survival Analysis based on gene signature
Prostate cancer patient gene expression data (GSE21034) was clustered and matched to clinical data and Kaplan-Meier plots were calculated. The probability of recurrence was calculated by using biochemical measurements defined as surge in PSA after treatment as defined in (Taylor et al., 2010).
Human Tumor Samples and Recurrence-free Survival Analysis
Specimens were collected at the Institute of Surgical Pathology, University of Zurich, Switzerland, and the Institute of Pathology, University of Regensburg, Germany (Mortezavi et al., 2011). The local scientific ethics committees approved all cohorts (Cantonal Scientific Ethics Committee Zurich, approval numbers: StV-Nr. 25/2007 and StV-Nr. 25-2008) and informed consent was obtained from all the patients. See the Supplemental Experimental Procedures for further details.
Recurrence-free survival curves were calculated using the Kaplan–Meier method. Patients were censored at the time of their last tumor-free clinical follow-up visit. Time to PSA recurrence (cutoff 0.1 ng/mL) was selected as clinical end point. Only patients with PPC undergoing radical prostatectomy and reaching the PSA nadir (<0.1 ng/mL) postoperatively were used for survival analysis.
Immunohistochemistry (IHC) and qPCR on patient samples
The IHC and scoring system for TRIM24 and NCOA3 have been described previously (Theurillat et al., 2014).
RNA was extracted out of paraffin-embedded tissue from normal prostate, PPC and CRPC according to manufacturer's instructions (Maxwell® 16 LEV RNA FFPE purification kit).
Supplementary Material
Significance.
Development of resistance to androgen deprivation therapy remains a major challenge in the treatment of advanced prostate cancer patients. Here, we identify TRIM24 as a critical factor that contributes to this process by stimulating androgen receptor signaling and thereby promoting proliferation under low androgen conditions. We show that TRIM24 promotes hormone independent growth in the context of driver mutations in SPOP and even more broadly in advanced, castration-resistant disease, where TRIM24 is essential to mediate cell proliferation. Our study supports a fundamental role for TRIM24 in prostate cancer and provides a rationale for the therapeutic targeting of TRIM24 in SPOP mutant and castration-resistant disease.
Highlights.
TRIM24 mediates tumor cell proliferation in SPOP-mutant PC and CRPC
TRIM24 activates pro-proliferative genes together with AR in CRPC
TRIM24 protein increases in CRPC and predicts disease recurrence
The TRIM24 bromodomain mediates CRPC growth and is a potential drug target
eTOC Blurb.
Groner et al. show that recurrent prostate cancer-driver mutations in SPOP stabilize the TRIM24 protein, which promotes proliferation under low androgen conditions. TRIM24 augments AR signaling; AR and TRIM24 coactivated genes are upregulated in castration-resistant state and predictive of disease recurrence.
Acknowledgments
We acknowledge Kexin Xu, Fugen Li, Jin Zhao, Jennifer Spangle and Nancy Hynes for fruitful advice, assistance and discussion. We thank Andre Fitsche and Martina Storz for excellent histology assistance. We also thank Quang-De Nguyen and Amy Saur Conway for carrying out the orthotopic in vivo studies. ACG is funded by an Advanced Postdoc.Mobility fellowship from the Swiss National Science Foundation (P300P3_151145) and a NRSA Institutional Training Grant (T32 CA009172-39). JPT is funded by a Swiss National Science Foundation Professorship (PP00P3_150645) and a grant by the Swiss Cancer League (KSL-3654-02-2015). PT is supported by an HFSP Long-term Fellowship. ACBC. is funded by Deutsche Krebshilfe 110237. QZ is supported by a Promedica foundation grant to Holger Moch. PJW is funded by a SystemX.ch grant (PhophoNet PPM) from the Swiss initiative in systems biology. LC is funded by NIH P50 CA090381-13 (DF/HCC SPORE). MB is funded by NIH 5P01 CA163227 and the DF/HCC SPORE in Prostate Cancer.
Footnotes
Accession Numbers: The GEO accession number for the data series is GSE69332. It includes the microarray data (GSE69330) and the ChIP-seq data (GSE69331).
Author Contributions: ACG, JPT, MB conceived and designed the experiments. ACG, LC, TB, HJ, DM performed the experiments. ACG, LC, JDTH, RH, PT, LG, CF, CF, QZ, CP, PJW, JPT analyzed the data. JDTH, LC, RH, ACBC, PT, LG, AC, CF, CF, QZ, CP, UW, TG, RA, PJW, LAG, JPT contributed reagents, materials and analysis tools. ACG, JPT, MB wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, 3rd, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL, Barton MC. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106:11612–11616. doi: 10.1073/pnas.0813177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–669. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Fedorov O, Tallant C, Monteiro O, Meier J, Gamble V, Savitsky P, Nunez-Alonso GA, Haendler B, Rogers C, et al. Discovery of a Chemical Tool Inhibitor Targeting the Bromodomains of TRIM24 and BRPF. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner M, Lee DJ, O'Reilly C, Park K, MacDonald TY, Khani F, Turner KR, Chiu YL, Wild PJ, Dolgalev I, et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavailles V, Dauvois S, L'Horset F, Lopez G, Hoare S, Kushner PJ, Parker MG. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J. 1995;14:3741–3751. doi: 10.1002/j.1460-2075.1995.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon M, Orsetti B, Berthe ML, Bascoul-Mollevi C, Rodriguez C, Duong V, Gleizes M, Thenot S, Bibeau F, Theillet C, et al. Prognostic significance of TRIM24/TIF-1alpha gene expression in breast cancer. Am J Pathol. 2011;178:1461–1469. doi: 10.1016/j.ajpath.2010.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Cao W, Li J, Song X, Mao L, Chen W. TRIM24 overexpression is common in locally advanced head and neck squamous cell carcinoma and correlates with aggressive malignant phenotypes. PLoS One. 2013;8:e63887. doi: 10.1371/journal.pone.0063887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999;81:242–251. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder IE, Culig Z, Ramoner R, Thurnher M, Putz T, Nessler-Menardi C, Tiefenthaler M, Bartsch G, Klocker H. Inhibition of LncaP prostate cancer cells by means of androgen receptor antisense oligonucleotides. Cancer Gene Ther. 2000;7:997–1007. doi: 10.1038/sj.cgt.7700202. [DOI] [PubMed] [Google Scholar]
- Elsadek B, Graeser R, Esser N, Schafer-Obodozie C, Tsurumi C, Abu Ajaj K, Warnecke A, Unger C, Saleem T, Kratz F. In vivo evaluation of a novel albumin-binding prodrug of doxorubicin in an orthotopic mouse model of prostate cancer (LNCaP) Prostate Cancer Prostatic Dis. 2011;14:14–21. doi: 10.1038/pcan.2010.43. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–5643. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- Hazelett DJ, Rhie SK, Gaddis M, Yan C, Lakeland DL, Coetzee SG, Henderson BE, Noushmehr H, Cozen W, Kote-Jarai Z, et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014;10:e1004102. doi: 10.1371/journal.pgen.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- Herquel B, Ouararhni K, Davidson I. The TIF1alpha-related TRIM cofactors couple chromatin modifications to transcriptional regulation, signaling and tumor suppression. Transcription. 2011;2:231–236. doi: 10.4161/trns.2.5.17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AK, Allton K, Duncan AD, Barton MC. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol Cell Biol. 2014;34:2695–2709. doi: 10.1128/MCB.01705-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle K, Cato L, Neeb A, Muhle-Goll C, Jung N, Smith EW, Buzon V, Carbo LR, Estebanez-Perpina E, Schmitz K, et al. Coregulator control of androgen receptor action by a novel nuclear receptor-binding motif. J Biol Chem. 2014;289:8839–8851. doi: 10.1074/jbc.M113.534859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Ji X, Li W, Song J, Wei L, Liu XS. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 2006;34:W551–554. doi: 10.1093/nar/gkl322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi M, Okumura F, Tsukiyama T, Watanabe M, Miyajima N, Tanaka J, Imamura M, Hatakeyama S. TRIM24 mediates ligand-dependent activation of androgen receptor and is repressed by a bromodomain-containing protein, BRD7, in prostate cancer cells. Biochim Biophys Acta. 2009;1793:1828–1836. doi: 10.1016/j.bbamcr.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996;15:6701–6715. [PMC free article] [PubMed] [Google Scholar]
- Li H, Sun L, Tang Z, Fu L, Xu Y, Li Z, Luo W, Qiu X, Wang E. Overexpression of TRIM24 correlates with tumor progression in non-small cell lung cancer. PLoS One. 2012;7:e37657. doi: 10.1371/journal.pone.0037657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linja MJ, Porkka KP, Kang Z, Savinainen KJ, Janne OA, Tammela TL, Vessella RL, Palvimo JJ, Visakorpi T. Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res. 2004;10:1032–1040. doi: 10.1158/1078-0432.ccr-0990-3. [DOI] [PubMed] [Google Scholar]
- Liu X, Huang Y, Yang D, Li X, Liang J, Lin L, Zhang M, Zhong K, Liang B, Li J. Overexpression of TRIM24 is associated with the onset and progress of human hepatocellular carcinoma. PLoS One. 2014;9:e85462. doi: 10.1371/journal.pone.0085462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Sakari M, Okada M, Yokoyama A, Takahashi S, Kouzmenko A, Kato S. The androgen receptor in health and disease. Annu Rev Physiol. 2013;75:201–224. doi: 10.1146/annurev-physiol-030212-183656. [DOI] [PubMed] [Google Scholar]
- Miao ZF, Wang ZN, Zhao TT, Xu YY, Wu JH, Liu XY, Xu H, You Y, Xu HM. TRIM24 is upregulated in human gastric cancer and promotes gastric cancer cell growth and chemoresistance. Virchows Arch. 2015;466:525–532. doi: 10.1007/s00428-015-1737-4. [DOI] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortezavi A, Hermanns T, Seifert HH, Baumgartner MK, Provenzano M, Sulser T, Burger M, Montani M, Ikenberg K, Hofstadter F, et al. KPNA2 expression is an independent adverse predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res. 2011;17:1111–1121. doi: 10.1158/1078-0432.CCR-10-0081. [DOI] [PubMed] [Google Scholar]
- Niraula S, Chi K, Joshua AM. Beyond castration-defining future directions in the hormonal treatment of prostate cancer. Horm Cancer. 2012;3:3–13. doi: 10.1007/s12672-011-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP, et al. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thenot S, Henriquet C, Rochefort H, Cavailles V. Differential interaction of nuclear receptors with the putative human transcriptional coactivator hTIF1. J Biol Chem. 1997;272:12062–12068. doi: 10.1074/jbc.272.18.12062. [DOI] [PubMed] [Google Scholar]
- Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–89. doi: 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- vom Baur E, Zechel C, Heery D, Heine MJ, Garnier JM, Vivat V, Le Douarin B, Gronemeyer H, Chambon P, Losson R. Differential ligand-dependent interactions between the AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J. 1996;15:110–124. [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Zhang X, Liu J, Li N, Liu C, Liu Y, Wang P. Clinical significance and biological roles of TRIM24 in human bladder carcinoma. Tumour Biol. 2015 doi: 10.1007/s13277-015-3393-3. [DOI] [PubMed] [Google Scholar]
- Zhang LH, Yin AA, Cheng JX, Huang HY, Li XM, Zhang YQ, Han N, Zhang X. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene. 2015;34:600–610. doi: 10.1038/onc.2013.593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.