

Abstract
Understanding molecular mechanisms that underlie the recent emergence of metabolic diseases such as diabetes and heart failure has revealed the need for a multi-disciplinary research integrating the key metabolic pathways which change the susceptibility to environmental or pathologic stress. At the physiological level these include the circadian control of metabolism which aligns metabolism with temporal demand. The mitochondria play an important role in integrating the redox signals and metabolic flux in response to the changing activities associated with chronobiology, exercise and diet. At the molecular level this involves dynamic post-translational modifications regulating transcription, metabolism and autophagy. In this review we will discuss different examples of mechanisms which link these processes together. An important pathway capable of linking signaling to metabolism is the post-translational modification of proteins by O-linked N-acetylglucosamine (O-GlcNAc). This is a nutrient regulated protein modification that plays an important role in impaired cellular stress responses. Circadian clocks have also emerged as critical regulators of numerous cardiometabolic processes, including glucose/lipid homeostasis, hormone secretion, redox status and cardiovascular function. Central to these pathways are the response of autophagy, bioenergetics to oxidative stress, regulated by Keap1/Nrf2 and mechanisms of metabolic control. The extension of these ideas to the emerging concept of bioenergetic health will be discussed.
Keywords: bioenergetic health, extracellular flux analysis, autophagy, signaling, reserve capacity, oxidative stress, metabolic shift, Keap1, Nrf2, chronobiology
Graphical abstract
1. Introduction
Studies of mitochondria were among the first to integrate redox biology into metabolism based on the observations that inhibition of mitochondrial respiration with toxic xenobiotics or hyperoxia increased hydrogen peroxide [1-3]. Since both were pathological models or scenarios this led to two postulates that a) mitochondria are a major source of hydrogen peroxide/superoxide in the cell and b) this was a pathological process that led to mitochondrial dysfunction [3, 4]. Naturally, over the subsequent 40 years these postulates, while still often quoted as dogma, have become much more nuanced. The discovery of the NADPH oxidases and their various isoforms now make it abundantly clear that mitochondria are by no means the major or only source of hydrogen peroxide in the cell [5]. Indeed, the NOX enzymes are generally much higher capacity generators of hydrogen peroxide/superoxide than mitochondria [6, 7]. It has also become clear that the data derived from mitochondrial toxins and hyperoxia are not broadly applicable to normal physiology and mitochondrial production of superoxide/hydrogen peroxide are a part of normal physiology and cell signaling [8]. Furthermore, the sites of production of hydrogen peroxide/superoxide from the mitochondria have rapidly expanded beyond the electron transport chain and are intimately linked to metabolism and also offering the potential for independent regulation [2, 8]. At first glance the early data which showed that partial deletion of SOD2 promoted cardiovascular and other pathologies also supported a damaging role for mitochondrial superoxide [9]. An alternative explanation is now gaining acceptance and in this paradigm the mitochondrial superoxide dismutase (SOD2) is essential for control of mitochondrial redox signaling [2, 10, 11]. The role of mitochondria in cell physiology clearly extends beyond simply providing ATP to the cell and involves a variety of metabolites which integrate the nuclear transcriptome with metabolic requirements [12-14]. We and others have proposed that among these pathways is the mitochondrial production of hydrogen peroxide/superoxide and is integrated by the regulatory effects of nitric oxide [11, 15, 16]. This appears to be a controlled process under physiological conditions and plays a central role in the signaling pathways from the organelle to the nucleus often referred to as the “retrograde” signaling pathway [6, 8, 14, 17, 18].
It has been known for some time that there are diseases which arise from mutations in mitochondrial DNA and these can present with a broad range of symptoms emphasizing both the capacity for metabolic plasticity and adaptation [17, 19]. Beyond direct mutations in mtDNA the impact of bioenergetic dysfunction is now recognized as a key contributor to diabetes, neurodegeneration, cancer and cardiovascular diseases [17, 20-26]. This has extended the range of what are considered metabolic or mitochondrial therapeutics which can function through different mechanisms. Application of screening technologies has also shown that established therapeutics, such as metformin, also modulate bioenergetics [27-33]. The mechanisms through which this occurs are still not clear but have been shown in several cases to involve the controlled generation of superoxide and hydrogen peroxide from the respiratory chain [14, 18, 34, 35]. Interestingly, among the 13 proteins coded for by mitochondrial DNA, are critical redox centers in the respiratory chain which offers a mechanism through which mutations in mtDNA could modulate superoxide levels in response to stress and so impact on pathological processes [36]. Taken together, these findings are resulting in the new field of redox bioenergetics.
If the antioxidant networks within the mitochondria are playing a redox regulatory role for the purposes of metabolic integration then what are the mechanisms used to mitigate the slow but progressive accumulation of damaged or aggregated proteins? The discovery of autophagy and the specialized form of the pathway known as mitophagy has given at least a partial answer to this question [37, 38]. Autophagy has become a broad field encompassing many aspects of metabolism including exercise, the response to starvation and oxidative stress [39, 40]. It has a high capacity for removing and detoxifying oxidatively damaged proteins and organelles including the mitochondrion and is emerging as an important antioxidant pathways [6, 12]. From this background it is clear that the regulatory role of redox-dependent pathways in metabolism is now taking on added importance. This also includes the findings that link circadian biology to redox networks, autophagy and mitochondrial function which will be discussed in a later section.
Temporal partitioning, a component of circadian biology (i.e., chronobiology), of metabolic processes occurs due to the combined actions of extrinsic and intrinsic modulators. Various neurohumoral factors fluctuate secondarily to sleep/wake and fasting/feeding cycles, such as cortisol, growth hormone, catecholamines, and insulin [41]. These extracellular (extrinsic) factors exert potent catabolic and anabolic actions on metabolically active tissues in a classic stimulus-response coupling manner. More recently, the existence of cell-autonomous circadian clocks that have the capacity to modulate processes such as metabolism over the course of the day has been exposed [42]. This transcriptionally-based intrinsic mechanism confers the selective advantage of anticipation, preparing the cell/organ for a predicted stimulus/stress prior to its onset [43]. Importantly, the circadian clock has been linked to daily rhythms in oxidative metabolism, antioxidant levels, protein synthesis, autophagy, and even mitochondrial biogenesis as will be discussed in more depth in a later section. It is noteworthy that an additional, non-transcriptional, highly evolutionarily conversed clock mechanism has been described, involving the redox proteins peroxiredoxins [44]. Underscoring further the interrelationship between redox biology and circadian rhythms is the crosstalk between circadian clocks, peroxiredoxins, and antioxidant potential [45].
Our perspective of mitochondrial function is also rapidly changing as it becomes possible to determine function in cells with low oxidative capacity compared to the traditionally studied highly active heart and liver cells. An important new area focusses on cells of the innate immune system which have an obligatory role for metabolic switching between glycolytic and oxidative metabolism in normal function [46, 47]. The long established paradigm in which cancer cells exhibit an altered bioenergetic metabolism characterized by aerobic glycolysis has now been extended to encompass lymphocytes and monocytes as they adapt to their changing biological functions in normal physiology [17, 46, 48, 49]. In this short overview we will highlight these emerging themes in redox bioenergetics with a primary emphasis on cardiovascular physiology and pathology since this is one of the fields in which studies linking redox biology, circadian biology, metabolism and autophagy are reasonably extensive.
2. Epigenetics and redox dependent metabolic regulation
An important new area of research in redox biology is that of long-term genetic regulation through the process known as epigenetics. This control of gene expression includes post-translational modifications of the histone proteins used to package DNA and modification of the DNA itself, all without changing the underlying genetic code [50]. Epigenetics has emerged as a contributing pathway in both normal development as well as in the pathogenesis of a number of chronic diseases including obesity, diabetes, cancer, neurological disorders, and heart failure [51-54]. In this section we will focus on recent advances directly linking changes in the redox status of the cellular environment to specific mechanisms by which genes are regulated and then expand upon direct redox regulation within multiple pathways (Figure 1A).
Figure 1. Redox and metabolic control of epigenetic modifications and molecular function.
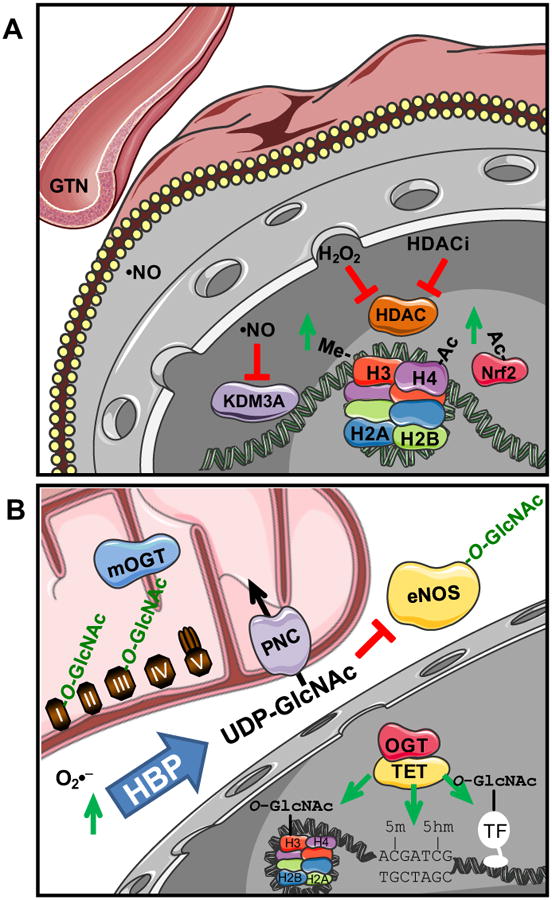
(A) Redox control of gene expression as explained in the text. Central focus is on cellular nitric oxide (•NO) signaling. (B). Metabolic control of gene expression through mitochondrial superoxide (O2•−) signaling via hexosamine biosynthesis pathway (HBP) flux increasing UDP-GlcNAc and protein O-GlcNAcylation (O-GlcNAc). Including both cytosolic endothelial cell, nitric oxide synthase 3 (eNOS) to reduce nitric oxide signaling and mitochondrial proteins of oxidative phosphorylation (I, II, III, IV, and V). Additional recent focus has been on the tet methylcytosine dioxygenases (TETs) to regulate DNA methylation (5-m) to hydroxymethylation (5-hm).
One common theme in epigenetics is the communication of environmental influences on molecular regulation. Much of this is controlled by metabolic flux through mitochondria and the direct utilization of metabolite intermediates and reactive species in the modification of transcription factors, histone core proteins, and DNA [55]. A new and interesting aspect is the direct role intermediates of the TCA cycle play in the regulation of gene expression [56]. In addition to the TCA cycle the role of metabolites of nitric oxide are particularly important and also the pharmacological modulators of the NO pathway (eg nitroglycerin) [57]. Interestingly, it appears that in patients with cardiovascular disease treated with organic nitrates epigenetic and post-translational regulation by downstream signaling occurs. This may be mediated by sustained changes in gene expression dependent on nitric oxide and peroxynitrite. Treatment of cells with nitric oxide regulates both histone H3 and H4 methylation and acetylation associated with changes in gene expression [58]. For changes in protein methylation, part of the mechanism of action is through the post-translational nitrosyliron complex formation in the catalytic pocket of the demethylase lysine demethylase 3A (KDM3A) [59]. Another important post-translational modification integrating metabolic redox signaling to epigenetic pathways is protein O-GlcNAcylation that will be discussed in the next section [60-62].
The Keap1-Nrf2 pathway is one of the best characterized redox signaling pathways and is regulated by epigenetics. Specifically, it was recently shown that in models of the inflammatory disease of osteoarthritis, HDAC inhibition (HDACi) reduced symptoms by directly increasing acetylation of Nrf2 and histone H3 [63]. These changes occurred in parallel with a decrease in inflammatory markers such as IL-6 and TNFα, potentially via direct transcriptional regulation. This is not limited to arthritis and a connection between HDACi and cancer treatment has identified the potential role for epigenetic regulation of Nrf2 and precision medicine for treating colon cancer [64]. The metabolic contribution to this combined pathway is further supported by studies where hyperglycemia could additionally alter histone H3 via methylation at key regions of Nrf2-mediated transcriptional regulation [65], suggesting a mechanistic link between nutrient utilization and epigenetic control of key antioxidant defense pathways. It is tempting to speculate that similar mechanisms are involved in regulation of obesity and inflammatory diseases with a potential therapeutic option of this transcriptional control via Nrf2, epigenetics, and its modulation by curcumin [66].
Similar to the link between histone acetylation and Nrf2-mediated regulation, growing evidence suggests that oxidative stress in the brain can contribute to Parkinson's and Alzheimer's disease pathway through an autophagy-mediated regulation of epigenetics. Specifically, histone acetylation is increased in neuronal cells following H2O2 treatment concurrent with increased Aβ production [67]. More recently it was found that the changing histone acetylation environment is directly regulated by autophagy and HDAC degradation likely contributing to disease progression [68]. Autophagy and the link to regulation by redox signaling beyond epigenetics will be discussed in more detail below.
3. The GlcNAc pathway and mitochondrial function under stress
The redox signaling mechanisms that integrate metabolism and cellular responses to stress are clearly important to understand from both a mechanistic aspect and also as new avenues for the development of therapeutics. In this respect the post-translational modification of proteins by the attachment of β-N-acetylglucosamine on serine and threonine residues via an O-linkage (O-GlcNAc) is achieving particular prominence because of its intimate relationship with glycolysis, response to oxidative stress and as a nexus linking nutrient signaling to gene expression [60-62, 69, 70]. In contrast to typical glycosylation, O-GlcNAcylation is a dynamic and reversible modification, regulated by O-GlcNAc transferase (OGT), a glycosyl transferase that uses UDP-GlcNAc to catalyze the attachment of O-GlcNAc to proteins and O-GlcNAcase, a hexosaminidase, which catalyzes its removal. The availability of UDP-GlcNAc is a major determinant of overall O-GlcNAc levels and it is the product of the hexosamine biosynthesis pathway (HBP), which requires glucose, glutamine, acetyl-CoA and high energy phosphates in the synthesis of UDP-GlcNAc.
As mentioned earlier, epigenetic control is central to the co-ordination of metabolic and stress responses. Although it has been well established that O-GlcNAc regulates the activity of transcription factors the newest discoveries in this area has been the O-GlcNAc modification of histone proteins and the physical interaction of the proteins that regulate O-GlcNAc cycling and that of DNA hydroxylation to 5-hydroxymethylcytosine (5-hmC) (Figure 1B). Specifically, O-GlcNAc transferase or OGT interacts with each of the three different methylcytosine dioxygenase ten-eleven translocation proteins (TETs) that are involved in addition of DNA 5-hmC [71-73]. Therefore, this novel interaction of OGT and TET provides a critical junction of nutrient and oxidative signaling to regulation of gene expression. A number of outstanding questions remain as to the location, temporal pattern, and regulation of 5-hmC in different tissues and disease states. The regulation of these 5-hmC levels by changing ascorbate levels links its relevance to redox signaling [74], suggesting areas of potential therapeutic intervention.
Increased flux through the HBP has long been implicated as a major factor mediating the adverse effects of hyperglycemia and thus the complications associated with diabetes. For example, in endothelial cells hyperglycemia induced increase in mitochondrial superoxide was shown to increase HBP flux and O-GlcNAc levels and subsequent activation of genes that are associated with diabetic complications [75]. Hyperglycemia has also been shown to decrease eNOS activity via direct O-GlcNAcylation of eNOS and a reciprocal decrease in phosphorylation of serine 1177, thereby potentially contributing to vascular dysfunction in diabetes [76]. Moreover, hyperglycemia induced mitochondrial dysfunction has been linked to O-GlcNAcylation of key mitochondrial proteins including subunit NDUFA9 of complex I subunits core 1 and core 2 of complex III, and the mitochondrial DNA-encoded subunit I of complex IV (COX I) [77]. Increased O-GlcNAc modification of Dynamin-related protein 1 (DRP1) at threonine 585 and 586 has been shown to induce its translocation to mitochondria resulting in reduction of mitochondrial membrane potential and increased mitochondrial fragmentation [78]. Mitochondrial dysfunction associated with increased O-GlcNAc levels has also been linked to O-GlcNAcylation of VDAC2 [79]. It should be noted however that there remains some controversy whether the adverse effects of hyperglycemia on mitochondrial function are mediated via increased O-GlcNAcylation [80]. On the other hand OGT has been shown to interact directly with complex IV in cardiac mitochondria from normal rats and this interaction is markedly decreased in mitochondria from diabetic rats [81].
While the above studies link increased O-GlcNAcylation to mitochondrial and cellular dysfunction, overall cellular O-GlcNAc levels increase in response to acute stress, including oxidant stress and this has been shown to be an endogenous pro-survival response [82]. Moreover both in vitro and in vivo acute augmentation of O-GlcNAc levels has been shown to be cardioprotective against ischemia/reperfusion injury. Increased O-GlcNAc levels have also shown to attenuate the loss of mitochondrial membrane potential induced by hydrogen peroxide [83]. While the specific mechanisms underlying the cytoprotective effects of O-GlcNAc remain open to debate, increased O-GlcNAc levels were associated with attenuation of mPTP opening [84, 85]. In the ischemia/reperfusion model increased O-GlcNAcylation of VDAC is associated with cytoprotection [85, 86]. Studies have also shown that enhanced mitochondrial Bcl-2 translocation occurred in an O-GlcNAc dependent manner [83] and Bcl-2 itself has been reported to be O-GlcNAcylated [87]. Interestingly, protein O-GlcNAcylation has also been shown to be an important prosurvival signal in cardiac stem cells; however, in these cells this appeared to be independent of any effects on mitochondrial function [88].
Despite growing evidence that O-GlcNAcylation of mitochondrial proteins contributed to cellular response to stress the question remained as to how mitochondrial proteins became O-GlcNAcylated? Although a mitochondrial specific isoform of OGT (mOGT) was identified in 2003 [89] a mitochondrial O-GlcNAcase had not been found and it was not known if UDP-GlcNAc could enter mitochondria. However, in 2015 it was shown that the pyrimidine nucleotide carrier transported UDP-GlcNAc from the cytosol into the mitochondria. A mitochondrial O-GlcNAcase activity has been identified demonstrating for the first time a functional mitochondrial O-GlcNAc cycle [81]. A recent O-GlcNAcome profiling study identified 88 mitochondrial proteins as O-GlcNAc targets of which nearly half were linked to oxidative phosphorylation [90]. It is becoming increasingly clear therefore that O-GlcNAc modification of mitochondrial proteins is important in regulating mitochondrial function and provides a critical link between nutrient and redox signaling (Figure 1B).
4. Bioenergetic reserve capacity and the response to oxidative stress
Although the impact of oxidative or nitrative stress on mitochondria have long been an active theme in redox biology research it has only recently become possible to assess the effects of reactive species on cellular bioenergetics [91]. It is now possible to measure the time course of effects of oxidative stress on other cellular processes, such as autophagy, and integrate these with bioenergetic function [6]. This integrative capability is important because metabolism encompasses multiple pathways outside the organelle, which are limiting to mitochondrial function by controlling substrate supply or signaling. Understanding these mechanisms greatly extends the potential targets through which oxidative or nitrative stress can modulate metabolic function which has obvious implications for both the development of therapeutics and the mechanisms of pathological processes. It has been known for some time that cellular bioenergetic capacity substantially exceeds that required for meeting ATP demand [92]. To define cellular bioenergetics, oxygen consumption rate is measured during the sequential addition pharmacological inhibitors of oxidative phosphorylation [93, 94]. The basic elements of this protocol and the interpretation are outlined in Figure 2. One feature of particular interest is the residual oxygen consumption rate after the addition of antimycin which is due to non-electron transport oxygen consumption and is generally termed non-mitochondrial. This measure is an integrated sum of a number of different enzymes and pathways some of which could also reside in the mitochondria and varies among cell types [6]. Interestingly, we have few quantitative measures of the capacity of non-mitochondrial pathways which consume oxygen and largely rely on extrapolated data obtained over 40 years ago. We now know that there are multiple controlled enzymatic processes which consume oxygen including the NADPH oxidases and nitric oxide synthases. For example, on the basis of experiments with isolated liver mitochondria and respiratory chain inhibitors it was calculated in the early 1970's that mitochondrial superoxide production was 1–2% of oxygen consumption [1, 95]. This was complemented with experiments in hepatocytes in which cell fractionation was coupled with measurements of hydrogen peroxide generation in each fraction. From this it was calculated that hydrogen peroxide production was 10% of the total oxygen consumption by the cell [4]. As we have recently shown these measures underestimates the contribution of oxygen consuming pathways beyond mitochondrial cytochrome c oxidase and is approximately 30-40% of the basal oxygen consumption in hepatocytes but varies dramatically among different cell types [6, 91]. The widely held view, based on these earlier studies, that mitochondria are responsible for over 95% of the oxygen consumption clearly need to be reconsidered.
Figure 2. The mitochondrial stress test.

A typical profile is shown in which basal oxygen consumption rate (OCR) is allowed to stabilize before the sequential addition of oligomycin which inhibits ATP synthase preventing protons returning to the mitochondria and so decreasing OCR (ATP linked). The OCR remaining after oligomycin is ascribed to movement of ions across the mitochondrial inner membrane and proton translocation not involving the ATP synthase-collectively termed proton leak. The addition of the uncoupler, FCCP allows protons to flow into the mitochondrion increasing OCR to the level which can be sustained by endogenous substrates. The final addition of antimycin A and/or rotenone results in a residual OCR is ascribed to oxygen consuming processes outside mitochondrial electron transport. The insert shows a simplified proton circuit for oxidative phosphorylation showing how the proton gradient controls OCR. This time course is annotated to show the relative contribution of non-respiratory chain oxygen consumption, ATP-linked oxygen consumption, the maximal OCR after the addition of FCCP, and the reserve capacity (Maximal – basal OCR) of the cells.
The bioenergetic reserve capacity can be estimated from the parameters using the cellular mitochondrial stress test as the difference between the basal and uncoupler stimulated oxygen consumption rate (Figure 2). The effects of redox stressors vary depending on the reactivity of the oxidants and also the cell type [91]. For example, in the comparison of the response of different cell lines to 4-hydroxynonenal we found that basal respiration was suppressed in SH-SY5Y cells but stimulated in rat ventricular cardiomyocytes. An uncoupling effect was evident in MES13 cells but not SH-SY5Y cells. In vascular smooth muscle cells the threshold at which 4-hydroxynonenal adducts increased over 5 fold and induction of heme oxygenase-1 was suppressed was the concentration at which reserve capacity was lost and bioenergetic dysfunction occurred.
The combination of the bioenergetic measurements shown in Figure 2 with measures of signaling or metabolic changes reveals the sequence of events in the response to mediators of oxidative stress. This is illustrated in Figure 3 in which the prosthetic group in hemoglobin, hemin, is exposed to endothelial cells and changes in oxygen consumption rate (OCR) monitored with time and correlated with measures of autophagy and apoptosis [96]. From these data it is evident that one of the earliest responses in response to hemin is a change in energetics followed by the initiation of autophagy then ultimately apoptosis. Hemin toxicity is important in the pathologies associated with blood transfusions and in hemolytic anemias [96]. In further support of the hypothesis that reserve capacity is important in combatting oxidative stress the response to stress can be modulated by controlling mitochondrial fuel supply [97]. Interestingly, it is the loss of bioenergetic reserve capacity which appears to establish the threshold at which protective pathways fail and cell death ensues. Reserve capacity is then a critical determinant of the ability of cells to respond and adapt to oxidative or nitrative stress perhaps through the energetic demands needed for repair pathways such as autophagy or perhaps through other signaling related mechanisms. The reserve capacity is sensitive to the substrate supply and can potentially be manipulated by changing the fuel availability [97, 98]. These data then establish a direct link between cellular bioenergetic function and the response to oxidative stress.
Figure 3. Integrating Bioenergetics and Cellular Responses to Stress.

These data have been adapted from [96] and show the injection of hemin (25 μM) onto bovine aortic endothelial cells. Panel A: measurement of OCR (oxygen consumption rate) following hemin injection at 24 minutes followed by the mitochondrial stress test following the protocol shown in Figure 2. Panel B: time dependent decreased in basal OCR showing detectable changes 30 min after hemin addition. Panel C: in a parallel plate protein samples were prepared at the times shown and the LC3 conversion to LC3-II determined. The arrow shows the 2 hour point at which time a detectable change in the levels of LC3-II were found. Panel D: Samples were also taken for measurement of the loss of Procaspase 9 as one marker of apoptosis and the arrow shows that these changes were not detectable until the 4 hour time point. Panel E: Reserve Capacity decreases dramatically after 4 hours exposure at the concentration which is the threshold for toxicity.
An emerging concept in the energetics field is that the mitochondrial stress profile can be used as an index of mitochondrial health [49, 99-104]. By indicating how close a cell is to operating at its bioenergetic limit, predictions can be made regarding the cellular response to stress or increased energy demand. This has been demonstrated in model systems in the cellular response to oxidative stress, as well as clinically in patients with diabetes, sickle cell disease, asthma, autism and following cardiac surgery [49, 99-107].
The extent to which changes in pathological conditions represent a bioenergetic dysfunction or metabolic adaptation is an area which will require more in depth research. This is particularly evident in the cancer field since metabolic plasticity is emerging as a potentially important mechanism in the development of resistance to chemotherapy. One of the clearest examples is in the development of resistance to telozomide, which is a front line treatment for glioma. Resistance to this drug is associated with reversal of the Warburg effect due to changes in the mitochondrial respiratory chain (predominantly at cytochrome c oxidase) and an increase in reserve capacity [108].
5. The regulatory role of the Keap1-Nrf2 (NF-E2–related factor) system in metabolism
Studies with isolated mitochondria and more recently with cellular bioenergetics have clearly shown that mitochondrial metabolism is a target, regulator, and generator of reactive species. In a cellular context, these findings imply that the regulation of redox dependent pathways should cross-talk with metabolic pathways. In support of this hypothesis, it is now becoming clear that the Keap1/Nrf2 pathway is intricately linked with metabolism. Keap1 has over 20 redox sensitive thiols [35, 109-111], and can form a complex with NF-E2-related factor (Nrf2) and directs its ubiquitination and degradation by the Cullin-3-dependent proteasome. Modification of the redox active thiols on Keap1 results in loss of this adaptor function for the ubiquitin ligase complex releasing Nrf2 from the complex ultimately facilitating its nuclear entry and activation of electrophile response element (EpRE-also called the antioxidant response element)-dependent transcription. The versatility of Keap1 as a redox sensor allows endogenous stress to be surveyed by phylogenetically conserved sensors responsive to nitric oxide, zinc and alkenals [112].
A key concept that is emerging in redox signaling is that the site for generation of the redox signal is in close proximity to the redox sensor. This appears to be the case for a sub-population of Keap1/Nrf2 which is associated with the outer membrane of the mitochondrion [113]. In addition, the redox regulation of mitochondrial thiols in the reduced state is required for the transcriptional regulation of heme oxygenase-1, an antioxidant protein regulated by the Keap1/Nrf2 system [114]. The increased production of mitochondrial hydrogen peroxide in response to exercise provides a potential component in the signaling pathways which integrate both mitochondrial and cytosolic redox status utilizing dynamic redox sensors such as Keap1/Nrf2 [115, 116].
Nrf2 dependent signaling varies with age and is important in cardiac remodeling in response to exercise. In the heart, extensive endurance exercise elevates the levels of NOX4, SOD2, NRF2, but not NOX-2 or SOD1 protein levels in WT young mice, and the mRNA of Nqo1, catalase, Gclm, Gclc, Gpx-1, Gsr, G6pdx without changing the mRNA of heme oxygenase-1. While the expression of these mRNAs are all similar between young Nrf2-/- and WT mice in the heart without exercise, their induction in response to exercise was attenuated in Nrf2-/- mice [117]. Consistent with these data G6PD, GCLM, heme oxygenase-1 but not catalase protein levels are elevated by exercise in the WT young heart. While no significant differences in GCS, G6PD, or SOD1 mRNA between young WT and Nrf2-/- skeletal muscles except NQO1 mRNA is significantly decreased and catalase mRNA is significantly increased in Nrf2-/- mice, GCS, G6PD, SOD1, NQO1 and catalase mRNA are all significantly decreased by 20-50%, GSR, catalase, GCS, G6PD and NQO1 proteins are all significantly decreased in old Nrf2-/- compared to WT skeletal muscles [118]. Importantly, the normal aging process is also accompanied by a similar reduction in Nrf2 transcriptional activity as the electrophile responsive element (EpRE) gene products are decreased in the skeletal muscle of aged WT mice [118, 119] highlighting the impact of age on pathologies with an oxidative stress component [118-120]. Although cardiac function is maintained in Nrf2-/- mice without exercise, these animals are highly susceptible to oxidative stress. For example, pathological cardiac remodeling occurs in the Nrf2-/- upon intense exercise stress [121].
Nrf2 has also been shown to be important for mitochondrial function and ROS production in cultured cells [115]. Although mechanisms unclear, Nrf2 knockout has been shown to impact mitochondrial membrane potential and ATP [122]. Interestingly it has been demonstrated that UCP3 has an EpRE and Nrf2 binds to UCP3 promoter after exposure to hydrogen peroxide. This suggests that UCP3-mediated proton leak in response to hydrogen peroxide may influence cell survival [123]. Furthermore, nuclear respiratory factor (NRF)-1 also has EpRE and is under Nrf2 regulation in cardiomyocytes. Carbon monoxide stimulates Nrf2 nuclear translocation, Nrf2 binding to the Nrf-1 promoter, and mitochondrial biogenesis [124]. With the potential for therapeutic intervention, the interplay between mitochondrial deficiencies with age related changes in Nrf2 activity warrants further investigation.
While endogenous Nrf2 function decreases with age, chronic exercise training can restore Nrf2 transcriptional activity. In addition to the many benefits of exercise, preservation of Nrf2 signaling may represent an important non-pharmacological strategy to combat the typical incidence of oxidative stress seen with cardiac aging [120]. However, exercise training variables are of critical importance when considering prescription of physical activity for pathological conditions characterized by oxidative stress. For example, aged WT mice exposed to high intensity acute exercise exhibit impairment in the antioxidant response, and fail to meet the demands of increased ROS production induced by exercise. Interestingly, this phenomenon is reversed when aged mice are subjected to chronic training regimen of moderate intensity [120]. As a direct test of the requirement for Nrf2 in the response to training we demonstrated that over-expression of Nrf2 protein in aged mice coupled with moderate training restored the exercise-dependent induction of EpRE regulated genes. Further, enhanced nuclear translocation of Nrf2 in these animals directly induces expression of antioxidant genes that are typically downregulated with advancing age [118, 120]. Interestingly, reports have demonstrated a relationship between heme oxygenase-1, Nrf2 and mitochondrial biogenesis in cardiac tissue [124]. The transcriptional co-activator PGC1-α is among the most widely studied factors mediating the beneficial mitochondrial adaptations following exercise. PGC1-α is a transcriptional target of Nrf2, elucidation of molecular mechanisms responsible for Nrf2 mediated PGC1-α induction and the resultant effects on mitochondrial parameters would be interesting in the context of adaptation to exercise in aged populations.
6. Autophagy, Redox Regulation and Metabolic Health
Recycling of cellular materials is an essential aspect of cell biology and keeping the cells mitochondria healthy is a complex and integrated process known in general as autophagy and mitophagy when referring to the mitochondrion. This pathway is a lysosomal mediated process that plays an essential role in the response to metabolic and oxidative stress in addition to clearance of damaged proteins and organelles. Indeed, it was first discovered during development and in response to nutrient starvation and later found to be responsive to epigenetic and post-translational regulation, redox regulation, cellular bioenergetic status and circadian rhythm [125-139]. More than 40 AuTophaGy related (ATG) genes have been identified. Part of the core machinery involved in the autophagy process includes the mTOR pathway which senses glucose and amino acid starvation and is intrinsically linked to metabolism and proteotoxic stress. In support of this overexpression of Atg5 in the whole body and Atg7 in the heart has been shown to prolong lifespan or confer cardiac protection against accumulation of protein aggregates, respectively [140, 141].
Furthermore, autophagy protein Atg5 deficiency in the heart led to accumulation of ubiquitinated proteins, increased ER stress and apoptosis, cardiac hypertrophy, left ventricular dilatation and contractile dysfunction [142]. We have in the past shown that autophagy is regulated by oxidants, nitric oxide, mitochondrial inhibitors and proteotoxic stress and inhibition of autophagy is detrimental to cell survival [143-150] (Figure 4). In beta cells with insulin 2 gene mutation and misfolding, decreased mitochondrial quality is associated with decreased p62 and PARKIN and accumulation of LC3-II in the mitochondria [145]. Similarly, exposure of endothelial cells to hemin resulted in mitochondrial dysfunction, protein modification by oxidized lipids, autophagosomal accumulation and increased LC3 localization to the mitochondria and inhibition of autophagy exacerbates hemin cytotoxicity [150].
Figure 4. Autophagy and the response to oxidative stress.

Redox, mitochondrial and proteotoxic stress damage cellular proteins and organelles and if not cleared by autophagy, cell death occurs and contributes to metabolic and cardiovascular pathologies. Upregulation of autophagy has been shown to provide beneficial effects on cell survival.
Cardiac transgenic mice overexpressing PINK1 exhibited attenuated mitochondrial fragmentation, decreased myocardial infarction, and preserved cardiac function in response to ischemia-reperfusion [151]. Parkin translocation to the mitochondrial has been demonstrated in HL-1 cells in response to simulated ischemia, in Langendorff-perfused rat hearts, and in mice subjected to regional ischemic preconditioning [152]. Parkin overexpression ameliorates mitochondrial and cardiac functional decline in aged hearts, decreases senescence-associated β–galactosidase activity and proinflammatory phenotypes [153]. In the heart, Parkin translocation and mitophagy may also occur independent of PINK1 [154] (Figure 5).
Figure 5. Mitophagy mechanisms have been shown to involve PINK1-PARKIN, DRP1, and MFN1/2 mediated mechanisms.

Mitophagy plays an important role in mitochondrial quality control and the involvement of PARKIN-PINK1, as well as fission/fusion proteins has been demonstrated. PINK1 stabilization in the mitochondria is facilitated by mitochondrial membrane depolarization. PINK1 phosphorylates ubiquitin and enables PARKIN translocation to the mitochondria, leading to ubiquitination of several mitochondrial proteins, resulting in p62 and LC3 recruitment and autophagosomal engulfment of the mitochondrion. Interaction between MFN2 and PARKIN may play a key role in integrating fusion machinery and PARKIN mediated mitophagy.
Controlling mitochondrial morphology is an essential element in the mitochondrial life cycle and required for mitophagy. Cardiomyocyte-specific deletion of Drp1 leads to decreased contractility and P9-P11 lethality with enlargement of mitochondria and decreased mitochondrial respiration [155]. Cardiac deletion of fission protein Drp1, or double deletion of fusion protein Mfn1 and Mfn2, leads to increased autophagy substrate P62, changed mitochondrial morphology and cardiomyopathies [156]. Cardiac inducible deletion of Drp1 leads to mitochondrial elongation, suppression of autophagic and mitophagic flux, left ventricle dysfunction, and enhanced sensitivity to fasting and ischemia-reperfusion [157]. Interestingly, Mfn1 deficient myocytes exhibit small spherical mitochondria and resistance to hydrogen peroxide induced cell death [158], while Mfn2 deficient cardiomyocytes have large mitochondria and better recovery following reperfusion injury [159]. One explanation for the enlargement of mitochondria in Mfn2 deficient cardiomyocytes is that Mfn2 is a receptor for Parkin and thus Mfn2 deficiency leads to decreased elimination of damaged mitochondria [160] (Figure 5).
Because of the importance to regulate autophagy in response to metabolic and redox stress, the level and activities of the autophagy proteins are highly regulated at epigenetic, transcriptional, post-transcriptional and post-translational levels. Histone acetylation and DNA methylation have been found to regulate the expression of various autophagy genes, including ATG16L2, LC3A, ULK2, BNIP3 and GABARAPL1 [161-168]. Histone deacetylase (HDAC) inhibitor trichostatin A (TSA) has been reported to attenuate cardiac hypertrophy and suppress LC3II increase in response to pressure overload induced by transverse aortic constriction (TAC) [169]. However, an increase in autophagy flux has been found to be associated with, and required in mediating, cardioprotection by histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) in ischemia-reperfusion [170]. However, the mechanisms of the involvement of histone deacetylase in autophagy may not always be related to epigenetic regulation of autophagy genes. Post-translational modification of cytosolic proteins may also play a role in mediating HDAC effects. In the cytosol, HDAC1 is involved in the conversion of LC3-I to LC3-II and HDAC6 is involved in autophagosome maturation [171, 172]. A key regulatory role of protein O-GlcNAcylation on autophagy has also been demonstrated by a recent study that SNAP-29, a protein regulating autophagosome-lysosome fusion is O-GlcNAcylated and decreasing its O-GlcNAc modification promotes autophagy [173]. As a central regulator of metabolism, bioenergetics and redox signaling, circadian rhythm also has a strong impact on autophagy, as key autophagy proteins, including ATG14, ULK, BNIP3, GABARAPL1, and LC3-II are regulated by the circadian clock, and transcription factors including BMAL1 [174, 175]. The cross regulation of autophagy and cellular redox status has been highlighted by findings demonstrating that autophagy adaptor protein p62 is transcriptionally regulated by Nrf2. Reciprocally, autophagy is important for degradation of KEAP1, a cytosolic sequester of Nrf2 and an important protein for Nrf2 degradation by the proteasomes [130] (Figure 6).
Figure 6. Autophagy is regulated by epigenetics, protein O-GlcNAcylation, circadian clock, and cellular redox status.

Histone deacetylases have been shown to participate in autophagy regulation although whether their activities in the nucleus or the cytosol are important for autophagy regulation is still being investigated. HDAC6 recruits an actin-remodeling machinery, and stimulates autophagosome-lysosome fusion and substrate degradation. P62 interaction with HDAC6 regulates its activity. Inhibition or disruption of HDAC1 leads to the conversion of LC3-I to LC3-II. Methylation of ATG16L2, ULK2, LC3A, BNIP3, and GABARAPL1 is associated with their downregulation. In addition, transcription regulation of p62 by antioxidant transcription factor Nrf2, and regulation of ATG14, ULK1, BNIP3, GABARAPL1, and LC3 by clock and BMAL1 have been demonstrated. Post-translational regulation of SNAP29 by O-GlcNAcylation has been shown to attenuate autophagosome-lysosome fusion.
The heart is one of the major organs that exhibit significant autophagic response in starvation. Vacuoles containing lysosomal cathepsin D appear in cardiomyocytes as early as 12 hours after starvation of 8-10 week old GFP-LC3 transgenic mice [176]. LC3II, cathepsin D and ubiquitin are increased [176]. The importance of the autophagic response to starvation has been demonstrated by the observation that inhibition of autophagy by bafilomycin has no effect in fed mice but depressed cardiac function and caused left ventricular dilation in starved mice [176]. Prolonged caloric restriction (40% for 30 weeks) also increased the LC3II/I ratio, but the increase was dampened by bafilomycin consistent with suppressed autophagic flux [177]. Akt2 knockout alleviated cardiac phenotypes and augmented mTOR inhibition induced by caloric restriction [177]. It should be noted that autophagy inhibition has also been associated with cardioprotection during starvation. For example, IGF-1 and salvianolic acid B are cardioprotective for cell survival despite their inhibition of autophagy [178, 179]. However, it is unclear in these studies whether inhibition of autophagy plays an active role in decreasing pathology or only reflects a consequence of decreased oxidative and metabolic stress in response to the cardioprotective agents.
Deterioration in mitochondrial quality is not simply a matter of the loss of energy producing capacity. Indeed, studies with exogenous stressors or models of diabetes have shown that ATP synthesis is maintained in the presence of mitochondrial DNA damage, increased hydrogen peroxide production, thiol oxidation and loss of bioenergetic efficiency [96, 180, 181]. It appears that it is a “gain of function” of the deteriorating mitochondrial population rather than simply the loss of ATP generating capacity that is contributing to the pathological effects of mitophagy failure. For example, it has been shown that mitochondrial DNA that escapes the mitophagic process leads to Toll-like receptor 9-mediated inflammation and cardiomyopathy, with a phenotype promoted by lysosomal DNase II deficiency and attenuated by TLR9 ablation [182]. The loss of control of the redox signals from the mitochondrion due to damaged complexes generating uncontrolled hydrogen peroxide or superoxide may also be a contributory element. If these dysfunctional mitochondria are not removed by the controlled process of mitophagy then release from damaged cells can exacerbate an underlying pathology.
7. Integration of Mitochondrial Function, Autophagy and Circadian Control of Metabolism
Metabolism is exceedingly dynamic in nature, allowing rapid and dramatic changes in flux in response to the biological and temporal variation in stimuli and physiological and pathological stressors[183]. Recent studies are beginning to define the temporal changes that occur in redox biology and how these interface with bioenergetics and metabolism in general. Indeed, a central concept in the contemporary view of metabolism is that cells/organs/organisms not only respond to changes in their environment, but also anticipate these fluctuations before they occur. In general, mammals have two primary behavioral oscillations to contend with on a daily basis; namely awake/sleep and feeding/fasting cycles. Foraging for food, avoidance of predation, and reproduction during the awake period are energetically demanding, and these energetic demands remain even if the animal in the wild is not successful in its forage for food. Organisms with the molecular machinery that can anticipate this scenario (i.e., physical activity/energetic demand rhythms independent of feeding status) have an evolutionary selective advantage.
At a cellular and molecular level, increased energetic demand could be anticipated by increased oxidative metabolism (for ATP generation) at that time and/or concomitant decreases in ATP utilization by processes that do not immediately benefit locomotion and cognitive function. One such process is cellular growth and repair. Protein and organelle turnover are energetically demanding processes, which could potentially compete with contractile/cognitive function during the active period, in terms of ATP utilization. It is noteworthy that during periods of increased physical activity, the likelihood of protein damage (e.g., by reactive oxygen/nitrogen species) is increased. One would therefore predict that cells would upregulate antioxidant defenses during the active period in anticipation of increased oxidative stress at this time. Replacement of damaged proteins/organelles immediately following physical activity (e.g., during the sleep phase) would likely facilitate preparation of the subsequent active/awake period. Accordingly, processes such as autophagy, proteolysis, mitochondrial fission/fusion and biogenesis, as well as protein synthesis would be predicted to be increased during the sleep phase. In this section will discuss the potential mechanisms orchestrating temporal partitioning of metabolic processes, and highlight their interplay with redox biology.
As mentioned above, oxidative metabolism and mitochondrial function are likely increased at the beginning/middle of the active period, in anticipation of increased energetic demand and oxidant production at this time. At the whole body level, oxygen consumption rises sharply at the onset of the sleep-to-wake transition, and remains elevated throughout the active period, concomitant with increased physical activity and food intake [184]. However, when oxidative metabolism capacity is assayed in isolated tissues, similar temporal patterns are often observed, suggesting that intrinsic properties fluctuate. For example, murine liver homogenates exhibit greatest fatty acid oxidation rates at the sleep-to-wake transition [185]. In the murine heart, rates of glucose oxidation peak in the middle of the active period [186]. Although few studies have directly assessed oscillations in mitochondrial function over the course of the day, Simon et al revealed a greater respiratory control ratio of rat brain mitochondria during the active period, indicating a better coupling of mitochondria at this time [187]. Interestingly, this can be interpreted as an increased efficiency for ATP production but also regulation of mitochondrial superoxide and hydrogen peroxide for cell signaling and the production of heat.
A number of studies of examined the oscillation of key redox sensitive pathways in metabolically active tissues, with respect to time-of-day. Lapenna et al reported increased glutathione levels and glutathione transferase activity in the rat heart at the beginning of the active period, although this was not sufficient to suppress oxidative stress induced cardiac damage at this time [188]. Different organs appear to have distinct oscillatory patterns. For example, glutathione levels, as well as the expression of glutathione peroxidase, glutathione transferase, and catalase are elevated during the light/sleep phase in the mouse liver consistent with a time-of-day dependent regulation of the Keap1-Nrf2 pathway [189]. In the liver this is functionally important and may explain the decreased acetaminophen hepatotoxicity during the light/sleep phase [190]. Interestingly, hepatic metallothionein levels peak at the beginning of the active period, which may afford some level of enhanced antioxidant defense at this time [191].
Mitochondrial oxidative metabolism is clearly elevated in tissues such as the liver and heart during the active period. During this period of time, there is an elevation in muscular contraction and blood pressure, as well as nutrient signals such as insulin and amino acids in an ad libitum fed animal, all of which are known to promote protein synthesis. Consistent with this milieu, skeletal muscle protein synthesis peaks during the active period [192]. However, liver protein synthesis is elevated at the beginning of the sleep phase, while cardiac protein synthesis increases closer to the middle of the sleep phase [192-194]. These observations suggest that that there is tissue-specific control of protein synthesis, in terms of time-of-day-dependent oscillations. Although little is known regarding daily oscillations in protein degradation, evidence exists in support of the concept that both proteasome activity and autophagy likely exhibit 24-hr rhythms [195, 196]. In the latter case, histologic and biochemical approaches suggest increased autophagic flux in the liver and heart during the sleep phase [197].
It is clear that an evidence-based model for the temporal partitioning of metabolic processes is emerging although the cross-talk to redox regulated pathways is less well understood. Studies in the heart have shown increased oxidative metabolism and antioxidant potential during/at the beginning of the active period, while protein synthesis and autophagy peak during the sleep phase. Moreover, these rhythms are likely orchestrated by the synchronization of extrinsic (e.g., neurohumoral factors) and intrinsic (e.g., circadian clock) influences. The selective advantage of circadian clocks is anticipation, thus preparing cells/organs prior to the onset of stimuli/stresses. Disruption of these rhythmic mechanisms invariably results in cardiometabolic diseases, such as obesity, type 2 diabetes mellitus, and cardiovascular disease.
8. Summary
Shown schematically in Figure 7 are the key elements discussed in this review linking together Redox Biology, Bioenergetics and Chronobiology. Taken together these findings suggest that the reversibility of redox-dependent deleterious changes in the reserve capacity through removal of damaged mitochondria by mitophagy and stimulation of biogenesis may be critical for recovery. These concepts are readily translatable to the human population. With the development of methods to measure bioenergetic health the impact of chronobiology and the response to oxidative stress could be integrated in human populations subject to sleep disturbance such as shift workers.
Figure 7. Interactions between Redox Signaling and Metabolic Networks.
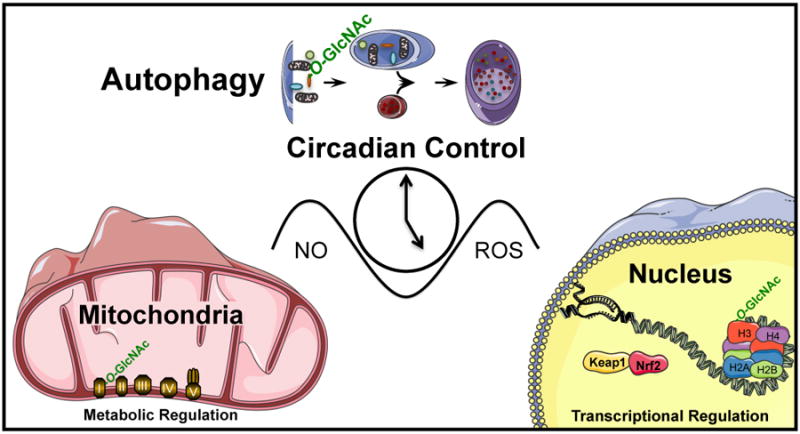
In this review we have described the interactions between redox dependent pathways encompassing the signaling node controlled by the GlcNAc pathway, regulation by biological clocks, mitochondrial metabolism and epigenetics.
Highlights.
Cellular Bioenergetics represents an integrated metabolic program.
Integration is mediated through epigenetics, autophagy and the Keap1/Nrf2 pathway.
Post-translational modification by O-GlcNAc modulates metabolic responses to stress.
Circadian control of metabolism is a critical element in maintaining physiological metabolism.
Acknowledgments
This work was supported by R00 HL111322 (AW), R01 HL122975 (MEY,JC), R01 HL123574, R01 HL122975 (MEW),R01 HL101192 (JC), R01 R01HL118067, NIA R03AG042860 and AHA-BGIA (RS). This work was also supported by UAB AMC21 reload multi-investigator grant (all authors). All authors reported that they have no relationships with industry relevant to the contents of this paper to disclose. Some figures were produced using free images modified from Servier Medical Art (www.servier.com).
Abbreviations
- ARE
Antioxidant reponse element
- ATP
Adenosine triphosphate
- 5-hmC
Hydroxymethylcytosine
- ATG
AuTophaGy related
- BNIP
Bcl-2/adenovirus E18 19-kDa-interacting protein
- Drp1
Dynamin related protein 1
- EpRE
electrophile response element
- eNOS
Endothelial Cell, Nitric Oxide Synthase 3
- G6PD
Glucose-6-phosphate dehydrogenase
- GCLC/GCLM
Glutamate cysteine ligase
- GCS
glutamylcysteine synthetase
- GPX1
Glutathione peroxidase 1
- GSR
glutathione-reductase
- GTN
Nitroglycerin
- HO-1
heme oxygenase-1
- HBP
Hexosamine Biosynthesis Pathway
- HDAC
Histone deacetylase
- H2O2
Hydrogen peroxide
- IGF
Insulin growth factor-1
- KDM3A
Lysine Demethylase 3A
- KEAP1
Kelch-like ECH-associated protein 1
- mTOR
Mammalian target of rapamycin
- LC3
Microtubule associated protein 1 light chain subunit 3
- mPTP
mitochondrial permeability transition pore
- •NO
Nitric Oxide
- eNOS
endothelial nitric oxide synthase
- NOX
NADPH oxidases
- NQO1
NAD(P)H:quinone oxidoreductase 1
- NRF-1
Nuclear respiratory factor-1
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- O-GlcNAc
O-linked N-acetylglucosamine
- OGT
O-GlcNAc Transferase
- PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PINK1
PTEN-induced kinase 1
- p62
Sequestosome 1
- SAHA
Suberoylanilide hydroxamic acid
- SOD
Superoxide dismutase
- TET
Methylcytosine Dioxygenase Ten-eleven Translocation
- TLR9
Tolllike receptor 9
- TAC
Transverse aortic constriction
- TSA
Trichostatin A
- UCP3
uncoupling protein 3
- ULK
Uncoordinated family member (unc)-51-Like Kinase
- VDAC
Voltage dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 4.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, Darley-Usmar V, Shiva S, Cifuentes-Pagano E, Freeman BA, Gladwin MT, Pagano PJ. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free radical biology & medicine. 2011;51:1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill BG, Benavides GA, Lancaster JR, Jr, Ballinger S, Dell'Italia L, Jianhua Z, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biological chemistry. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, Darley-Usmar VM. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Laboratory investigation; a journal of technical methods and pathology. 2013;93:690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox biology. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballinger SW. Mitochondrial dysfunction in cardiovascular disease. Free radical biology & medicine. 2005;38:1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Levonen AL, Patel RP, Brookes P, Go YM, Jo H, Parthasarathy S, Anderson PG, Darley-Usmar VM. Mechanisms of cell signaling by nitric oxide and peroxynitrite: from mitochondria to MAP kinases. Antioxidants & redox signaling. 2001;3:215–229. doi: 10.1089/152308601300185188. [DOI] [PubMed] [Google Scholar]
- 11.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free radical biology & medicine. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 12.Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free radical biology & medicine. 2013;63:207–221. doi: 10.1016/j.freeradbiomed.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Molecular cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 14.Cadenas E. Mitochondrial free radical production and cell signaling. Mol Aspects Med. 2004;25:17–26. doi: 10.1016/j.mam.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 16.Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. American journal of physiology Cell physiology. 2007;292:C1993–2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 17.Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. The international journal of biochemistry & cell biology. 2015;59:167–181. doi: 10.1016/j.biocel.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell metabolism. 2015;22:207–218. doi: 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Rahman S. Emerging aspects of treatment in mitochondrial disorders. J Inherit Metab Dis. 2015;38:641–653. doi: 10.1007/s10545-015-9855-3. [DOI] [PubMed] [Google Scholar]
- 20.Narendra DP, Youle RJ. Neurodegeneration: Trouble in the cell's powerhouse. Nature. 2012;483:418–419. doi: 10.1038/nature10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santos RX, Correia SC, Carvalho C, Cardoso S, Santos MS, Moreira PI. Mitophagy in neurodegeneration: an opportunity for therapy? Current drug targets. 2011;12:790–799. doi: 10.2174/138945011795528813. [DOI] [PubMed] [Google Scholar]
- 22.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annual review of pathology. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krzywanski DM, Moellering DR, Fetterman JL, Dunham-Snary KJ, Sammy MJ, Ballinger SW. The mitochondrial paradigm for cardiovascular disease susceptibility and cellular function: a complementary concept to Mendelian genetics. Laboratory investigation; a journal of technical methods and pathology. 2011;91:1122–1135. doi: 10.1038/labinvest.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison CM, Pompilius M, Pinkerton KE, Ballinger SW. Mitochondrial oxidative stress significantly influences atherogenic risk and cytokine-induced oxidant production. Environmental health perspectives. 2011;119:676–681. doi: 10.1289/ehp.1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circulation research. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell T, Johnson MS, Ouyang X, Chacko BK, Mitra K, Lei X, Gai Y, Moore DR, Barnes S, Zhang J, Koizumi A, Ramanadham S, Darley-Usmar VM. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita(+/Ins2)-derived beta-cells. American journal of physiology Endocrinology and metabolism. 2013;305:E585–599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chemistry & biology. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 28.Rehman H, Ramshesh VK, Theruvath TP, Kim I, Currin RT, Giri S, Lemasters JJ, Zhong Z. NIM811 (N-methyl-4-isoleucine cyclosporine), a mitochondrial permeability transition inhibitor, attenuates cholestatic liver injury but not fibrosis in mice. The Journal of pharmacology and experimental therapeutics. 2008;327:699–706. doi: 10.1124/jpet.108.143578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part II. Expert opinion on therapeutic targets. 2010;14:497–511. doi: 10.1517/14728221003730434. [DOI] [PubMed] [Google Scholar]
- 30.Carreira RS, Lee P, Gottlieb RA. Mitochondrial therapeutics for cardioprotection. Current pharmaceutical design. 2011;17:2017–2035. doi: 10.2174/138161211796904777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy MP. Targeting lipophilic cations to mitochondria. Biochimica et biophysica acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 32.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP, Sotgia F. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell cycle. 2011;10:4047–4064. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free radical biology & medicine. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 35.Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. The Biochemical journal. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fetterman JL, Zelickson BR, Johnson LW, Moellering DR, Westbrook DG, Pompilius M, Sammy MJ, Johnson M, Dunham-Snary KJ, Cao X, Bradley WE, Zhang J, Wei CC, Chacko B, Schurr TG, Kesterson RA, Dell'italia LJ, Darley-Usmar VM, Welch DR, Ballinger SW. Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. The Biochemical journal. 2013;455:157–167. doi: 10.1042/BJ20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3) Redox biology. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox biology. 2015;4:242–259. doi: 10.1016/j.redox.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. The Biochemical journal. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox biology. 2014;2:82–90. doi: 10.1016/j.redox.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nature reviews Endocrinology. 2014;10:466–475. doi: 10.1038/nrendo.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA, Maywood ES, Hastings MH, Baliga NS, Merrow M, Millar AJ, Johnson CH, Kyriacou CP, O'Neill JS, Reddy AB. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stangherlin A, Reddy AB. Regulation of circadian clocks by redox homeostasis. The Journal of biological chemistry. 2013;288:26505–26511. doi: 10.1074/jbc.R113.457564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tavakoli S, Zamora D, Ullevig S, Asmis R. Bioenergetic Profiles Diverge During Macrophage Polarization: Implications for the Interpretation of 18F-FDG PET Imaging of Atherosclerosis. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2013;54:1661–1667. doi: 10.2967/jnumed.112.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravi S, Mitchell T, Kramer PA, Chacko B, Darley-Usmar VM. Mitochondria in monocytes and macrophages-implications for translational and basic research. The international journal of biochemistry & cell biology. 2014;53C:202–207. doi: 10.1016/j.biocel.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 51.Salminen A, Haapasalo A, Kauppinen A, Kaarniranta K, Soininen H, Hiltunen M. Impaired mitochondrial energy metabolism in Alzheimer's disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog Neurobiol. 2015;131:1–20. doi: 10.1016/j.pneurobio.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Kumar R, Li DQ, Muller S, Knapp S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene. 2016 doi: 10.1038/onc.2015.513. [DOI] [PubMed] [Google Scholar]
- 53.Wende AR. Post-translational modifications of the cardiac proteome in diabetes and heart failure. Proteomics Clin Appl. 2016;10:25–38. doi: 10.1002/prca.201500052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schones DE, Leung A, Natarajan R. Chromatin modifications associated with diabetes and obesity. Arterioscler Thromb Vasc Biol. 2015;35:1557–1561. doi: 10.1161/ATVBAHA.115.305041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 56.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122:1271–1278. doi: 10.1289/ehp.1408418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Daiber A, Münzel T. Organic nitrate therapy, nitrate tolerance, and nitrate-induced endothelial dysfunction: Emphasis on redox biology and oxidative stress. Antioxidants & Redox Signaling. 2015;23:899–942. doi: 10.1089/ars.2015.6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vasudevan D, Hickok JR, Bovee RC, Pham V, Mantell LL, Bahroos N, Kanabar P, Cao XJ, Maienschein-Cline M, Garcia BA, Thomas DD. Nitric oxide regulates gene expression in cancers by controlling histone posttranslational modifications. Cancer Res. 2015;75:5299–5308. doi: 10.1158/0008-5472.CAN-15-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem. 2013;288:16004–16015. doi: 10.1074/jbc.M112.432294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olivier-Van Stichelen S, Hanover JA. You are what you eat: O-linked N-acetylglucosamine in disease, development and epigenetics. Curr Opin Clin Nutr Metab Care. 2015;18:339–345. doi: 10.1097/MCO.0000000000000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hardivillé S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014;20:208–213. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Darley-Usmar VM, Ball LE, Chatham JC. Protein O-linked beta-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. Journal of molecular and cellular cardiology. 2012;52:538–549. doi: 10.1016/j.yjmcc.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai D, Yin S, Yang J, Jiang Q, Cao W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res Ther. 2015;17:1–11. doi: 10.1186/s13075-015-0774-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Fischer KA, Löhr CV, Williams DE, Ho E, Yamamoto M, Lieberman DA, Dashwood RH. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clinical Epigenetics. 2015;7:1–12. doi: 10.1186/s13148-015-0132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mishra M, Zhong Q, Kowluru RA. Epigenetic modifications of Nrf2-mediated glutamate–cysteine ligase: Implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic Biol Med. 2014;75:129–139. doi: 10.1016/j.freeradbiomed.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boyanapalli SS, Tony Kong AN. “Curcumin, the King of Spices”: Epigenetic Regulatory Mechanisms in the Prevention of Cancer, Neurological, and Inflammatory Diseases. Curr Pharmacol Rep. 2015;1:129–139. doi: 10.1007/s40495-015-0018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu X, Sun J, Li S, Wu X, Li L. Oxidative stress induces DNA demethylation and histone acetylation in SH-SY5Y cells: potential epigenetic mechanisms in gene transcription in Aβ production. Neurobiol Aging. 2013;34:1069–1079. doi: 10.1016/j.neurobiolaging.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 68.Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J Biol Chem. 2016;291:3531–3540. doi: 10.1074/jbc.M115.675488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z. Ten-eleven translocation 1 (Tet1) is regulated by O-Linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem. 2013;288:20776–20784. doi: 10.1074/jbc.M113.460386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–13674. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–555. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates J, Hoshijima M, Dillmann WH. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. The Journal of biological chemistry. 2012 doi: 10.1074/jbc.M112.390682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palaniappan KK, Hangauer MJ, Smith TJ, Smart BP, Pitcher AA, Cheng EH, Bertozzi CR, Boyce M. A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell reports. 2013;5:546–552. doi: 10.1016/j.celrep.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dassanayaka S, Readnower RD, Salabei JK, Long BW, Aird AL, Zheng YT, Muthusamy S, Facundo HT, Hill BG, Jones SP. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem J. 2015;467:115–126. doi: 10.1042/BJ20141018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A. 2015;112:6050–6055. doi: 10.1073/pnas.1424017112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zachara NE, O'Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 83.Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. American journal of physiology Cell physiology. 2008;294:C1509–1520. doi: 10.1152/ajpcell.00456.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ngoh GA, Watson LJ, Facundo HT, Jones SP. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids. 2011;40:895–911. doi: 10.1007/s00726-010-0728-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. Journal of molecular and cellular cardiology. 2008;45:313–325. doi: 10.1016/j.yjmcc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, Jones SP. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ Res. 2009;104:41–49. doi: 10.1161/CIRCRESAHA.108.189431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marsh SA, Powell PC, Dell'italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci. 2013;92:648–656. doi: 10.1016/j.lfs.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, Cummins TD, Li Q, Hill BG, Bhatnagar A, Prabhu SD, Bolli R, Jones SP. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem cells. 2013;31:765–775. doi: 10.1002/stem.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci. 2003;116:647–654. doi: 10.1242/jcs.00246. [DOI] [PubMed] [Google Scholar]
- 90.Ma J, Liu T, Wei AC, Banerjee P, O'Rourke B, Hart GW. O-GlcNAcomic Profiling Identifies Widespread O-Linked beta-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function. J Biol Chem. 2015;290:29141–29153. doi: 10.1074/jbc.M115.691741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou LY, Chatham JC, Hill BG, Zhang JH, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biology and Medicine. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. The Biochemical journal. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free radical biology & medicine. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, Zhang J, Darley-Usmar VM. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. American journal of physiology Heart and circulatory physiology. 2012;302:H1394–1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chemico-biological interactions. 2011;191:288–295. doi: 10.1016/j.cbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Diers AR, Broniowska KA, Chang CF, Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. The Biochemical journal. 2012;444:561–571. doi: 10.1042/BJ20120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chacko BK, Kramer PA, Ravi S, Benavides GA, Mitchell T, Dranka BP, Ferrick D, Singal AK, Ballinger SW, Bailey SM, Hardy RW, Zhang J, Zhi D, Darley-Usmar VM. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond) 2014;127:367–373. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kramer PA, Chacko BK, George DJ, Zhi D, Wei CC, Dell'Italia LJ, Melby SJ, George JF, Darley-Usmar V. Decreased Bioenergetic Health Index in monocytes isolated from the pericardial fluid and blood of post-operative cardiac surgery patients. Biosci Rep. 2015 doi: 10.1042/BSR20150161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Czajka A, Ajaz S, Gnudi L, Parsade CK, Jones P, Reid F, Malik AN. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine. doi: 10.1016/j.ebiom.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cardenes N, Corey C, Geary L, Jain S, Zharikov S, Barge S, Novelli EM, Shiva S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123:2864–2872. doi: 10.1182/blood-2013-09-529420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zharikov S, Shiva S. Platelet mitochondrial function: from regulation of thrombosis to biomarker of disease. Biochemical Society transactions. 2013;41:118–123. doi: 10.1042/BST20120327. [DOI] [PubMed] [Google Scholar]
- 104.Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox biology. 2014;2:206–210. doi: 10.1016/j.redox.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xu W, Cardenes N, Corey C, Erzurum SC, Shiva S. Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PloS one. 2015;10:e0132007. doi: 10.1371/journal.pone.0132007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Molecular psychiatry. 2012;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Frye RE, Melnyk S, Macfabe DF. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Translational psychiatry. 2013;3:e220. doi: 10.1038/tp.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oliva CR, Nozell SE, Diers A, Mc Clugage SG, 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, Griguer CE. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. The Journal of biological chemistry. 2010;285:39759–39767. doi: 10.1074/jbc.M110.147504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual review of pharmacology and toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 110.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox biology. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. The Journal of biological chemistry. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 112.McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ricart KC, Bolisetty S, Johnson MS, Perez J, Agarwal A, Murphy MP, Landar A. The permissive role of mitochondria in the induction of haem oxygenase-1 in endothelial cells. The Biochemical journal. 2009;419:427–436. doi: 10.1042/BJ20081350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radical Biology and Medicine. 2015;88, Part B:179–188. doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annual review of pharmacology and toxicology. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Muthusamy VR, Kannan S, Sadhaasivam K, Gounder SS, Davidson CJ, Boeheme C, Hoidal JR, Wang L, Rajasekaran NS. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free radical biology & medicine. 2012;52 doi: 10.1016/j.freeradbiomed.2011.10.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller CJ, Gounder SS, Kannan S, Goutam K, Muthusamy VR, Firpo MA, Symons JD, Paine R, 3rd, Hoidal JR, Rajasekaran NS. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochimica et biophysica acta. 2012;1822:1038–1050. doi: 10.1016/j.bbadis.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 119.Narasimhan M, Hong J, Atieno N, Muthusamy VR, Davidson CJ, Abu-Rmaileh N, Richardson RS, Gomes AV, Hoidal JR, Rajasekaran NS. Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free radical biology & medicine. 2014;71:402–414. doi: 10.1016/j.freeradbiomed.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gounder SS, Kannan S, Devadoss D, Miller CJ, Whitehead KJ, Odelberg SJ, Firpo MA, Paine R, Hoidal JR, Abel ED, Rajasekaran NS. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PloS one. 2012;7 doi: 10.1371/journal.pone.0045697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kumar RR, Narasimhan M, Shanmugam G, Hong J, Devarajan A, Palaniappan S, Zhang J, Halade GV, Darley-Usmar VM, Hoidal JR, Rajasekaran NS. Abrogation of Nrf2 impairs antioxidant signaling and promotes atrial hypertrophy in response to high-intensity exercise stress. Journal of Translational Medicine. 2016;14:1–16. doi: 10.1186/s12967-016-0839-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT, Abramov AY. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biology open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Anedda A, Lopez-Bernardo E, Acosta-Iborra B, Saadeh Suleiman M, Landazuri MO, Cadenas S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free radical biology & medicine. 2013;61:395–407. doi: 10.1016/j.freeradbiomed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 124.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circulation research. 2008;103:1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 126.Zhang J. Teaching the basics of autophagy and mitophagy to redox biologists-Mechanisms and experimental approaches. Redox Biol. 2015;4C:242–259. doi: 10.1016/j.redox.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol. 2013;1:19–23. doi: 10.1016/j.redox.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wani WY, Boyer-Guittaut M, Dodson M, Chatham J, Darley-Usmar V, Zhang J. Regulation of autophagy by protein post-translational modification. Lab Invest. 2015;95:14–25. doi: 10.1038/labinvest.2014.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J. 2015;469:347–355. doi: 10.1042/BJ20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sachdeva UM, Thompson CB. Diurnal rhythms of autophagy: implications for cell biology and human disease. Autophagy. 2008;4:581–589. doi: 10.4161/auto.6141. [DOI] [PubMed] [Google Scholar]
- 132.Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, Hattori N, Wang D, Du Y, Song B, Cao LL, Shen C, Wang L, Wang H, Yang Y, Xie D, Wang F, Ushijima T, Zhao Y, Zhu WG. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy. 2015;11:2309–2322. doi: 10.1080/15548627.2015.1117734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlalla D, Amer AO. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics. 2016 doi: 10.1080/15592294.2016.1144007. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bernard A, Jin M, Gonzalez-Rodriguez P, Fullgrabe J, orme-Axford E, Backues SK, Joseph B, Klionsky DJ. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr Biol. 2015;25:546–555. doi: 10.1016/j.cub.2014.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fullgrabe J, Heldring N, Hermanson O, Joseph B. Cracking the survival code: autophagy-related histone modifications. Autophagy. 2014;10:556–561. doi: 10.4161/auto.27280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15:65–74. doi: 10.1038/nrm3716. [DOI] [PubMed] [Google Scholar]
- 137.Medina DL, Di PS, Peluso I, Armani A, De SD, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Settembre C, Di MC, Polito VA, Garcia AM, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S, Jung YK. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 143.Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, Zhang J, Darley-Usmar VM, Matalon S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med. 2015;85:83–94. doi: 10.1016/j.freeradbiomed.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J, Thannickal VJ. Novel Mechanisms for the Anti-Fibrotic Action of Nintedanib. Am J Respir Cell Mol BioL. 2015 doi: 10.1165/rcmb.2014-0445OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mitchell T, Johnson MS, Ouyang X, Chacko BK, Mitra K, Lei X, Gai Y, Moore DR, Barnes S, Zhang J, Koizumi A, Ramanadham S, Darley-Usmar VM. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita+/Ins2-derived beta-cells. Am J Physiol Endocrinol Metab. 2013;305:E585–E599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Benavides GA, Liang Q, Dodson M, Darley-Usmar V, Zhang J. Inhibition of autophagy and glycolysis by nitric oxide during hypoxia-reoxygenation impairs cellular bioenergetics and promotes cell death in primary neurons. Free Radic Biol Med. 2013;65:1215–1228. doi: 10.1016/j.freeradbiomed.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liang Q, Benavides GA, Vassilopoulos A, Gius D, Darley-Usmar V, Zhang J. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem J. 2013;454:249–257. doi: 10.1042/BJ20130414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dodson M, Liang Q, Johnson MS, Redmann M, Fineberg N, Darley-Usmar VM, Zhang J. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy. 2013;9:1996–2008. doi: 10.4161/auto.26094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Higdon AN, Benavides GA, Chacko BK, Ouyang X, Johnson MS, Landar A, Zhang J, Darley-Usmar VM. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol. 2012;302:H1394–H1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang K, Zhou LY, Wang JX, Wang Y, Sun T, Zhao B, Yang YJ, An T, Long B, Li N, Liu CY, Gong Y, Gao JN, Dong YH, Zhang J, Li PF. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat Commun. 2015;6:7619. doi: 10.1038/ncomms8619. [DOI] [PubMed] [Google Scholar]
- 152.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning Involves Selective Mitophagy Mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 154.Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, Gustafsson AB. PINK1 Is Dispensable for Mitochondrial Recruitment of Parkin and Activation of Mitophagy in Cardiac Myocytes. PLoS ONE. 2015;10:e0130707. doi: 10.1371/journal.pone.0130707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 158.Papanicolaou KN, Ngoh GA, Dabkowski ER, O'Connell KA, Ribeiro RF, Jr, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen Y, Dorn GW. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hervouet E, Claude-Taupin A, Gauthier T, Perez V, Fraichard A, Adami P, Despouy G, Monnien F, Algros MP, Jouvenot M, age-Mourroux R, Boyer-Guittaut M. The autophagy GABARAPL1 gene is epigenetically regulated in breast cancer models. BMC Cancer. 2015;15:729. doi: 10.1186/s12885-015-1761-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bai H, Inoue J, Kawano T, Inazawa J. A transcriptional variant of the LC3A gene is involved in autophagy and frequently inactivated in human cancers. Oncogene. 2012;31:4397–4408. doi: 10.1038/onc.2011.613. [DOI] [PubMed] [Google Scholar]
- 163.Dunwell T, Hesson L, Rauch TA, Wang L, Clark RE, Dallol A, Gentle D, Catchpoole D, Maher ER, Pfeifer GP, Latif F. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer. 2010;9:44. doi: 10.1186/1476-4598-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li Z, Chen B, Wu Y, Jin F, Xia Y, Liu X. Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer. 2010;10:98. doi: 10.1186/1471-2407-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shukla S, Patric IR, Patil V, Shwetha SD, Hegde AS, Chandramouli BA, Arivazhagan A, Santosh V, Somasundaram K. Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth. J Biol Chem. 2014;289:22306–22318. doi: 10.1074/jbc.M114.567032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Swiderek E, Kalas W, Wysokinska E, Pawlak A, Rak J, Strzadala L. The interplay between epigenetic silencing, oncogenic KRas and HIF-1 regulatory pathways in control of BNIP3 expression in human colorectal cancer cells. Biochem Biophys Res Commun. 2013;441:707–712. doi: 10.1016/j.bbrc.2013.10.098. [DOI] [PubMed] [Google Scholar]
- 167.Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468–471. doi: 10.1038/nature12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fullgrabe J, Klionsky DJ, Joseph B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy. 2013;9:1621–1623. doi: 10.4161/auto.25803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, Jiang N, Jessen ME, Warner JJ, Lavandero S, Gillette TG, Turer AT, Hill JA. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Oh M, Choi IK, Kwon HJ. Inhibition of histone deacetylase1 induces autophagy. Biochem Biophys Res Commun. 2008;369:1179–1183. doi: 10.1016/j.bbrc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 172.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, Feng D, Chen S, Zhang H. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16:1215–1226. doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- 174.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 2011;30:4642–4651. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, Nagashima K, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol. 2009;174:1705–1714. doi: 10.2353/ajpath.2009.080875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang Y, Han X, Hu N, Huff AF, Gao F, Ren J. Akt2 knockout alleviates prolonged caloric restriction-induced change in cardiac contractile function through regulation of autophagy. J Mol Cell Cardiol. 2014;71:81–91. doi: 10.1016/j.yjmcc.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 178.Han X, Liu JX, Li XZ. Salvianolic acid B inhibits autophagy and protects starving cardiac myocytes. Acta Pharmacol Sin. 2011;32:38–44. doi: 10.1038/aps.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del CA, Toro B, Battiprolu PK, Aranguiz P, Chiong M, Yakar S, Gillette TG, Hill JA, Abel ED, Leroith D, Lavandero S. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mitchell T, Johnson MS, Ouyang X, Chacko BK, Mitra K, Lei X, Gai Y, Moore DR, Barnes S, Zhang J, Koizumi A, Ramanadham S, Darley-Usmar VM. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita(+/Ins2)-derived beta-cells. American journal of physiology Endocrinology and metabolism. 2013;305:E585–599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mitchell T, Chacko B, Ballinger SW, Bailey SM, Zhang J, Darley-Usmar V. Convergent mechanisms for dysregulation of mitochondrial quality control in metabolic disease: implications for mitochondrial therapeutics. Biochemical Society transactions. 2013;41:127–133. doi: 10.1042/BST20120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Qian J, Scheer FA. Circadian System and Glucose Metabolism: Implications for Physiology and Disease. Trends in endocrinology and metabolism: TEM. 2016;27:282–293. doi: 10.1016/j.tem.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Simonson DC, DeFronzo RA. Indirect calorimetry: methodological and interpretative problems. The American journal of physiology. 1990;258:E399–412. doi: 10.1152/ajpendo.1990.258.3.E399. [DOI] [PubMed] [Google Scholar]
- 185.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J. Circadian Clock NAD+ Cycle Drives Mitochondrial Oxidative Metabolism in Mice. Science. 2013;342 doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, Ratcliffe WF, Brewer RA, Nagendran J, Villegas-Montoya C, Zou C, Zou L, Johnson RL, Jr, Dyck JR, Bray MS, Gamble KL, Chatham JC, Young ME. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–44619. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Simon N, Papa K, Vidal J, Boulamery A, Bruguerolle B. Circadian rhythms of oxidative phosphorylation: effects of rotenone and melatonin on isolated rat brain mitochondria. Chronobiol Int. 2003;20:451–461. doi: 10.1081/cbi-120021385. [DOI] [PubMed] [Google Scholar]
- 188.Lapenna D, De Gioia S, Mezzetti A, Porreca E, Ciofani G, Marzio L, Capani F, Di Ilio C, Cuccurullo F. Circadian variations in antioxidant defences and lipid peroxidation in the rat heart. Free radical research communications. 1992;17:187–194. doi: 10.3109/10715769209068165. [DOI] [PubMed] [Google Scholar]
- 189.Xu YQ, Zhang D, Jin T, Cai DJ, Wu Q, Lu Y, Liu J, Klaassen CD. Diurnal variation of hepatic antioxidant gene expression in mice. PloS one. 2012;7:e44237. doi: 10.1371/journal.pone.0044237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kakan X, Chen P, Zhang J. Clock gene mPer2 functions in diurnal variation of acetaminophen induced hepatotoxicity in mice. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2011;63:581–585. doi: 10.1016/j.etp.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 191.Zhang D, Jin T, Xu YQ, Lu YF, Wu Q, Zhang YK, Liu J. Diurnal-and sex-related difference of metallothionein expression in mice. Journal of circadian rhythms. 2012;10:5. doi: 10.1186/1740-3391-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Garlick PJ, Millward DJ, James WP. The diurnal response of muscle and liver protein synthesis in vivo in meal-fed rats. The Biochemical journal. 1973;136:935–945. doi: 10.1042/bj1360935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, Sahin M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell. 2015;161:1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rau E, Meyer DK. A diurnal rhythm of incorporation of L-[3H] leucine in myocardium of the rat. Recent advances in studies on cardiac structure and metabolism. 1975;7:105–110. [PubMed] [Google Scholar]
- 195.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 2011;30:4642–4651. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Desvergne A, Ugarte N, Radjei S, Gareil M, Petropoulos I, Friguet B. Circadian modulation of proteasome activity and accumulation of oxidized protein in human embryonic kidney HEK 293 cells and primary dermal fibroblasts. Free radical biology & medicine. 2016;94:195–207. doi: 10.1016/j.freeradbiomed.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 197.Pfeifer U, Strauss P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. Journal of molecular and cellular cardiology. 1981;13:37–49. doi: 10.1016/0022-2828(81)90227-3. [DOI] [PubMed] [Google Scholar]