

Abstract
Clostridium difficile infection (CDI) is a significant healthcare concern worldwide, and C. difficile is recognised as the most frequent aetiological agent of infectious healthcare-associated diarrhoea in hospitalised adult patients. The clinical manifestation of CDI varies from self-limited diarrhoea to life-threatening colitis. Such a broad disease spectrum can be explained by the impact of host factors. Currently, a complex CDI aetiology is widely accepted, acknowledging the interaction between bacteria and the host. Clostridium difficile strains producing clostridial toxins A and B are considered toxigenic and can cause disease; those not producing the toxins are non-pathogenic. A person colonised with a toxigenic strain will not necessarily develop CDI. It is imperative to recognise patients with active disease from those only colonised with this pathogen and to implement appropriate treatment. This can be achieved by diagnostics that rely on host factors specific to CDI. This review will focus on major aspects of CDI pathogenesis and molecular mechanisms, describing host factors in disease progression and assessment of the host response in order to facilitate the development of CDI-specific diagnostics.
Keywords: Clostridium difficile infection, CDI, Pathogenesis, Host response, Biomarker, Diagnostics
Graphical Abstract
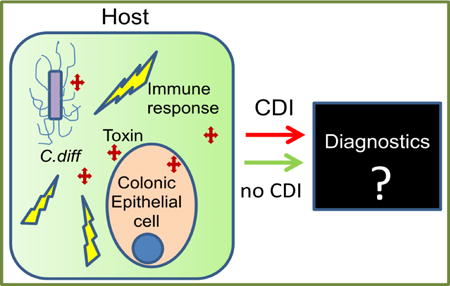
1. Introduction
Clostridium difficile infection (CDI) results from infection of the bowel by C. difficile, a Gram-positive, spore-forming, obligate anaerobic bacterium. Complications of CDI include severe infection, hypotension, shock and sepsis, ileus, megacolon and perforation, or death secondary to CDI [1,2]. Traditional risk factors for CDI include age >65 years, recent hospitalisation, increased length of hospital stay, long-term healthcare facility residence and antibiotic exposure [3–5]. However, only 25% of all cases of antibiotic-associated diarrhoea are associated with CDI [6]. This infectious disease is presently diagnosed at a rate of several hundred thousand cases per year in the USA [7,8]. It has been estimated that for every additional year of age after age 18 years, the risk of healthcare-associated CDI increases by ca. 2% [5].
For the last two decades, CDI has re-emerged in healthcare facilities with nearly a 10-fold increase in mortality [9,10]. It is now rivalling Staphylococcus aureus and vancomycin-resistant enterococci as a cause of nosocomial infection [11]. In the USA, C. difficile was associated with ca. 29 000 deaths in 2011 [4]. In addition to the morbidity and mortality associated with CDI, the US healthcare system expends considerable financial resources for care of this disease [12].
Traditionally, CDI has been considered as a hospital-acquired disease. Currently, however, only 20–25% of all CDI represents disease associated with healthcare exposure [12]. Recent epidemiology suggests the emergence of CDI into new populations having virtually no contact with healthcare settings, including healthy adults, children, pregnant women and patients who have not been subjected to antibiotic therapy [3,13,14]. An additional main challenge in CDI management is the rate of disease recurrence of 15–30% [15].
Critically, there is no specific diagnostic test for CDI. The current diagnostic strategy relies on combining clinical symptoms and signs (such as frequency of diarrhoea, antibiotic exposure and elevated white blood cell count) with a positive diagnostic test for toxigenic C. difficile. Remarkably, this organism can colonise 10 times as many patients asymptomatically than actually develop infection [16]. Combining a high colonisation rate with the fact that 20% of hospitalised patients can have diarrhoea from any cause [17–21] makes it imperative to develop CDI-specific diagnostics that goes beyond simply detecting C. difficile toxin(s)/gene(s). Here we briefly review the current state of understanding of the molecular bases of CDI and efforts to develop a CDI-specific test.
2. Clostridium difficile infection and the host microbiome
CDI is characterised by severe alterations in the normal colonic bacterial flora [22]. The human colon is a complex and diverse ecosystem lined by a mucous membrane [23]. It has been postulated that the normal colonic microbiome provides some degree of protection against pathogenic organisms. The mechanism of this protection is incompletely understood but it has been described as ‘colonisation resistance’, with a healthy microbiome making it more difficult for C. difficile to colonise and infect the colon [24].
One of the mechanisms is competition for essential nutrients and attachment sites to the gut wall [25]. The other mechanism refers to bacterial populations within the gastrointestinal tract existing in two forms—free-floating cells and sessile bacteria within mucosa-associated biofilm communities. The sessile bacteria are often comprised of multispecies populations [23,26]. Mucosal communities interact closely with host epithelial cells and may have a greater influence on disease pathogenesis. Alteration of the normal gut microflora or colon microbial diversity (by antibiotic therapy) decreases the level of protection and represents the main risk factor for CDI [21,27]. However, not all classes of antimicrobials an carry equal risk for CDI predisposition [6].
Typically, disease onset occurs 4–9 days after the beginning of antimicrobial treatment [28]. Antibiotics having the highest CDI risk include clindamycin, cephalosporins, penicillins and, more recently, fluoroquinolones. Their frequent usage can increase cumulative antibiotic exposure over time leading to alteration of the indigenous gut microbiota, therefore providing a possibility of C. difficile colonisation and subsequent disease [6,18,29].
There is a belief that failure to restore the normally diverse microbial intestinal community may be related to disease recurrence in patients recovering from CDI [30,31]. Interestingly, a small study showed no difference between the faecal microbiota of asymptomatic C. difficile carriers and healthy subjects, but lower bacterial diversity in patients with C. difficile-related diarrhoea [32]. Animal studies demonstrated that certain bacterial species are present at low frequencies under gut homeostasis but they become more frequent and exhibit many opportunistic properties during dysbiosis [33].
3. Colonisation
Carriage of toxigenic C. difficile in asymptomatic patients is increasingly common. Numerous reports demonstrated a prevalence of asymptomatic C. difficile colonisation during hospital admission as high as 10–15% [19,34–37]. One explanation for such high rates compared with earlier reports is application of more sensitive methods for detection.
Clearly, there are some patient populations (infants and the elderly) prone to high rates of C. difficile colonisation [10]. Thus, C. difficile is a frequent component of the faecal microbiota of newborn infants not causing disease [38]. Clostridium difficile colonisation in children is common but severe infection and death are much less frequent than in adults [39]. This might be explained by the fact that in infants the microbiota is insufficiently developed and colonisation resistance is not yet established [40,41]. In one study, the microbial flora composition of infants colonised with C. difficile had increased frequencies of Klebsiella pneumoniae, Ruminococcus gnavus and Clostridium nexile [40]. Their healthy non-colonised counterparts exhibited higher levels of Staphylococcus epidermidis, Escherichia coli and Bifidobacterium longum. It was suggested that specific microflora composition may promote C. difficile colonisation. Interestingly, the study reported that infants were colonised with a single clone of C. difficile for several months [40], but the clone could change as new infant food is introduced.
High colonisation rates of 10–50% were also reported in elderly institutionalised adults [42]. Such observations may reflect the comprehensive influence of host factors such as age, co-morbidity, co-administered medications and functional status for disease severity [43].
4. Pathogenesis of Clostridium difficile infection
Clostridium difficile toxin(s) can cause disease within 1 day of the inciting event, usually initial antibiotic therapy, and for up to 2 months after discontinuation of treatment [18,29]. Clostridium difficile spores, the main mode of transmission due to strict anaerobic requirements of the organism, must interact with host epithelial tissue, germinate following interactions with small molecule germinants resulting in vegetative growth of the pathogen, and produce toxin(s) during the cycle of sporulation of a new bacterial generation [44] (Fig. 1). Toxin production and secretion increases after vegetative cells enter into the stationary growth phase [33,39].
Fig. 1.

Complexity of the host response to Clostridium difficile infection (CDI). (A) Intoxication of host epithelial cells by C. difficile toxins produced by vegetative cells is primary to inflammatory response. Toxin production and secretion increases after vegetative cells enter into the stationary growth phase [22,39]. During cell infection, the toxins are subjects to time-dependent degradation due to proteolysis and pH effects. The toxin’s entry into the intestinal epithelial cells is one of the earliest pathogenic events. It leads to loss of structural integrity (actin skeleton disruption, disruption of tight junctions, reduced cell–cell contact), cell death and epithelium disruption. TLR, Toll-like receptor. (B) Inflammatory response by two mechanisms: (i) secondary to toxin intoxication (within a few hours after toxin exposure); and (ii) activation of intracellular cascades by non-toxin virulence factors such as surface layer proteins (SLPs), flagellar proteins (FliC and FliD), adhesins (cwp66, cwpV), fibronectin-binding proteins and cell surface polysaccharides [22]. (C) CDI clinical manifestation. The inflammatory response causes tissue damage: neutrophil accumulation (one of the major mechanisms) is responsible for pseudomembrane formation seen in severe colitis [38]; diarrhoea, toxic megacolon. Toxin B can also cause multiple organ dysfunction syndrome due to systemic toxin damage [50,68].
A mouse model study demonstrated that during disease, CDI was localised in the large intestine and did not occur in the small intestine [45]. However, it was confirmed that germination of C. difficile spores occurred in the small intestine regardless of antibiotic pre-treatment. The cecum was determined as the site for optimal C. difficile growth, toxin production and disease after antibiotic treatment [45].
It appears that two interconnected events cause manifestation of clinical CDI [46]: (i) epithelial cell intoxication by C. difficile toxins (Fig. 1A); and (ii) inflammation-associated colon tissue damage (Fig. 1B). The latter, in turn, is simultaneously modulated by: (i) toxin–receptor interaction through activation of mucosal immune cells; and (ii) adhesion of C. difficile vegetative cells to the epithelium through non-toxin virulence factors.
4.1. Toxin aetiology of Clostridium difficile infection
The main C. difficile toxins are monoglucosyltransferases. Toxin A (TcdA) was classically considered an important enterotoxin but not essential for virulence, whereas toxin B (TcdB) had more potent cytotoxic activity. Whilst some studies re-established the importance of the both toxins suggesting a synergised effect between them [47–49], recent studies clearly indicated that TcdB was the major virulence factor of C. difficile and did not require the presence of TcdA [50,51].
At a clinical level, variant strains of TcdA-negative, TcdB-positive C. difficile were indistinguishable from strains producing both toxins. Patients infected with such strains exhibited the full spectrum of CDI symptoms [50]. Alternatively, variant toxins have alterations in their substrate specificity that may have an impact on disease severity and outcome [50]. To date there have been no confirmed C. difficile strains that solely produce TcdA known to cause CDI [52]. However, there is evidence that the prevalence of clinically relevant CDI cases due to TcdA-negative, TcdB-positive strains has increased globally [53,54].
During cell infection, the toxins are subject to time-dependent degradation due to proteolysis and pH effects [50]. They modify the activity of members of the host Rho family of small GTPases [55], key cellular regulatory proteins, leading to actin filament depolymerisation, cytoskeletal disruption and subsequent intestinal epithelial cell death (Fig. 1A) [56–58]. Recent histopathological analysis of caecal and colonic tissues collected from infected mice showed that TcdB caused the majority of intestinal damage during infection, with TcdA causing more superficial and localised damage [51].
Besides the well characterised TcdA and TcdB toxins, a binary toxin CDT has been identified [59,60]. CDT is an ADP-ribosyltransferase that has been shown to disrupt the cytoskeleton of the cell, leading to cell rounding, loss of fluids and cell death [61,62]. It is a suspected C. difficile virulence factor shown to be involved in the formation of long microtubule protrusions in the host cell, facilitating bacterial attachment [63]. CDT is produced by some but not all toxigenic C. difficile strains and, in contrast to TcdA and TcdB, plays a minor role in CDI [51]. The CDT-positive but TcdA- and TcdB-negative strains were avirulent in the hamster infection model [64]. The contribution of the binary toxin to human disease is still being elucidated.
4.2. Inflammatory response
An essential first step for CDI is colonisation of host mucosal surfaces [65]. The disease is characterised by tissue injury and an acute intestinal inflammatory response highlighted by neutrophil infiltration [66] (Fig. 1C). A study in mice demonstrated that the primary cellular immune response to toxin was oedema and polymorphonuclear cell infiltration [67]. The intense immune activation results in the endoscopic findings of the ‘volcano lesion’ and pseudomembranes.
Inflammation-associated tissue damage is thought to be secondary to the intoxication of intestinal epithelial cells in CDI pathogenesis [52,68]. Following breakdown of the intestinal epithelial barrier, immune cells within the mucosa are activated by TcdA and TcdB, eventually leading to the release of inflammatory mediators [pro-inflammatory cytokines interleukin-1β (IL-1β), tumour necrosis factor-α (TNFα) and IL-8 from activated macrophages] [52,69] (Fig. 1B). The central role of inflammation in CDI pathogenesis is highlighted by the fact that the magnitude of the inflammatory response is the best predictor of CDI poor outcome, but not the overall bacteria/toxin burden within the intestine [70].
Of importance is the host immune system, as evidenced by the higher rates of infection and worsening disease severity among the elderly and other persons who lack the ability to mount an effective humoral immune response [5,66]. Therefore, all current data on CDI are moving towards emphasising a link between bacteria–host cellular immune interaction, homeostasis and the gut microbiome [69].
5. Clostridium difficile infection diagnostics
There is renewed interest in the development and validation of clinical prediction tools for CDI. Currently, laboratory testing available for CDI identification include sigmoidoscopy and colonoscopy, toxigenic culture (TC), cell cytotoxicity assay (CCTA), enzyme immunoassay (EIA), glutamate dehydrogenase (GDH) EIA, as well as real-time PCR (rtPCR) and loop-mediated isothermal amplification (LAMP) assay, which is less costly but uncommon in the clinical laboratory environment [71]. Together, clinical prediction algorithms and reliable laboratory diagnostics will greatly facilitate early diagnosis. Yet a number of CDI studies demonstrated that host response determines the character of the intestinal inflammation and clinical severity.
5.1. Bacteria detection
Currently there is no ‘gold standard’ for the diagnosis of CDI, other than possible direct visualisation of characteristic lesions using colonoscopy [72]. The two laboratory reference methods most commonly cited are the stool CCTA, which detects the presence of ‘free’ C. difficile toxins, and TC, which detects C. difficile isolates producing toxins in vitro with the potential for producing toxins in vivo [20,73]. However, it must be noted that these methods only detect the presence of a disease-causing organism and when positive are not diagnostic of clinical infection.
Of note, a recent large multicentre study of CDI reported CCTA as the best diagnostic indicator for CDI disease [20,22], however the sensitivity of this test can be as low as 50% when TC is used as the reference method [74]. CCTA appears to have a sensitivity of only 89% in cases where pseudomembranes are seen on colonoscopy [29]. A positive TC may still indicate a patient who is an infection risk to others [20]. Yet it should be clear that the current diagnosis requires clinical judgement along with the positive result of a diagnostic test. Also, TC and CCTA are laborious, time consuming, and suffer from suboptimal specificity and/or sensitivity [38,75].
Newer rapid nucleic acid amplification tests (NAATs) formatted in rtPCR and LAMP for detection of TcdB and TcdA genes offer improved sensitivity over immunoassays [76,77]. However, concerns relate to the biology of C. difficile and how detection of the genes correlates with expression of the toxins [38]. NAATs yield more positives than CCTA [20]. A number of studies confirmed that molecular detection of CDI is very sensitive although less specific, therefore leading to overdiagnosis of CDI [74,78].
Two meta-analyses of the performance of rtPCR reported its high sensitivity and specificity, however they were highly dependent on CDI prevalence [71,79]. According to Deshpande et al., the negative predictive value (NPV) of rtPCR was acceptable at a C. difficile prevalence of <10% [79]. This suggests the assay may serve as an effective screening test in endemic situations, with emphasis on a negative test result. However, Lloyd et al. reported the possibility that the LAMP assay may be more sensitive and specific than rtPCR [80]. The positive predictive value (PPV) and NPV for LAMP were better than rtPCR in settings where the CDI prevalence was <15%. It was suggested that rtPCR may be more suitable in epidemic conditions with a higher prevalence, and LAMP for settings with a lower C. difficile prevalence [80]. Nevertheless, the diagnosis of CDI either with rtPCR or LAMP should only be made in the presence of strong clinical symptoms consistent with CDI. This is highlighted by the fact that stool PCR tests remain positive for C. difficile for up to 30 days after successful treatment [81]. Whilst a negative result is adequate to rule out the presence of the disease [71], a positive stool test does not distinguish colonised patients from those with symptomatic disease [82].
Currently, 13 commercial NAATs have been approved by the US Food and Drug Administration (FDA) [83]. Some of these use PCR techniques, whilst the others utilise LAMP or helicase-dependent amplification method for detection of C. difficile toxins or regulatory genes [77]. The TcdB gene is usually chosen as a NAAT target since it is produced by almost all toxigenic C. difficile strains [60]. The TcdA gene is less frequently used because ca. 3% of European toxigenic strains have been reported to be TcdA-negative [84]. A higher prevalence of such strains has been reported throughout Asia [54,77]. However, a discussion of the NAATs for C. difficile target detection is beyond the scope of this review.
The NAATs fail to discriminate between CDI and C. difficile asymptomatic colonisation. Therefore, colonised individuals may test falsely positive for CDI when evaluated for community-acquired diarrhoea caused by other enteric pathogens [16]. Also, the emergence of new C. difficile strains with altered toxins or genes can impact all currently existing CDI diagnostics [85]. This highlights the need for a test beyond simply detecting toxin(s) or gene(s). Of note, there is great variation between studies in the characteristics of C. difficile tests, suggesting possible, yet unidentified, human-related or microbe-related factors affecting test performance [20]. Therefore, it is invaluable to develop CDI diagnostics that include a biomarker correlated with active infection [38,82], and the most effective would be the one that relies on specific human response to CDI.
5.2. Host response
There is growing evidence demonstrating the contribution of the host response (immune and inflammatory) to CDI outcome. Yet to date an assay for CDI host response has not been established, as well as an optimal assay to supplant or be used with other rapid testing.
5.2.1. Biological markers
As noted earlier, the presence of C. difficile toxins stimulates a multifaceted immune response involving cytokines, chemokines and mucosal immune cells [65], eventually activating antibody production. The levels of such antibodies in serum or stool could be potential CDI biomarkers, however study results to date are complex.
Approximately 60% of healthy older children and adults have detectable serum immunoglobulin G (IgG) and IgA antibodies to TcdA and TcdB even in the absence of C. difficile colonisation or active infection [66]. The level of anti-TcdA and anti-TcdB antibodies has been considered important in determining whether colonisation or clinical infection follows C. difficile spore acquisition [86]. Reduced anti-TcdA levels at the start of infection have been linked both to recurrence and increased 30-day mortality. However, Loo et al. did not find a significant association between levels of antibodies against TcdA and TcdB at the time of admission and subsequent healthcare-associated CDI [5,35].
The surface layer proteins (SLPs) are the outermost protein component of C. difficile responsible for adhesion to host tissue, known to be variable between strains, and may play an important role in intestinal colonisation and the persistence of CDI [65]. A study revealed that the levels of antibody to SLPs were similar in patients with CDI, asymptomatic carriers and controls [87]. In another study, IgG levels to SLPs were similar in cases and carriers but were higher compared with controls [88]. Of note, it has been shown that strains that adhere better to human intestinal cell lines proved to be more virulent in hamsters [89].
Concentrations of a systemic inflammation biomarker (C-reactive protein), white blood cells, serum creatinine, serum 25-hydroxyvitamin D and albumin have been also reported to be associated with CDI severity and possible independent mortality predictors [90–92]. However, these markers are non-specific to C. difficile disease [39,92].
It has been suggested that C. difficile is initially recognised in a Toll-like receptor 4- and MyD88-dependent manner, resulting in low serum expression of IL-23 [46]. Intoxication of primed host cells by TcdA and TcdB leads to robust IL-1β secretion that further enhances IL-23 production. However, increased serum IL-1β was also noted in patients with diarrhoea from other causes [46]. Therefore, these do not appear to be specific for CDI.
Procalcitonin, a biomarker for bacterial infection, was evaluated in association with CDI severity [93]. In this small study, serum procalcitonin levels did show some promise as a biomarker for CDI severity. Further studies in a larger cohort need to be done.
5.2.2. Faecal biomarkers
Faecal proteins are ideal biomarkers for gastrointestinal inflammation owing to direct contact with the intestinal mucosa (containing a large number of neutrophils) and the non-invasive sampling mode [94]. Faecal markers include a biologically heterogeneous group of substances that either leak from or are actively released by the inflamed mucosa [95]. Over the last several decades, a few faecal inflammation biomarkers have been investigated. Some of these markers have been shown to be produced in response to C. difficile toxins and therefore have been investigated as predictors of CDI disease severity (Table 1).
Table 1.
Summary of faecal inflammatory biomarkers as possible predictors of Clostridium difficile infection (CDI) disease
| Biomarker | Clinical indication/prediction | Role in immunopathogenesis | Specific/sensitive to CDI | References |
|---|---|---|---|---|
| Lactoferrin | Colonic inflammation, CDI severity (when level is elevated) | Innate inflammatory response; related to level of neutrophil translocation | No/no | [97,99–102,107] |
| Calprotectin | Intestinal inflammatory conditions (when level is elevated) | Innate inflammatory response; correlates with level of released neutrophils | No/no | [103,105,106] |
| IL-8 | CDI severity (when elevated) | Involved in the recruitment of neutrophils to sites of infection | no/yes | [73,99,109,107] |
| IL-23 | May relate to CDI recurrence (when level is decreased) | Lack of a robust immunological response | No/no | [109] |
| pMK2 | Presence of toxigenic C. difficile (when level is elevated) | Key mediator of p38-dependent inflammation | No/– | [107] |
| pp38 | Symptomatic CDI in paediatrics (when level is elevated) | Activation of p38 protein pathway | Yes/no | [99] |
A few publications have indicated that levels of faecal cytokines could be indicators of CDI severity. El Feghaly reported that the cytokines CXCL-5 and IL-8 as well as lactoferrin (LF) were more sensitive in identifying patients at risk than clinical parameter or score [70,96], but they were not specific for disease caused solely by C. difficile. The conclusion in children was that faecal inflammatory cytokines differentiate asymptomatic C. difficile colonisation from disease and are associated with CDI poor outcome [96]. To date, however, there are no commercial assays available for measurement of cytokines in stool.
5.2.2.1. Lactoferrin (LF)
Multiple studies have validated faecal LF measurement as an accurate marker of intestinal inflammation and its usefulness for inflammatory diarrhoea [94,97–99]. This glycoprotein is resistant to proteolysis, is not degraded by C. difficile toxins, and is released following neutrophil activation. LF concentrations in stool and other fluids are proportional to the number of neutrophils recruited [98,100]. Van Langerberg et al. showed that a normal LF level effectively excludes inflammatory diarrhoea, therefore it was proposed as a screening test prior to microbiological assessment of faeces [98]. Faecal LF is not useful in discriminating between inflammatory diarrhoea caused by bacterial infection and non-inflammatory diarrhoea in children as well. A study of 1000 stools submitted for diagnostic testing found the sensitivity and specificity of LF detection to be only 75% and 60%, respectively [85], indicating that it is not a useful test for clinical evaluation of patients with potential CDI.
5.2.2.2. Calprotectin (CP)
CP, a protein resistant to bacterial degradation, has been found within the cytosol of neutrophils, accounting for ca. 60% of their cytoplasmic proteins [101]. Under inflammatory conditions of the intestinal tract, CP is excreted in stool [100,101] and can be measured by commercial assays. To differentiate bowel diseases, some cut-off values of faecal CP have been established. However, other causes of inflammation, such as infection with enteric pathogens or disease from non-steroidal anti-inflammatory agents, have to be considered since they may raise the CP level [100]. Currently there is no reference method or standard for faecal CP [100] other than endoscopy, the ‘gold standard’ for assessing intestinal inflammation.
Overall, CP or LF as faecal markers have emerged as new diagnostic tools to detect and monitor intestinal inflammation. Evaluation of the accuracy of faecal LF and CP as well as faecal occult blood testing showed statistically enhanced specificity and positive and negative likelihood ratios only for CP in predicting infectious diarrhoea [102]. Of note, LF and CP have been reported be able to differentiate inflammatory disease from functional bowel disorders [94]. Measuring both faecal LF and CP did not benefit detection [100]. Neither of these assays would differentiate CDI from other causes of diarrhoea.
5.2.2.3. pMK2
Clostridium difficile toxins activate the p38 pathway and its downstream kinase target MK2 [103]. Whilst p38 protein was suggested as a major regulator and essential to C. difficile toxin-induced inflammation, MK2 kinase (the p38 substrate) was proposed to be specifically involved in stress-induced inflammation. Phosphorylated MK2 (pMK2) phosphorylates specific molecules that regulate the actin cytoskeleton and stabilise cytokine mRNA transcripts. Tested stool specimens collected from CDI patients demonstrated that an elevated pMK2 level was significantly associated with the presence of toxigenic C. difficile [103]. However, like inflammatory cytokine levels, faecal pMK2 may be a general indicator of intestinal inflammation. It was also shown that MK2 can be activated by Shiga toxin and during influenza A virus infection [104].
5.2.2.4. Phosphorylated p38 (pp38)
pp38 has been tested as a biomarker for symptomatic CDI in the paediatric population [96]. Elevation of pp38 in the stools of children with CDI compared with their symptomatic controls was demonstrated. Consistent with previous data [103], the p38 pathway was suggested as specific for C. difficile-associated injury. Of note, in this study C. difficile bacterial burden was not associated with any clinical outcomes. Yet despite the observation that pp38 might be specific for C. difficile infection, it lacked sensitivity [96].
5.2.2.5. Interleukin-23
Thirty-six major inflammatory markers present in the stools were examined in CDI and non-CDI patients [105]. CDI-positive stools exhibited significantly higher relative amounts of C5a, CD40L, granulocyte colony-stimulating factor (G-CSF), I-309, IL-13, IL-16, IL-27, monocyte chemoattractant protein 1 (MCP-1), TNFα and IL-8. However, the study suggested the importance of IL-8 and IL-23 in CDI immunopathogenesis. The relative amount of IL-23 was significantly higher in CDI-negative stools than in CDI-positive stools. On the other hand, the average concentration of IL-8 in CDI-positive stools was significantly higher than in CDI-negative stools. These two cytokines were detected in more CDI-positive stools than CDI-negative stools [105].
A novel proposed marker is justified by the fact that destruction of the actin cytoskeleton by C. difficile toxins results in accelerated dissociation of colonic epithelial cells leading to cell death (Fig. 1) [106]. The hypothesis is that the release of cytoskeleton contents of colon epithelial cells could serve as a specific indicator for the host response in CDI. To demonstrate this novel approach, we have obtained preliminary results showing the feasibility of detecting human non-muscle tropomyosin, a major cytoskeleton protein of colon epithelial cells, released into patients’ stool samples. The preliminary results presented a promising correlation with the presence of CDI [107]. The biomarker is currently under investigation, supported by the National Institutes of Health (NIH) (grant R21AI116659).
6. ‘Gold standard’ for Clostridium difficile infection host response assay
Currently, there is no reliable single standing test for CDI diagnostics. The question raises what could be the choice of reference standard assay in development of a new CDI diagnostic test. The standard’s performance is crucial for defining true positives and true negatives. Use of suboptimal tests can blur CDI diagnosis owing to difficulty of distinguishing C. difficile and other infective or non-infective causes of diarrhoea [108].
CCTA has been traditionally chosen as the ‘gold standard’ assay for CDI confirmation, providing the final result in a day from sample submission [109,110], albeit with a sensitivity of <90% even in the presence of documented colitis [29]. Some investigators consider TC as more reliable for CDI although it takes 4–5 days. From a number of studies, the conclusion is drawn that TC detects more positive samples than CCTA [108], however the specificity is diminished.
Planche and Wilcox discussed the importance of the fact that these two methods detect different targets [108]. CCTA detects the presence of C. difficile toxins, whilst TC detects C. difficile bacteria or spores. A faecal sample may be CCTA-negative but TC-positive. Conversely, a TC-positive result may occur in the absence of CDI. Interestingly, studies reported that only 90% [111] or 50% [112] of pseudomembranous colitis patients were CCTA-positive. Therefore, the use of a single laboratory test should not be considered sufficient as a reference standard for new testing to detect CDI.
Ideally, the accuracy of laboratory diagnostic tests should be measured using reference assays utilising the same or equivalent targets [108]. On discrepant samples the use of assays with different targets likely will not improve assessment of the true accuracy of a diagnostic test. On note, a meta-analysis of the accuracy of rtPCR for CDI did not reveal differences in diagnostic sensitivity and specificity by the type of reference standard (CCTA/TC) [79].
In general, patients who are TC-positive but CCTA-negative appear different from those who are CCTA-positive [29]. Whilst a significant increase in the sensitivity of CDI detection is observed when TC is used, such advantage is diminished in regard to poor specificity of culture-based diagnosis secondary to C. difficile carriage [108]. A positive TC does not necessarily confirm CDI as the cause of diarrhoea. Therefore, none of these two methods are absolute for CDI diagnostics. If both reference tests were performed, a positive CCTA would confirm CDI and might be useful for the positive reference standard. Faecal samples negative by CCTA but positive by culture may be difficult to categorise, but it appears that many of these cases do not have CDI and it would be reasonable to exclude these from consideration of validation for a new diagnostic test. Therefore, clinical assessment of such cases would be important.
Is rtPCR an alternative? The rtPCR-based tests potentially yield false-positive results, demonstrating moderate specificity and PPVs owing to their high sensitivity and the potential for detecting colonised, but not clinically infected, persons leading to overdiagnosis of CDI. The reported sensitivity and specificity has ranged from 83% to 94% and from 97% to 98%, respectively. Importantly, this approach is not designed to detect the disease but rather the causative agent, and performance depends on the disease prevalence. Therefore, it is not a reliable choice for ‘gold standard’ as a stand-alone test.
For years, rapid and simple commercial EIAs detecting clostridial toxin(s) were the most frequently used CDI assays in clinical laboratories. However, their well confirmed characteristic is low sensitivity. The sensitivity and specificity of EIAs ranged from 32% to 83% and from 84% to 100%, respectively [20,74,113]. Thus, EIA is suboptimal for diagnosis of CDI. In addition, it has been demonstrated that EIAs are unable to detect some newer C. difficile strains, including epidemic clones [114]. The performance of toxin immunoassays varies markedly across manufacturers [110]. Therefore, the overall poor performance of toxin EIAs led to the recommendation to use them only as a part of a two- or three-stage algorithm [58,110]. Thus, these tests also are not a reliable choice for ‘gold standard’ as a stand-alone test.
Another EIA, for GDH (a protein found on most C. difficile isolates), is more sensitive (76–100%) but less specific. For this reason, GDH is only recommended to be used in combination with other assays, such as a toxin immunoassay. Taken together, the conclusion is that EIAs cannot serve as ‘gold standard’ as well.
Summarising from above, it is reasonable to hypothesise that validation of a new diagnostic test for true CDI, when linked to host response target, should rely on the combined outcome of two currently available laboratory diagnostic tests and relevant clinical signs. The CCTA should be the first choice as reference method for determining CDI cases, and a positive assay should be consistent with the diagnosis. Since the assay for detection of host response does not have an equivalent target as in CCTA, samples positive for TC and negative by CCTA should be excluded in the analysis. Those patients with both TC and CCTA tests negative should be considered as not having CDI. This will provide a reference standard where true positive and negative patients are recognised and those with an uncertain diagnosis are excluded.
7. Conclusion
CDI is a globally important yet poorly diagnosed infectious disease. Current CDI diagnostics is limited to detection of the organism and/or its toxin product(s) in conjunction with clinical symptoms, not differentiating infected from colonised patients, thus leading to inaccurate diagnosis and antibiotic mis/overuse. This review outlines major knowledge of CDI molecular aetiology to form a holistic view of the disease and to advocate the development of specific and accurate diagnostics for CDI.
It is now clear that there is a close link between pathogen and host response that controls disease progression and outcome. Therefore, two clear needs seem obvious for moving forward with this disease. One is the identification of a biomarker that is able to measure the effects of C. difficile toxin(s) on human colonic tissue. In addition to clinical signs and symptoms, the utility of a biomarker will significantly enhance the existing CDI diagnostic tools that entirely rely on organism detection. The second is to establish a reasonable set of criteria for the ‘gold standard’ of CDI diagnosis.
In conclusion, in addition to pathogenic toxin production, the composition and function of the intestinal microbiome and host immune factors have direct impacts on C. difficile pathogenesis. To improve the care of patients with potential CDI, there remains a critical need for the optimal diagnostic test approach to this persistent infectious disease.
Highlights.
High rate of colonisation with Clostridium difficile.
Toxin aetiology and inflammatory response are interconnected events in C. difficile infection (CDI).
Bacteria detection versus biological marker for CDI diagnostics.
Host response to CDI.
Acknowledgments
Funding: This work was supported by an investigator-initiated grant from the NIH National Institute of Allergy and Infectious Diseases [NIH/NIAID grant R21AI116659].
Footnotes
Competing interests: None declared.
Ethical approval: Not required.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31:431–55. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 2.Burke KE, Lamont JT. Clostridium difficile infection: a worldwide disease. Gut Liver. 2014;8:1–6. doi: 10.5009/gnl.2014.8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khanna S, Pardi DS. Clostridium difficile infection: management strategies for a difficult disease. Therap Adv Gastroenterol. 2014;7:72–86. doi: 10.1177/1756283X13508519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–34. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loo VG, Bourgault AM, Poirier L, Lamothe F, Michaud S, Turgeon N, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365:1693–703. doi: 10.1056/NEJMoa1012413. [DOI] [PubMed] [Google Scholar]
- 6.Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012;20:313–9. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karas JA, Enoch DA, Aliyu SH. A review of mortality due to Clostridium difficile infection. J Infect. 2010;61:1–8. doi: 10.1016/j.jinf.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Steiner C, Barrett M, Weiss A. HCUP projections: Clostridium difficile hospitalizations 2001 to 2013. HCUP projections report # 2014-01. US Agency for Healthcare Research and Quality; 2014. [Google Scholar]
- 9.Kochanek KD, Murphy SL, Xu J. Deaths: final data for 2011. National Vital Statistics Reports. 2015:63. [PubMed] [Google Scholar]
- 10.Lessa FC, Gould CV, McDonald LC. Current status of Clostridium difficile infection epidemiology. Clin Infect Dis. 2012;55(Suppl 2):S65–70. doi: 10.1093/cid/cis319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller BA, Chen LF, Sexton DJ, Anderson DJ. Comparison of the burdens of hospital-onset, healthcare facility-associated Clostridium difficile infection and of healthcare-associated infection due to methicillin-resistant Staphylococcus aureus in community hospitals. Infect Control Hosp Epidemiol. 2011;32:387–90. doi: 10.1086/659156. [DOI] [PubMed] [Google Scholar]
- 12.Tabak YP, Zilberberg MD, Johannes RS, Sun X, McDonald LC. Attributable burden of hospital-onset Clostridium difficile infection: a propensity score matching study. Infect Control Hosp Epidemiol. 2013;34:588–96. doi: 10.1086/670621. [DOI] [PubMed] [Google Scholar]
- 13.Jarvis WR, Schlosser J, Jarvis AA, Chinn RY. National point prevalence of Clostridium difficile in US health care facility inpatients, 2008. Am J Infect Control. 2009;37:263–70. doi: 10.1016/j.ajic.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Zilberberg MD, Tillotson GS, McDonald C. Clostridium difficile infections among hospitalized children, United States, 1997–2006. Emerg Infect Dis. 2010;16:604–9. doi: 10.3201/eid1604.090680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drekonja D, Reich J, Gezahegn S, Greer N, Shaukat A, MacDonald R, et al. Fecal microbiota transplantation for Clostridium difficile infection: a systematic review. Ann Intern Med. 2015;162:630–8. doi: 10.7326/M14-2693. [DOI] [PubMed] [Google Scholar]
- 16.Galdys AL, Nelson JS, Shutt KA, Schlackman JL, Pakstis DL, Pasculle AW, et al. Prevalence and duration of asymptomatic Clostridium difficile carriage among healthy subjects in Pittsburgh, Pennsylvania. J Clin Microbiol. 2014;52:2406–9. doi: 10.1128/JCM.00222-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teasley DG, Gerding DN, Olson MM, Peterson LR, Gebhard RL, Schwartz MJ, et al. Prospective randomised trial of metronidazole versus vancomycin for Clostridium-difficile-associated diarrhoea and colitis. Lancet. 1983;2:1043–6. doi: 10.1016/s0140-6736(83)91036-x. [DOI] [PubMed] [Google Scholar]
- 18.Johnson S, Clabots CR, Linn FV, Olson MM, Peterson LR, Gerding DN. Nosocomial Clostridium difficile colonisation and disease. Lancet. 1990;336:97–100. doi: 10.1016/0140-6736(90)91605-a. [DOI] [PubMed] [Google Scholar]
- 19.Alasmari F, Seiler SM, Hink T, Burnham CA, Dubberke ER. Prevalence and risk factors for asymptomatic Clostridium difficile carriage. Clin Infect Dis. 2014;59:216–22. doi: 10.1093/cid/ciu258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Planche T, Wilcox MH. Diagnostic pitfalls in Clostridium difficile infection. Infect Dis Clin North Am. 2015;29:63–82. doi: 10.1016/j.idc.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Young VB, Schmidt TM. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J Clin Microbiol. 2004;42:1203–6. doi: 10.1128/JCM.42.3.1203-1206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Planche TD, Davies KA, Coen PG, Finney JM, Monahan IM, Morris KA, et al. Differences in outcome according to Clostridium difficile testing method: a prospective multicentre diagnostic validation study of C difficile infection. Lancet Infect Dis. 2013;13:936–45. doi: 10.1016/S1473-3099(13)70200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Waaij LA, Harmsen HJ, Madjipour M, Kroese FG, Zwiers M, van Dullemen HM, et al. Bacterial population analysis of human colon and terminal ileum biopsies with 16S rRNA-based fluorescent probes: commensal bacteria live in suspension and have no direct contact with epithelial cells. Inflamm Bowel Dis. 2005;11:865–71. doi: 10.1097/01.mib.0000179212.80778.d3. [DOI] [PubMed] [Google Scholar]
- 24.Bakken JS. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe. 2009;15:285–9. doi: 10.1016/j.anaerobe.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Nord CE, Kager L, Heimdahl A. Impact of antimicrobial agents on the gastrointestinal microflora and the risk of infections. Am J Med. 1984;76:99–106. doi: 10.1016/0002-9343(84)90250-x. [DOI] [PubMed] [Google Scholar]
- 26.Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol. 2002;68:3401–7. doi: 10.1128/AEM.68.7.3401-3407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owens RC, Jr, Donskey CJ, Gaynes RP, Loo VG, Muto CA. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S19–31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 28.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458:1176–9. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerding DN, Olson MM, Peterson LR, Teasley DG, Gebhard RL, Schwartz ML, et al. Clostridium difficile-associated diarrhea and colitis in adults. A prospective case-controlled epidemiologic study. Arch Intern Med. 1986;146:95–100. [PubMed] [Google Scholar]
- 30.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–61. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rea MC, O’Sullivan O, Shanahan F, O’Toole PW, Stanton C, Ross RP, et al. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol. 2012;50:867–75. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peniche AG, Savidge TC, Dann SM. Recent insights into Clostridium difficile pathogenesis. Curr Opin Infect Dis. 2013;26:447–53. doi: 10.1097/01.qco.0000433318.82618.c6. [DOI] [PubMed] [Google Scholar]
- 34.Hutin Y, Casin I, Lesprit P, Welker Y, Decazes JM, Lagrange P, et al. Prevalence of and risk factors for Clostridium difficile colonization at admission to an infectious diseases ward. Clin Infect Dis. 1997;24:920–4. doi: 10.1093/clinids/24.5.920. [DOI] [PubMed] [Google Scholar]
- 35.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342:390–7. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 36.Leekha S, Aronhalt KC, Sloan LM, Patel R, Orenstein R. Asymptomatic Clostridium difficile colonization in a tertiary care hospital: admission prevalence and risk factors. Am J Infect Control. 2013;41:390–3. doi: 10.1016/j.ajic.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 37.Samore MH, DeGirolami PC, Tlucko A, Lichtenberg DA, Melvin ZA, Karchmer AW. Clostridium difficile colonization and diarrhea at a tertiary care hospital. Clin Infect Dis. 1994;18:181–7. doi: 10.1093/clinids/18.2.181. [DOI] [PubMed] [Google Scholar]
- 38.Burnham CA, Carroll KC. Diagnosis of Clostridium difficile infection: an ongoing conundrum for clinicians and for clinical laboratories. Clin Microbiol Rev. 2013;26:604–30. doi: 10.1128/CMR.00016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin J. The contribution of strains and hosts to outcomes in Clostridium difficile infection. Infect Dis Clin North Am. 2015;29:51–61. doi: 10.1016/j.idc.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Rousseau C, Poilane I, De Pontual L, Maherault AC, Le Monnier A, Collignon A. Clostridium difficile carriage in healthy infants in the community: a potential reservoir for pathogenic strains. Clin Infect Dis. 2012;55:1209–15. doi: 10.1093/cid/cis637. [DOI] [PubMed] [Google Scholar]
- 41.Donskey CJ, Kundrapu S, Deshpande A. Colonization versus carriage of Clostridium difficile. Infect Dis Clin North Am. 2015;29:13–28. doi: 10.1016/j.idc.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RL, Donskey CJ. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis. 2007;45:992–8. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 43.Aronoff DM. Editorial commentary: host–pathogen interactions in Clostridium difficile infection: it takes two to tango. Clin Infect Dis. 2014;58:1401–3. doi: 10.1093/cid/ciu141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paredes-Sabja D, Shen A, Sorg JA. Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 2014;22:406–16. doi: 10.1016/j.tim.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, Young VB. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun. 2015;83:934–41. doi: 10.1128/IAI.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowardin CA, Kuehne SA, Buonomo EL, Marie CS, Minton NP, Petri WA., Jr Inflammasome activation contributes to interleukin-23 production in response to Clostridium difficile. MBio. 2015;6 doi: 10.1128/mBio.02386-14. pii: e02386-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467:711–3. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 48.Hirota SA, Iablokov V, Tulk SE, Schenck LP, Becker H, Nguyen J, et al. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun. 2012;80:4474–84. doi: 10.1128/IAI.00933-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuehne SA, Collery MM, Kelly ML, Cartman ST, Cockayne A, Minton NP. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis. 2014;209:83–6. doi: 10.1093/infdis/jit426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter GP, Rood JI, Lyras D. The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol. 2012;20:21–9. doi: 10.1016/j.tim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Carter GP, Chakravorty A, Pham Nguyen TA, Mileto S, Schreiber F, Li L, et al. Defining the roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile infections. MBio. 2015;6:e00551. doi: 10.1128/mBio.00551-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun X, Hirota SA. The roles of host and pathogen factors and the innate immune response in the pathogenesis of Clostridium difficile infection. Mol Immunol. 2015;63:193–202. doi: 10.1016/j.molimm.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cairns MD, Preston MD, Lawley TD, Clark TG, Stabler RA, Wren BW. Genomic epidemiology of a protracted hospital outbreak caused by a toxin A-negative Clostridium difficile sublineage PCR ribotype 017 strain in London, England. J Clin Microbiol. 2015;53:3141–7. doi: 10.1128/JCM.00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin BM, Kuak EY, Yoo HM, Kim EC, Lee K, Kang JO, et al. Multicentre study of the prevalence of toxigenic Clostridium difficile in Korea: results of a retrospective study 2000–2005. J Med Microbiol. 2008;57:697–701. doi: 10.1099/jmm.0.47771-0. [DOI] [PubMed] [Google Scholar]
- 55.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–3. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 56.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–63. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, et al. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 2012;8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rupnik M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev. 2008;32:541–55. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- 59.Stubbs S, Rupnik M, Gibert M, Brazier J, Duerden B, Popoff M. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett. 2000;186:307–12. doi: 10.1111/j.1574-6968.2000.tb09122.x. [DOI] [PubMed] [Google Scholar]
- 60.Geric B, Rupnik M, Gerding DN, Grabnar M, Johnson S. Distribution of Clostridium difficile variant toxinotypes and strains with binary toxin genes among clinical isolates in an American hospital. J Med Microbiol. 2004;53:887–94. doi: 10.1099/jmm.0.45610-0. [DOI] [PubMed] [Google Scholar]
- 61.Shen A. Clostridium difficile toxins: mediators of inflammation. J Innate Immun. 2012;4:149–58. doi: 10.1159/000332946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sundriyal A, Roberts AK, Ling R, McGlashan J, Shone CC, Acharya KR. Expression, purification and cell cytotoxicity of actin-modifying binary toxin from Clostridium difficile. Protein Expr Purif. 2010;74:42–8. doi: 10.1016/j.pep.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 63.Schwan C, Stecher B, Tzivelekidis T, van Ham M, Rohde M, Hardt WD, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009;5:e1000626. doi: 10.1371/journal.ppat.1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geric B, Carman RJ, Rupnik M, Genheimer CW, Sambol SP, Lyerly DM, et al. Binary toxin-producing, large clostridial toxin-negative Clostridium difficile strains are enterotoxic but do not cause disease in hamsters. J Infect Dis. 2006;193:1143–50. doi: 10.1086/501368. [DOI] [PubMed] [Google Scholar]
- 65.Vedantam G, Clark A, Chu M, McQuade R, Mallozzi M, Viswanathan VK. Clostridium difficile infection: toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes. 2012;3:121–34. doi: 10.4161/gmic.19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60:1070–9. doi: 10.1099/jmm.0.030015-0. [DOI] [PubMed] [Google Scholar]
- 67.Onderdonk AB, Cisneros RL, Bartlett JG. Clostridium difficile in gnotobiotic mice. Infect Immun. 1980;28:277–82. doi: 10.1128/iai.28.1.277-282.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobson G, Hickey C, Trinder J. Clostridium difficile colitis causing toxic megacolon, severe sepsis and multiple organ dysfunction syndrome. Intensive Care Med. 2003;29:1030. doi: 10.1007/s00134-003-1754-7. [DOI] [PubMed] [Google Scholar]
- 69.Yacyshyn MB, Yacyshyn B. The role of gut inflammation in recurrent Clostridium difficile-associated disease. Clin Infect Dis. 2013;56:1722–3. doi: 10.1093/cid/cit151. [DOI] [PubMed] [Google Scholar]
- 70.El Feghaly RE, Stauber JL, Deych E, Gonzalez C, Tarr PI, Haslam DB. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis. 2013;56:1713–21. doi: 10.1093/cid/cit147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Horo JC, Jones A, Sternke M, Harper C, Safdar N. Molecular techniques for diagnosis of Clostridium difficile infection: systematic review and meta-analysis. Mayo Clin Proc. 2012;87:643–51. doi: 10.1016/j.mayocp.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gebhard RL, Gerding DN, Olson MM, Peterson LR, McClain CJ, Ansel HJ, et al. Clinical and endoscopic findings in patients early in the course of Clostridium difficile-associated pseudomembranous colitis. Am J Med. 1985;78:45–8. doi: 10.1016/0002-9343(85)90460-7. [DOI] [PubMed] [Google Scholar]
- 73.Barbut F, Surgers L, Eckert C, Visseaux B, Cuingnet M, Mesquita C, et al. Does a rapid diagnosis of Clostridium difficile infection impact on quality of patient management? Clin Microbiol Infect. 2014;20:136–44. doi: 10.1111/1469-0691.12221. [DOI] [PubMed] [Google Scholar]
- 74.Peterson LR, Mehta MS, Patel PA, Hacek DM, Harazin M, Nagwekar PP, et al. Laboratory testing for Clostridium difficile infection: light at the end of the tunnel. Am J Clin Pathol. 2011;136:372–80. doi: 10.1309/AJCPTP5XKRSNXVIL. [DOI] [PubMed] [Google Scholar]
- 75.Wilcox MH. Overcoming barriers to effective recognition and diagnosis of Clostridium difficile infection. Clin Microbiol Infect. 2012;18(Suppl 6):13–20. doi: 10.1111/1469-0691.12057. [DOI] [PubMed] [Google Scholar]
- 76.Peterson LR, Robicsek A. Does my patient have Clostridium difficile infection? Ann Intern Med. 2009;151:176–9. doi: 10.7326/0003-4819-151-3-200908040-00005. [DOI] [PubMed] [Google Scholar]
- 77.Shin BM, Yoo SM, Shin WC. Evaluation of Xpert C. difficile, BD MAX Cdiff, IMDx C. difficile for Abbott m2000, and Illumigene C difficile assays for direct detection of toxigenic Clostridium difficile in stool specimens. Ann Lab Med. 2016;36:131–7. doi: 10.3343/alm.2016.36.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith B, Peterson L. Molecular diagnostic testing for Clostridium difficile infection: a double edged sword. Medscape; Nov 5, 2015. http://www.medscape.com/viewarticle/853529. [Google Scholar]
- 79.Deshpande A, Pasupuleti V, Rolston DD, Jain A, Deshpande N, Pant C, et al. Diagnostic accuracy of real-time polymerase chain reaction in detection of Clostridium difficile in the stool samples of patients with suspected Clostridium difficile infection: a meta-analysis. Clin Infect Dis. 2011;53:e81–90. doi: 10.1093/cid/cir505. [DOI] [PubMed] [Google Scholar]
- 80.Lloyd A, Pasupuleti V, Thota P, Pant C, Rolston DD, Hernandez AV, et al. Accuracy of loop-mediated isothermal amplification for the diagnosis of Clostridium difficile infection: a systematic review. Diagn Microbiol Infect Dis. 2015;82:4–10. doi: 10.1016/j.diagmicrobio.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 81.Surawicz CM, McFarland LV, Greenberg RN, Rubin M, Fekety R, Mulligan ME, et al. The search for a better treatment for recurrent Clostridium difficile disease: use of high-dose vancomycin combined with Saccharomyces boulardii. Clin Infect Dis. 2000;31:1012–7. doi: 10.1086/318130. [DOI] [PubMed] [Google Scholar]
- 82.Koo HL, Van JN, Zhao M, Ye X, Revell PA, Jiang ZD, et al. Real-time polymerase chain reaction detection of asymptomatic Clostridium difficile colonization and rising C. difficile -associated disease rates. Infect Control Hosp Epidemiol. 2014;35:667–73. doi: 10.1086/676433. [DOI] [PubMed] [Google Scholar]
- 83.US Food and Drug Administration. [accessed 12 September 2016];Medical devices: products and medical procedures: in vitro diagnostics. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm.
- 84.Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 85.Tenover FC, Baron EJ, Peterson LR, Persing DH. Laboratory diagnosis of Clostridium difficile infection. Can molecular amplification methods move us out of uncertainty? J Mol Diagn. 2011;13:573–82. doi: 10.1016/j.jmoldx.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Solomon K, Martin AJ, O’Donoghue C, Chen X, Fenelon L, Fanning S, et al. Mortality in patients with Clostridium difficile infection correlates with host pro-inflammatory and humoral immune responses. J Med Microbiol. 2013;62:1453–60. doi: 10.1099/jmm.0.058479-0. [DOI] [PubMed] [Google Scholar]
- 87.Drudy D, Calabi E, Kyne L, Sougioultzis S, Kelly E, Fairweather N, et al. Human antibody response to surface layer proteins in Clostridium difficile infection. FEMS Immunol Med Microbiol. 2004;41:237–42. doi: 10.1016/j.femsim.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 88.Sanchez-Hurtado K, Corretge M, Mutlu E, McIlhagger R, Starr JM, Poxton IR. Systemic antibody response to Clostridium difficile in colonized patients with and without symptoms and matched controls. J Med Microbiol. 2008;57:717–24. doi: 10.1099/jmm.0.47713-0. [DOI] [PubMed] [Google Scholar]
- 89.Dingle TC, Mulvey GL, Armstrong GD. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect Immun. 2011;79:4061–7. doi: 10.1128/IAI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abou Chakra CN, Pepin J, Valiquette L. Prediction tools for unfavourable outcomes in Clostridium difficile infection: a systematic review. PLoS One. 2012;7:e30258. doi: 10.1371/journal.pone.0030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walker AS, Eyre DW, Wyllie DH, Dingle KE, Griffiths D, Shine B, et al. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis. 2013;56:1589–600. doi: 10.1093/cid/cit127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Micic D, Rao K, Trindade BC, Walk ST, Chenoweth E, Jain R, et al. Serum 25-hydroxyvitamin D levels are not associated with adverse outcomes in Clostridium difficile infection. Infect Dis Rep. 2015;7:5979. doi: 10.4081/idr.2015.5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rao K, Walk ST, Micic D, Chenoweth E, Deng L, Galecki AT, et al. Procalcitonin levels associate with severity of Clostridium difficile infection. PLoS One. 2013;8:e58265. doi: 10.1371/journal.pone.0058265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abraham BP, Kane S. Fecal markers: calprotectin and lactoferrin. Gastroenterol Clin North Am. 2012;41:483–95. doi: 10.1016/j.gtc.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 95.Dabritz J, Musci J, Foell D. Diagnostic utility of faecal biomarkers in patients with irritable bowel syndrome. World J Gastroenterol. 2014;20:363–75. doi: 10.3748/wjg.v20.i2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.El Feghaly RE, Stauber JL, Tarr PI, Haslam DB. Intestinal inflammatory biomarkers and outcome in pediatric Clostridium difficile infections. J Pediatr. 2013;163:1697–704. e2. doi: 10.1016/j.jpeds.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kane SV, Sandborn WJ, Rufo PA, Zholudev A, Boone J, Lyerly D, et al. Fecal lactoferrin is a sensitive and specific marker in identifying intestinal inflammation. Am J Gastroenterol. 2003;98:1309–14. doi: 10.1111/j.1572-0241.2003.07458.x. [DOI] [PubMed] [Google Scholar]
- 98.van Langenberg DR, Gearry RB, Wong HL, Ward M, Gibson PR. The potential value of faecal lactoferrin as a screening test in hospitalized patients with diarrhoea. Intern Med J. 2010;40:819–27. doi: 10.1111/j.1445-5994.2009.02102.x. [DOI] [PubMed] [Google Scholar]
- 99.Vaishnavi C, Bhasin D, Kochhar R, Singh K. Clostridium difficile toxin and faecal lactoferrin assays in adult patients. Microbes Infect. 2000;2:1827–30. doi: 10.1016/s1286-4579(00)01343-5. [DOI] [PubMed] [Google Scholar]
- 100.Sherwood RA. Faecal markers of gastrointestinal inflammation. J Clin Pathol. 2012;65:981–5. doi: 10.1136/jclinpath-2012-200901. [DOI] [PubMed] [Google Scholar]
- 101.Roseth AG, Fagerhol MK, Aadland E, Schjonsby H. Assessment of the neutrophil dominating protein calprotectin in feces. A methodologic study. Scand J Gastroenterol. 1992;27:793–8. doi: 10.3109/00365529209011186. [DOI] [PubMed] [Google Scholar]
- 102.Shastri YM, Bergis D, Povse N, Schafer V, Shastri S, Weindel M, et al. Prospective multicenter study evaluating fecal calprotectin in adult acute bacterial diarrhea. Am J Med. 2008;121:1099–106. doi: 10.1016/j.amjmed.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 103.Bobo LD, El Feghaly RE, Chen YS, Dubberke ER, Han Z, Baker AH, et al. MAPK-activated protein kinase 2 contributes to Clostridium difficile-associated inflammation. Infect Immun. 2013;81:713–22. doi: 10.1128/IAI.00186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saenz JB, Li J, Haslam DB. The MAP kinase-activated protein kinase 2 (MK2) contributes to the Shiga toxin-induced inflammatory response. Cell Microbiol. 2010;12:516–29. doi: 10.1111/j.1462-5822.2009.01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Darkoh C, Turnwald BP, Koo HL, Garey KW, Jiang ZD, Aitken SL, et al. Colonic immunopathogenesis of Clostridium difficile infections. Clin Vaccine Immunol. 2014;21:509–17. doi: 10.1128/CVI.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buda A, Pignatelli M. Cytoskeletal network in colon cancer: from genes to clinical application. Int J Biochem Cell Biol. 2004;36:759–65. doi: 10.1016/j.biocel.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 107.Jin J-P, Peterson L. Host response in C. difficile infection: release of a cytoskeleton protein and its diagnostic application. 2nd ASMET—The ASM Emerging Technologies Conference; 8–11 March 2011; San Juan, Puerto Rico. Washington, DC: ASM Press; 2011. [Google Scholar]
- 108.Planche T, Wilcox M. Reference assays for Clostridium difficile infection: one or two gold standards? J Clin Pathol. 2011;64:1–5. doi: 10.1136/jcp.2010.080135. [DOI] [PubMed] [Google Scholar]
- 109.de Boer RF, Wijma JJ, Schuurman T, Moedt J, Dijk-Alberts BG, Ott A, et al. Evaluation of a rapid molecular screening approach for the detection of toxigenic Clostridium difficile in general and subsequent identification of the tcdC 117 mutation in human stools. J Microbiol Methods. 2010;83:59–65. doi: 10.1016/j.mimet.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 110.Eastwood K, Else P, Charlett A, Wilcox M. Comparison of nine commercially available Clostridium difficile toxin detection assays, a real-time PCR assay for C. difficile tcdB, and a glutamate dehydrogenase detection assay to cytotoxin testing and cytotoxigenic culture methods. J Clin Microbiol. 2009;47:3211–7. doi: 10.1128/JCM.01082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.George WL, Rolfe RD, Harding GK, Klein R, Putnam CW, Finegold SM. Clostridium difficile and cytotoxin in feces of patients with antimicrobial agent-associated pseudomembranous colitis. Infection. 1982;10:205–8. doi: 10.1007/BF01666910. [DOI] [PubMed] [Google Scholar]
- 112.Johal SS, Hammond J, Solomon K, James PD, Mahida YR. Clostridium difficile associated diarrhoea in hospitalised patients: onset in the community and hospital and role of flexible sigmoidoscopy. Gut. 2004;53:673–7. doi: 10.1136/gut.2003.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Planche T, Aghaizu A, Holliman R, Riley P, Poloniecki J, Breathnach A, et al. Diagnosis of Clostridium difficile infection by toxin detection kits: a systematic review. Lancet Infect Dis. 2008;8:777–84. doi: 10.1016/S1473-3099(08)70233-0. [DOI] [PubMed] [Google Scholar]
- 114.Tenover FC, Novak-Weekley S, Woods CW, Peterson LR, Davis T, Schreckenberger P, et al. Impact of strain type on detection of toxigenic Clostridium difficile: comparison of molecular diagnostic and enzyme immunoassay approaches. J Clin Microbiol. 2010;48:3719–24. doi: 10.1128/JCM.00427-10. [DOI] [PMC free article] [PubMed] [Google Scholar]