

Abstract
Recent work from our laboratory has shown that hyperlipidemia promotes accelerated rejection of vascularized cardiac allografts in mice by inducing anti-donor Th17 reactivity and production of IL-17. Here, we show that hyperlipidemia also affects FoxP3+ regulatory T cells (Tregs). Hyperlipidemia promotes the development of Tregs that express low levels of CD25. Hyperlipidemia also promotes a decrease in central Tregs and an increase in effector Tregs that appears to account for the increase in the frequency of CD25low Tregs. Alterations in Treg subsets also appear to lead to alterations in Treg function. The ability of FoxP3+, CD25high, CD4+ Tregs from hyperlipidemic mice to inhibit proliferation of effector T cells stimulated with anti-CD3 and CD28 was reduced when compared with Tregs from control mice. Regulatory T cells isolated from hyperlipidemic recipients exhibit increased activation of Akt, and a reduction in Bim levels that permits the expansion of FoxP3+CD25lowCD4+ T cells. Hyperlipidemic mice were also resistant to tolerance induction using costimulatory molecule blockade consisting of anti-CD154 and CTLA4Ig, a strategy that requires Tregs. Together, our data suggest that hyperlipidemia profoundly affects Treg subsets and function as well as the ability to induce tolerance.
Introduction
Approaches to prevent rejection have been based on an understanding that has been formed over many years based on animal models of rejection. These studies have led to the canonical understanding that T cell–mediated rejection results from recognition of donor antigen through the direct and indirect pathways of antigen recognition and is mediated by alloreactive T-helper type 1 (Th1) CD4 and CD8 T cells (1–5). Rejection is balanced by peripheral tolerance mechanisms involving regulatory T cells (Tregs). Based on this paradigm, a considerable amount of effort has been placed on inhibiting activation of alloreactive T cells while increasing the activity of Tregs through the use of approaches such as costimulatory blockade that requires regulatory T cells (6–10) in order to promote long-term allograft survival. These efforts along with modern immunosuppression regimens have led to increases in the first year of transplant survival (11,12). However, mortality rates beyond the first year of heart transplantation have not significantly improved in the last 2 decades (13). Moreover, approaches such as costimulatory molecule blockade while successful in mice have not exhibited successful tolerance induction in the clinic (14–19), and improving long-term outcomes remains a major problem. These startling facts underscore the need to better our understanding of transplant rejection and the factors that contribute to graft loss.
We hypothesized that the inability to improve long-term transplant survival might be due to limitations in our understanding of transplant rejection and acceptance that result from the use of animal models that do not capture health conditions present in the human transplant population. A major recipient characteristic associated with cardiac allograft rejection after the first year is a history of ischemic heart disease resulting from coronary artery disease caused by hyperlipidemia (13). Hyperlipidemia, increased cholesterol and triglycerides, leads to atherosclerosis primarily as a result of increased serum cholesterol levels; however, it also now known to cause systemic inflammation that contributes to disease progression in atherosclerosis and has the potential to contribute to other disease processes (20–29). Hyperlipidemic humans and mice exhibit increased levels of inflammatory cytokines in their serum, and exhibit increased inflammatory T cell responses (21,27–46). Hyperlipidemia is a comorbidity that is highly relevant in transplantation, causing end-stage heart disease in approximately 40% of all patients requiring a heart transplant (13). Hyperlipidemia also develops in 50% of heart transplant patients after the first year of transplant, and 95% of patients within 5 years. In spite of treatment, two-thirds of patients remain dyslipidemic and a significant number are statin intolerant (47,48). A major recipient characteristic associated with cardiac allograft rejection after the first year is a history of ischemic heart disease resulting from coronary artery disease caused by hyperlipidemia (13).
Limited reports have suggested that increased cholesterol contributes to neointimal proliferation in artery grafts (49), and atherosclerosis in cardiac allografts (50). However, little is known about how hyperlipidemia affects rejection and an effect of hyperlipidemia on anti-donor adaptive immune responses has not been described. We hypothesized that the ability of hyperlipidemia to promote inflammation may alter anti-donor responses or their regulation and thereby influence transplant outcome. In an accompanying manuscript, we describe that hyperlipidemia profoundly alters rejection of cardiac allografts (51). Cardiac allografts that normally undergo chronic rejection are acutely rejected when transplanted into hyperlipidemic recipients. Accelerated rejection involved the induction of a strong anti-donor Th17 response; this is not observed in mice with normal lipid levels. While production of IL-17 is clearly involved in driving accelerated rejection, IL-17 deficiency was not sufficient to overcome the changes in rejection observed in hyperlipidemic mice. We, therefore, hypothesized that hyperlipidemia results in additional changes that alter anti-donor responses and promote strong rejection responses. Our data indicate that in addition to altering the effector T cell subsets involved in rejection, hyperlipidemia results in alterations in regulatory T cells (Tregs) and the ability to induce tolerance.
Materials and Methods
Mice
Apoe<tm1Unc>/J (ApoE−/−) mice, B6.C-H2-Ab1bm12/KhEg (bm12), BALB/cJ and C57BL/6 controls were obtained from Jackson Laboratories (Bar Harbor, ME). FoxP3hCD2 knock-in mice (52) were kindly provided by Dr. Shohei Hori (RIKEN Research Center for Allergy and Immunology, Yokohama, Kanagawa, Japan). Mice were maintained on either high-fat diet, Western Diet, Teklad—21% fat, or normal chow LabDiet, JL mouse breeder/auto, 12% fat obtained from Harlan Laboratories (Indianapolis, IN). Our Institutional Animal Care and Use Committee (IACUC) approved all procedures in accordance with NIH guidelines for the care and use of animals.
Heterotopic heart transplantation
Vascularized heterotopic cardiac transplants were performed into the abdomen as previously described (53). Hearts were considered rejected when a heartbeat was no longer detectable by palpation. Rejection was verified by autopsy and visual inspection. Transplants that stopped beating within 48 h were assumed to be technical failures and eliminated from further analysis. All transplant recipients were 10–14 weeks of age.
Flow cytometry
Cell surface staining was performed using standard methods with the following antibodies: anti-CD4 (GK1.5, Biolegend, San Diego, CA), anti-CD8 (53-6.7, BD Pharmingen, San Jose, CA), anti-B220 (RA3-6B2, BD Pharmingen), anti-CD11b (M1/70, BD Pharmingen), anti-CD11c HL3, (BD Pharmingen) and CD25 (PC61, BD Pharmingen). Staining for FoxP3 was performed using the FoxP3 buffer set (eBioscience, San Diego, CA) according to the manufacturers instructions, anti-FoxP3 antibody (FJK16A, eBioscience), anti-Bim antibody (C34C5, Cell Signaling Technologies, Beverly, MA) and anti-AKT pS473 (M89-61, BD Pharmingen). Central (cTregs) and effector Tregs (eTregs) were analyzed as described in reference (54). All flow cytometry were performed on a FacsCanto II cytometer (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo v X 10.0.7 software (Tree Star, Inc., Ashland, OR).
Costimulatory molecule blockade
Costimulatory molecule blockade was performed as described in reference (55). Briefly, 0.5 mg of anti-CD154L mAb (MR1, BioXcell, West Lebanon, NH) and CTLA4Ig were administered intraperitoneally on the day of transplant. An additional 0.25 mg of each was administered 2, 4 and 6 days after transplantation. CTLA4Ig was the kind gift of Dr. Mohamed Sayegh.
Regulatory T cell function
CD4 T cells were purified using the Dynabeads FlowComp mouse CD4 kit (Life Technologies, Carlsbad, CA). Cells were then stained for CD25 expression (BD Pharmingen) and sorted by the Tufts Flow Cytometry Core. In indicated experiments, anti-human CD2 (RPA-2.10, BD Pharmingen) was used as a pseudomarker for FoxP3 expression. Isolated cells were counted and cultured with CD4+ T cells from naïve ApoE−/− mice on high-fat diet, or control C57BL/6 mice on normal chow labeled with CFSE. Cells were stimulated with 1 mg/mL anti-CD3 and anti-CD28 for 72 h and proliferation was assessed by CFSE dilution detected by flow cytometry and expressed as the Division Index for responding cells. Division Index is the average number of cell divisions that a cell in the original population has undergone and includes for cells that never divided (i.e. includes the undivided peak). Thus, it is a measure of proliferation that accurately accounts for cells that fail to divide.
Statistical analysis
The Kaplan and Meier method with a 95% confidence interval was used for the calculation of survival curves. Survival curves were compared using the log rank test. p-Values <0.05 using Student’s t-test were considered statistically significant.
Results
Hyperlipidemia leads to alterations in FoxP3+ T cell populations
Hearts from bm12 mice transplanted into H-2b mice normally undergo chronic rejection (56). However, recent data from our laboratory have shown that in hyperlipidemic recipients, bm12 hearts are acutely rejected suggesting that hyperlipidemia alters anti-donor rejection responses (51). This accelerated rejection response involves the induction of anti-donor Th17 alloreactivity that is not observed in mice with normal lipid levels. However, we noted that although IL-17 production is clearly involved in driving accelerated rejection in hyperlipidemic mice, ablation of IL-17 is not sufficient to fully prevent accelerated rejection. Since Tregs play a dominant role in preventing rejection of bm12 hearts (14), we hypothesized that hyperlipidemia may also result in defects in Tregs.
We examined Treg populations in apolipoprotein E (ApoE)-deficient and C57BL/6 mice made hyperlipidemic by feeding them a high-fat diet. We chose to use ApoE-deficient mice because the degree of hyperlipidemia can be manipulated in these mice by feeding them a high-fat diet. ApoE−/− mice fed a high-fat diet become profoundly hyperlipidemic, but maintain ratios and levels of plasma cholesterol, LDLs, VLDL, HDL, and triglycerides that are observed in patients (21,57–59). ApoE−/− mice maintained on normal chow also become hyperlipidemic, although to a lesser extent then mice fed a high-fat diet, and exhibit lipid levels similar to humans with hyperlipidemia (60). In either case, hyperlipidemia does not lead to elevated plasma glucose levels or obesity in ApoE−/− mice even when they are fed a high-fat diet (60,61). This eliminates the confounding factors of diabetes and obesity, which are known to have independent effects on immunity. Similarly, it is possible to induce hyperlipidemia in C57BL/6 mice, although to lesser levels than in ApoE−/− mice. The use of C57BL/6 mice eliminates the possibility that ApoE deficiency could explain any effects observed.
Feeding ApoE−/− mice a high-fat diet resulted in an increase in the frequency of FoxP3+, CD4+ cells in the spleen when compared with age-matched mice with normal lipid levels (Figure 1A, Figure S1). This increase was also apparent in C57BL/6 mice fed a high-fat diet when examined after 10 weeks when compared with age-matched controls fed normal chow (Figure 1A, Figure S1). ApoE−/− mice fed normal chow also had an increased frequency of FoxP3+, CD4+ cells compared to age-matched controls (Figure 1A). There was no significant difference in total splenocyte number, or in the frequency of CD4 T cells in hyperlipidemic mice (51). We also analyzed FoxP3+ CD4 T cells in the blood of hyperlipidemic mice in order to follow changes prospectively. Feeding ApoE−/− mice a high-fat diet for 5 weeks resulted in a significant increase in the frequency of FoxP3+, CD4+ cells in the blood compared with levels prior to initiation of high-fat diet (Figure 1B). This change was not apparent in the blood of C57BL/6 mice fed normal chow (Figure 1B). In addition, ApoE−/− mice fed a high-fat diet for 5 weeks contained a higher frequency of FoxP3+, CD4+ cells than age-matched ApoE−/− controls (Figure 1C), indicating that the rise in FoxP3+, CD4+ cells was attributable to lipid levels. The frequency of FoxP3+, CD4+ cells in the blood of C57BL/6 mice fed a high-fat diet for 5 weeks tended to be greater than in age-matched C57BL/6 controls fed normal chow, although the difference did not reach statistical significance at this time point. Age-matched ApoE−/− mice fed normal chow contained a significantly higher frequency of FoxP3+, CD4+ cells than C57BL/6 controls, consistent with their elevated lipid levels. Thus, hyperlipidemic mice experience a progressive increase in the frequency of FoxP3+ cells consistent with their degree of hyperlipidemia.
Figure 1. Hyperlipidemia promotes changes in FoxP3+ T cell populations.
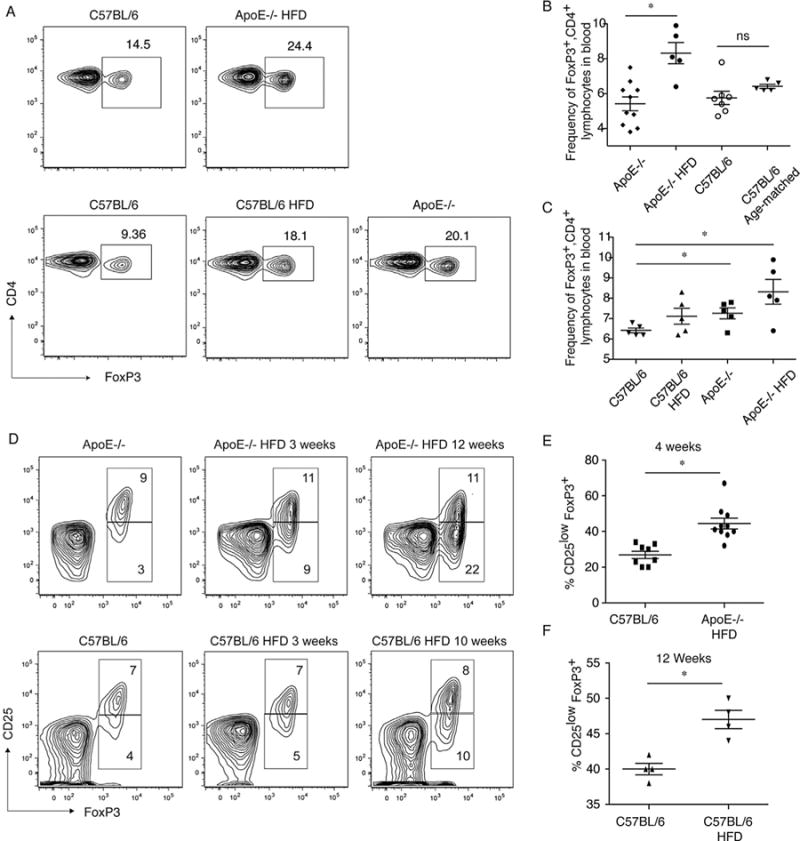
(A) Hyperlipidemia results in an increase in the frequency of CD4 T cells expressing FoxP3. Top panel, analysis of CD4 and FoxP3 in splenocytes from ApoE−/− mice fed a high-fat diet (HFD, total cholesterol [TC] 1821±321 mg/dL) for 6 weeks and age-matched C57BL/6 controls fed normal chow (TC 60±20 mg/dL). Bottom panel, CD4 and FoxP3 on C57BL/6 mice fed a high-fat diet for 10 weeks (TC 132±18 mg/dL) and age-matched C57BL/6 and ApoE−/− controls fed normal chow (494±95 mg/dL). Total cholesterol values are provided from (51) for reference. (B) Frequencies of FoxP3+, CD4+ lymphocytes increase over time after induction of hyperlipidemia, but not in mice with normal lipid levels. Frequencies of FoxP3+, CD4+ lymphocytes in the blood of ApoE−/− mice were examined prior to (diamonds), and after being fed a high-fat diet for 5 weeks (closed circles) as well as in the blood of 4-week-old C57BL/6 mice prior to (open circles), and after being fed a normal chow for 5 weeks (triangles). (C) Frequencies of FoxP3+, CD4+ lymphocytes in the blood of age-matched (9-week-old) C57BL/6 mice fed normal chow, C57BL/6 mice fed a high-fat diet for 5 weeks, starting at 4 weeks of age, aged-matched ApoE−/− mice fed normal chow (9 weeks old) and ApoE−/− mice fed a high-fat diet for 5 weeks, starting at 4 weeks of age. (D) ApoE−/− and C57BL/6 mice were fed high-fat diet (HFD) for 3 or 12 weeks. Animals were sacrificed and CD25 expression in CD4+ FoxP3+ cells was examined by intracellular staining followed by flow cytometry. Age-matched ApoE−/− and C57BL/6 mice fed a normal diet are shown as controls. Representative flow cytometry images are shown. (E) The percentage of CD4+ FoxP3+ cells expressing low levels of CD25 in ApoE−/− mice fed high-fat diet for four weeks (circles), together with age-matched C57BL/6 controls (squares). Combined results of three experiments are shown (p = 0.0003 between groups). (F) The percentage of CD4+ FoxP3+ cells expressing low levels of CD25 in C57BL/6 mice fed high-fat diet for 12 weeks (inverted triangles), together with age-matched C57BL/6 controls (triangles). Combined results of three experiments are shown (p = 0.004 between groups). ApoE, apolipoproteinE.
Further analysis revealed that in both ApoE−/− and C57BL/6 mice fed a high-fat diet, the frequency of FoxP3+, CD25high, CD4+ cells remained constant over time when compared to age-matched controls (Figure 1D). In contrast, in both ApoE−/− and C57BL/6 mice fed a high-fat diet, the frequency of FoxP3+, CD25low/− CD4+ cells increased over time (Figure 1D–F). Together, these data suggest that the increase in total FoxP3+ cells observed in hyper-lipidemic mice resulted from an increase in the frequency of FoxP3+, CD25low/−, CD4+ cells and did not reflect an increase in the FoxP3+, CD25high,CD4+ population that is known to contain functional Tregs (62).
Hyperlipidemia leads to alterations in central and effector Tregs
Tregs can be divided into cTregs and eTregs based on expression of FoxP3, CD62L, and CD44 (54,63,64). FoxP3+ cTregs are surface CD44low and CD62Lhigh. These cells recirculate through lymphoid tissue and are able to inhibit T cell priming in mice and humans (63–65). FoxP3+ eTreg are surface CD44high and CD62Llow, do not recirculate efficiently and comprise the major Treg population in non-lymphoid tissue. In ApoE−/− mice fed a high-fat diet, we observed a reduction in the frequency of cTregs compared to ApoE−/− controls fed normal chow (Figure 2A and B). Similarly, we observed a reduction in the frequency of cTregs in C57BL/6 mice fed a high-fat diet compared to controls (Figure 2A and B). The frequency of cTregs in C57BL/6 controls tended to be higher than that observed in ApoE−/− controls (Figure 2B). Although this difference did not reach statistical significance, it appears that the reduction in the frequency of cTregs observed tends to be proportionate to the degree of hyperlipidemia.
Figure 2. Hyperlipidemia leads to a decrease in cTregs and an increase in eTregs.
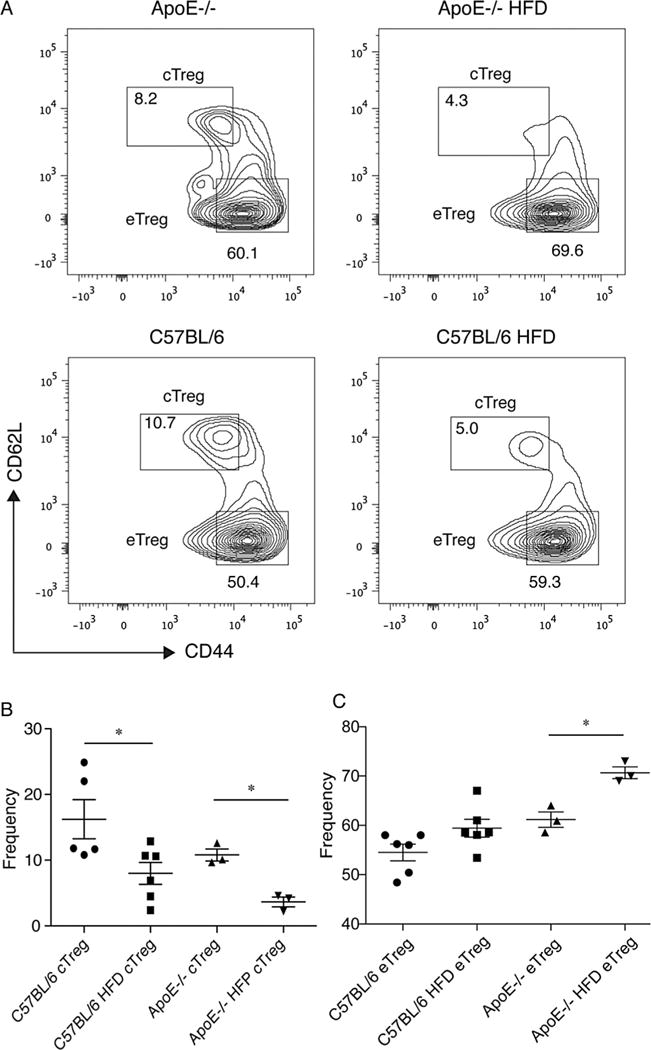
(A) Expression of CD44 and CD62L on FoxP3+, CD4+ T cells from ApoE−/− controls, ApoE−/− fed a high-fat diet (HFD) for 4 weeks, C57BL/6 controls and C57BL/6 mice fed a high-fat diet. Numbers shown represent the frequency after gating on FoxP3+ CD4+ T lymphocytes. Data are representative of at least two experiments containing three to six mice per group. (B) Frequencies of cTregs for mice in each group after gating on FoxP3+, CD4+ lymphocytes. (C) Frequencies of eTregs for mice in each group after gating on FoxP3+, CD4+ lymphocytes. ApoE, apolipoproteinE; CFSE, carboxyfluorescein succinimidyl ester; FoxP3, forkhead box P3; Treg, regulatory T cell.
In ApoE−/− mice fed a high-fat diet, we observed an increase in the frequency of eTregs compared to ApoE−/− controls fed normal chow (Figure 2A). Similarly, we observed a trend toward an increase in the frequency of eTregs in C57BL/6 mice fed a high-fat diet compared to controls (Figure 2A and C). We observed that eTregs expressed lower levels of CD25 than cTregs, consistent with other reports (54). Thus, while cTregs were decreased in hyperlipidemic mice, frequencies of eTregs were increased. Thus, it appears that the increase in FoxP3+, CD4+ T cells observed in hyperlipidemic mice is due at least in part to an increase in eTregs.
Hyperlipidemia alters Treg function
Alterations in Treg subsets prompted us to examine whether hyperlipidemia resulted in functional changes in Tregs. We initially observed that on a per cell basis, CD25high, CD4+ cells from hyperlipidemic mice, a population known to contain functional Tregs (62), inhibited proliferation of CD25− CD4+ T cells stimulated with anti-CD3 and CD28 less effectively than CD25high, CD4+ T cells purified from control mice (Figure 3A and B). To determine whether this defect was the result of alterations in bona fide Tregs, we examined the ability of FoxP3+, CD25high, CD4+ T cells from hyperlipidemic mice to inhibit proliferation. To this end, we made use of FoxP3hCD2 knock-in mice in which Foxp3+ T cells express a GPI-anchored human CD2-CD52 fusion protein on their surface that serves as a surrogate for Foxp3 expression allowing detection and purification of Foxp3+ and Foxp3− T cells based on hCD2 expression (52). hCD2(FoxP3)+, CD25high, CD4+ T cells purified from mice fed a high-fat diet showed a reduced ability to inhibit proliferation of T cells stimulated with anti-CD3 and CD28 on a per cell basis when compared to the same population isolated from control mice resulting in an increased division index when compared with Tregs from control mice (Figure 3C). Taken together, these data strongly suggest that hyperlipidemia leads to alterations in Treg function.
Figure 3. Decreased regulatory T cell function in hyperlipidemic mice.
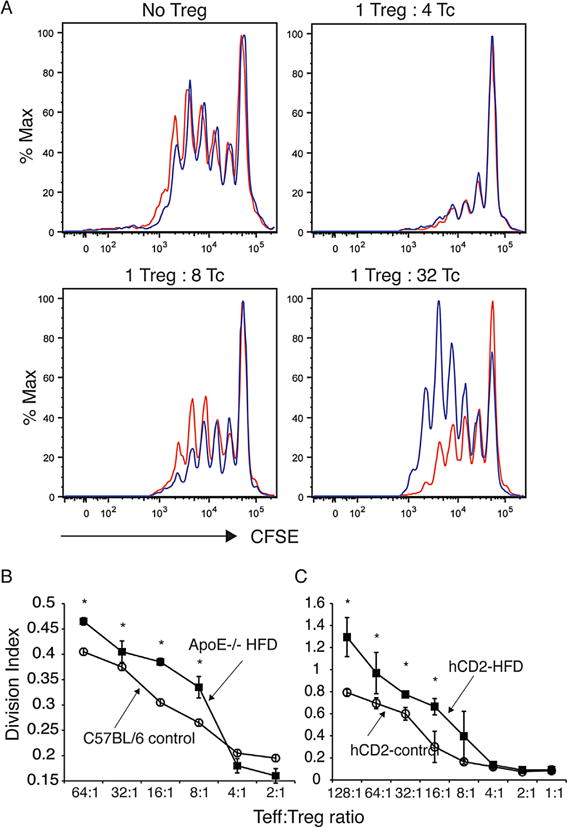
(A) Shown is the ability of CD4+CD25+ T cells from ApoE−/− mice fed a high-fat diet for 4 weeks (blue line) or C57BL/6 controls fed normal chow (red line) to inhibit the proliferation of CD4+ cells based on CFSE dilution following stimulation with anti-CD3 and CD28. (B) CD4+CD25+ cells were isolated from ApoE−/− mice fed a high-fat diet for 4 weeks or C57BL/6 controls fed normal chow and tested for the ability to inhibit proliferation of CD4+ cells stimulated with anti-CD3 and CD28 based on CFSE dilution. Division index is reported. Asterisk indicates p < 0.05. (C) FoxP3+CD4+CD25high cells were isolated from hCD2-FoxP3 mice on a C57BL/6 background that were fed a high-fat diet or normal chow and tested for the ability to inhibit proliferation of CD4+ cells stimulated with anti-CD3 and CD28 based on CFSE dilution. For panels B and C, division index is reported. Asterisk indicates p < 0.05. Shown is one representative experiment of three, with standard error. ApoE, apolipoproteinE; CFSE, carboxyfluorescein succinimidyl ester; FoxP3, forkhead box P3; HFD, high-fat diet; TC, total cholesterol; Treg, regulatory T cell.
Hyperlipidemia increase Akt activation in Tregs
In order to examine possible mechanisms that may promote reduced Treg function in hyperlipidemic mice, we examined signaling pathways that control Treg development and function. Akt signaling negatively regulates Treg development and function (66–69). Analysis of hCD2+ Tregs from control mice and mice fed a high-fat diet revealed that Tregs from hyperlipidemic mice contain increased levels of phosphorylated Akt (Ser473, Figure 4). Increased levels of phosphorylated Akt were observed in both ApoE−/− mice fed a high-fat diet (Figure 4A) and C57BL/6 mice fed a high-fat diet when compared to controls (Figure 4B and C). Given that increased levels of Akt phosphorylation are observed in mice that have not been challenged with antigen suggests that hyperlipidemia is sufficient to drive this change in Tregs.
Figure 4. Hyperlipidemia leads to Akt activation.
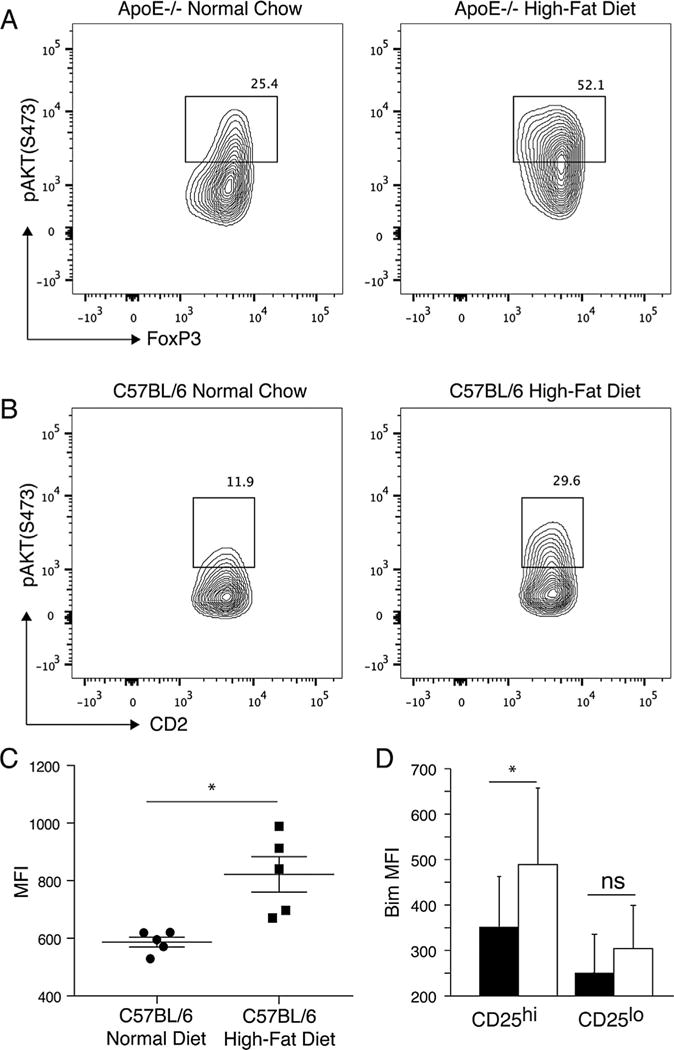
(A) Phospho-Akt levels in ApoE−/− controls fed normal chow and ApoE−/− mice fed a high-fat diet. ApoE−/− mice were fed either high-fat diet or normal chow for 10 weeks. Splenocytes were examined for expression of CD4, FoxP3 and phospho-AKT directly ex vivo. Shown gated on CD4+FoxP3+ lymphocytes. (B) Phospho-Akt levels in C57BL/6 controls fed normal chow and C57BL/6 mice fed a high-fat diet. C57BL/6 mice were fed either high-fat diet or normal chow for 10 weeks. Splenocytes were examined for expression of CD4, hCD2 and phospho-AKT directly ex vivo. Shown gated on CD4+CD2+ lymphocytes. (C) Median fluorescent intensity (MFI) of phospho-Akt levels in C57BL/6 controls fed normal chow and C57BL/6 mice fed a high-fat diet. (D) Expression of Bim in CD4+FoxP3+ cells expressing high levels (CD25hi) or low levels (CD25lo) of CD25 in ApoE−/− mice fed a high-fat diet for 4 weeks (black bars) or age-matched C57BL/6 mice fed normal chow (white bars) are shown. Combined results of three experiments are shown. Asterisk indicates p < 0.05. ApoE, apolipoproteinE; CFSE, carboxyfluorescein succinimidyl ester; FoxP3, forkhead box P3.
We next analyzed expression of potential targets of Akt that might affect Treg function, and the expansion of FoxP3+, CD25low/−, CD4+ cells. Akt has been previously reported to be able to phosphorylate BCL-2 protein family member Bim leading to its degradation (70). Downregulation of the BCL-2 protein family member Bim has also been reported to lead to expansion of FoxP3+, CD25 low/−, CD4+ cells that do not exert Treg function or exhibit reduced regulatory ability (71–75). We observed reduced levels of Bim in FoxP3+, CD25high, CD4+ Tregs from hyperlipidemic mice when compared to cells from controls (Figure 4D). Bim was expressed at low levels in FoxP3+, CD25 low/−, CD4+ cells from both hyperlipidemic and control mice (Figure 4D). We suggest that increased activation of Akt in Tregs from hyperlipidemic mice leads to defects in Treg function and leads to a reduction in Bim levels that may permit the expansion of FoxP3+, CD25low/−, CD4+ cells.
Hyperlipidemia prevents tolerance induction through costimulatory molecule blockade
To further assess defects in Treg function, we examined the ability to induce tolerance in hyperlipidemic mice. The ability to induce tolerance using costimulatory molecule blockade is dependent on Tregs (6–10). We, therefore, examined the ability to induce tolerance to fully allogeneic hearts in hyperlipidemic mice using costimulatory molecule blockade in order to examine the impact of hyperlipidemia on Treg function in vivo. Hyperlipidemic mice were transplanted with a fully mismatched allogeneic BALB/c heart and treated with anti-CD154 (MR1) and CTLA4-Ig to block costimulation though CD28 and CD40. This protocol routinely leads to long-term survival of fully allogeneic heart transplants (76). Control C57BL/6 mice treated with anti-CD154 (MR1) and CTLA4-Ig accepted fully allogeneic BALB/c hearts long-term (Figure 5). In contrast, hyperlipidemic mice treated with MR1 and CTLA4-Ig rejected fully allogeneic heart transplants (Figure 5D). Although transplant survival was prolonged relative to controls, costimulatory molecule blockade was not sufficient to induce operational tolerance in hyperlipidemic mice (Figure 5D). We have also analyzed the effect of hyperlipidemia on resistance to costimulatory molecule blockade induced tolerance in C57BL/6 mice fed a high-fat diet. These mice, like ApoE−/− mice fed a high-fat diet, also exhibit resistance to tolerance induction using MR1 and CTLA4-Ig rejecting fully allogeneic BALB/c hearts between day 41 and 56 (n = 4). These results support our data in Figure 5 showing that hyperlipidemia leads to resistance to tolerance induction. Thus, hyperlipidemia prevents tolerance induction using approaches that require Treg activity such as costimulatory molecule blockade.
Figure 5. Hyperlipidemia prevents the induction of operational tolerance through costimulatory blockade.
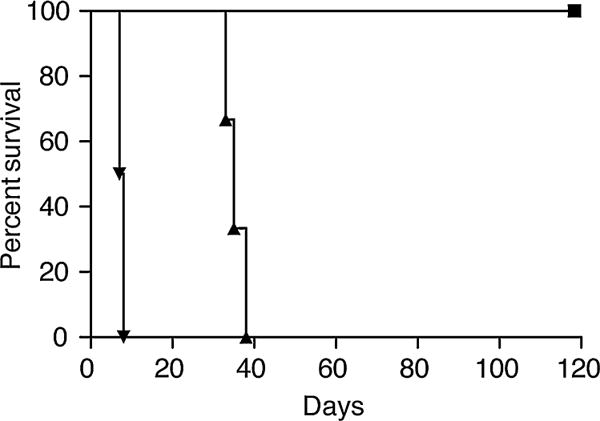
ApoE−/− mice were fed a high-fat diet and transplanted with fully mismatched BALB/c cardiac grafts. Mice were treated with a combination of CTLA-4Ig, and MR1 (triangles), or received no treatment (inverted triangles). Prolongation of BALB/c heart grafts in control C57BL/6 recipients fed normal chow and treated with CTLA-4Ig and MR1 (squares). ApoE, apolipoproteinE.
Discussion
Our data suggest that the common condition of hyperlipidemia can powerfully accelerate the rejection of cardiac allografts (51). Here, we show that hyperlipidemia mediates these effects on rejection at least in part by promoting changes in Tregs that reduce their function. These alterations, together with changes in the T cell subsets that mediated rejection, appear to promote resistance to tolerance induction using costimulatory molecule blockade. Thus, hyperlipidemia fundamentally alters anti-donor effector and regulatory responses. Our data, therefore, reveal a previously unappreciated effect of hyperlipidemia on rejection, its regulation and the ability to induce tolerance.
In an apparent contradiction to the observation that hyperlipidemic mice contain an increased frequency of alloreactive T cells, anti-donor Th17 responses and exhibit accelerated rejection (51), we observed that hyperlipidemia leads to an increase in total FoxP3+ CD4 T cells. These observations were consistent with another study showing that the total number of FoxP3+ CD4 T cells is increased in hyperlipidemic low-density lipoprotein null mice (77). Further examination revealed that the increase in the frequency of FoxP3+ cells was primarily the result of an increase in FoxP3+ CD4 T cells expressing low levels of CD25 rather than CD25high Tregs. We also observed that hyperlipidemia leads to alterations in Treg subsets, promoting a decrease in the frequency of cTregs and an increase in eTregs. Since eTregs express lower levels of CD25 (54), we suggest that the increase in FoxP3+ cells is the result of an increase this population. Based on the inverse relationship between changes in cTregs and eTreg numbers in hyperlipidemic mice, it is tempting to suggest that hyperlipidemia promotes the conversion of cTregs to eTregs. Alternatively, hyperlipidemia may alter the ability of naïve CD4 T cells to be induced to become cTregs favoring commitment to an eTreg phenotype, or allow for increased expansion of eTregs. We are currently examining these issues.
Since cTregs are able to inhibit T cell priming in mice and humans (63–65), we hypothesize that a decrease in this population would be likely to lead to an overall decrease in Treg function. Indeed, we found that the ability of CD4+ CD25+ and CD4+FoxP3+CD25high Tregs from hyperlipidemic C57BL/6 mice to suppress proliferation of CD4 T cells was reduced compared to cells from controls. Thus, our data demonstrate that hyperlipidemia is sufficient to mediate the changes we observed in Tregs. We are unaware of any previous reports demonstrating that induction of hyperlipidemia is sufficient lead to alterations in Treg subsets or function.
Analysis of signaling pathways that control Treg development and function revealed that Tregs from hyperlipidemic mice contain higher levels of activated Akt. Given that Akt signaling negatively regulates Treg development and function (66–69), we suggest that alterations in Akt play a role in altering the regulatory function of Tregs in hyperlipidemic mice. Akt has been previously reported to be able to phosphorylate BCL-2 protein family member Bim leading to its degradation (70). We observed reduced Bim levels in FoxP3+ CD25high CD4 Tregs from hyperlipidemic ApoE−/− mice, as well as C57BL/6 mice (data not shown). Reduced Bim levels have been shown to lead to an increase in FoxP3+ CD25low/− CD4 T cells (71–75). Our data suggest that reduced Bim levels in CD25high FoxP3+ CD4 T cells may promote the expansion of CD25low/−, FoxP3+ cells, a possibility we are currently examining. Together, these data suggest that altering the balance between activating signals mediated through Akt and cell-death via Bim affects Treg homeostasis leading to a reduction in Treg function and expansion of FoxP3+ CD25low/− CD4+ cells.
A key issue is whether the alterations observed in Tregs resulting from hyperlipidemia have in vivo consequences. Given the importance of Tregs in tolerance induction, we reasoned that alterations in Tregs in hyperlipidemic mice might affect the ability to induce tolerance for approaches that require Tregs, such as costimulatory molecule blockade (8,9,78–80). Costimulatory molecule blockade using CTLA4-Ig and anti-CD154 failed to induce tolerance to fully allogeneic cardiac transplants in hyperlipidemic ApoE−/− mice. These data support the notion that hyperlipidemia alters Treg function, although the alterations we observed in T cell subsets observed in hyperlipidemic mice (51) may also play a role in tolerance resistance. Like transplant patients, ApoE−/− mice develop hyperlipidemia, and elevated levels of low-density lipoprotein as a result of alterations in lipid metabolism, rather than as a result of dietary changes resulting in obesity and elevated blood glucose. Thus, these data suggest that hyperlipidemia in transplant patients may similarly be impeded by hyperlipidemia. Similar results were observed in hyperlipidemic C57BL/6 mice that received a bm12 cardiac allograft (not shown). Given that survival of bm12 hearts transplanted into C57BL/6 mice is strictly controlled by Tregs (14), accelerated rejection of bm12 hearts in hyperlipidemic mice also supports the idea that hyperlipidemia alters Tregs. Given that the majority of transplant patients become hyperlipidemic, we hypothesize that hyperlipidemia represents a barrier to costimulatory molecule blockade induced tolerance through effects on Tregs.
Several studies have suggested that ApoE-deficient mice have changes in immune responses (81). However, it is not clear whether these changes are due to the effect of lipid levels on cells of the immune system, or some undefined role for ApoE in immunity. We demonstrate that ApoE−/− mice exhibit changed Treg frequency and phenotype when compared with ApoE−/− controls fed normal diet. Thus, the effect observed is increased in mice fed a high-fat diet and proportional to the degree of hyperlipidemia. Furthermore, we observed that in wild-type C57BL/6 mice, raising lipid levels through feeding a high-fat diet results in changes to Tregs. These data suggest that the observed effects are, therefore, a direct result of increased lipid levels and are dependent on the degree of recipient hyperlipidemia. Indeed, we wish to point out that unlike the LdL receptor, ApoE is a component of the lipid transport system and is not expressed on T cells, or other components of the immune system. This together with the fact that ApoE mice do not become obese of hyperglycemic after inducing hyperlipidemia suggest that the effects observed are directly related to increase lipid levels.
The data presented here demonstrate that hyperlipidemia by diet profoundly changes the host’s ability to regulate immune responses by affecting Tregs. These studies provide the proof-of-principle for examining whether similar changes are observed in humans with hyperlipidemia. If hyperlipidemia leads to similar alterations in humans, it may be possible to improve transplant outcomes by developing therapies that allow for maintenance of full Treg function. Indeed, it is possible that the inability to induce tolerance or long-term allograft survival in humans using costimulatory molecule blockade may be due, at least in part, to the effects of hyperlipidemia on anti-donor responses and the ability to regulate them.
Supplementary Material
Figure S1: Frequency of FoxP3+CD4+ cells in the spleen of ApoE−/− mice fed high-fat diet (HFD) for 4 weeks, and age-matched controls fed normal chow (NC), or in C57BL/6 mice fed high-fat diet for 10 weeks, and age-matched C57BL/6 mice fed normal chow. Asterisk indicates p < 0.05.
Acknowledgments
The authors thank Philip W. Hinds (Department of Developmental, Molecular and Chemical Biology, Tufts University School of Medicine) for critical review of the manuscript and Mohamed H. Sayegh (American University of Beirut) for helpful discussions and providing critical reagents. We also thank Wayne Hancock (The Children’s Hospital of Philadelphia and University of Pennsylvania) for providing helpful insight and critical review of the manuscript, and Daniel J. Campbell (Benaroya Research Institute, Seattle) for helpful discussions. This work was supported in part by a grant from a Grant in Aid from the American Heart Association to J.I., an American Heart Association Scientist Development Grant to J.B. and an American Society of Transplantation/Genentech Basic Science Fellowship Grant to J.Y.
Abbreviations
- ApoE
apolipoproteinE
- CFSE
carboxyfluorescein succinimidyl ester
- cTregs
central regulatory T cell
- eTregs
effector regulatory T cell
- FoxP3
forkhead box P3
- HDL
high density lipoprotein
- IFN-γ
interferon gamma
- Ig
immunoglobulin
- IL
interleukin
- LDL
low density lipoprotein
- MST
median survival time
- PMA
phorbol myristate acetate
- TC
total cholesterol
- Th1
T-helper type 1
- Th17
T-helper type 17
- Treg
regulatory T cell
- VLDL
very low density lipoprotein
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Author Contributions
J.B. planned experiments, performed all flow cytometry, ELISpot and suppression assays, analyzed data, and wrote the manuscript. J.Y. performed all transplants. A.C. prepared and supplied CTLA-4 Ig and provided guidance on experiments using this reagent. J.I. oversaw the study, analyzed the data, and wrote the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- 1.Strom TB, Roy-Chaudhury P, Manfro R, et al. The Th1/Th2 paradigm and the allograft response. Curr Opin Immunol. 1996;8:688–693. doi: 10.1016/s0952-7915(96)80087-2. [DOI] [PubMed] [Google Scholar]
- 2.Le Moine A, Goldman M, Abramowicz D. Multiple pathways to allograft rejection. Transplantation. 2002;73:1373–1381. doi: 10.1097/00007890-200205150-00001. [DOI] [PubMed] [Google Scholar]
- 3.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100:550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood KJ, Goto R. Mechanisms of rejection: Current perspectives. Transplantation. 2012;93:1–10. doi: 10.1097/TP.0b013e31823cab44. [DOI] [PubMed] [Google Scholar]
- 5.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev. 2003;196:51–64. doi: 10.1046/j.1600-065x.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 6.Verbinnen B, Billiau AD, Vermeiren J, et al. Contribution of regulatory T cells and effector T cell deletion in tolerance induction by costimulation blockade. J Immunol. 2008;181:1034–1042. doi: 10.4049/jimmunol.181.2.1034. [DOI] [PubMed] [Google Scholar]
- 7.Vogel I, Verbinnen B, Maes W, Boon L, Van Gool SW, Ceuppens JL. Foxp3+ regulatory T cells are activated in spite of B7-CD28 and CD40-CD40L blockade. Eur J Immunol. 2013;43:1013–1023. doi: 10.1002/eji.201242737. [DOI] [PubMed] [Google Scholar]
- 8.Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med. 2001;193:1311–1318. doi: 10.1084/jem.193.11.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrer IR, Wagener ME, Song M, Kirk AD, Larsen CP, Ford ML. Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proc Natl Acad Sci USA. 2011;108:20701–20706. doi: 10.1073/pnas.1105500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 11.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: A critical reappraisal. Am J Transplant. 2011;11:450–462. doi: 10.1111/j.1600-6143.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 12.Lodhi SA, Lamb KE, Meier-Kriesche HU. Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant. 2011;11:1226–1235. doi: 10.1111/j.1600-6143.2011.03539.x. [DOI] [PubMed] [Google Scholar]
- 13.Stehlik J, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Twenty-eighth Adult Heart Transplant Report—2011. J Heart Lung Transplant. 2011;30:1078–1094. doi: 10.1016/j.healun.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Schenk S, Kish DD, He C, et al. Alloreactive T cell responses and acute rejection of single class II MHC-disparate heart allografts are under strict regulation by CD4+ CD25+ T cells. J Immunol. 2005;174:3741–3748. doi: 10.4049/jimmunol.174.6.3741. [DOI] [PubMed] [Google Scholar]
- 15.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 16.Cobbold SP, Waldmann H. Regulatory cells and transplantation tolerance. Cold Spring Harb Perspect Med. 2013;3 doi: 10.1101/cshperspect.a015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrer IR, Hester J, Bushell A, Wood KJ. Induction of transplantation tolerance through regulatory cells: From mice to men. Immunol Rev. 2014;258:102–116. doi: 10.1111/imr.12158. [DOI] [PubMed] [Google Scholar]
- 18.Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. 2014;5:46. doi: 10.3389/fimmu.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ford ML, Larsen CP. Translating costimulation blockade to the clinic: Lessons learned from three pathways. Immunol Rev. 2009;229:294–306. doi: 10.1111/j.1600-065X.2009.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18:4266–4288. doi: 10.2174/138161212802481237. [DOI] [PubMed] [Google Scholar]
- 21.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 22.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 23.Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138(5 Pt 2):S419–S420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- 24.Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 25.Tedgui A, Mallat Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 26.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 27.Gao Q, Jiang Y, Ma T, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. 2010;185:5820–5827. doi: 10.4049/jimmunol.1000116. [DOI] [PubMed] [Google Scholar]
- 28.Ng HP, Burris RL, Nagarajan S. Attenuated atherosclerotic lesions in apoE-Fcgamma-chain-deficient hyperlipidemic mouse model is associated with inhibition of Th17 cells and promotion of regulatory T cells. J Immunol. 2011;187:6082–6093. doi: 10.4049/jimmunol.1004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith E, Prasad K-MR, Butcher M, et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2010;121:1746–1755. doi: 10.1161/CIRCULATIONAHA.109.924886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eid RE, Rao DA, Zhou J, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119:1424–1432. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jafarzadeh A, Nemati M, Rezayati MT. Serum levels of interleukin (IL)-27 in patients with ischemic heart disease. Cytokine. 2011;56:153–156. doi: 10.1016/j.cyto.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Jefferis BJ, Papacosta O, Owen CG, et al. Interleukin 18 and coronary heart disease: prospective study and systematic review. Atherosclerosis. 2011;217:227–233. doi: 10.1016/j.atherosclerosis.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 34.Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet. 1998;351:88–92. doi: 10.1016/S0140-6736(97)09032-6. [DOI] [PubMed] [Google Scholar]
- 35.Rosso R, Roth A, Herz I, Miller H, Keren G, George J. Serum levels of interleukin-18 in patients with stable and unstable angina pectoris. Int J Cardiol. 2005;98:45–48. doi: 10.1016/j.ijcard.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 36.Tzoulaki I, Murray GD, Lee AJ, Rumley A, Lowe GD, Fowkes FG. C-reactive protein, interleukin-6, and soluble adhesion molecules as predictors of progressive peripheral atherosclerosis in the general population: Edinburgh Artery Study. Circulation. 2005;112:976–983. doi: 10.1161/CIRCULATIONAHA.104.513085. [DOI] [PubMed] [Google Scholar]
- 37.Correa CR, Dias-Melicio LA, Calvi SA, Lastoria S, Soares AM. Activation of monocytes and cytokine production in patients with peripheral atherosclerosis obliterans. J Inflamm (Lond) 2011;8:23. doi: 10.1186/1476-9255-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris TB, Ferrucci L, Tracy RP, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–512. doi: 10.1016/s0002-9343(99)00066-2. [DOI] [PubMed] [Google Scholar]
- 39.Biasucci LM, Vitelli A, Liuzzo G, et al. Elevated levels of interleukin-6 in unstable angina. Circulation. 1996;94:874–877. doi: 10.1161/01.cir.94.5.874. [DOI] [PubMed] [Google Scholar]
- 40.Yamashita H, Shimada K, Seki E, Mokuno H, Daida H. Concentrations of interleukins, interferon, and C-reactive protein in stable and unstable angina pectoris. Am J Cardiol. 2003;91:133–136. doi: 10.1016/s0002-9149(02)03097-7. [DOI] [PubMed] [Google Scholar]
- 41.Narins CR, Lin DA, Burton PB, Jin ZG, Berk BC. Interleukin-18 and interleukin-18 binding protein levels before and after percutaneous coronary intervention in patients with and without recent myocardial infarction. Am J Cardiol. 2004;94:1285–1287. doi: 10.1016/j.amjcard.2004.07.114. [DOI] [PubMed] [Google Scholar]
- 42.Mallat Z, Henry P, Fressonnet R, et al. Increased plasma concentrations of interleukin-18 in acute coronary syndromes. Heart. 2002;88:467–469. doi: 10.1136/heart.88.5.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie JJ, Wang J, Tang TT, et al. The Th17/Treg functional imbalance during atherogenesis in ApoE(−/−) mice. Cytokine. 2010;49:185–193. doi: 10.1016/j.cyto.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Yin M, Zhang L, Sun XM, Mao LF, Pan J. Lack of apoE causes alteration of cytokines expression in young mice liver. Mol Biol Rep. 2010;37:2049–2054. doi: 10.1007/s11033-009-9660-x. [DOI] [PubMed] [Google Scholar]
- 45.Lim H, Kim YU, Sun H, et al. Proatherogenic conditions promote autoimmune T helper 17 cell responses in vivo. Immunity. 2014;40:153–165. doi: 10.1016/j.immuni.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Y, Wang Z, Deng L, et al. Osteopontin promotes inflammation in patients with acute coronary syndrome through its activity on IL-17 producing cells. Eur J Immunol. 2012;42:2803–2814. doi: 10.1002/eji.201242475. [DOI] [PubMed] [Google Scholar]
- 47.Pflugfelder PW, Huff M, Oskalns R, Rudas L, Kostuk WJ. Cholesterol-lowering therapy after heart transplantation: A 12-month randomized trial. J Heart Lung Transplant. 1995;14:613–622. [PubMed] [Google Scholar]
- 48.Zakliczynski M, Boguslawska J, Wojniak E, et al. In the era of the universal use of statins dyslipidemia’s are still common in heart transplant recipients: A cross-sectional study. Transplant Proc. 2011;43:3071–3073. doi: 10.1016/j.transproceed.2011.08.052. [DOI] [PubMed] [Google Scholar]
- 49.Shi C, Lee W-S, Russell ME, et al. Hypercholesterolemia exacerbates transplant arteriosclerosis via increased neointimal smooth muscle cell accumulation. Circulation. 1997;96:2722–2728. doi: 10.1161/01.cir.96.8.2722. [DOI] [PubMed] [Google Scholar]
- 50.Russell PS, Chase CM, Colvin RB. Accelerated atheromatous lesions in mouse hearts transplanted to apolipoprotein-E-deficient recipients. Am J Pathol. 1996;149:91–99. [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan J, Bagley J, Iacomini J. Hyperlipidemia promotes anti-donor Th17 responses that accelerate allograft rejection. Am J Transplant. 2015;15:2336–2345. doi: 10.1111/ajt.13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyao T, Floess S, Setoguchi R, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 53.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 54.Smigiel KS, Richards E, Srivastava S, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211:121–136. doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan X, Ansari MJ, D’Addio F, et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci USA. 2009;106:10734–10739. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sayegh MH, Wu Z, Hancock WW, et al. Allograft rejection in a new allospecific CD4+ TCR transgenic mouse. Am J Transplant. 2003;3:381–389. doi: 10.1034/j.1600-6143.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 57.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitman SC. A practical approach to using mice in atherosclerosis research. Clin Biochem Rev. 2004;25:81–93. [PMC free article] [PubMed] [Google Scholar]
- 59.Plump AS, Breslow JL. Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr. 1995;15:495–518. doi: 10.1146/annurev.nu.15.070195.002431. [DOI] [PubMed] [Google Scholar]
- 60.Moghadasian MH, McManus BM, Nguyen LB, et al. Pathophysiology of apolipoprotein E deficiency in mice: Relevance to apo E-related disorders in humans. FASEB J. 2001;15:2623–2630. doi: 10.1096/fj.01-0463com. [DOI] [PubMed] [Google Scholar]
- 61.Davis HR, Jr, Hoos LM, Tetzloff G, et al. Deficiency of Niemann-Pick C1 Like 1 prevents atherosclerosis in ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2007;27:841–849. doi: 10.1161/01.ATV.0000257627.40486.46. [DOI] [PubMed] [Google Scholar]
- 62.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol. 2013;34:74–80. doi: 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 64.Huehn J, Siegmund K, Hamann A. Migration rules: Functional properties of naive and effector/memory-like regulatory T cell subsets. Curr Top Microbiol Immunol. 2005;293:89–114. doi: 10.1007/3-540-27702-1_5. [DOI] [PubMed] [Google Scholar]
- 65.Lange CM, Tran TY, Farnik H, et al. Increased frequency of regulatory T cells and selection of highly potent CD62L+ cells during treatment of human lung transplant recipients with rapamycin. Transpl Int. 2010;23:266–276. doi: 10.1111/j.1432-2277.2009.00973.x. [DOI] [PubMed] [Google Scholar]
- 66.Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109:2014–2022. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- 67.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sauer S, Bruno L, Hertweck A, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 70.Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813–823. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 71.Zhan Y, Zhang Y, Gray D, et al. Defects in the Bcl-2-regulated apoptotic pathway lead to preferential increase of CD25 low Foxp3+ anergic CD4+ T cells. J Immunol. 2011;187:1566–1577. doi: 10.4049/jimmunol.1100027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Szymczak-Workman AL, Gravano DM, Workman CJ, Green DR, Vignali DA. Preferential control of induced regulatory T cell homeostasis via a Bim/Bcl-2 axis. Cell Death Dis. 2012;3:e270. doi: 10.1038/cddis.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chougnet CA, Tripathi P, Lages CS, et al. A major role for Bim in regulatory T cell homeostasis. J Immunol. 2011;186:156–163. doi: 10.4049/jimmunol.1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tischner D, Gaggl I, Peschel I, et al. Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and limits their functionality in mice. J Autoimmun. 2012;38:59–69. doi: 10.1016/j.jaut.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kashiwakura Y, Sakurai D, Kanno Y, et al. CD2-mediated regulation of peripheral CD4(+) CD25(+) regulatory T-cell apoptosis accompanied by down-regulation of Bim. Immunology. 2013;139:48–60. doi: 10.1111/imm.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 77.Maganto-Garcia E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation. 2011;124:185–195. doi: 10.1161/CIRCULATIONAHA.110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ochando JC, Homma C, Yang Y, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. 2006;7:652–662. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 79.Taylor PA, Lees CJ, Waldmann H, Noelle RJ, Blazar BR. Requirements for the promotion of allogeneic engraftment by anti-CD154 (anti-CD40L) monoclonal antibody under nonmyeloablative conditions. Blood. 2001;98:467–474. doi: 10.1182/blood.v98.2.467. [DOI] [PubMed] [Google Scholar]
- 80.Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: Transplantation and autoimmunity. Nat Rev Nephrol. 2014;10:14–24. doi: 10.1038/nrneph.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang HL, Wu J, Zhu J. The immune-modulatory role of apolipoprotein E with emphasis on multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Dev Immunol. 2010;2010:186813. doi: 10.1155/2010/186813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Frequency of FoxP3+CD4+ cells in the spleen of ApoE−/− mice fed high-fat diet (HFD) for 4 weeks, and age-matched controls fed normal chow (NC), or in C57BL/6 mice fed high-fat diet for 10 weeks, and age-matched C57BL/6 mice fed normal chow. Asterisk indicates p < 0.05.