

Abstract
Background
Outcome of low-grade glioma (LGG, WHO grade II) is highly variable reflecting molecular heterogeneity of the disease. We compared two different single modality treatment strategies: standard radiotherapy (RT) versus primary temozolomide (TMZ) chemotherapy with the aim of tailoring treatment and identifying predictive molecular factors.
Methods
477 patients (2005 – 2012, median FU 48 months) with a low-grade glioma (astrocytoma, oligoastrocytoma, oligodendroglioma, WHO grade II) with at least one high-risk feature (age > 40 years, progressive disease, tumor > 5 cm or crossing the midline, neurological symptoms (e.g. focal or mental deficits, increased intracranial pressure or intractable seizures)) were, after stratification by chromosome 1p-status, randomized to either conformal RT (50.4 Gy/28 fractions) or dose-dense TMZ (75 mg/m2 daily × 21 days, q28 days, max. 12 cycles). Random treatment allocation was performed online using a minimization technique. A planned analysis was performed after 246 progression events. All analyses are intent to treat. Primary clinical endpoint was progression-free survival (PFS), correlative analyses included molecular markers (1p/19q co-deletion, MGMT methylation status, IDH1+2 mutations). The trial has been registered at the European Trials Registry (EudraCT 2004-002714-11) and at ClinicalTrials.gov (NCT00182819).
Findings
Four hundred seventy-seven patients were randomized. Severe hematological toxicity occurred in 14% of TMZ-treated patients, infections in 3% of TMZ-treated patients, and 1% of RT-treated patients. Moderate to severe fatigue was recorded in 3% of patients in the RT group and 7% in the TMZ group.
At a median follow-up of 48 months (IQR:31–56), median PFS was 39 months (IQR:16–46) in the TMZ arm and 46 months (IQR:19–48) in the RT group (hazard ratio 1.16, 95% CI, 0.9–1.5; p=0.22). Median OS has not been reached. Exploratory analyses identified treatment-dependent variation in outcome of molecular LGG subgroups (n=318).
Interpretation
There was no significant difference in outcome of the overall patient population treated with either radiotherapy alone or TMZ chemotherapy alone. Further data maturation is needed for overall survival analyses and evaluation of the full predictive impact of the molecular subtypes for individualized treatment choices.
Funding
Merck & Co, Swiss-Bridge Award 2011, Swiss Cancer League.
Research in context
Evidence before this study
Management of patients suffering from low grade glioma (LGG, WHO grade II) has been based mainly on clinical prognostic factors. Median survival is highly variable from 3.2 (high-risk disease) to 7.8 years (low risk), with a large range within each clinical subgroup. Known favorable molecular prognostic factors comprise co-deletion of chromosome 1p/19q, and more recently mutation of isocitrate dehydrogenase 1 or 2 (IDH1/2) that was discovered to be present in most LGG (>80%). The optimal treatment modality and sequence of LGG patients is highly controversial, aiming at balancing a favorable effect on progression-free survival versus long-term toxicity in this overall young patient population (median <45 years). We searched PubMed between June 1993 and April 2016 using the search terms, “randomized” “low grade glioma” “chemotherapy” “radiotherapy” and identified a total of only 4 conclusive trials, including reports on delaying treatment initiation, or the optimal dose of radiotherapy (EORTC 22844; EORTC 22845/MRC BR04; NCCTG/RTOG/ECOG; RTOG9802). The most recent report of RTOG 9802 demonstrates an impressive 5.5-year improvement in median overall survival when up to 6 cycles of adjuvant PCV-chemotherapy (procarbazine, CCNU, vincristine) were added after standard RT. However, no data is available on the prognostically highly relevant molecular subtype displaying 1p/19q co-deletion. The lack of this molecular information and the availability of less toxic drugs encumber the translation into clinical practice.
Added value of this study
Our study aimed after central histological confirmation at molecular characterization, and stratification before randomization, and we thus have tumor tissue available for most patients. It is the largest trial of prospectively treated patients allowing for molecular tumor characterization and correlation with outcome. Although overall, no significant difference in outcome (PFS) could be demonstrated, it demonstrates the value of molecular characterization and subgrouping of the disease. Patients with an IDH mutation without co-deletion of 1p/19q, displayed a significantly longer PFS when treated with RT, while patients with wild type IDH belong to different categories of glioma often with a much more aggressive course.
Implications of all the available evidence
This is first study in the era of modern and molecular neuro-oncology that successfully achieved its recruitment goal. The availability of tumor tissue from most patients allowed for confirming the prognostic value of recently identified molecular subgroups that are now an integral part of the WHO classification of LGG. For future clinical trials, treatment strategies shall be adapted to the risk of tumor progression or recurrence. Ultimately this should lead individually and risk-adapted customized treatments. Most patients with a low-grade glioma without mutation of IDH1/2 fare substantially worse under either single modality treatment. Wild-type IDH1/2 tumors may molecularly be attributed to other tumor entities including indolent tumors such as pilocytic astrocytoma, or most frequently to different subtypes of glioblastoma. The latter patients may benefit from a more intensive combined modality approach. Similarly, patients with IDH mutated tumors without 1p/19q co-deletion are best to be treated with radiotherapy and derive little benefit from chemotherapy (be it PCV or TMZ). On the other hand, patients with co-deleted tumors carry the best prognosis, and may benefit from adjuvant chemotherapy after irradiation; whether treatment with chemotherapy alone is as effective will require further maturation of the data of the current trial.
Ongoing exploratory analyses may allow for identifying putative predictive molecular markers for further refinement of the prognostic value of the molecular subtyping, and importantly may identify novel therapeutic targets.
Introduction
Low-grade glioma (LGG) encompasses a diverse group of diffusely infiltrative, slowly growing glial brain tumors. They frequently affect adults in their third or fourth decade of life. The natural history varies greatly and optimal management remains controversial. Treatment options include watchful waiting, radical surgery, radiotherapy, chemotherapy or a combination thereof.1 Individual management decisions depend on clinical and molecular prognostic factors, extent of symptoms, estimated risk of malignant transformation, and risk of acute and late treatment-associated toxicity. We previously derived a prognostic score with estimated median survival times varying from 3.2 to 7.8 years. Age ≥ 40 years, astrocytic histology, tumor size ≥ 6 cm, tumors crossing the midline and presence of neurological deficits were associated with an inferior prognosis.2 More recently, molecular characteristics particularly co-deletion of chromosomal arms 1p/19q (codel) that are associated with oligodendroglial histology and mutations of isocitrate dehydrogenase (IDH) 1 or 2 genes (IDHmt) have been associated with a more favorable prognosis and better response to both chemotherapy and radiation.3,4
Radiation therapy (RT) has been the accepted standard treatment for progressive and inoperable LGG for over 3 decades, established at a time when neither modern imaging technology nor alternative treatment modalities were available.2,5–8 Temozolomide (TMZ), an alkylating agent was specifically developed with chemical properties to cross the blood-brain-barrier, and it was first approved for the treatment of recurrent anaplastic astrocytoma in 1999. Exquisite sensitivity to chemotherapy had been demonstrated for oligodendroglioma with 1p/19q co-deletion, and uncontrolled trials suggested activity of TMZ also against LGG.9–12 Dose-dense regimens allowing for increased doses and prolonged exposure were considered conceptually attractive in particular for slow proliferating tumors like LGG.13–15 We addressed the question whether initial TMZ chemotherapy confers an advantage in outcome compared to standard RT aiming at prolonging time to progression, reduction in long-term toxicity and better quality of life. An equally important secondary objective was to identify molecular predictive factors, thus allowing tailoring treatment decisions based on individual tumor characteristics.
Methods
Study design and participants
This prospective randomized phase III study was comprised of 2 steps, an initial registration step at any time after initial diagnosis, allowing for tissue collection and molecular analyses required for stratification; and a randomization step at the time point when treatment was considered clinically indicated.
Adult patients (age ≥ 18 years) with histologically confirmed supratentorial diffusely infiltrating WHO grade II gliomas (astrocytoma, oligoastrocytoma and oligodendroglioma) were eligible for the study. Availability of paraffin-embedded tumor tissue, and a heparin blood sample was required for central pathology review and molecular testing for 1p. For patients with a substantial interval between initial tissue diagnosis and treatment start, confirmation of the histology by repeat biopsy was recommended. Patients whose tumour had transformed into a higher grade were excluded. Before randomization (2nd step) a separate informed consent was obtained. To be eligible for step 2, patients had to require active treatment other than surgery, defined by at least one of the following criteria: (1) age ≥ 40 years, (2) radiologic tumor progression, (3) new or worsening symptoms and (4) refractory seizures. Other eligibility criteria included: WHO performance status ≤ 2, Radiation Therapy Oncology Group (RTOG) neurological function score of 0–3, adequate hematologic, renal, and hepatic function (absolute neutrophil count, ≥1500/mm3; platelet count, ≥100,000/mm3; serum creatinine level ≤1.5 times the upper limit of normal (ULN); total serum bilirubin level ≤1.5 times ULN; and liver-function values <2.5 times ULN), results of genetic testing available and not candidate for treatment exclusively by surgery. Patients who had received prior chemotherapy or RT were excluded.
Organization of the trial
The trial was developed by the principal investigators in collaboration with the investigators from the participating groups and the EORTC Headquarters. All data were reviewed by EORTC collaborators and Drs. Stupp or Baumert, where appropriate. Statistical analyses were performed by Thierry Gorlia, PhD. Translational research and molecular marker evaluation were coordinated by Monika E. Hegi, PhD.
Trial sponsors were EORTC (22033/22036) in Europe, (some responsibilities delegated to the Medical Research Council Clinical Trial Unit (MRC-CTU) in the UK (BR13)), the NCIC Clinical Trials Group (NCIC CTG) in Canada (CE.5), and the Tasman Radiation Oncology Group (TROG) in Australia. The protocol was approved by the ethics committees and competent authorities of all participating centers and countries. All patients gave written informed consent for pathology review and molecular testing, and participation in the trial.
Randomisation and masking
Patients were stratified before randomization according to World Health Association (WHO) performance status (0 or 1 versus 2), age (<40 vs ≥ 40 years), presence of contrast enhancement on magnetic resonance tomography (MRI), the 1p status (deleted versus normal versus indeterminate) and the treating medical center. Randomization was performed online by the European Organisation for Research and Treatment of Cancer (EORTC) using a minimization technique. A minimization technique was used for random treatment allocation with prospective stratification by institution, 1p deletion (yes/no/undetermined), contrast enhancement (yes/no), age (<40/≥40 years), WHO performance status (0 or ≥1). The assigned treatment had to begin within six weeks after randomization. By the nature of the intervention, the trial was open-labeled and both patients, treating physicians and researchers were aware of the assigned intervention.
Procedures
Standard treatment consisted of 3D-conformal RT up to 50.4 Gy (28 × 1.8 Gy daily, 5 days per week over a period of 5–6 weeks). Intensity-modulated and stereotactically-guided RT was allowed if same dose prescription as for non-stereotactic RT was used. RT treatment volumes were defined based on T2 or FLAIR magnetic resonance imaging (MRI). In case of tumor resection, postoperative imaging was used. All participating sites had to comply with an extensive quality assurance program including dosimetry, dummy run and individual patient radiotherapy plan review (level III).16–18
Patients randomized to the experimental arm were to receive TMZ (Temodal®, Temodar®, Merck & Co, White House Station, NJ) in a dose-dense schedule of 75 mg/m2 per day for 21 days, repeated every 28 days for up to 12 cycles, or until tumor progression or unacceptable toxicity. Antibiotic prophylaxis against opportunistic Pneumocystis jiroveci infections was recommended for lymphocyte counts < 500/mm3. Anti-emetic prophylaxis was administered at the local investigators’ discretion.
The baseline evaluation (within 6 weeks prior to randomization) included MRI, a health-related quality-of-life questionnaire (EORTC QLQ-30 and Brain Cancer ModuleBN-20), full clinical and neurological evaluation including assessment of seizure frequency (if applicable), as well as complete blood counts and blood chemistry. The extent of tumor resection was assessed by the neurosurgeon per local practice based on postoperative imaging. All patients were evaluated clinically and neurologically every 3 months including quality of life, tumor assessment with MRI was performed every 6 months. Radiological progression was defined as an increase of 25% in bi-dimensional perpendicular product of signal hyperintensity on MRI T2 weighted images, or appearance of an area of new contrast enhancement, or a 25% increase in contrast enhancement on T1 weighted MR images with or without an increase in the area of T2-weighted signal hyperintensity. Clinical progression was defined as deterioration due to increasing neurological signs or symptoms with no other explanation or deterioration of WHO performance status or RTOG neurologic function. Quality of life results will be reported separately.
Pathology review and molecular testing was performed centrally (AvD; CH, DC, SK, JMK; JPR for Canada). Deletion of chromosomal arms 1p and 19q was tested by microsatellite markers or using FISH as described.19,20 The mutation status of IDH1 and 2 was determined by immunohistochemistry for the most common mutation IDH1-R132H, complemented by DNA sequencing of negative cases.21 The MGMT promoter methylation status was determined by the MGMT-STP27 model.22,23
Outcomes
The primary endpoint was progression-free survival (PFS) defined as the time between the date of randomization and the date of clinical or radiological progression or death. Secondary and translational endpoints were overall survival (OS), quality-of-life, mini-mental state examination (MMSE) and the association of molecular markers with outcome. Markers pre-specified in the protocol comprised 1p/19q status, and MGMT promotor methylation status (amended protocol version 2.2, October 2007). The IDH1/2 mutations in glioma have been published in 2009 and were included subsequently.4,24
Statistical analysis
Observation of 216 PFS events among the 466 randomized patients are needed for an 80% power and a significance level of 5% (two-sided) in order to detect a 13% increase in 5-year progression-free survival rate i.e. from 45% in the RT arm to 58%, corresponding to a treatment hazard ratio for progression (HR) equal to 0.68. Estimated PFS is calculated according to Kaplan–Meier from the date of randomization until event; for comparison a 2-sided log-rank test at 5% significance was used. All analyses were conducted on an intention-to-treat basis. A Cox model with the treatment as only variable was fitted to measure unadjusted treatment effect (HR). For sensitivity analysis, HR was also computed adjusted by the stratification factors (1p deleted versus 1p normal versus undeterminable; contrast enhancement, +/− contrast on MRI; age, <40 years versus ≥40 years; WHO performance status, 0 or 1 versus 2) and other possible confounding factors — tumor crossing the midline (No/Yes), and baseline RTOG neurological function score (0,1,2,3,4). The markers’ predictive effect was assessed by logrank interaction test. Markers prognostic value was assessed by Kaplan–Meier, log-rank test and Cox models (to measure HR) stratified by the treatment. These exploratory analyses were performed at 5% significance level. No proportionality of hazards assumptions check was planned by the protocol. The relationship between IDHmt and the extent of primary resection (biopsy versus partial resection versus total resection) was tested with the Cochran-Armitage Trend Test. Treatment duration was different between the 2 arms, and adverse effects are reported separately for the duration of TMZ chemotherapy (over 1 year) and for the duration of radiotherapy, defined as day 1 of RT until 28 days after the last administration of RT. All statistical analyses were done using SAS version 9.4. For this analysis a clinical cut-off date of 17. January was used and the database was locked on 7. August 2013.
Role of the funding source
The trial was supported by an unrestricted educational grant and free supply of TMZ by Merck & Co (formerly Schering-Plough). The drug manufacturer was neither involved in the trial design nor analysis. Translational research was funded in part by the Swiss Cancer League and the Swiss Bridge Award (to MEH). The corresponding author, MEH and RS had full access to all of the data and the final responsibility to submit for publication.
The study was registered at EudraCT#2004-002714-11 and ClinicalTrials.gov NCT#00182819, and was performed according to the declaration of Helsinki. The trial completed its enrollment, patients continue to be followed as per protocol.
Results
From September 23, 2005 until March 26, 2010, 707 patients (78 institutions/19 countries) were registered, and from December 6, 2005 until December 21, 2012 477 patients were randomized to receive either RT (n=240) or TMZ (n=237, figure 1). Twelve institutions randomized about half of the patients. The patient characteristics were well balanced with a median age of 45 years, 189/477 (40%) had undergone a diagnostic biopsy only, 206/477 (43%) a partial tumor debulking and in 81/477(17%) the surgeon considered the resection to be complete (table 1). The institutional diagnosis of a WHO grade II glioma was confirmed in 438/477 tumor samples (92%). The diagnosis of the histologic subtypes, astrocytoma, oligodendroglioma, or oligoastrocytoma, and tumor grade revealed poor interobserver agreement between the local and central, as well as the two central pathologists with kappa values between 0.3 and 0.4 (see appendix of supplemental data, appendix table S1 and figure S1).
Figure 1.
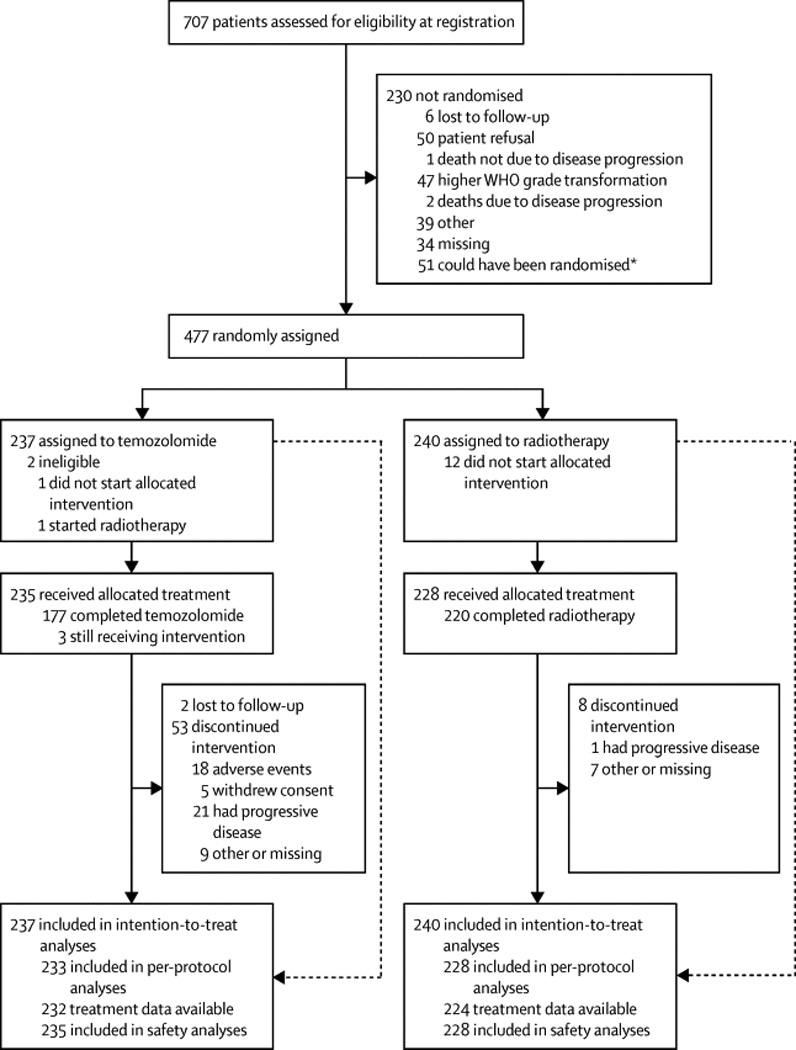
Consort statement. The numbers of patients registered, eligibility and allocation per treatment arms, including reasons for exclusion per treatment arm. At the date of database lock, 51 registered patients did not meet the criteria to be eligible for randomization.
Table 1.
Baseline characteristics
| RT N=240 (%) |
TMZ N=237 (%) |
Total N=477 (%) |
|
|---|---|---|---|
| Age [yrs], median (range) | 44 (18–72) | 45 (19–75) | 45 (18–75) |
| < 40 | 92 (38) | 85 (36) | 177 (37.1) |
| >= 40 | 148 (62) | 152 (64) | 300 (62.9) |
| Sex | |||
| Male | 138 (58) | 137 (58) | 275 (58) |
| Female | 102 (43) | 100 (42) | 202 (42) |
| WHO Performance Status | |||
| 0 | 151 (63) | 143 (60) | 294 (62) |
| 1 | 79 (33) | 86 (36) | 165 (35) |
| 2 | 10 (4) | 8 (3) | 18 (4) |
|
RTOG Neurologic Function Status |
|||
| 0 | 126 (53) | 127 (54) | 253 (53) |
| 1 | 90 (38) | 82 (35) | 172 (36) |
| 2 | 17 (7) | 22 (9) | 39 (8) |
| 3 | 7 (3) | 6 (3) | 13 (3) |
| Contrast enhancement on MRI | |||
| No | 119 (50) | 119 (50) | 238 (50) |
| Yes | 121 (50) | 118 (50) | 239 (50) |
|
Initial resection status (by investigator) |
|||
| Biopsy | 96 (40) | 93 (39) | 189 (40) |
| Partial removal | 106 (44) | 100 (42) | 206 (43) |
| Total removal | 37 (15.4) | 44 (18.6) | 81 (17) |
| Missing | 1(<1) | 0(0) | 1 (<1) |
| Time between last biopsy or surgery and treatment start (months) a | |||
| Median (range) | 4.8 (0.8–116.6) | 4.8 (0–151.5) | 4.8 (0–151.5) |
| Time between first histological diagnosis and treatment start (months) a | |||
| Median (range) | 5.2 (0.7–124.6) | 5.9 (0.9–151.5) | 5.6 (0.7–151.5) |
| Reason for treatmentb | |||
| Refractory seizuresc | |||
| No | 210 (88) | 202 (85) | 412 (86) |
| Yes | 28 (12) | 34 (14) | 62 (13) |
| Radiological progression | |||
| No | 90 (38) | 83 (35) | 173 (36) |
| Yes | 148 (62) | 154 (65) | 302 (63) |
|
New/worsening symptoms other than seizures |
|||
| No | 183 (76) | 187 (79) | 370 (78) |
| Yes | 55 (23) | 49 (21) | 104 (22) |
| Tumor involving midline | |||
| No | 173 (72) | 186 (79) | 359 (75) |
| Midline shift | 29 (12) | 23 (10) | 52 (11) |
| Midline infiltration | 24 (10) | 20 (8) | 44 (9) |
| Both | 10 (4) | 8 (3) | 18 (4) |
| Histology (local pathology) | |||
| Astrocytoma WHO II | 88 (37) | 79 (33) | 167 (35) |
| Oligoastrocytoma WHO II | 58 (24) | 60 (25) | 118 (25) |
| Oligodendroglioma WHO II | 94 (39) | 98 (41) | 192 (40) |
| Histology (central review)d | |||
| *LGG confirmed | 227 (95) | 211 (89) | 438 (92) |
| **LGG not confirmed | 8(3) | 12 (5) | 20 (4) |
| Missing review | 5 (2) | 14 (6) | 19 (4) |
| Molecular markers | |||
| 1p | |||
| 1p deleted | 98 (41) | 97 (41) | 195 (41) |
| 1p normal | 107 (45) | 106 (45) | 213 (45) |
| Missing | 35 (15) | 34 (14) | 69 (15) |
| 1p/19q status | |||
| 1p/19q co-deleted | 55 (23) | 62 (26) | 117 (25) |
| 1p/19q non-co-deleted | 125 (52) | 115 (49) | 240 (50) |
| Missing | 60 (25) | 60 (25) | 120(25) |
| IDH 1/2 mutation status | |||
| IDH 1/2 mutated | 164 (68) | 163 (69) | 327 (69) |
| IDH 1/2 wild-type | 35 (15) | 30 (13) | 65 (14) |
| Undetermined | 41 (17) | 44 (19) | 85 (18) |
| MGMT status | |||
| MGMT unmethylated | 6 (3) | 9 (4) | 15 (33) |
| MGMT methylated | 72 (30) | 63 (27) | 135 (28) |
| Missing | 162 (68) | 165 (70) | 327 (69) |
For patients who did not start randomized treatment, date of randomization was used.
Multiple mention/reasons
Refractory seizures defined as: Suffering from persistent seizures, defined as having both: persistent seizures interfering with every day life activities other than driving a car and failed three lines of anti-epileptic drug regimen including at least one combination regimen
* agreement with at least one of the central reviewers
** discordance mainly glioma grade III
- WHO Performance Status: a performance status of 0 denotes asymptomatic, 1 symptomatic and fully ambulatory, and 2 symptomatic and in bed less than 50 % of the day.
- RTOG NFS: a neurologic functions status of 0 denotes asymptomatic, a status of 1 minor neurologic symptoms, 2 moderate symptomatic, ambulatory with assistance, 3 moderate symptomatic; not fully ambulatory and requires assistance, 4 severely symptomatic; complete assistance at home or in institution-unable to work.
Treatment could be administered in both arms at the planned intensity and without undue major interruptions or delays in most patients. Ninety-one percent of the patients completed RT as planned, 75% completed 12 cycles of TMZ chemotherapy (range 1–14), 4 patients (1.8%) continued TMZ for more than 12 cycles for unspecified reasons. In the TMZ arm, 21 patients (9%) discontinued treatment due to progression, 18 patients (8%) due to toxicity, and 8 (3%) for other reasons. A total of 34/232 patients (154%) had at least one dose reduction due to haematotoxicity in 19 patients (8%), non-haematological toxicity in 9 (4%) patients and for other reasons in 11 patients (5%, in some patients multiple reasons). Two patients were reported to have died several months after the end of treatment possibly due to complications of the therapy; one in the RT arm due to encephalopathy and progressive disease and one due to bone marrow failure in the TMZ arm (Appendix table S2).
Overall, grade 3 / 4 hematological toxicity was observed in 13.7% (n=32) patients in the TMZ group (table S2). Infections were reported in 2 patients during RT (0.9%), and in 8 patients while on TMZ therapy (3.4%), including 2 patients with Pneumocystis jiroveci pneumonia (1 in each treatment arm). The most common non-hematologic adverse events were neurologic – (i.e. sensory deficits, mood alteration, ischemia, seizures and neurocognitive disturbances), occurring in both groups and likely related to the underlying brain disease. Moderate to severe fatigue was recorded in 8 (3.3%) patients in the RT group (grade 2) and 16 (6.8%) in the TMZ group (one reported as grade 3). Thromboembolic events were reported in 3 patients (1.2%, 2 in RT, 1 in TMZ arm).
At a median follow-up of 4 years (48 months, (IQR:31–56)), 262 (55%) progression events (126/52.5% with RT, 136/57.4% with TMZ) (for details see appendix of supplemental data, appendix table S3) and 119 (25%) deaths (63/26.3% with RT, 56/23.6% with TMZ) were recorded. The median PFS was 46 months (95 percent confidence interval [CI], 40 to 56) with RT alone and 39 months (CI, 35 to 44) with TMZ alone (Figure 2). The unadjusted HR for progression was 1.16 for patients treated with TMZ versus RT (CI, 0.9 to 1.5; log-rank p=0.22). Adjusted HR were computed (see appendix of supplemental data, appendix table S4) and provided the same conclusions without significant differences between groups. With only 25% of patients having died to date, no meaningful analyses of OS are possible.
Figure 2.

Progression-free survival per treatment group. The difference between per log rank test was not significant (HR: 1.16; 95 percent confidence interval, 0.9 to 1.5; P=0.22).
In 407 of the 477 patients (85%) randomized, sufficient material was available for successfully assessing the 1p status (stratification factor). Molecular data was missing in particular in patients who underwent a stereotactic biopsy only. IDH1 or 2 mutations were detected in 327/392 (83%) of the tumors, of which 30 (9.4%) had IDH1 mutations other than the common IDH1-R132H, and 9 (2.8%) were identified in IDH2. Co-deletions of 1p/19q (codel) were identified in 117/357 tumors (33%) and the MGMT promoter was methylated in 135/150 tumors (90%). The associations of distribution of molecular markers and histological glioma subtype (as classified by the local or the two reference pathologists, respectively) are shown in the appendix of supplemental data, figure S1. For a representative subgroup of 318 patients (for characteristics, see appendix of supplemental data, appendix table S5) the IDH mutation status and 1p/19q co-deletion was available: 269 (85%) were IDHmt, of which 104 (39%) were IDHmt/codel and 49 (15%) were IDH wild-type (IDHwt). These 3 molecular subgroups differed in clinical characteristics, namely age, time between initial histological diagnosis and start of treatment, extent of resection and MGMT status (table 3), in line with recent reports of several WHO grade II and III glioma datasets.4,24–26 Interestingly, time from initial surgery/histology to treatment was longer in IDHmt/non-codel (9.0 months) compared to IDHmt/codel (5.6 months) and IDHwt (3.4 months) (p = 0.0057). The MGMT promoter status was methylated in all IDHmt/codel (45/45), most but not all (86%, 62/72) of the IDHmt/non-codel, and in 56% (5/9) of the IDHwt cases (table 3).
Table 3.
Molecular subtypes: Clinical parameters and outcome
| IDHmt/codel (n = 104) |
IDHmt/non-codel (n = 165) |
IDHwt (n = 49) |
Total (n=318)† |
p-values | |
|---|---|---|---|---|---|
|
Median age at diagnosis (range) |
47 (22–69) | 41 (20–69) | 52 (18–75) | 45 (18–75) | 0.0001 |
| Age [yrs] <40 | 25 (24%) | 80 (48%) | 10 (20%) | 115 (36%) | 0.0001 |
| Age [yrs] >=40 | 79 (76%) | 85 (52%) | 39(80%) | 203(64%) | |
| Time between last biopsy or surgery and treatment start (months)$ | |||||
| Median (range) | 4.5 (1.2–89.5) | 5.5 (0.0–136.1) | 3.4 (1.0–78.7) | 4.4 (0.0–136.1) | 0.07 |
| Time between first histological diagnosis and treatment start (months)$ | |||||
| Median (range) | 5.6 (1.0–124.6) | 9.0 (1.3–136.0) | 3.4 (1.0–78.7) | 5.6 (1.0–136.0) | 0.006 |
| Extent of resection | |||||
| Biopsy | 37 (36%) | 44 (27%) | 32 (65%) | 113 (36%) | <0.0001 |
| Partial removal | 51 (49%) | 86 (52%) | 11 (22%) | 148 (47%) | |
| Total removal | 16 (15%) | 34 (21%) | 6 (12%) | 56 (18%) | |
| Missing | 0 (0%) | 1 (<1%) | 0 (0%) | 1 (<1%) | |
| Female/Male | 39 / 65 (37%) | 74 / 91 (45%) | 21 / 28 (43%) | 134/184 (42%) | 0.50 |
| *MGMTSTP27meth | (n=45) | (n=72) | (n=9) | (n=126) | |
| Unmethylated | 0 (0%) | 10 (14%) | 4 (44%) | 14 (11%) | 0.0003 |
| Methylated | 45 (100%) | 62 (86%) | 5 (56%) | 112 (89%) | |
| Median Progression Free Survival, months (95% CI) | Interaction test p=0.03 |
||||
| TMZ | 55.03 (37.95, N) | 36.01 (28.42, 46.95) |
23.69 (5.55, 42.25) |
40.48 (35.25, 46.95) |
|
| RT | 61.63 (42.32, N) | 55.36 (47.87, 65.87) |
19.09 (11.27, 25.69) |
50.99 (39.79, 61.63) |
|
| Progression Free Survival at 5 years, % (95% CI) | |||||
| TMZ | 47.39(30.71, 62.35) |
19.43(8.87, 33.00) | 17.78(3.69, 40.48) |
28.92(19.78, 38.69) |
|
| RT | 58.49(39.43, 73.41) |
42.50(27.38, 56.83) |
0.00(0.00, 0.00) | 40.18(29.94, 50.19) |
|
| Hazard ratio | |||||
| TMZ vs RT | 1.04 (0.56, 1.93) |
1.86 (1.21, 2.87) | 0.67 (0.34, 1.32) | 1.18 (0.87, 1.60) |
|
| p-values | p=0.91 | p=0.004 | p=0.24 | p=0.30 | |
Test only available for a subgroup.
For patients who did not start randomized treatment, date of randomization was used. N is for median not reached.
Subgroup representative of the overall population with respect to baseline characteristics (except for the more frequent debulking surgery) and overall outcome (PFS).
The interaction test showed a significant predictive value of the molecular subtypes for PFS in the 318 molecularly characterized patients (table 3, p=0.03). Pairwise analyses revealed a favorable prognosis for patients with IDHmt tumors, with a median PFS of 62 months (95% CI, 41- upper limit not yet determined) for IDHmt/codel patients; of 48 months (95% CI, 41–55) for IDHmt/non-codel patients compared to only 20 months (95% CI, 12 −26) for IDHwt patients (figure 3). Only patients with IDHmt/non-codel tumors had a longer PFS when treated with RT than with TMZ (median PFS 55 months (95% CI, 48 to 66) versus 36 months (95% CI, 28.4 to 47) translating into a HR of 0.53 (0.35 – 0.82; logrank p= 0.0043) (see appendix of supplemental data, appendix table S6). No treatment related differences were observed for patients of the other two molecular subgroups (IDHmt/codel and IDHwt tumors, respectively).
Figure 3.




Molecular subgroups, PFS and treatment. Kaplan-Meier curves are shown for the 3 molecular subgroups, IDH mutated and 1p/19q co-deleted (IDHmt/codel), IDHmt/non-codel, and IDH wild-type (IDHwt). (A) prognostic value of the molecular subgroups is observed. Pairwise comparisons of the two treatment arms RT and TMZ are shown by molecular subtype: IDHmt/non-codel (B), IDHmt/codel (C), and IDHwt (D).
Discussion
To the best of knowledge, this is the first randomized trial testing the use of chemotherapy alone as initial treatment and the first prospective randomized trial in LGG to molecularly stratify tumor subgroups before randomization. The study has been designed over 15 years ago based on the insights available at that time. Yet, the current analysis includes also recently defined molecular markers and subgroups for their prognostic and importantly, also predictive value. With a median follow-up of 4 years (IQR:2.6–4.7), there was no significant difference in outcome between the two treatment modalities (PFS HR (TMZ vs RT): 1.16, 95% (0.91–1.48)). Patients with IDHwt tumors had the worst prognosis independent of therapy, while patients IDHmt/codel fared best, consistent with other datasets of lower grade glioma.4,24–26 Younger age was associated with worse outcome in this trial (table 3). This is likely due to molecular subtype dependent differences in age at diagnosis, and age (>40) that was one of the criteria for randomization, hence younger patients had to have clinical symptoms or unfavorable characteristics to enter the trial (the statistical difference is lost after adjustment for molecular subtype, data not shown).
Nowadays lower grade gliomas (WHO grade II/III) can readily be subclassified into prognostic molecular subgroups using an integrative diagnostic approach.21 IDH mutations that were discovered after trial implementation are characteristic for LGG and are present in over 80%. By today’s understanding, IDHwt tumors may actually belong molecularly to other tumor entities, mostly different subtypes of glioblastoma, but also indolent tumors such as ganglioglioma, or pilocytic astrocytoma.24,27 Recent retrospective reports suggest that among IDHmt gliomas, molecular characterization may be a stronger predictor for outcome than tumor grade and other prognostic markers.4,24,26,28 However, the initial treatment concept varied and was influenced by the tumor grade; for example in the prospective German Glioma Network registry less than 10% of WHO grade II gliomas received adjuvant radiotherapy and/or chemotherapy after initial surgery, in contrast to over 90% of the WHO grade III population.26 Furthermore, the criteria and indication for treatment has likely varied over time and between institutions.
For LGG an expectative approach with watchful waiting or surgery when feasible and risk-adapted adjuvant treatment are commonly the initial management.1 Treatment is initiated based on clinical symptoms, patient’s age at presentation, tumor size and radiological characteristics as well as tumor growth rate. This leads to a variable time point of treatment initiation depending on local practice and judgment, histological tumor characteristics and grade may have changed over time, adding to heterogeneity of patient population included. In our trial the wide time range from initial histological diagnosis to treatment (1–152 months, median 6) reflects both the variation in local practice and the heterogeneous natural history of the disease. In order to account for the variability between local practice and investigators, we stratified patients before randomization according to the treating institution. Molecular tumor characterization, correlation with outcome and subsequent rational treatment decisions will be of great benefit in individualizing and standardizing management of patients with LGG.
We interrogated our data for the predictive value of molecular subgroups. Of note, albeit the largest trial ever conducted in this disease, the study was not powered for the molecular subgroup analyses. In order to save statistical power, our data were only used to test the predictive value of known molecular subgroups and not for identifying new markers. Our analyses suggest that primary chemotherapy in patients with IDHmt/non-codel tumors may be deleterious; patients treated with TMZ had a significantly shorter PFS. Conversely, PFS was comparable between RT and TMZ treated patients in both subgroups of IDHmt/codel, and of IDHwt tumors (interaction test p=0.03). However, the IDHwt subgroup is a small (n=49), potentially heterogeneous and molecularly ill-defined group of patients.
All IDHmt/codel and 90% of the IDHmt/non-codel tumors have a methylated MGMT promoter, this implies that its clinical impact cannot be differentiated from the IDH mutation alone, or from its associated CpG island methylator phenotype (CIMP),29 due to the nested dependency of these alterations.22 Hence, MGMT testing does not provide additional prognostic or predictive value in the IDHmt subgroup. In IDHwt tumors, MGMT may be of a predictive value, however the rarity of this constellation in our cohort does not allow for formal testing. Nevertheless, other reports by our and other groups in IDHwt grade III gliomas strongly suggest a predictive effect for benefit from chemotherapy for patients with a methylated MGMT status.30,31
Accurate molecular prognostication is of particular importance in a disease with such a highly variable outcome. While intensive therapy is warranted for rapidly progressive tumors, acute and long-term toxicity and deficits may impair quality of life and neurological function in patients with indolent tumors.32–34 Quality of life and MMSE are reported in detail separately.35 The median PFS with RT alone in our trial is 46 months, comparable to the 48 months reported for the RT only arm of the randomized RTOG98-02 trial.8 In that trial, patients were randomized between RT alone, or RT followed by adjuvant PCV-chemotherapy (procarbazine, lomustine [CCNU], vincristine). Recently, and only after a long-term follow-up (12 years) the RTOG98-02 trial demonstrates a survival benefit (HR 0.59, p=0.003) favoring adjuvant chemotherapy,8 almost exclusively in patients with oligodendroglioma, the subtype that commonly harbors the 1p/19q co-deletion. However, RTOG98-02 and our trial are not readily comparable; the median age in RTOG98-02 was 4 years younger (median 40/41 years, treatment arms vs 44/45 years, this trial), the distribution of the histologic subtypes was different, and molecular subgroup analyses for 1p/19q codel are not available in RTOG98-02.8 The high interobserver difference even among expert neuropathologists observed in our and other studies precludes reliable comparisons at present. Whether combined modality therapy should be used for initial treatment of LGG was not addressed by our trial as we were aiming at minimizing the potential risk of late toxicity in a patient population that may live for > 10–20 years.
Our trial also identified molecular subgroups (IDHwt or IDHmt/non-codel tumors) that carry a clearly inferior prognosis. Here novel or more intensive treatment strategies are warranted even at the price of some toxicity, while the optimal treatment strategy for favorable-prognosis patients with IDHmt/codel tumors remains controversial. Our results failed to demonstrate improvement of PFS when treating patients upfront with chemotherapy alone, but substituting irradiation by equally effective TMZ chemotherapy may prevent or delay from long-term radiation-induced side effects. In the German NOA04-trial in patients with anaplastic glioma the treatment sequence (chemo first →RT at progression or vice versa) did not matter with regards to overall survival.36 Long-term follow-up of EORTC and RTOG trials on anaplastic (WHO grade III) glioma suggests an improved outcome for patients with 1p/19q codel tumors treated with radiotherapy and early adjuvant chemotherapy.37,38
We set out to evaluate a well-tolerated and at the time of trial initiation novel alkylating agent as primary treatment for LGG. Still, there are some inherent limitations to our trial. The primary endpoint is progression-free survival, thus dependent on imaging technique, methodology of tumor assessment and the frequency of imaging.39 Observer experience and potential imaging changes induced by radiotherapy that would be absent in the chemotherapy arm may be further confounding factors. In the most favorable subgroups, patients in either arm may not yet have experienced tumor progression, and data is thus still somewhat immature. The striking differences in outcome for the distinct molecular subgroups underline the need for integration of the molecular markers into tumor diagnosis for future treatment stratification. Based on our results, the search for IDH mutation as well as 1p/19q deletion should be part of the initial diagnostic workup for all patients with low-grade glioma as now implemented in the updated WHO classification 2016,40 while the MGMT promoter methylation status may have an added value only in IDHwt tumors. Our trial contributes to molecularly defining the individual therapeutic strategy, intensive treatments for poor-prognosis patients while avoiding overtreatment in indolent disease. While recent mature results and subgroup analyses from a randomized trial conducted in the 1990’s demonstrates a superior outcome with radiotherapy followed by chemotherapy both in grade II and grade III glioma, our results may support the option of initial chemotherapy alone in good-prognosis IDHmt and codeleted tumors.8,41 Only very long-term follow-up will determine the role of single modality chemotherapy in patients with chemotherapy-sensitive disease.
Supplementary Material
Table 2.
Toxicity and treatment emerging adverse events [TEAE]
| RT | TMZ | |||||
|---|---|---|---|---|---|---|
| (N=228) | (N=235) | |||||
| Hematological toxicity, N(%) | Gr1–2 | Gr3 | Gr4 | Gr1–2 | Gr3 | Gr4 |
| Leukopenia | 12(5.3) | 120(51.1) | 8(3.4) | 1(0.4) | ||
| Neutropenia | 6(2.6) | 1(0.4) | 80(34) | 6(2.6) | 4(1.7) | |
| Thrombocytopenia | 8(3.4) | 4(1.7) | 7(3.0) | |||
| Anemia | 7(3.1) | 58(24.7) | 1(0.4) | 1(0.4) | ||
| Other TEAE, N(%) | ||||||
| Allergy/Immunology | 3(1.3) | 11(4.7) | 1(0.4) | |||
| Auditory/ear | 37(16.2) | 4(1.8) | 35(14.9) | 1(0.4) | ||
| Cardiac(general) | 10(4.4) | 11(4.7) | 2(0.9) | |||
| Constitutional symptoms | 141(61.8) | 8(3.5) | 159(67.7) | 15(6.4) | 1(0.4) | |
| Dermatology/skin | 112(49.1) | 73(31.1) | 4(1.7) | |||
| Endocrine | 9(3.9) | 12(5.1) | ||||
| Gastrointestinal | 66(28.9) | 4(1.8) | 158(67.2) | 10(4.3) | ||
| Hemorrhage/bleeding | 8(3.4) | |||||
| Hepatobiliar/pancreas | 2(0.9) | |||||
| Infection | 23(10.1) | 2(0.9) | 66(28.1) | 7(3.0) | 1(0.4) | |
| Lymphatics | 1(0.4) | 1(0.4) | 9(3.8) | |||
| Metabolic/laboratory | 1(0.4) | 2(0.9) | 4(1.7) | |||
| Musculoskeletal/soft tissue | 20(8.8) | 2(0.9) | 21(8.9) | 2(0.9) | ||
| Neurology | 134(58.8) | 25(11.0) | 3(1.3) | 125(53.2) | 36(15.3) | 5(2.1) |
| Ocular/visual | 36(15.8) | 46(19.6) | ||||
| Pain | 107(46.9) | 6(2.6) | 121(51.5) | 7(3.0) | ||
| Pulmonary/upper respiratory | 13(5.7) | 1(0.4) | 42(17.9) | 1(0.4) | ||
| Renal/genitourinary | 4(1.8) | 2(0.9) | 16(6.8) | |||
| Secondary malignancy | 1(0.4) | 1(0.4) | 4(1.7) | |||
| Sexual/reproductive function | 7(3.1) | 11(4.7) | 4(1.7) | |||
| Surgery/intra-operative injury | 2(0.9) | |||||
| Syndromes (flu-like, somnolence) | 3(1.3) | 12(5.1) | ||||
| Vascular | 2(0.9) | 1(0.4) | 1(0.4) | 1(0.4) | ||
An individual patient may have experienced diverse and multiple toxicities, thus the number of patients affected by toxicity is lower.
Acknowledgments
Supported in part by an unrestricted educational grant and drug by MSD-Merck&Co. Molecular analyses were supported by the Swiss-Bridge Award 2011 and the Swiss Cancer League (KFS-2949-02-2012, MEH, SK). The EORTC is supported by NCI grants 5U10 CA011488-35 through 2U10 CA011488-41. The UK participation was supported by the National Institute for Health Research through the National Cancer Research Network, and a grant to the MRC Clinical Trials Unit at UCL from Cancer Research UK (trial reference CRUK/07/032). The NCIC CTG participation has been supported by the Canadian Cancer Society Research Institute (grant #015469 and #021039). The TROG participation has been supported by a National Health and Medical Research Council (NHMRC) project grant (ID 509094). This publication was supported by the EORTC Cancer Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors:
BGB, MEH and RS designed the trial concept and protocol, with contributions from MJvdB, AvD, TG, GR, DL and WPM.
BGB, MEH and RS acted as principal investigators for the trial coordination (BGB for radiation oncology, RS for medical oncology and MEH for molecular and translational research coordination), and reviewed the data and data quality. Quality assurance for radiation therapy was performed with the guidance of BGB. MEH coordinated tissue collection, translational research and molecular analyses.
Pathology review were performed by AvD, CH, DC, JMK and JPR.
Molecular analyses were performed by AvD, CH, C, SK, MEH.
MT, JR and MK were responsible for the quality-of-life and neurocognitive component of the trial.
GR, WM, and JR were responsible in setting up the trial and getting ethical approval in their national organisations of TROG, NCI-C and MRC UK.
TG assumes responsibility for the statistical analyses.
The trial was coordinated by the EORTC headquarters staff, under the responsibility of ND and DL.
All authors provided administrative support and contributed to trial activation, patient inclusion, care, treatment and follow-up (patient-related activities only clinicians), data collection, data interpretation, manuscript writing or reviewing and commenting, and approval of the final version of the manuscript.
Declaration of interests:
BGB reports personal fees from Merck & Co (MSD), outside the submitted work.
MEH reports grants from Swiss Bridge Award, grants from Swiss Cancer League, during the conduct of the study; other from MSD, other from MDxHealth, outside the submitted work.
MJvdB reports grants and personal fees from Roche, grants and personal fees from Abbvie, personal fees from Merck AG, personal fees from Novocure, personal fees from cavion, personal fees from BMS, personal fees from Novartis, personal fees from Actelion, outside the submitted work.
AvD reports a patent US 8,367,347 B2: "Methods for the diagnosis and prognosis of a brain Tumor" with royalties paid to Dianova GmbH, Hamburg, Germany.
TG has nothing to disclose.
KHX has nothing to disclose.
AAB has nothing to disclose.
GK has nothing to disclose.
MJBT reports personal fees from Hoffmann La Roche, outside the submitted work.
MBH has nothing to disclose.
CH has a patent IDH1 R132H specific antibody with royalties paid to Company Dianova.
GR has nothing to disclose.
DC reports a patent US 8,367,347 B2: "Methods for the diagnosis and prognosis of a brain Tumor" with royalties paid to Dianova GmbH, Hamburg, Germany.
JMK has nothing to disclose.
SK has nothing to disclose.
WW reports grants and personal fees from MSD, grants and personal fees from Roche, grants from Boehringer Ingelheim, grants from Apogenix, grants from Vaximm, outside the submitted work. In addition, Dr. Wick has a patent IDH1 R132H specific antibody with royalties paid to Company Dianova
RE has nothing to disclose.
MR reports grants and personal fees from Celgene personal fees from Boehringer, personal fees from Genentech, personal fees from Lilly, personal fees from Merck-Serono, grants from Novartis, grants from Pharmamar, outside the submitted work.
BT acknowledges financial support from NCIC-CTG, during the conduct of the study
FD has nothing to disclose.
JEB has nothing to disclose.
LF has nothing to disclose.
JCR has nothing to disclose.
OC reports grants, personal fees and non-financial support from Roche, personal fees from Ipsen, personal fees from Astra-Zeneca, outside the submitted work.
JMMG has nothing to disclose.
JPR has nothing to disclose.
ND has nothing to disclose.
CB has nothing to disclose.
JBM acknowledges support from EORTC, during the conduct of the study.
PMC reports grants from MSD, outside the submitted work.
CM has nothing to disclose.
TTS has nothing to disclose.
RAN has nothing to disclose.
JR has nothing to disclose.
DL has nothing to disclose.
WPM has nothing to disclose.
RS has served on advisory boards for Abbvie, Merck KGaA, Merck & Co/MSD (outside the submitted work), Novartis, Novocure, Pfizer and Roche, fees (when applicable) to institution.
References
- 1.Soffietti R, Baumert BG, Bello L, et al. Guidelines on management of low-grade gliomas: report of an EFNS-EANO Task Force. Eur J Neurol. 2010;17(9):1124–1133. doi: 10.1111/j.1468-1331.2010.03151.x. [DOI] [PubMed] [Google Scholar]
- 2.Pignatti F, van den Bent M, Curran D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20(8):2076–2084. doi: 10.1200/JCO.2002.08.121. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66(20):9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 5.Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36(3):549–556. doi: 10.1016/s0360-3016(96)00352-5. [DOI] [PubMed] [Google Scholar]
- 6.van den Bent MJ, Afra D, de Witte O, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366(9490):985–990. doi: 10.1016/S0140-6736(05)67070-5. [DOI] [PubMed] [Google Scholar]
- 7.Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low-versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20(9):2267–2276. doi: 10.1200/JCO.2002.09.126. [DOI] [PubMed] [Google Scholar]
- 8.Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med. 2016;374(14):1344–1355. doi: 10.1056/NEJMoa1500925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoang-Xuan K, Capelle L, Kujas M, et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions. J Clin Oncol. 2004;22(15):3133–3138. doi: 10.1200/JCO.2004.10.169. [DOI] [PubMed] [Google Scholar]
- 10.Quinn JA, Reardon DA, Friedman AH, et al. Phase II trial of temozolomide in patients with progressive low-grade glioma. J Clin Oncol. 2003;21(4):646–651. doi: 10.1200/JCO.2003.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Brada M, Vivers L, Abson C, et al. Phase II study of primary temozolomide chemotherapy in patients with WHO grade II gliomas. Ann Oncol. 2003;14(11):1715–1721. doi: 10.1093/annonc/mdg371. [DOI] [PubMed] [Google Scholar]
- 12.Ricard D, Kaloshi G, Amiel-Benouaich A, et al. Dynamic history of low-grade gliomas before and after temozolomide treatment. Ann Neurol. 2007;61(5):484–490. doi: 10.1002/ana.21125. [DOI] [PubMed] [Google Scholar]
- 13.Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88(7):1004–1011. doi: 10.1038/sj.bjc.6600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandes AA, Tosoni A, Cavallo G, et al. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: phase II study from gruppo italiano cooperativo di neuro-oncologia (GICNO) Br J Cancer. 2006;95(9):1155–1160. doi: 10.1038/sj.bjc.6603376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wick A, Felsberg J, Steinbach JP, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25(22):3357–3361. doi: 10.1200/JCO.2007.10.7722. [DOI] [PubMed] [Google Scholar]
- 16.Weber DC, Poortmans PM, Hurkmans CW, Aird E, Gulyban A, Fairchild A. Quality assurance for prospective EORTC radiation oncology trials: the challenges of advanced technology in a multicenter international setting. Radiother Oncol. 2011;100(1):150–156. doi: 10.1016/j.radonc.2011.05.073. [DOI] [PubMed] [Google Scholar]
- 17.Musat E, Roelofs E, Bar-Deroma R, et al. Dummy run and conformity indices in the ongoing EORTC low-grade glioma trial 22033–26033: First evaluation of quality of radiotherapy planning. Radiother Oncol. 2010;95(2):218–224. doi: 10.1016/j.radonc.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Fairchild A, Weber DC, Bar-Deroma R, et al. Quality assurance in the EORTC 22033–26033/CE5 phase III randomized trial for low grade glioma: the digital individual case review. Radiother Oncol. 2012;103(3):287–292. doi: 10.1016/j.radonc.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Hartmann C, Mueller W, Lass U, Kamel-Reid S, von Deimling A. Molecular genetic analysis of oligodendroglial tumors. J Neuropathol Exp Neurol. 2005;64(1):10–14. doi: 10.1093/jnen/64.1.10. [DOI] [PubMed] [Google Scholar]
- 20.Seiz M, Tuettenberg J, Meyer J, et al. Detection of IDH1 mutations in gliomatosis cerebri, but only in tumors with additional solid component: evidence for molecular subtypes. Acta Neuropathol. 2010;120(2):261–267. doi: 10.1007/s00401-010-0701-2. [DOI] [PubMed] [Google Scholar]
- 21.Reuss DE, Sahm F, Schrimpf D, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an "integrated" diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129:133–146. doi: 10.1007/s00401-014-1370-3. [DOI] [PubMed] [Google Scholar]
- 22.Bady P, Sciuscio D, Diserens AC, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124(4):547–560. doi: 10.1007/s00401-012-1016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bady P, Delorenzi M, Hegi ME. Sensitivity Analysis of the MGMT-STP27 Model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J Mol Diagn. 2016;18(3):350–361. doi: 10.1016/j.jmoldx.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Ceccarelli M, Barthel FP, Malta TM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiestler B, Capper D, Sill M, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014;128(4):561–571. doi: 10.1007/s00401-014-1315-x. [DOI] [PubMed] [Google Scholar]
- 26.Weller M, Weber RG, Willscher E, et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015;129(5):679–693. doi: 10.1007/s00401-015-1409-0. [DOI] [PubMed] [Google Scholar]
- 27.Reuss DE, Kratz A, Sahm F, et al. Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol. 2015;129:407–417. doi: 10.1007/s00401-015-1454-8. [DOI] [PubMed] [Google Scholar]
- 28.Reuss DE, Mamatjan Y, Schrimpf D, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 2015;129(6):867–873. doi: 10.1007/s00401-015-1438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Bent MJ, Erdem Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513–5522. doi: 10.1158/1078-0432.CCR-13-1157. [DOI] [PubMed] [Google Scholar]
- 31.Wick W, Meisner C, Hentschel B, et al. Prognostic or predictive value of MGMT promoter methylation in gliomas depends on IDH1 mutation. Neurology. 2013;81(17):1515–1522. doi: 10.1212/WNL.0b013e3182a95680. [DOI] [PubMed] [Google Scholar]
- 32.Douw L, Klein M, Fagel SS, et al. Cognitive and radiological effects of radiotherapy in patients with low-grade glioma: long-term follow-up. Lancet Neurol. 2009;8(9):810–818. doi: 10.1016/S1474-4422(09)70204-2. [DOI] [PubMed] [Google Scholar]
- 33.Boele FW, Douw L, Reijneveld JC, et al. Health-related quality of life in stable, long-term survivors of low-grade glioma. J Clin Oncol. 2015;33(9):1023–1029. doi: 10.1200/JCO.2014.56.9079. [DOI] [PubMed] [Google Scholar]
- 34.Correa DD, DeAngelis LM, Shi W, Thaler HT, Lin M, Abrey LE. Cognitive functions in low-grade gliomas: disease and treatment effects. J Neurooncol. 2007;81(2):175–184. doi: 10.1007/s11060-006-9212-3. [DOI] [PubMed] [Google Scholar]
- 35.Reijneveld JC, Martin J, Taphoorn MJ, Coens C, et al. Health-related quality of life in high-risk low-grade glioma; results of a randomized controlled trial. Lancet Oncol. 2016 in press. [Google Scholar]
- 36.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27(35):5874–5880. doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 37.van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. doi: 10.1200/JCO.2012.43.2229. [DOI] [PubMed] [Google Scholar]
- 38.Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. doi: 10.1200/JCO.2012.43.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pallud J, Llitjos JF, Dhermain F, et al. Dynamic imaging response following radiation therapy predicts long-term outcomes for diffuse low-grade gliomas. Neuro Oncol. 2012;14(4):496–505. doi: 10.1093/neuonc/nos069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of tumours of the central nervous system. Lyon: IARC; 2016. Revised 4th edition ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32(8):783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.