

Abstract
Radiation-induced lung fibrosis (RILF) is a common side effect of thoracic irradiation therapy and leads to high mortality rates after cancer treatment. Radiation injury induces inflammatory M1 macrophage polarization leading to radiation pneumonitis, the first stage of RILF progression. Fibrosis occurs due to the transition of M1 macrophages to the anti-inflammatory pro-fibrotic M2 phenotype, and the resulting imbalance of macrophage regulated inflammatory signaling. Non-coding RNA signaling has been shown to play a large role in the regulation of the M2 mediated signaling pathways that are associated with the development and progression of fibrosis. While many studies show the link between M2 macrophages and fibrosis, there are only a few that explore their distinct role and the regulation of their signaling by non-coding RNA in RILF. In this review we summarize the current body of knowledge describing the roles of M2 macrophages in RILF, with an emphasis on the expression and functions of non-coding RNAs.
Keywords: Macrophages, M1, M2, Non-coding RNA, MicroRNA, Long-noncoding RNAs, Radiation-induced lung fibrosis, Fibrosis
Core tip: We discuss the mechanisms of initiation and progression of radiation-induced lung fibrosis. First we summarize the role of M2 macrophages in the initiation and development of pulmonary fibrosis with an emphasis on their function in radiation-induced lung fibrosis. We then examine the growing evidence describing non-coding RNAs in the development and progression of radiation-induced lung fibrosis.
INTRODUCTION
Ionizing radiation (IR) is one of the most common methods of treatment for breast and lung cancer[1]. While effective at treating thoracic cancers, IR damages the cells of the lung epithelium, resulting in the chronic accumulation of inflammatory cytokines. In 10%-20% of patients who receive thoracic radiotherapy, the dysregulation of the immune microenvironment results in radiation-induced lung fibrosis (RILF)[2]. RILF is a progressive disease that occurs in two distinct phases. The first stage of RILF is pneumonitis, which occurs within the first six-months post-treatment. Pneumonitis is characterized by shortness of breath, cough, and fever. Patients with severe radiation pneumonitis experience an almost 50% mortality rate[3]. After 6-12 mo (up to two years after irradiation) pneumonitis progresses to fibrosis[4]. Fibrosis is the result of excess wound healing, characterized by the accumulation of fibroblasts and myofibroblasts, and high levels of extracellular matrix (ECM) deposition and ECM remodeling[5].
Many factors affect the development and progression of RILF. These include radiation dose, volume of the irradiated parenchyma, adjuvant use of chemotherapy, age, and genetic predisposition of the patient. Furthermore, IR induces the activation of several cell types which affect RILF in a variety of ways, including pneumocytes, endothelial cells, myofibroblasts and immune cells[4]. The most important cells for the development of RILF are immune cells, which secrete fibrosis activating cytokines, and fibroblasts, which are the predominant cell type of the stromal microenvironment and are necessary for the production and maintenance of the extracellular membrane and wound healing response[6]. While the mechanisms of RILF development and progression remain poorly understood, research into general fibrosis has laid a strong foundation for further work. Significant evidence demonstrates the critical role that macrophage polarization plays in the development of a pro-fibrotic microenvironment, and early studies have demonstrated the impact that radiation has on the balance of M1 and M2 macrophages. Moreover, noncoding RNAs have been shown to be critical regulators of most signaling pathways, including in fibrosis development and RILF.
Macrophage polarization
Macrophages are phagocytic white blood cells that have important roles in inflammation and wound healing. They are necessary in the innate immune response, and function in adaptive immunity as well through recruitment of other immune cells. When dysregulated, macrophages and macrophage activated signaling pathways play a key role in the development of RILF[7]. There are independent macrophage populations in most tissues, including alveolar macrophages in the lungs. The origin of tissue resident macrophages is not fully understood. Initial reports described tissue resident macrophage populations that were renewed through the constant recruitment and differentiation of bone-marrow derived circulating monocytes[8]. However, recent data has shown that mature macrophage populations may also originate during fetal development and are maintained through self-renewal within each tissue[9,10]. The true origin of tissue specific macrophages is likely a combination of the two. Primitive macrophages are derived from the yolk-sac, and are directly formed through a rapid differentiation mechanism without monocyte formation. However, after maturation the primary macrophage source switches to embryonic progenitor cells, which dilute the population of original yolk-sac derived macrophages[11,12].
Macrophages are known for their heterogeneity and plasticity, and classified based on their phenotypic differences in response to stimuli. They are separated into two general groups, classically activated M1 macrophages and alternatively activated M2 macrophages[13]. M1 macrophages are inflammatory, antigen presenting cells that are activated by stimulation with interferon-γ (IFN-γ), toll-like receptor (TLR) ligands and microbial products such as lipopolysaccharide (LPS)[14]. They can be identified by their expression of CD80, CD86, IL-1R, TLR2, TLR4 and iNOS, and secrete inflammatory cytokines and chemokines including TNF-α, IL-1β, IL-6, IL-12, IL-23, CCL2-CCL5, and CCL8-CCL11. M1 macrophages also produce toxic intermediates such as nitric oxide (NO) and reactive oxygen intermediates[15]. There are three sub-groups of M2 macrophages. M2a are induced by IL4 and IL13, M2b by antigen-antibody immune complexes and LPS, and M2c by IL-10 and transforming growth factor beta (TGF-β). M2a and M2c macrophages secrete low levels of inflammatory cytokines, while M2b produce inflammatory cytokines while still protecting from LPS. Th2 cytokines IL-4 and IL-13 preferentially stimulate arginase-1 activity in pro-fibrotic M2 macrophages[16]. Expression of mannose receptor-1 (CD206), the lectin-binding protein Ym1, the resistin-like protein Fizz1, and the chemokine CCL18 are also associated with M2 macrophages. All M2 macrophages promote angiogenesis and tissue remodeling, and regulate inflammation and immune responses[15].
M2 macrophages secrete pro-fibrotic growth factors including TGF-β, fibroblast growth factor, platelet derived growth factor (PDGFα), and vascular endothelial growth factor (VEGF). Stimulation by these cytokines enhances fibroblast proliferation, and can induce differentiation into myofibroblasts, a highly active fibroblast-like cell. Myofibroblasts’ rapid proliferation, excessive deposition of ECM proteins such as collagen and fibronectin, and high contractile ability are critical to the development of fibrosis[17].
Macrophage polarization is predominantly governed by several signaling pathways: The C-Jun N-terminal kinase signaling pathway, PI3K/AKT signaling pathway, Notch signaling pathway and the JAK/STAT signaling pathway[18]. While these signaling pathways are activated by their respective ligands and cytokines, non-coding RNA signaling also plays a large role in their regulation[19].
Non-coding RNAs
When the human genome project was completed, researchers found that less than 2% of the human genome is composed of protein-coding genes[20]. The large majority of non-protein coding genes are still actively transcribed and make up a new class of molecule, non-coding RNAs (ncRNA). There are many different species of ncRNA that are commonly divided based on size into short noncoding RNA and long-noncoding RNAs (lncRNA).
To date, the most commonly studied ncRNA are microRNAs, a short (approximately 22 nucleotides) noncoding RNA molecules that binds specifically to a 6-9 base pair seed sequence in the 3’ untranslated region (UTR) of messenger RNAs. miRNAs are transcribed primarily by RNA polymerase II, and after nuclear processing and cleavage are exported to the cytosol. In the cytosol miRNA precursors undergo further cleaving before being incorporated into the RNA induced silencing complex (RISC). miRNAs then guide the RISC to specific mRNAs through binding to the 3’UTR seed sequence, and silence the mRNA through degradation or inhibition of translation via steric hindrance[21]. Since their discovery, miRNAs have been widely studied in cancer and disease, and have been shown to regulate numerous physiological processes at the post-transcriptional level.
LncRNAs are an abundant type of ncRNA that is increasingly well studied. lncRNA are defined as any non-coding RNA that is > 200 base pairs in length. The majority of lncRNAs are transcribed by RNA polymerase II, followed by polyadenylation, splicing, and the addition of a 5’ Cap. lncRNA have great functional diversity, and interact with genomic DNA, other non-coding RNAs (including miRNA), mRNA and proteins while including functioning as epigenetic modulators, scaffold molecules, or as competitive endogenous sponges[22].
Despite the prevalence and high morbidity of RILF, very few studies have interrogated the processes of RILF development. It is increasingly evident that RILF occurs through a model of progressive macrophage polarization and temporal regulation of cytokines and ncRNAs in complex signaling pathways. Therefore, it is critical that further studies are performed using in vivo models of RILF to determine the intricate mechanisms behind RILF. Here, we review the function of macrophages in RILF with a focus on the roles of alternatively activated M2 macrophages and discuss the involvement of ncRNAs in RILF development and progression.
MACROPHAGES PROMOTE LUNG FIBROSIS
Lung fibrosis is caused by repetitive injury to the epithelial cells lining the alveoli, resulting in scarring of the lung tissue. A delicate balance between pro- and anti-inflammatory macrophages maintains the physiological roles of wound healing and inflammation, and is necessary to avoid fibrosis. Animal studies and clinical data suggest that M1 macrophages are associated with pneumonitis, and alternatively activated M2 and M2-like macrophages are associated with lung fibrosis.
Role of M2 macrophages in lung fibrosis in animal models
Animal models have been commonly used to demonstrate the involvement of alternatively activated alveolar macrophages in the development and progression of lung fibrosis. These studies provide insights into the macrophage polarization pathways and identify the crucial roles of M2 and M2-like macrophages in lung fibrosis.
One animal model used to study the mechanisms of lung fibrosis is Cu,Zn-SOD-/- mice, which do not develop fibrosis[23]. In wild-type populations, mitochondrial Cu,Zn-superoxide dismutase (Cu,Zn-SOD)-mediated production of H2O2 leads to DNA damage and the development of lung fibrosis. Cu,Zn-SOD is able to facilitate the nuclear translocation and enhance the transcriptional activation of STAT6 through the modulation of the redox status of Cys528 in the SH2 domain of STAT6, resulting in polarization of macrophages towards the M2 phenotype. Furthermore, CuZnSOD-transgenic (Cu,Zn-SODTg) mice, which have more than a 3-fold increase of Cu,Zn-SOD protein expression and activity, had high activation of pro-fibrotic signaling pathways in their lungs. This resulted in a pro-fibrotic microenvironment and enhanced development of lung fibrosis. These data describe the role of Cu,Zn-SOD in M2 polarization, and suggest that polarization of macrophages towards the M2 phenotype in a Th2-independent manner contributes to the development of lung fibrosis[24].
In a recent study, conditional Src homology domain-containing tyrosine phosphatase (Shp2) mutant mice were generated by selective removal of Shp2 in monocytes/macrophages through lysozyme M promoter-driven Cre recombinase. These mice were used to investigate the role of Shp2 in M2 polarization and the development of lung fibrosis. When Shp2 knockout was induced in mature mouse macrophages, IL-4 mediated M2 polarization was increased. Moreover, arginase activity, a marker of M2 polarization, was enhanced in Shp2 conditional mutant mice after intraperitoneal (i.p.) injection of chitin, an inducer of M2 macrophage polarization. Finally, when treated with bleomycin, Shp2 mutant mice were more susceptible to develop inflammation and lung fibrosis compared to control mice. Further mechanistic studies showed that loss of Shp2 leads to the association of JAK1 with IL-4Rα, causing IL-4-mediated JAK1/STAT6 activation and eventually M2 polarization. These data suggest that Shp2 is necessary to prevent the development of lung fibrosis through the inhibition of M2 polarization[25].
To investigate the function of increased IL-9 in lung fibrosis, Arras et al[26] compared the development of fibrosis in transgenic IL-9 overexpressing C57BL/6 mice vs their wild-type counterparts after 5 mg silica (DQ12) was implanted intratracheally. When the mice were sacrificed after 4 and 6 mo of DQ12 implantation, the transgenic mice had a decreased level of silica-induced lung fibrosis compared to their wild-type counterparts as evidenced by the histologic examination and measurement of lung hydroxyproline content. To further confirm the effect of increased IL-9 production in fibrosis, they compared wild-type C57BL/6 mice that were i.p. injected either with recombinant IL-9 or with PBS three times a week for 2 mo to mice that had been implanted with DQ12 and also received IL-9 during the second month of implantation. Mice were sacrificed after 2, 4 and 6 mo of DQ12 implantation and the results showed that IL-9 dosed mice had a decreased level of silica-induced lung fibrosis compared to their non-dosed counterparts. Moreover, the silica-induced fibrosis that developed in wild-type mice was associated with Th2 markers including the upregulation of IL-4 in inflammatory alveolar macrophages. On the other hand, in the IL-9 transgenic mouse model IL-4 levels were not changed significantly in macrophage populations. These results suggest that upregulation of IL-9 exerts an anti-fibrogenic effect in silica-induced lung fibrosis through the inhibition of M2 macrophage polarization[26].
Studies of a mouse model with long-term overexpression of IL-10 provided evidence for the role of IL-10 in the induction of lung fibrosis. Bronchoalveolar lavage (BAL) samples of mice overexpressing IL-10 had enhanced infiltration of T and B-lymphocytes as well as increased number of collagen-producing cells and fibrocytes. Additionally, CCR2 and its ligand, CCL2 were found to be significantly upregulated in IL-10 overexpressing mice. Interestingly, the proportion of M2 macrophages was significantly higher in both BAL and whole lung tissue of IL-10 overexpressing mice compared to wild-type ones, and administration of anti-CCL2 antiserum decreased fibrosis. These results suggest that chronic overexpression of IL-10 in mice causes the induction of lung fibrosis, increased M2 macrophages and high levels of CCR2 and CCL2[27].
Clinical data supporting the role of M2 macrophages in lung fibrosis
Clinical data also support the role of alternatively activated alveolar macrophages in lung fibrosis. Alveolar macrophages of patients with lung fibrosis had increased expression of CD206 as well as enhanced production of pro-inflammatory cytokines CCL17, CCL18 and CCL22. To confirm M2 polarization during lung inflammation, they stimulated normal human alveolar macrophages in vitro with Th2 cytokine IL-4 by itself or with both IL-4 and IL-10. Stimulation with IL-4 induced the polarization towards the M2-phenotype, demonstrating that healthy alveolar macrophages could be induced for M2 polarization. Furthermore, co-stimulation with IL-10 increased the IL-4 mediated expression of CCL18 and IL-1RA and decreased the production of CCL17 and CCL22. Additional data showed that IL-10 differentially regulates CCL18 and IL-1RA expression in vitro, suggesting that it effects IL-4 polarization of M2 macrophages[28].
Patients with lung fibrosis have increased CCL18 gene and protein expression, which is a marker of alternative activation. Expression of CCL18 is increased in normal alveolar macrophages when they were stimulated with Th2 cytokines or co-cultured with human lung fibroblasts. In addition, co-culture of normal lung fibroblasts with cell supernatants from alternatively activated alveolar macrophages of lung fibrosis patients increased collagen production compared to co-culture of cell supernatants from alveolar macrophages of healthy individuals. These results indicate that alveolar macrophages of patients with lung fibrosis display an alternative activation phenotype[29].
A recent study investigated the role of macrophages in acute exacerbation (AE) of idiopathic pulmonary fibrosis (IPF). The levels of several cytokines including IL-1ra, CCL2, CCL17, CCL18, CCL22, TNF-α, IL-1β, CXCL1 and IL-8 were measured in BAL fluids of IPF patients with or without AE. The results showed that pro-inflammatory cytokines CXCL1 and IL-8 as well as all tested M2 cytokines were upregulated in BAL-cells from IPF patients. Furthermore, they found that increased production of CCL18 in BAL-cells were a strong indicator for the development of AE. Their data suggest that M2 macrophage activation has a large impact on the development of IPF acute exacerbation[30].
M2 macrophages in RILF
To test the hypothesis that classically activated macrophages (M1) are associated with radiation-induced pneumonitis and alternatively activated macrophages (M2) are associated with RILF, C57BL/6 mice were treated with 12 Gy of X-rays to their thoracic region. Control and irradiated mice were sacrificed at different time-points ranging from 1 h to 24 wk, and the expression of M1 and M2 markers, inducible nitric oxide synthase (iNOS) and arginase type 1 (Arg-1) respectively, were assessed at the mRNA and protein level. The analyses showed the enhanced expression of iNOS at the early time-points post irradiation (the pneumonitis stage), and enhanced expression of Arg-1 at later time points (the fibrotic phase). Further experiments confirmed that these markers were differentially expressed and were specific to macrophages[31]. The same group in a similar study showed that the expression of factors that are associated with Th2-immune response including GATA-3, hydroxyproline, IL-3, and Arg-1 reached their maximum levels at week 16 post-irradiation. These data indicate that thoracic irradiation first induces Th1 polarization during pneumonitis, followed by transition to Th2 polarization at later time points that correspond to fibrosis. While the immediate response to irradiation involves classically activated macrophages, alternatively activated macrophages are predominant during fibrosis and likely have an important role in its development and progression[32].
Ym-1 positive M2 macrophages were shown to be elevated in response to radiation in murine lungs. Furthermore, fibrotic lungs had higher level of gal-3 expression, which is mainly expressed by M2 macrophages. The proliferation of fibroblasts and expression of α-SMA was induced by mouse recombinant gal-3. In vitro studies showed that when gal-3 was knocked-down the radiation-induced expression of YM-1 in Raw 264.7 cells was also decreased, indicating that gal-3 might be involved in the development of RILF through the regulation of the alternative activation of M2 macrophages[33].
The Th2-type immune system response has been suggested to be involved in the development of lung fibrosis[34,35]. IL-4, which is a Th2 cytokine, is induced in fibrosis and enhances extracellular matrix protein synthesis, including collagen and fibronectin[36], polarization of T cells toward the Th2 phenotype, and alternative activation of macrophages[37]. Furthermore, when rats were treated with a single 20 Gy dose of IR to the thoracic region, IL-4 levels increased and reached their plateau 3 wk after irradiation. The cellular origin of IL-4 was determined to be mainly the macrophages[38]. To study the role of radiation in the activation of type-2 immune response, mice with Lewis lung cancer received radiation therapy alone or radiation combined with Th1 immunomodulator unmethylated cytosine-phosphorothioate-guanine containing oligodeoxynucleotide (CpG-ODN). The mice were sacrificed 3-wk post-irradiation and the immune status of tumors as well as the histological grade of lung fibrosis was assessed. Lung fibrosis in tumor-bearing mice was more progressed compared to normal mice after radiation treatment. Th2-immune response associated markers hydroxyproline and gata-3 had higher expression in the lung tissues of tumor-bearing mice post-radiation. Furthermore, CpG-ODN weakened fibrosis by significantly decreasing gata-3 expression. The levels of IL-13 and IL-5 were increased, while the levels of INF-γ and IL-12 expression was decreased in irradiated tumor-bearing mice, which was reversed with CpG-ODN treatment. These results indicate that Type-2 immunity in tumors effects the development and progress of RILF[39] and provides further evidence for the association of M2 macrophages, Th2 immune system and development of RILF.
To investigate the role of fractionated irradiation (FIR) on macrophage recruitment and phenotype, human monocyte derived-macrophages were treated with a FIR of 2 Gy/d, mimicking the radiotherapy that cancer patients would go through. Although their DNA was damaged upon irradiation, the macrophages remained viable and metabolically active and at 10 Gy cumulative dose a significant increase in pro-inflammatory macrophage markers CD80, CD86 and HLA-DR was observed, while anti-inflammatory markers CD163, MRC1, VCAN and IL-10 decreased. These results suggest that FIR facilitates the polarization of macrophages towards pro-inflammatory phenotype[40].
Accumulating evidence clearly indicate the role of alternative activation of M2 and M2-like macrophages in development and progress of lung fibrosis; however, the mechanisms leading to M2 polarization are poorly identified and further work is necessary to detail the role of M2 macrophages in RILF.
NON-CODING RNA IN FIBROSIS
While non-coding RNAs have been well characterized in cancer, relatively few have been shown to directly influence RILF. In addition to directly affecting known fibrotic signaling pathways, it is likely that non-coding RNA regulation of macrophage polarization and induction of pro-fibrotic alternatively activated macrophages affects RILF. Therefore, we will focus on miRNAs and lncRNAs with defined roles in RILF, pulmonary fibrosis and alternative activation of alveolar macrophages (Figure 1). Please refer to the cited reviews for a broader review of the roles of non-coding RNAs in fibrosis[41-43] and macrophage polarization[19].
Figure 1.
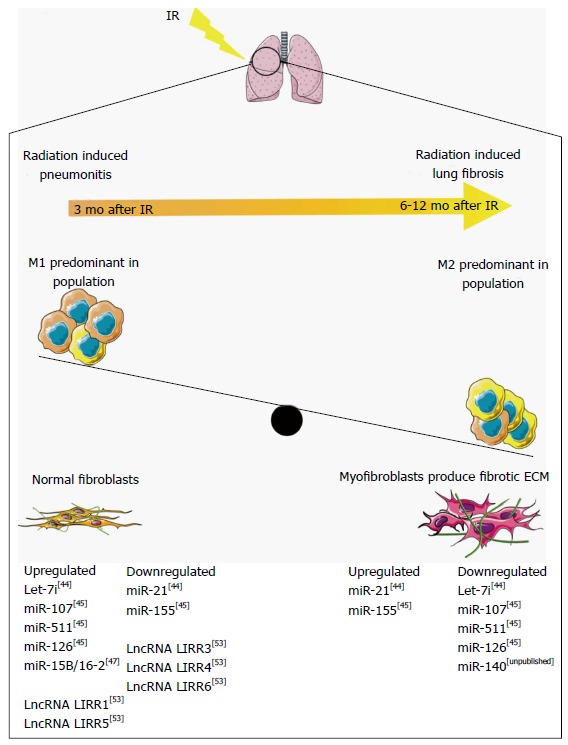
Non-coding RNAs regulation of macrophage polarization in radiation-induced lung fibrosis. Radiation pneumonitis develops approximately 3 mo after ionizing radiation, resulting from the accumulation of M1 macrophages. Six to twelve months after ionizing radiation (IR) the accumulation of M2 macrophages activates myofibroblast differentiation, resulting in a fibrotic microenvironment and radiation-induced lung fibrosis (RILF). Non-coding RNAs regulate the pathways involved in RILF, and are temporally regulated through RILF development and progression. ECM: Extracellular matrix. This figure was produced using Servier medical art, available from http://www.servier.com/Powerpoint-image-bank.
miRNA in RILF
To study the roles of miRNAs in RILF, Xie et al[44] treated adult male rats with 20 Gy of radiation. Using miRNA microarray analysis, they examined the differential miRNA expression at 3, 12, and 26 wk post-irradiation. While the authors observed minor physiological changes after 3 wk, significant fibrosis was not seen until the 26-wk time point. At the 3-wk time point, the authors found 7 upregulated miRNAs and 4 downregulated, at 12 wk 10 upregulated and 4 downregulated, and at 26 wk 7 upregulated and 4 downregulated. The authors validated the expression levels of only two miRNAs, miR-21 and let-7i and identified novel temporal regulation of miRNAs during fibrosis initiation and progression. At the 3-wk time-point let-7i was highly expressed and miR-21 had low expression, whereas after 26 wk let-7i expression decreased and miR-21 was upregulated. Furthermore, let-7i targets TGFBR1, inhibiting TGF-β signaling, while miR-21 degrades the TGF-β inhibitor SMAD7. Moreover, miR-21 has been shown to be upregulated in several models of fibrosis[45,46], and to be activated by pro-fibrotic TGF-β. The authors postulate that the temporal regulation of let-7i and miR-21 is a mechanism for regulating TGF-β signaling. Early inhibition of TGF-β allows for lung regeneration and healing, followed by an increase in TGF-β signaling that leads to lung fibrosis[44].
Kalash et al[47] performed similar animal model time-point studies. They irradiated C67BL/6NTac mice with 20 Gy to the thoracic area, and at 2, 14, 28, 75, 110, 150 and 200 d post-irradiation isolated RNA from the lungs of irradiated and control mice. Using RT-PCR, they examined miRNA expression, focusing on miRNAs known to target toll-like receptor 4 (Tlr4, an activator of the innate immune system), vascular endothelial growth factor α (Vegfa, a pro-angiogenic factor), and TGF-β, three transcripts that they had observed to be differentially expressed in irradiated tissues. They found that miR-107 and miR-511, both of which are known to target Tlr4, were upregulated in the first 50 day after irradiation, followed by downregulation during the later time points that correspond with fibrosis. miR-126 has been shown to stimulate expression of Vegfa, and was upregulated during the immediate post-irradiation inflammatory phase, but expression decreased during later stages. Interestingly, the levels of Vegfa remained elevated throughout the entire time course, indicating that miR-126 regulation of Vegfa is not important during RILF, and that miR-126 may have some other action in the development of RILF. Finally, miR-155, which targets Tgfb, was decreased during the early inflammatory phase, but starting at day 75 miR-155 levels were increased. While Tgfb expression matches that of miR-155 before day 75, it is upregulated in conjunction with miR-155 in the late fibrotic phase. This study demonstrates the significant differences in several distinct miRNAs that happens both in the inflammatory phase immediately post-irradiation, and in the late term fibrotic phases. It also demonstrates complexity of miRNA-mRNA regulatory networks in RILF. As the expression changes of mRNA and miRNA do not always match their previously defined functions, this points to the involvement of other currently unknown factors.
Ezzie et al[48] directly used irradiated cell models to examine miRNA function. miR-15b/16-2 was previously shown to be upregulated in fibrotic regions of human lung tissue, and to target TGF-β signaling inhibitor SMAD7. To examine the role of miR-15b/16-2 in radiation-induced fibrosis, Rahman et al[49] treated human lung epithelial cells with 1 Gy of radiation, and after 24 h isolated RNA for RT-PCR. In contrast to previous findings showing chronic upregulation of miR-15b/16-2 in fibrotic tissue, they showed that miR-15b/16-2 was induced directly after irradiation followed by a gradual decrease. Moreover, they identified Wip1 as a novel target of miR-15b/16-2. Wip1 inactivates the DNA damage response through dephosphorylation of p53, γ-H2AX, and ATM. Upregulation of miR-15b/16-2 therefore activates the DNA damage response, protecting cells from radiation damage. It is likely that while miR-15b/16-2 is initially activated to protect the cell from radiation-induced fibrosis, it’s activation results in increased TGF-β signaling through degradation of SMAD7.
We have recently shown that miR-140 plays a key role in protection of RILF[50]. We irradiated the thoracic region of C57BL/6 mice with 13 Gy, and after 1 year sacrificed them for histological examination. RNA in situ staining using a DIG labeled probe revealed that irradiated lung tissue had higher expression of miR-140, and through in vitro characterization we found that miR-140 expression is activated by nuclear factor-erythroid2-related factor 2 (NRF2). NRF2 is a transcription factor that activates antioxidant and anti-inflammatory genes, and is upregulated in response to the free radicals created after IR[50]. Recently we have continued these studies, and found that lung tissue with RILF isolated from C57BL/6 mice one year after 13 Gy of radiation had significantly abrogated miR-140 expression. Moreover, we treated lung fibroblasts isolated from wild-type and miR-140 knock-out mice[51] with 20 Gy of radiation, and demonstrated that miR-140 knock-out fibroblasts were more susceptible to radiation-induced fibrosis as evidenced by increased myofibroblast population, TGF-β expression and enhanced contraction ability (unpublished data). miR-140 has been demonstrated to target the TGF-β pathway through degradation of SMAD3 in chondrocytes and TGF-β receptor TGFBR1 in preadipocytes[52]. We have also identified that miR-140 also targets fibronectin, a key member of the extracellular matrix, in pulmonary fibroblasts (unpublished data). This indicates that miR-140 regulates RILF in multiple ways, both through inhibition of myofibroblast differentiation and through degradation of fibronectin, which is highly upregulated in fibrotic tissue and a key factor in development of the stiff extracellular matrix.
lncRNAs in RILF
One study has examined the expression changes of lncRNAs in response to lung irradiation. Twenty-four hours after exposing C57BL/6 mice to 12 Gy of whole body irradiation, lncRNAs LIRR1 and LIRR5 were upregulated whereas LIRR2, 3, 4, and 6 were downregulated. The authors chose lncRNA LIRR1 for further study. LIRR1 is on chromosome 1, and is co-expressed with 27 coding genes. Through in vitro studies using the human bronchial epithelial cell line BEAS-2B, lncRNA LIRR1 upregulation was validated, and they demonstrated that overexpression of LIRR1 affects the radiosensitivity of BEAS-2B cells. After exposing BEAS-2B to 4 Gy of IR, they found decreased cell survival, and an increased number of cells in the G1 phase. To further interrogate DNA repair, they examined γ-H2AX foci formation and found it was significantly increased in irradiated cells that overexpressed LIRR1. Finally, LIRR1 overexpression decreased the protein expression of several DNA Repair proteins, including KU70, KU80, and RAD50. While the authors suggest that LIRR1 mediates the cellular DNA damage response after irradiation, the more specific mechanisms of this regulation remain unknown[53].
CONCLUSION
It is clear that both macrophages and ncRNA are critical for the initiation and progression of radiation induced lung fibrosis. While radiation first induces inflammatory M1 macrophages leading to pneumonitis, dysregulation of M1 inflammatory signaling pathways leads to the transition to M2 macrophages, prompting the development of fibrosis. Non-coding RNAs appear to have distinct and critical roles in this transition and the development of RILF through regulation of the key inflammatory and fibrotic signaling pathways. However, while fibrosis continues to be thoroughly investigated, there are few published studies examining the specific mechanisms occurring due to exposure to radiation that lead to fibrosis. RILF is an important and deadly pathology of thoracic irradiation, however the lack of mechanistic information means there are no effective treatments. Due to the difficulty and length of in vivo radiation studies, most investigations of the mechanisms of fibrosis use methods other than radiation to induction fibrosis. However, long-term radiation work is necessary to truly investigate the mechanisms of RILF. These studies will likely lead to the discovery of key molecular targets in the protection and treatment of RILF.
Footnotes
Conflict-of-interest statement: There are no conflicts of interest.
Manuscript source: Invited manuscript
Specialty type: Biochemistry and molecular biology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: July 31, 2016
First decision: September 2, 2016
Article in press: October 27, 2016
P- Reviewer: Freire-De-Lima CG, Kitagawa S, Kuan YH, Lee TS, Liu ZH, Vetvicka V S- Editor: Kong JX L- Editor: A E- Editor: Wu HL
References
- 1.Choi YW, Munden RF, Erasmus JJ, Park KJ, Chung WK, Jeon SC, Park CK. Effects of radiation therapy on the lung: radiologic appearances and differential diagnosis. Radiographics. 2004;24:985–997; discussion 998. doi: 10.1148/rg.244035160. [DOI] [PubMed] [Google Scholar]
- 2.Roach M, Gandara DR, Yuo HS, Swift PS, Kroll S, Shrieve DC, Wara WM, Margolis L, Phillips TL. Radiation pneumonitis following combined modality therapy for lung cancer: analysis of prognostic factors. J Clin Oncol. 1995;13:2606–2612. doi: 10.1200/JCO.1995.13.10.2606. [DOI] [PubMed] [Google Scholar]
- 3.Mehta V. Radiation pneumonitis and pulmonary fibrosis in non-small-cell lung cancer: pulmonary function, prediction, and prevention. Int J Radiat Oncol Biol Phys. 2005;63:5–24. doi: 10.1016/j.ijrobp.2005.03.047. [DOI] [PubMed] [Google Scholar]
- 4.Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets. 2013;14:1347–1356. doi: 10.2174/13894501113149990198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 7.Wermuth PJ, Jimenez SA. The significance of macrophage polarization subtypes for animal models of tissue fibrosis and human fibrotic diseases. Clin Transl Med. 2015;4:2. doi: 10.1186/s40169-015-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128:415–435. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 13.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J Immunol. 2005;175:5–14. [PubMed] [Google Scholar]
- 14.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 17.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 18.Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, Li J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014;26:192–197. doi: 10.1016/j.cellsig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Cui H, Liu G. MicroRNAs and Other Non-Coding RNAs in Inflammation. Berlin: Springer International Publishing; 2015. How Noncoding RNAs Contribute to Macrophage Polarization; pp. 59–84. [Google Scholar]
- 20.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 21.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 22.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He C, Murthy S, McCormick ML, Spitz DR, Ryan AJ, Carter AB. Mitochondrial Cu,Zn-superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J Biol Chem. 2011;286:15597–15607. doi: 10.1074/jbc.M110.187377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem. 2013;288:20745–20757. doi: 10.1074/jbc.M112.410720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y, Wang K, Cheng H, Zhang X, Ke Y. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol. 2014;193:2801–2811. doi: 10.4049/jimmunol.1303463. [DOI] [PubMed] [Google Scholar]
- 26.Arras M, Huaux F, Vink A, Delos M, Coutelier JP, Many MC, Barbarin V, Renauld JC, Lison D. Interleukin-9 reduces lung fibrosis and type 2 immune polarization induced by silica particles in a murine model. Am J Respir Cell Mol Biol. 2001;24:368–375. doi: 10.1165/ajrcmb.24.4.4249. [DOI] [PubMed] [Google Scholar]
- 27.Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341–L353. doi: 10.1152/ajplung.00122.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, Friedrich K, Müller-Quernheim J, Zissel G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. 2010;137:89–101. doi: 10.1016/j.clim.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, Vollmer E, Müller-Quernheim J, Zissel G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–792. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 30.Schupp JC, Binder H, Jäger B, Cillis G, Zissel G, Müller-Quernheim J, Prasse A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS One. 2015;10:e0116775. doi: 10.1371/journal.pone.0116775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Han G, Liu H, Chen J, Ji X, Zhou F, Zhou Y, Xie C. The development of classically and alternatively activated macrophages has different effects on the varied stages of radiation-induced pulmonary injury in mice. J Radiat Res. 2011;52:717–726. doi: 10.1269/jrr.11054. [DOI] [PubMed] [Google Scholar]
- 32.Han G, Zhang H, Xie CH, Zhou YF. Th2-like immune response in radiation-induced lung fibrosis. Oncol Rep. 2011;26:383–388. doi: 10.3892/or.2011.1300. [DOI] [PubMed] [Google Scholar]
- 33.Zhao W, Zhang JH, Lopes IJ. Enhanced expression of galectin-3 isassociated with the alternative activation of macrophages and development oflung fibrosis following radiation. Cancer Research. 2015;75(Suppl 15):3308. [Google Scholar]
- 34.Sime PJ. The antifibrogenic potential of PPARgamma ligands in pulmonary fibrosis. J Investig Med. 2008;56:534–538. doi: 10.2310/JIM.0b013e31816464e9. [DOI] [PubMed] [Google Scholar]
- 35.Brush J, Lipnick SL, Phillips T, Sitko J, McDonald JT, McBride WH. Molecular mechanisms of late normal tissue injury. Semin Radiat Oncol. 2007;17:121–130. doi: 10.1016/j.semradonc.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. Role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol. 1994;152:3606–3614. [PubMed] [Google Scholar]
- 37.Lee E, Yook J, Haa K, Chang HW. Induction of Ym1/2 in mouse bone marrow-derived mast cells by IL-4 and identification of Ym1/2 in connective tissue type-like mast cells derived from bone marrow cells cultured with IL-4 and stem cell factor. Immunol Cell Biol. 2005;83:468–474. doi: 10.1111/j.1440-1711.2005.01352.x. [DOI] [PubMed] [Google Scholar]
- 38.Büttner C, Skupin A, Reimann T, Rieber EP, Unteregger G, Geyer P, Frank KH. Local production of interleukin-4 during radiation-induced pneumonitis and pulmonary fibrosis in rats: macrophages as a prominent source of interleukin-4. Am J Respir Cell Mol Biol. 1997;17:315–325. doi: 10.1165/ajrcmb.17.3.2279. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Wang Y, Mei Z, Zhang S, Yang J, Li X, Yao Y, Xie C. Radiation-induced lung fibrosis in a tumor-bearing mouse model is associated with enhanced Type-2 immunity. J Radiat Res. 2016;57:133–141. doi: 10.1093/jrr/rrv077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teresa Pinto A, Laranjeiro Pinto M, Patrícia Cardoso A, Monteiro C, Teixeira Pinto M, Filipe Maia A, Castro P, Figueira R, Monteiro A, Marques M, et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci Rep. 2016;6:18765. doi: 10.1038/srep18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel V, Noureddine L. MicroRNAs and fibrosis. Curr Opin Nephrol Hypertens. 2012;21:410–416. doi: 10.1097/MNH.0b013e328354e559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Tsitsiou E, Herrick SE, Lindsay MA. MicroRNAs and the regulation of fibrosis. FEBS J. 2010;277:2015–2021. doi: 10.1111/j.1742-4658.2010.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Reilly S. MicroRNAs in fibrosis: opportunities and challenges. Arthritis Res Ther. 2016;18:11. doi: 10.1186/s13075-016-0929-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie L, Zhou J, Zhang S, Chen Q, Lai R, Ding W, Song C, Meng X, Wu J. Integrating microRNA and mRNA expression profiles in response to radiation-induced injury in rat lung. Radiat Oncol. 2014;9:111. doi: 10.1186/1748-717X-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honeyman L, Bazett M, Tomko TG, Haston CK. MicroRNA profiling implicates the insulin-like growth factor pathway in bleomycin-induced pulmonary fibrosis in mice. Fibrogenesis Tissue Repair. 2013;6:16. doi: 10.1186/1755-1536-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu H, Luo H, Li Y, Zhou Y, Jiang Y, Chai J, Xiao X, You Y, Zuo X. MicroRNA-21 in scleroderma fibrosis and its function in TGF-β-regulated fibrosis-related genes expression. J Clin Immunol. 2013;33:1100–1109. doi: 10.1007/s10875-013-9896-z. [DOI] [PubMed] [Google Scholar]
- 47.Kalash R, Berhane H, Goff J, Houghton F, Epperly MW, Dixon T, Zhang X, Sprachman MM, Wipf P, Franicola D, et al. Effects of thoracic irradiation on pulmonary endothelial compared to alveolar type-II cells in fibrosis-prone C57BL/6NTac mice. In Vivo. 2013;27:291–297. [PMC free article] [PubMed] [Google Scholar]
- 48.Ezzie ME, Crawford M, Cho JH, Orellana R, Zhang S, Gelinas R, Batte K, Yu L, Nuovo G, Galas D, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67:122–131. doi: 10.1136/thoraxjnl-2011-200089. [DOI] [PubMed] [Google Scholar]
- 49.Rahman M, Lovat F, Romano G, Calore F, Acunzo M, Bell EH, Nana-Sinkam P. miR-15b/16-2 regulates factors that promote p53 phosphorylation and augments the DNA damage response following radiation in the lung. J Biol Chem. 2014;289:26406–26416. doi: 10.1074/jbc.M114.573592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duru N, Gernapudi R, Zhang Y, Yao Y, Lo PK, Wolfson B, Zhou Q. NRF2/miR-140 signaling confers radioprotection to human lung fibroblasts. Cancer Lett. 2015;369:184–191. doi: 10.1016/j.canlet.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyaki S, Sato T, Inoue A, Otsuki S, Ito Y, Yokoyama S, Kato Y, Takemoto F, Nakasa T, Yamashita S, et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010;24:1173–1185. doi: 10.1101/gad.1915510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X, Chang A, Li Y, Gao Y, Wang H, Ma Z, Li X, Wang B. miR-140-5p regulates adipocyte differentiation by targeting transforming growth factor-β signaling. Sci Rep. 2015;5:18118. doi: 10.1038/srep18118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiao Y, Liu C, Cui FM, Xu JY, Tong J, Qi XF, Wang LL, Zhu W. Long intergenic non coding RNA induced by X ray irradiation regulates DNA damage response signaling in the human bronchial epithelial BEAS 2B cell line. Oncol Lett. 2014;9:169–176. doi: 10.3892/ol.2014.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]