

Abstract
Targeted therapy has proven to be beneficial at producing significant responses in patients with a wide variety of cancers. Despite initially impressive responses, most individuals ultimately fail these therapies and show signs of drug resistance. Very few patients are ever cured. Emerging evidence suggests that treatment of cancer cells with kinase inhibitors leads a minor population of cells to undergo a phenotypic switch to a more embryonic-like state. The adoption of this state, which is analogous to an epithelial-to-mesenchymal transition, is associated with drug resistance and increased tumor aggressiveness. In this commentary we will provide a comprehensive analysis of the mechanisms that underlie the embryonic reversion that occurs on targeted cancer therapy and will review potential novel therapeutic strategies designed to eradicate the escaping cells.
Keywords: melanoma, BRAF, EMT, lung cancer, EGFR
Graphical Abstract
1. Introduction
For decades, the chemotherapeutic treatment of cancer has relied upon non-specific cytotoxic drugs which target rapidly dividing cells through the initiation of DNA damage, the stabilization of microtubules or the inhibition of nucleotide metabolism [1]. While being effective at killing cells with high proliferative rates, these therapies also target normal cell populations with rapid turnover, such as the cells of the hair follicles, the bone marrow and linings of the gut, leading to significant toxicity. A more effective approach to cancer therapy is the direct targeting of the genetic aberrations in the oncogenes that drive cancer development and progression - a strategy termed targeted therapy. As tumors typically have complex mutational profiles and a high level of redundancy in the pathways that drive growth and survival, targeted therapies work best in cases where the cancer is addicted to the activity of one oncogene for their growth and survival.
One of the first targeted therapies to be developed was imatinib, a Bcr-Abl kinase inhibitor/c-KIT inhibitor, with activity in chronic myeloid leukemia (CML) harboring the Bcr-Abl fusion protein and c-KIT mutant gastrointestinal stromal tumors (GIST), respectively[2, 3]. Following this, targeted therapies have since been developed for many other cancers where driver oncogenes have been identified including EGFR mutant/expressing lung cancer/breast cancer/colorectal carcinoma, BRAF-mutant melanoma/hairy cell leukemia and Alk-mutant non-small cell lung cancer (NSCLC) [4–6]. Although drugs developed against these targets have revolutionized the treatment of many cancers, therapy failure is common with resistance usually associated with reactivation of the initial oncogenic pathway. The reactivation of signaling can be mediated through secondary mutations in the target gene of interest e.g. T790M mutations in EGFR, T315 mutations in Abl and L1196M mutations in Alk [7–9]. Escape from therapy can also be driven through the activation of bypass pathways, e.g. NRAS, MEK and BRAF-splice mutations in the case of BRAF inhibitor failure in melanoma and increased c-MET signaling in the case of erlotinib resistant lung cancer [10–12]. Although resistance is often genetically mediated, it does not always arise from pre-existing mutation bearing clones and often takes time to develop. The exact sequence of events that occur in between initial drug response and the emergence of fully resistant cells are not well understood. There is a growing body of evidence that suggests acquired resistance is non-genetically mediated, and is independent of secondary genetic events. This process of phenotypic adaptation, which allows tumor cells to dedifferentiate and adopt a more primitive embryonic state, is likely to contribute to therapeutic escape and may also increase the dissemination of the resistant tumor cells to other sites. There also seems to be some overlap between the therapeutically adapted state and that of so-called cancer “stem cells” - which also show phenotypic plasticity, slow growth and drug resistance. Defining and therapeutically targeting this phenotypic state is likely to be critical to the effective long-term management of many advanced cancers in which targeted therapy is used. In this review we will outline some of the mechanisms that underlie the phenotypic switch in cancer cells and will describe how this contributes both to therapy escape and drug resistance. The majority of our focus will be upon BRAF mutant melanoma and EGFR driven lung cancer.
2. The phenomenon of phenotype switching in normal physiology
Recent work has suggested that the phenotypic adaptation of cancer cells in the escape from therapy is akin to that of an epithelial-to-mesenchymal (EMT) transition, the widely conserved developmental process that permits the conversion of epithelial cells to a more mesenchymal-like state[13]. Under normal physiology, epithelial cells exist as organized sheets of cells that serve both as a protective barrier as well as well performing important secretory functions. Polarity and organization within the epithelial cell layers is achieved through E-cadherin based adherens junctions, tight junctions and connexins. The adhesion mediated through E-cadherin and tight junctions maintain the architecture and integrity of the epithelium and allow for the directed flow and transport of ions, nutrients and growth factors [14]. Mesenchymal cells, in contrast, are more supportive and play a role in extracellular matrix (ECM)-deposition, serve as tissue scaffolds and have critical roles in the repair of tissue damage and wound healing [14]. During embryonic development, some polarized epithelial cells dedifferentiate, downregulate their tight E-cadherin mediated adhesion and alter their morphology. These changes allow the cells to exit the epithelial environment and migrate to new locations, after which they redifferentiate, reacquire their ordered epithelial morphology and restore their cell-cell adhesions with other neighboring cells, a process known as mesenchymal to epithelial transition (MET).
The induction of the EMT is dependent upon complex functional networks involving at least 3 families of transcription factors which alter the expression of many genes important for decreased cell adhesion, increased cell migration and mesenchymal differentiation. One family, the Snail Zinc-finger transcription factors, including Snail1 and Slug, play a key role in EMT induction through their repressive effects upon E-cadherin through direct binding to the E-boxes of the CDH1 promoter [15]. Snail1 has also been shown to upregulate the mesenchymal markers fibronectin and vimentin[16] while both Snail1 and Slug have been implicated in metastasis[17]. Another group of EMT-associated transcription factors are the basic helix–loop–helix (bHLH) family members Twist1 and Twist2. Both Twist1 and Twist2 have been implicated in EMT through their ability to induce a stem-like state and to repress E-cadherin expression through the induction of Snail family transcription factors[18]. The final group of EMT-associated transcription factors is the homeobox family, comprising ZEB1, ZEB2 and NANOG. Both ZEB1 and ZEB2 decrease the transcription of E-cadherin[19], with ZEB1 being further shown to repress miRNA-203 and miRNA-200 expression[20]. ZEB2 also has a role in the downregulation of transcripts encoding P-cadherin, claudin 4, and the tight junction protein 3 (ZO-3)[21]. NANOG regulates EMT through decreasing levels of E-cadherin expression while increasing the expression of vimentin, β-catenin and Snail[22, 23].
The role of EMT in cancer is well known and represents one mechanism by which non-motile, polarized, epithelial cells dedifferentiate and acquire a more invasive and mobile phenotype[24]. Induction of these EMT-associated transcription factors can also arise following microenvironmental signals including transforming growth factor-β (TGF-β) superfamily, the WNT family, and growth factors from the fibroblast growth factor (FGF) family. Among these, the TGF-β pathway is a primary driver of EMT[25] with the ability to induce all three EMT transcription factor families including Snail1/Slug, ZEB1/2, and Twist1[26]. TGF-β family members often remain in a latent form through their binding to several binding factors and their activation occurs by binding to membrane receptor serine/threonine kinases I/II, serving as a connector to Smad proteins[27]. TGF-β/Smad signaling induces EMT in cooperation with other signaling pathways, which can include Ras activation [28] and Wnt/β-catenin[29]. TGF-β itself is secreted by nearby stromal fibroblasts in response to tumor-derived signals or other stresses [30]. The fibroblasts are important regulators in this context and they can modulate the expression of smooth muscle actin (α-SMA), FGF and collagen which also lead to EMT induction[31]. While TNFα plays a vital role in the regulation and homeostasis of immune cells, it has also been shown to induce expression of EMT transcription factors such as Snail1 via the activation of NF-κB[32] and Twist1 via activation of IKK-β[33]. TNFα also has been shown to decrease the expression of E-cadherin and upregulate vimentin [34]. Within a tumor, EMT does not occur uniformly, with a greater proportion of mesenchymal cells being observed at the leading edge and in areas of hypoxia. The role of EMT in tumor progression has been widely discussed elsewhere, and the reader is directed to the following excellent articles for a more thorough discussion of this topic[35–38].
3. Phenotypic adaptation as a mediator of drug resistance
One of the major unanswered questions in the study of drug resistance is whether therapeutic escape result from the selection of pre-exiting rare clones of cells harboring resistance mutations or instead emerges from an evolutionary process in the cells that escape the initial therapeutic insult, allowing the later emergence of resistance conferring mutations. As the majority of patients receiving targeted therapy show at least some level of response, it seems likely that the second “adaptation dependent-resistance” process is critical for the emergence of resistance in most cases and that phenotypic adaptation is a key part of this.
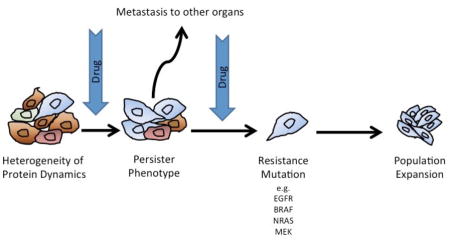
Baseline or stochastic variability exists even within genetically homogeneous populations of cancer cells. This heterogeneity is often non-genetically mediated and instead results from the normal cell-to-cell fluctuations in mRNA and protein expression. Within a large population of cells this variability often results in a wide range of cell states and phenotypes that are not fixed and readily switchable. At the simplest level, the basal level of phenotypic diversity likely explains why some cancer cells always evade therapy. This phenomenon has been elegantly demonstrated in a study in which multiple proteins were fluorescently tagged in individual cells [39]. The cells were then treated with drug and the temporal and spatial behavior of individual proteins followed by time-lapse microscopy. As expected, this initial step of therapeutic escape was dependent upon the protein expression at the single cell level, with examples given where two (presumably genetically identical) daughter cells from the same parent experienced vastly different fates (apoptosis vs. survival) following drug treatment. A number of proteins were identified whose level of expression determined outcome including BAG2/BAG3, calmodulin and ribosomal protein RPS3 [39]. A second study, again using time-lapse microscopy, also demonstrated that the initial fate of drug treated cells was not genetically mediated and resulted instead from differences in protein dynamics between individual cells. In this instance, two potential protein networks, one caspase-driven and the other Cyclin B1-dependent, were identified that determined cell fate [40].
It is likely that baseline variability in mRNA and protein expression within a population defines the mix of cellular states prior to therapy and therefore which cells best tolerate drug exposure. This transient state, while important for initial escape, is not likely to be sufficient for prolonged persistence in the presence of drug. There is growing evidence that long-term persistence is instead dependent upon an altered epigenetic state characterized by a slow-growth phenotype that can be maintained for prolonged periods of time [41, 42]. In this state, limited numbers of cancer cells remain quiescent before beginning to regrow in the presence of drug. The emergence of this persister phenotype has been demonstrated across multiple cancer cell lines following treatment with all major classes of anti-cancer drugs, including kinase inhibitors and more traditional chemotherapeutic drugs. The persister state is reversible and non-genetically mediated with studies showing single cells to give rise to both persister daughter cells and non-persister daughter cells [41] (Figure 1).
Figure 1.

Summary of mechanisms that contribute to therapeutic adaptation
The role of the persister state and its relationship to the eventual emergence of genetically mediated drug resistance remained obscure for many years. Recent work in NSCLC has shed light on this and has provided new insights linking phenotypic adaptation to acquired drug resistance [43, 44]. Initial studies showed that the treatment of individual PC9 cells with the EGFR gefinitib was associated with two modes of drug resistance, one arising rapidly and the other being very slow to emerge [43]. Use of a highly sensitive quantitative PCR assay revealed the clones developing early resistance to harbor a pre-existing T790M resistance-conferring EGFR mutation whereas the slower growing clones did not. Instead, it was found that the slower growing persister clones acquired the EGFR T790M at a much later time point (34–47 weeks after treatment initiation) and that this clone then rapidly took over the population [43]. Gene set enrichment analysis revealed the persister population to have an expression profile that was EMT-like [43]. Surprisingly, it was found that the late-onset resistance cells maintained their drug tolerance following the acquisition of the EGFR T790M mutation and had an impaired apoptotic response to the third generation EGFR inhibitor WZ4002, whereas the early onset resistant cells did not [43].
Further support for the role of the persister state in the emergence of resistance came from other work in NSCLC, in which 17 persister-derived drug resistant cell lines were derived from one single PC-9 cell [44]. It was found that in addition to developing EGFR inhibitor resistance the cells also exhibited cross-tolerance to other drugs such as aurora kinase inhibitors and chemotherapeutics. Whole exome sequencing of the 17 cell lines revealed a diverse array of resistance mechanisms that ranged from the EGFR T790M mutation, to MET amplification, NRAS mutations and CRAF amplification [44] (Figure 1). For many of the clones, resistance mechanisms could not be identified. Unlike the prior study the cells did not appear to undergo a switch to an EMT-like state, nor was any activation of the receptor tyrosine kinases (RTK) IGF1R and Axl or the NF-κB signaling pathway noted. The authors concluded that the persister state constituted a pool of cells from which diverse resistance mechanisms could emerge[44]. It still remains to be determined whether the persister cell population constitutes a pool of cells with diverse mutations that eventually grow out in the presence of drug or whether this represents a “mutatable” state that permits clones to adapt to continuous drug exposure through the acquisition of novel genetic mutations.
The role of phenotypic adaptations, such as the induction of an EMT, in therapeutic escape and drug resistance has long been suspected. Anaylsis of circulating tumor cells (CTCs) from breast cancer patients, where cells were scored according to whether they were more epithelial-like or mesenchymal–like, revealed a strong association between therapy response and an epithelial-like CTC population and therapy failure and a more mesenchymal state [45]. These findings are also supported by multiple studies on chemotherapy drugs. It is known in breast cancer that EMT-induction mediates resistance through increased AKT signaling and decreased estrogen receptor expression secondary to Twist-1 upregulation [46, 47]. In prostate cancer, EMT can mediate gemcitabine resistance as a result of increased Notch signaling [48]. Morevover, chemoresistance, secondary to an impaired apoptotic response to paclitaxel, is known to be associated with increased Snail1/Slug expression in ovarian carcinoma[49] (Figure 1).
Mechanisms underlying phenotype switching
Although phenotypic switching and the adoption of a persister state appears to be a common response to drugs across multiple cancer types, the mechanisms of induction can often be lineage specific. Much of the important work in this area has been done on BRAF-mutant melanomas following treatment with BRAF inhibitors. For many years, melanoma was thought to be refractory to all therapeutic interventions and survival rates for patients with disseminated disease were historically low [50]. In 2002 it was discovered that ~50% of all cutaneous melanoma harbored activating mutations in the serine-threonine kinase BRAF, with clinical studies showing mutant BRAF to be a bona fide therapeutic target for patients with this disease [51–53]. Despite initially impressive responses, most patients eventually failed on therapy [54]. Acquired BRAF inhibitor resistance was associated with reactivation of the mitogen activated protein kinase (MAPK) pathway mediated through multiple mechanisms including BRAF-splice mutations, NRAS mutations and MEK1/2 mutations [10–12]. Although great progress has been made determining the genetic mechanisms underlying acquired resistance, the role of phenotypic adaptation in the onset of resistance was determined only recently. Some of the initial insights into the process of adaptation came from the observation that BRAF and MEK inhibitor treatment led to a rapid recovery of phospho-ERK levels and an increase in signaling through RTKs such as EGFR, ERBB3, c-MET, PDGFR and IGF1R [10, 55–58]. It was noted that although levels of RTK expression increased, the melanoma cells did not necessarily produce their own autocrine neuregulin and HGF, which instead came from host cells, such as fibroblasts [59, 60]. Although the RTK signaling was found to be important for survival, the persisting cells adopted a slow-growth phenotype that showed similarities to oncogene-induced senescence [61, 62]. Further analysis of the persister or drug-tolerant state showed melanoma cells to exhibit chromatin remodeling and increased histone demethylase activity, and a loss of cell differentiation associated with decreased expression of melanoma markers such as MART-1 [63]. At the same time, an increased expression of stemness genes, such as CD271 was noted.
In one study, the quiescent phenotype led to the increased expression of EGFR, and was mediated through the suppression of the transcription factor SRY (sex determining region)-box 10 (SOX10) [61]. An RNA-seq study revealed SOX10 to be suppressed through a TGF-β dependent pathway and further showed that SOX10 suppression led to increased TGF-β receptor expression, in addition to a number of TGF-β target genes. This EMT-like effect, and BRAF inhibitor resistance, could be recapitulated when the melanoma cells were treated with exogenous TGF-β [61]. A role for TGF-β in driving the phenotypic switch was suggested by the observation that melanoma cells and tumor samples frequently show increased TGF-β expression following BRAF inhibitor treatment and the observation that exogenous TGF-β treatment increased the expression of the RTKs EGFR and PDGFR in melanoma cells [59, 61] (Figure 1).
Further evidence for melanoma cells experiencing an EMT-like switch following BRAF inhibitor treatment came from proteomic studies [64]. It was found that acute treatment with the BRAF inhibitor vemurafenib led to an increase in EMT associated proteins including FAK, integrin α5β1, vimentin and fibronectin [64]. The induction of fibronectin within the melanoma cells was important for therapeutic escape, with knockdown of either fibronectin or integrin α5β1 found to increase BRAF-inhibitor mediated apoptosis. Mechanistically, it was found that melanoma cells created their own protective ECM-derived niche that led to increased signaling through AKT and the maintenance of expression of the anti-apoptotic protein Mcl-1 [64].
Other work has pointed to a role for miRNAs and the EMT-like state associated with BRAF inhibitor resistance. A recent analysis of BRAF inhibitor resistant cell lines showed a role for miR-200C suppression in the upregulation of EMT-associated transcription factors such as Bmi1, Zeb2, Tubb3, ABCG5 and MDR1 [65]. Changes in these proteins were observed both in melanoma cell lines and in specimens from patients failing BRAF inhibitor therapy. Overexpression of MiR-200C or knockdown of Bmi1 reverse the EMT-like phenotype associated with BRAF inhibitor resistance and was accompanied by an recovery of MAPK and PI3K/AKT signaling as well as an increase in E-cadherin expression, and a decrease in N-cadherin, ABCG5 and MDR1 expression[65] (Figure 1).
Similar findings have been reported in NSCLC with the induction of an EMT found to mediate EGFR inhibitor resistance in a manner that was independent of both the T790M EGFR mutation and Met amplification[66]. Gene expression analysis showed the acquired resistance to be associated with TGF-β/SMAD signaling and an increase in the expression of ZEB1. Further analysis showed the increased ZEB1 expression to be associated with an EMT and concomitant alterations in EMT-associated genes such as E-cadherin, ST14 and vimentin. Knockdown of ZEB1 reversed the EMT-like phenotype and resensitized the cells to EGFR inhibition – a clear indication that the resistance was entirely phenotypic. In line with the data on melanoma, EGFR resistance was associated with decreased expressed of the miRNA miR200[66]. Downregulation of E-cadherin and upregulation of vimentin and TGF-β is also associated with ALK inhibitor resistance in NSCLC[67, 68].
4. MITF and phenotype switching
Melanoma is a tumor showing significant phenotypic plasticity with its cells existing in multiple cellular states. One of the factors most frequently associated with phenotype switching in melanoma is Microphthalmia associated transcription factor (MITF), a master regulator of the melanocytic lineage. MITF controls many of the genes that are essential to melanocyte function, including those involved in pigmentation and survival following exposure to ultraviolet radiation. MITF can also function as a melanoma oncogene, with studies identifying a subset of the tumors with high level MITF amplification[69]. The role of MITF in melanoma is complex however, and contradictory data has been reported, e.g. pro-tumorigenic vs tumor suppressive. The best explanation for this conflicting data is the “rheostat model” which posits that MITF expression levels determine the identity of the melanoma sub-populations. It is known that high levels of MITF block cell proliferation and drive a differentiated state associated with high levels of tyrosinase expression and melanosome production [70, 71]. In contrast, low levels of MITF lead to the adoption of a slow-growth arrested phenotype that is highly invasive and stem-like in nature [71, 72]. Within a tumor, MITF expression levels vary significantly and help to drive heterogeneity by regulating the switch between a proliferative, noninvasive state and slower growing, more invasive state.
Although it has been rarely stated, there is good evidence that the phenotypic state characterized by MITF overlaps significantly with the EMT-like state regulated through TGF-β. It is known that exogenous TGF-β inhibits MITF transcription through a pathway involving cyclic adenosine monophosphate (cAMP) and GLI2 [73]. There is also evidence that GLI2 directly downregulates E-cadherin expression in melanoma[74]. In melanocytes, MITF levels are controlled in through direct phosphorylation by components of the MAPK pathway [75]. It is perhaps comes as little surprise that BRAF inhibition has been associated with changes in MITF levels and this in turn has been implicated in BRAF inhibitor resistance. Again, the picture is complex and dependent upon the baseline cell state with reports outlining a role for both increased MITF expression and MITF loss in de novo and adaptive BRAF inhibitor resistance [44, 76–79].
Evidence for the role of MITF in the initial adaptation to BRAF inhibitors comes from studies demonstrating that inhibition of the MAPK pathway led to increased MITF expression in some melanoma cell lines [78]. The cells exhibiting high MITF expression were drug tolerant and had a gene expression profile associated with an EMT e.g. Snail, ZEB2 and EGF [78]. Interestingly, this early adaptive state was reversible, with the levels of MITF declining following drug removal [78]. Further evidence for increased MITF expression in phenotypic drug adaptation came from a high throughput gain-of-function genomic study that identified a novel melanocyte-specific G-protein coupled receptor/protein-kinase A, cAMP, cAMP response element binding protein (CREB)-driven pathway [79]. A number of transcription factors were increased in this adaptive cell state including MITF, c-FOS, NR4A1 and NR4A2, with functional studies showing each of these to confer BRAF inhibitor resistance [79].
In a converse fashion, melanomas with acquired BRAF inhibitor resistance and de novo resistance have also been identified with very low MITF expression levels [76, 77]. In these studies, MITF-low resistant cell lines were generally drug tolerant and exhibited cross-resistance to multiple MAPK pathway inhibitors including BRAF, MEK and ERK inhibitors and the BRAF-MEK inhibitor combination[77]. In these cells resistance was associated with increased signaling through NFκB and the RTK Axl, with increased NFκB activity having a direct suppressive effect upon MITF activation [76]. Although Axl expression was found to be associated with the MITF-low, NFκB driven transcriptional state, Axl inhibitors alone did not reverse BRAF inhibitor resistance. This work demonstrated that under baseline conditions melanoma cells could potentially exist in two distinct transcriptional states, with the ability to switch between an NFκB-high/MITF-low phenotype and a NFκB low/MITF-high phenotype [77]. Treatment of the cells with BRAF inhibitor led the MITF high cells to switch to the NFκB-high/MITF-low state. The MITF-mediated phenotypic switch also occurred following treatment with exogenous TNF-α [77] (Figure 2). These studies provided a link between immune infiltration, the “inflammatory” immune microenvironment and therapy escape. A second study, which also explored the relationship between MITF levels and BRAF inhibitor resistance, showed the MITF-low state to be associated with resistance and an increased expression of the RTKs Axl, EGFR and PDGFR-β. Like before, the inhibition of RTK signaling alone did not resensitize resistant cells to MAPK-targeted drugs. It was however noted that resistance could be reversed by treating the MITF-low cells with RTK inhibitors in combination with either BRAF inhibitors or the BRAF-MEK inhibitor combination [77]. A potential role for Axl inhibition in targeting the drug tolerant phenotype was suggested by the observation that the Axl inhibitor R428 restored sensitivity to BRAF inhibitor resistant cells in long-term colony formation assays [77]. Increased Axl expression has previously been implicated in both EMT and chemoresistance in many other cancers including breast cancer and NSCLC [80, 81]. In lung cancer, increased Axl signaling was identified as a mechanism of acquired erlotinib resistance in the absence of T790M EGFR mutations or MET upregulation. In this instance, the resistance phenotype was also associated with the adoption of an EMT[80]. In triple negative breast cancer, Axl receptor expression was associated both with EMT induction and acquired resistance to EGFR inhibitors, in part by co-opting other RTKs including c-MET, PDGFR and HER family kinases [81]. A number of small molecule Axl inhibitors, including TP-0903, MGCD516 and BPI 9016M, are currently undergoing phase I clinical evaluation in NSCLC and other solid tumors as single agents.
Figure 2. Signaling scheme showing some of the overlapping mechanisms behind the adaptive regulation of MITF expression following BRAF inhibitor treatment.

The adaptive signaling state in BRAF mutant melanoma cells can be driven through Wnt5a, TGF-β or a lineage specific program involving G-protein coupled receptor signaling and protein kinase A.
The role of MITF in therapeutic adaptation to BRAF inhibitors is complex, with the potential for its levels to be differentially regulated following MAPK pathway inhibition. There is evidence that two classes of MITF-expressing melanoma cell lines exist that show differential requirements for the transcription factor. This phenomenon was recently defined in a quantitative manner with studies showing that the baseline transcriptional state of melanoma cells (differentiated vs neural-crest like) determined the eventual phenotypic adaptation (and dependency upon MITF) of the cells to therapy [82].
5. The role of the drug adapted phenotype in metastasis
The persister/EMT phenotype is often associated with a switch to fibroblast-like features, reorganization of the actin cytoskeleton, an increase in FAK signaling and the ability to degrade the ECM, through increased expression of enzymes such as matrix metalloproteinases (MMPs) [64, 83, 84]. Together, these phenotypic changes suggest that the drug persistent state also confers metastatic potential. This indeed seems to be the case, with a recent comprehensive proteomic analysis of BRAF inhibitor sensitive and resistant melanoma cells demonstrating the emergence of a resistance associated signaling interactome based upon EGFR, ligand-independent EphA2 signaling, FAK, fibronectin, paxillin, vimentin, and integrin signaling [84]. The top pathways showing enrichment in the resistant cells were involved in cytoskeletal reorganization and EMT-like processes. In this instance, ligand-independent Epha2 signaling was identified as a key driver of the invasive phenotype and was strongly upregulated in mouse xenografts and human melanoma specimens from patients failing on BRAF inhibitor therapy[84]. Proteomic analyses by other groups have also suggested the BRAF inhibitor resistant is associated with invasion and metastasis. In these studies, the resistant cells had an emergent EGFR/Src-family kinases(SFK)/STAT3 network that both increased cell growth and enhanced the invasive and metastatic potential [56]. The translational potential of these findings was indicated by studies showing that the SFK/multi-RTK inhibitor dasatanib prevented BRAF inhibitor treated melanoma cells from metastasizing to the lung and lymph nodes in a mouse xenograft model[56]. Other work has also suggested acquired BRAF inhibitor resistance leads to a more aggressive metastatic phenotype; this time associated with increased expression of cancer cell markers (JARID1B, CD271, fibronectin) and invasion (MMP-1 and MMP-2). Phenotypically, the resistant cells showed an increased rate of migration through confluent layers of lung endothelial cells [63, 85].
MITF has long been implicated in melanoma cell invasion and metastasis, with loss of MITF expression increasing invasive potential [72]. Recent studies have pointed to a role for MITF downregulation in the context of BRAF inhibitor adaptation as the driver of a metastatic phenotype[77]. In melanoma cell lines that initially expressed E-cadherin, loss of MITF and acquisition of BRAF inhibitor resistance was associated with a decrease in E-cadherin at both the mRNA and protein level. Heterogeneous effects were also seen with regards to expression of the EMT-associated transcription factors ZEB1, ZEB2 and Twist [77]. Another transcription factor, FRA-1, which has also been implicated in the development of breast cancer metastasis was also upregulated in some of the resistant cultures in which MITF was suppressed. A role for FRA-1 in the invasive phenotype was demonstrated through shRNA studies with knockdown reversing the invasive phenotype associated with MITF loss [77].
There are likely to be multiple upstream regulators of MITF expression in the context of BRAF inhibitor inhibition. Recent studies have pointed to a role for non-canonical WNT signaling (via Wnt5A) in both the drug adapted phenotype and melanoma cell invasion[62, 86]. The effects of Wnt5A signaling were found to be mediated in part through regulation of MITF, with high Wnt5A expression levels correlated with reduced expression of both MITF and its downstream target MART1 (Figure 2). Increased levels of Wnt5A induced a slow-growing senescence-like phenotype that showed increased invasion. The Wnt5A-mediated phenotypic switch was associated with high p21 expression levels, heterochromatin foci and the adoption a secretory phenotype characterized by increased expression of IL-1A, CXCL2, CCL20 and MMP-3 [62]. Wnt5A is a known EMT inducer in many cancers, in part through its stimulation of PKC signaling and the induction of invasion through Rac1 [87, 88]. In line with the observation that cells undergoing an EMT are less differentiated it was also found that Wnt5A treatment decreased the expression of melanoma antigens, leading to impaired recognition by and activation of cytotoxic T-lymphocytes [89].
Although much work on the link between drug induced phenotype switching and metastatic behavior has focused on BRAF inhibitors, there is evidence that other anti-cancer agents lead to the adoption of similar cellular states. Recent studies have shown that PI3K inhibition in glioblastoma leads to a transcriptional reprogramming that led to increased RTK signaling, reactivation of AKT and mTOR signaling and increased tumor invasion and motility[90]. The invasive phenotype was associated with the appearance of persistent membrane ruffles and was mediated through a novel mechanism that was dependent upon the formation of elongated mitochondria that co-localized with the cortical cytoskeleton and the FAK-containing membrane protrusions[90]. The adoption of a persister, metastatic phenotype is common to many cancer types with increased cell invasion being observed in NSCLC cells treated with EGFR inhibitors and breast cancer cell lines with resistance to pan-HER kinase inhibitors[91, 92].
Despite there being ample evidence that targeted therapies increase metastatic potential in vitro and in animal xenograft studies, there is only limited evidence that this occurs in the clinic. This is in part due to difficulties associated with performing equivalent retrospective analyses of contemporary targeted therapy trials vs historical cohorts of patients on cytotoxic chemotherapy. There is however some suggestion that melanoma patients failing BRAF inhibitor therapy often present with new metastases, with studies showing 50% patients on BRAF inhibitor to progress at new sites, 44% at existing sites and 6% at both new and existing sites [93]. A further analysis showed 68% of patients failing vemurafenib therapy to relapse at new sites compared to only 20% of patients failing dacarbazine, despite the time on therapy being similar [84]. Further prospective analyses will be required to determine whether drug resistance leads to the development of new metastases.
6. Conclusion: Targeting and eradicating the escaper phenotypes through combination therapy strategies
The achievement of long-term clinical responses to chemotherapy and targeted therapy depends upon the successful targeting of adaptive phenotypes. These adaptations are likely to apply to both targeted therapies (as outlined here) and to immune therapies. There is already good evidence that failure on targeted therapy reduces the effectiveness of subsequent immune therapies, and this may be in part due to the adoption of a less differentiated (less immunogenic) phenotype [94]. One unifying idea is that the phenotypic adaptations to therapy are epigenetically mediated and various strategies have been proposed to overcome this. Some of the earliest work in this area demonstrated that the persister phenotype was driven through IGFR signaling and showed a dependency upon an epigenetic reprogramming mediated through the histone demethylase RBP2/KDM5A/Jarid1A [41] (Table 1). In these instances, pan-HDAC inhibitors or selective IGF1R inhibitors suppressed the persister phenotype and prevented the onset of resistance [41]. A similar epigenetic state also exists in melanoma, with studies identifying the presence of minor populations of slowly proliferating cells that undergo expansion following treatment with BRAF inhibitors and cytotoxic chemotherapy. These cells, which are characterized by the expression of the H3K4 histone demethylase JARID1B, show a marked rearrangement of their cell metabolism characterized by an increased dependency upon oxidative phosphorylation [42]. In this instance, the altered metabolic state represented a therapeutic vulnerability and could be effectively targeted with inhibitors of the mitochondrial respiratory chain [42] (Table 1). Work by a number of other groups has also suggested that persister cells have an altered metabolic state that could be targeted pharmacologically [95–97]. One drawback of metabolic inhibitors is their very narrow therapeutic index, potentially limiting their future translational potential.
Table 1.
Potential therapeutic strategies for the reversal of the drug-induced adaptive/persister phenotype
| Phenotype | Molecular target | Cancer type | Drug class | Compound | Ref |
|---|---|---|---|---|---|
| Epigenetic switching | JARID1A | Multiple | HDAC inhibitors | SAHA TSA IGF1R |
[41] |
| Epigenetic switching | JARID1B | Melanoma | Metabolic inhibitors | Oligomycin | [42] |
| Impaired apoptosis | Bcl-XL/Bcl-2 | NSCLC | BH3 antagonist | Navitoclax | [43] |
| Impaired apoptosis | BIM BCL-2 BMF |
Melanoma | HDAC inhibitors | SAHA | [99] |
| BET domain inhibitors | JQ-1 | [100] | |||
| BH3 antagonist | Navitoclax | [98] | |||
| Phenotypic switching | PAX3 MITF |
Melanoma | HIV protease inhibitor | Nelfinavir | [78] |
| cAMP/MITF/SOX10 | HDAC inhibitors | [79] | |||
| Adaptive phenotype | Axl | Melanoma | RTK inhibitor | R428 | [77] |
| NSCLC | [80] | ||||
| Breast Cancer | [81] |
There is evidence that the adapted phenotypic state confers secondary resistance that limits their response to drugs targeted against actionable genetic mutations. In the case of NSCLC, the persister/adapted cells show a reduced sensitivity to third generation EGFR inhibitors, even when they express the T790M EGFR mutant [43]. The observation that these cells show a reduced sensitivity to EGFR inhibitors, in particular through the suppression of BIM-mediated apoptosis, suggested an impairment of the entire apoptotic program. A small-scale drug screen performed on NSCLC cells adapted to gefitinib revealed sensitivity to the dual Bcl-2/Bcl-XL inhibitor navitoclax [43] (Table 1). Preclinical studies showed that the combination of the third-generation EGFR inhibitor WZ4002 with navitoclax restored sensitivity to xenografts of NSCLC cells with the T790M mutation that had undergone therapeutic adaptation [43]. Similar findings have also been reported in melanoma with the combination of a BRAF inhibitor and a BH3 antagonist showing enhanced efficacy compared to single agent BRAF inhibitor [98]. Epigenetic silencing of the pro-apoptotic BH3 family proteins Bcl-2, BMF and BIM is seen in melanoma cells following chronic BRAF inhibitor treatment [99]. In this instance the epigenetic suppression was reversed following treatment with HDAC inhibitors leading to the restoration of BRAF inhibitor sensitivity [99]. Other epigenetic modulators such as BET domain inhibitors are also known to synergize with BRAF inhibitors, potentially through regulating levels of pro-apoptotic proteins [100].
In melanoma cells, where the EMT-like, drug tolerant state is driven through MITF, tolerance to BRAF inhibitors can be reversed through use of the HIV protease inhibitor nelfinavir [78]. Mechanistically, nelfinavir decreases MITF expression through suppression of the transcriptional activator PAX3. In cells of melanocytic lineage, PAX3 expression is controlled by the transcriptional co-repressor SKI which is in turn regulated by TGF-β signaling and SMAD2 (Figure 2). As melanomas frequently show constitutive levels of TGF-β signaling, levels of SMAD2 are typically high [78]. Use of nelfinavir increased levels of SMAD2/SMAD4 and led to the recruitment of SKI to the PAX3 promoter and the suppression of expression of both PAX3 and MITF (Table 1). It was thus found that the combination of the BRAF inhibitor and nelfinavir suppressed phenotypic adaptation and abrogated the onset of resistance [78]. Other strategies have been proposed to target MITF including the use of methotrexate (to increase MITF levels and reduce invasion) following by the use of a tyrosinase processed anti-folate drug (3-O(3,4,5 trimethoxybenzoyl)-(−)-epicatechin to deplete intracellular thymidine pools and to induce DNA strand breaks[101]. MITF levels can also be regulated epigenetically and are sensitive to HDAC inhibition. The lineage-dependent resistant state mediated through GPCR signaling/ PKA/cAMP/MITF resistance was reversed following treatment with pan-HDAC inhibitors, indicating that this escape pathway was also epigenetically regulated [79, 102] (Figure 2).
Oncogenically transformed cells show a high level of signaling and phenotypic plasticity and can rapidly adapt their cellular state following treatment with drugs. The evidence to date suggests that these persister or adapted phenotypes are epigenetically mediated and are associated with an altered transcriptional state. This opens up numerous therapeutic opportunities with a large number of epigenetically targeted drugs becoming available. Specific therapeutic approaches directed against individual tumor histologies also seem likely. Potential options here include strategies to target MITF expression in melanoma, and approaches to inhibit BRAF/BH3 proteins and EGFR/BH3 proteins in melanoma and NSCLC, respectively. The feasibility of targeting RTKs such as Axl remains uncertain as cancer cells typically express multiple RTKs with overlapping functions. It is likely that the targeting of these adaptive phenotypes could have the added benefit of preventing further tumor dissemination on therapy. Unified approaches to abrogate therapy escape and metastasis will continue to improve outcomes for cancer patients, hopefully one day reducing cancer to the level of a chronic, manageable disease.
Acknowledgments
This work was supported by grants from the National Institutes of Health, R01 CA161107, R21 CA198550 and SPORE grant CA168536.
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mukherjee S. The emperor of all maladies: a biography of cancer. New York: Scribner; 2010. [Google Scholar]
- 2.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 3.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida T, Zhang G, Haura EB. Targeting epidermal growth factor receptor: central signaling kinase in lung cancer. Biochem Pharmacol. 2010;80:613–23. doi: 10.1016/j.bcp.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 5.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF Mutations in Hairy-Cell Leukemia. New Engl J Med. 2011;364:2305–15. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–30. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 9.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 10.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011 doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hay ED. An overview of epithelio-mesenchymal transformation. Acta anatomica. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 14.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature cell biology. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 16.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 17.Zheng M, Jiang YP, Chen W, Li KD, Liu X, Gao SY, et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget. 2015;6:6797–810. doi: 10.18632/oncotarget.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell stem cell. 2015;16:67–79. doi: 10.1016/j.stem.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Molecular cell. 2001;7:1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 20.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO reports. 2008;9:582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cellcell junctions. Nucleic acids research. 2005;33:6566–78. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Sun J, Cai B, Xi X, Yang L, Zhang Z, et al. NANOG regulates epithelial-mesenchymal transition and chemoresistance through activation of the STAT3 pathway in epithelial ovarian cancer. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016 doi: 10.1007/s13277-016-4848-x. [DOI] [PubMed] [Google Scholar]
- 23.Sun C, Sun L, Jiang K, Gao DM, Kang XN, Wang C, et al. NANOG promotes liver cancer cell invasion by inducing epithelial-mesenchymal transition through NODAL/SMAD3 signaling pathway. The international journal of biochemistry & cell biology. 2013;45:1099–108. doi: 10.1016/j.biocel.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Liu J, Ying X, Lin PC, Zhou BP. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Scientific reports. 2016;6:24606. doi: 10.1038/srep24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Current opinion in oncology. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 26.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes & development. 2005;19:2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 28.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nature reviews Molecular cell biology. 2003;4:657–65. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 29.Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells, tissues, organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. Journal of clinical medicine. 2016:5. doi: 10.3390/jcm5020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer cell. 2009;15:416–28. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer research. 2012;72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, et al. TNF-alpha induces epithelial-mesenchymal transition of renal cell carcinoma cells via a GSK3beta-dependent mechanism. Molecular cancer research : MCR. 2012;10:1109–19. doi: 10.1158/1541-7786.MCR-12-0160. [DOI] [PubMed] [Google Scholar]
- 35.Boyer B, Valles AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochemical pharmacology. 2000;60:1091–9. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 36.Bates RC, Mercurio AM. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer biology & therapy. 2005;4:365–70. doi: 10.4161/cbt.4.4.1655. [DOI] [PubMed] [Google Scholar]
- 37.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–60. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y, Elble RC. Homeostatic Signaling by Cell-Cell Junctions and Its Dysregulation during Cancer Progression. Journal of clinical medicine. 2016:5. doi: 10.3390/jcm5020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–6. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 40.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B(high) Cells. Cancer Cell. 2013;23:811–25. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–9. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7:10690. doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–4. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer research. 2007;67:1979–87. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- 47.Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P, et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012;31:3223–34. doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKeithen D, Graham T, Chung LW, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. The Prostate. 2010;70:982–92. doi: 10.1002/pros.21132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem cells. 2009;27:2059–68. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- 50.Rebecca VW, Sondak VK, Smalley KSM. A brief history of melanoma: from mummies to mutations. Melanoma Research. 2012;22:114–22. doi: 10.1097/CMR.0b013e328351fa4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 52.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011 doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82:201–9. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–30. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, et al. Inhibiting EGF Receptor or SRC Family Kinase Signaling Overcomes BRAF Inhibitor Resistance in Melanoma. Cancer Discov. 2013;3:158–67. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abel EV, Basile KJ, Kugel CH, 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. The Journal of clinical investigation. 2013;123:2155–68. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fedorenko IV, Wargo JA, Flaherty KT, Messina JL, Smalley KS. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J Invest Dermatol. 2015;135:3115–24. doi: 10.1038/jid.2015.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capparelli C, Rosenbaum S, Berger AC, Aplin AE. Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J Biol Chem. 2015;290:24267–77. doi: 10.1074/jbc.M115.657270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 62.Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment cell & melanoma research. 2015;28:184–95. doi: 10.1111/pcmr.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravindran Menon D, Das S, Krepler C, Vultur A, Rinner B, Schauer S, et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene. 2015;34:4448–59. doi: 10.1038/onc.2014.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fedorenko IV, Abel EV, Koomen JM, Fang B, Wood ER, Chen YA, et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene. 2016;35:1225–35. doi: 10.1038/onc.2015.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu S, Tetzlaff MT, Wang T, Yang R, Xie L, Zhang G, et al. miR-200c/Bmi1 axis and epithelial-mesenchymal transition contribute to acquired resistance to BRAF inhibitor treatment. Pigment cell & melanoma research. 2015;28:431–41. doi: 10.1111/pcmr.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoshida T, Song L, Bai Y, Kinose F, Li J, Ohaegbulam KC, et al. ZEB1 Mediates Acquired Resistance to the Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PloS one. 2016;11:e0147344. doi: 10.1371/journal.pone.0147344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paraiso KH, Smalley KS. Fibroblast-mediated drug resistance in cancer. Biochemical pharmacology. 2013;85:1033–41. doi: 10.1016/j.bcp.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H, Liu J, Yue D, Gao L, Wang D, Zhang H, et al. Clinical significance of E-cadherin, beta-catenin, vimentin and S100A4 expression in completely resected squamous cell lung carcinoma. Journal of clinical pathology. 2013;66:937–45. doi: 10.1136/jclinpath-2013-201467. [DOI] [PubMed] [Google Scholar]
- 69.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 70.Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, et al. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–9. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- 71.Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, et al. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30:2307–18. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- 72.Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–39. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pierrat MJ, Marsaud V, Mauviel A, Javelaud D. Expression of microphthalmia-associated transcription factor (MITF), which is critical for melanoma progression, is inhibited by both transcription factor GLI2 and transforming growth factor-beta. The Journal of biological chemistry. 2012;287:17996–8004. doi: 10.1074/jbc.M112.358341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perrot CY, Gilbert C, Marsaud V, Postigo A, Javelaud D, Mauviel A. GLI2 cooperates with ZEB1 for transcriptional repression of CDH1 expression in human melanoma cells. Pigment cell & melanoma research. 2013;26:861–73. doi: 10.1111/pcmr.12149. [DOI] [PubMed] [Google Scholar]
- 75.Goding CR. Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000;14:1712–28. [PubMed] [Google Scholar]
- 76.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. 2014;5:5712. doi: 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell. 2016;29:270–84. doi: 10.1016/j.ccell.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–42. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robert L, Tsoi J, Garcia-Diaz A, Homet-Moreno B, Truong N, Graeber TG, et al. Melanoma phenotype switching to adapt to BRAF inhibition. Cancer Research. 2015;75 Abstract 2682. [Google Scholar]
- 83.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–49. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paraiso KH, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer discovery. 2015;5:264–73. doi: 10.1158/2159-8290.CD-14-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zubrilov I, Sagi-Assif O, Izraely S, Meshel T, Ben-Menahem S, Ginat R, et al. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer letters. 2015;361:86–96. doi: 10.1016/j.canlet.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 86.O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer discovery. 2013;3:1378–93. doi: 10.1158/2159-8290.CD-13-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. 2007;282:17259–71. doi: 10.1074/jbc.M700075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T, et al. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006;66:10439–48. doi: 10.1158/0008-5472.CAN-06-2359. [DOI] [PubMed] [Google Scholar]
- 89.Dissanayake SK, Olkhanud PB, O’Connell MP, Carter A, French AD, Camilli TC, et al. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008;68:10205–14. doi: 10.1158/0008-5472.CAN-08-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8638–43. doi: 10.1073/pnas.1500722112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chang TH, Tsai MF, Su KY, Wu SG, Huang CP, Yu SL, et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. American journal of respiratory and critical care medicine. 2011;183:1071–9. doi: 10.1164/rccm.201009-1440OC. [DOI] [PubMed] [Google Scholar]
- 92.Creedon H, Gomez-Cuadrado L, Tarnauskaite Z, Balla J, Canel M, MacLeod KG, et al. Identification of novel pathways linking epithelial-to-mesenchymal transition with resistance to HER2-targeted therapy. Oncotarget. 2016;7:11539–52. doi: 10.18632/oncotarget.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Menzies AM, Haydu LE, Carlino MS, Azer MW, Carr PJ, Kefford RF, et al. Inter-and intra-patient heterogeneity of response and progression to targeted therapy in metastatic melanoma. PLoS ONE. 2014;9:e85004. doi: 10.1371/journal.pone.0085004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cooper ZA, Reuben A, Spencer CN, Prieto PA, Austin-Breneman JL, Jiang H, et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. Oncoimmunology. 2016;5:e1136044. doi: 10.1080/2162402X.2015.1136044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–15. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gopal YNV, Rizos H, Chen G, Deng WL, Frederick DT, Cooper ZA, et al. Inhibition of mTORC1/2 Overcomes Resistance to MAPK Pathway Inhibitors Mediated by PGC1 alpha and Oxidative Phosphorylation in Melanoma. Cancer Research. 2014;74:7037–47. doi: 10.1158/0008-5472.CAN-14-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, et al. Response of BRAF-Mutant Melanoma to BRAF Inhibition Is Mediated by a Network of Transcriptional Regulators of Glycolysis. Cancer Discov. 2014;4:423–33. doi: 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frederick DT, Salas Fragomeni RA, Schalck A, Ferreiro-Neira I, Hoff T, Cooper ZA, et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS One. 2014;9:e101286. doi: 10.1371/journal.pone.0101286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shao Y, Aplin AE. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012 doi: 10.1038/cdd.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paoluzzi L, Hanniford D, Sokolova E, Osman I, Darvishian F, Wang J, et al. BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med. 2016 doi: 10.1002/cam4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saez-Ayala M, Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, Chazarra S, Freter R, et al. Directed phenotype switching as an effective antimelanoma strategy. Cancer cell. 2013;24:105–19. doi: 10.1016/j.ccr.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 102.Yokoyama S, Feige E, Poling LL, Levy C, Widlund HR, Khaled M, et al. Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment Cell Melanoma Res. 2008;21:457–63. doi: 10.1111/j.1755-148X.2008.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]