

Abstract
Mushroom dendritic spine structures are essential for memory storage and the loss of mushroom spines may explain memory defects in aging and Alzheimer's disease (AD). The stability of mushroom spines depends on stromal interaction molecule 2 (STIM2)-mediated neuronal-store-operated Ca2+ influx (nSOC) pathway, which is compromised in AD mouse models, in aging neurons, and in sporadic AD patients. Here, we demonstrate that the Transient Receptor Potential Canonical 6 (TRPC6) and Orai2 channels form a STIM2-regulated nSOC Ca2+ channel complex in hippocampal mushroom spines. We further demonstrate that a known TRPC6 activator, hyperforin, and a novel nSOC positive modulator, NSN21778 (NSN), can stimulate activity of nSOC pathway in the spines and rescue mushroom spine loss in both presenilin and APP knock-in mouse models of AD. We further show that NSN rescues hippocampal long-term potentiation impairment in APP knock-in mouse model. We conclude that the STIM2-regulated TRPC6/Orai2 nSOC channel complex in dendritic mushroom spines is a new therapeutic target for the treatment of memory loss in aging and AD and that NSN is a potential candidate molecule for therapeutic intervention in brain aging and AD.
SIGNIFICANCE STATEMENT Mushroom dendritic spine structures are essential for memory storage and the loss of mushroom spines may explain memory defects in Alzheimer's disease (AD). This study demonstrated that Transient Receptor Potential Canonical 6 (TRPC6) and Orai2 form stromal interaction molecule 2 (STIM2)-regulated neuronal-store-operated Ca2+ influx (nSOC) channel complex in hippocampal synapse and the resulting Ca2+ influx is critical for long-term maintenance of mushroom spines in hippocampal neurons. A novel nSOC-positive modulator, NSN21778 (NSN), rescues mushroom spine loss and synaptic plasticity impairment in AD mice models. The TRPC6/Orai2 nSOC channel complex is a new therapeutic target and NSN is a potential candidate molecule for therapeutic intervention in brain aging and AD.
Keywords: calcium, channels, dendrites, hippocampus, imaging, synapse
Introduction
Alzheimer's disease (AD) is the most common reason for elderly dementia in the world. Despite extensive studies of AD pathology for >100 years, there are no disease-modifying therapies for AD. Memory loss in AD results from “synaptic failure” (Selkoe, 2002; Koffie et al., 2011; Tu et al., 2014). Postsynaptic dendritic spines play an important role in learning and memory (Kasai et al., 2003; Bourne and Harris, 2008). Postsynaptic spines are usually classified into three groups according to their morphological structure: mushroom spines, thin spines, and stubby spines (Kasai et al., 2003; Bourne and Harris, 2008). It has been proposed that the mushroom spines are stable “memory spines” that make functionally stronger synapses that are responsible for memory storage (Bourne and Harris, 2007). We and others have proposed previously that mushroom spines are destabilized selectively in AD and that the loss of mushroom spines may underlie cognitive decline during progression of the disease (Tackenberg et al., 2009; Popugaeva et al., 2012; Bezprozvanny and Hiesinger, 2013; Popugaeva and Bezprozvanny, 2013). However, the cell biological mechanisms that may lead to the loss of mushroom spines in AD are poorly understood.
In studies with hippocampal neurons neuronal-store-operated calcium entry (nSOC) pathway has been implicated previously in the modulation of synaptic plasticity (Baba et al., 2003) and in the control of neuronal growth cone mobility (Mitchell et al., 2012). We demonstrated recently that nSOC Ca2+ entry in postsynaptic spines plays a key role in stability of mushroom spines by activating synaptic CaMKII constitutively (Sun et al., 2014a). We further demonstrated that dendritic spine nSOC is controlled by stromal interaction molecule 2 (STIM2) and that the STIM2-nSOC-CaMKII pathway is compromised in PS1M146V knock-in (PS1KI) neurons, in aging neurons, and in sporadic AD brains due to downregulation of the STIM2 protein (Sun et al., 2014a). Moreover, we have demonstrated that the expression of STIM2 protein rescues spine nSOC and mushroom spine loss in PS1KI hippocampal neurons (Sun et al., 2014a). In the follow-up studies, we demonstrated that the STIM2-nSOC pathway is downregulated in conditions of amyloid toxicity and that overexpression of STIM2 protects hippocampal mushroom spines from amyloid-induced loss (Popugaeva et al., 2015; Zhang et al., 2015b). These studies suggested that the STIM2-nSOC pathway is a potentially important AD therapeutic target; however, the molecular identity of the STIM2-regulated nSOC channel in the spines is unknown.
In the present study, we used a candidate approach to demonstrate that STIM2-gated nSOC channels in the spines are formed by a complex of Transient Receptor Potential Canonical 6 (TRPC6) and Orai2. We further show that the known TRPC6 activator hyperforin (Hyp) (Leuner et al., 2007) and a novel nSOC-positive modulator, NSN21778 (NSN) (Wu et al., 2011), can stimulate STIM2-nSOC pathway in the spines and rescue mushroom spine loss in both PS1KI mice (Guo et al., 1999) and APPNL-F/NL-F knock-in mice (APPKI) mice (Saito et al., 2014) hippocampal neurons. Furthermore, we demonstrate that NSN rescues hippocampal long-term potentiation (LTP) impairment in APPKI mice. We conclude that the STIM2-regulated TRPC6/Orai2 nSOC channel complex in dendritic mushroom spines is a new therapeutic target for the treatment of memory loss in aging and AD and that NSN is a potential candidate molecule for therapeutic intervention in brain aging and AD.
Materials and Methods
Materials.
NSN (N-{4-[2-(6-amino-quinazolin-4-ylamino)-ethyl]-phenyl}-acetamide) was synthesized and purified by Nanosyn. YFP-STIM2 was kindly provided by Dr. Jen Liou. Human TRPC6 cDNA and mouse Orai2 cDNA clones were purchased from Open Biosystems and used to generate HA-TRPC6 and HA-Orai2 lentiviral expression constructs by PCR. YFP-TRPC6 was kindly provided by Dr. Craig Montell. HA-TRPC1, FLAG-TRPC3, FLAG-TRPC4, FLAG-TRPC5, FLAG-TRPC6, and FLAG-TRPC7 were kindly provided by Dr. Joseph Yuan. GST-S2-SOAR (aa 348–450) and GST-S2-CT (aa 248-C terminal) were generated by PCR and cloned into the PGEX-KG vector. STIM2-LASS (L377S, A380S) mutation was generated by Q5 mutagenesis Kit (Sigma-Aldrich), control short-hairpin RNA interference (Ctrl-shRNAi) (SHC002), mouse TRPC6-shRNAi (SHCLNG-NM_013838, TRCN0000068394), mouse Orai1-shRNAi (SHCLNG-NM_175423, TRCN0000125405), and mouse Orai2-shRNAi (SHCLNG-NM_178751, TRCN0000126314) lentivirus shuttle constructs were obtained from Sigma-Aldrich. Lentiviruses were generated by cotransfection of two helper plasmids (pVSVg and pCMVΔ8.9) into the packaging cell line HEK293T, as we described previously (Zhang et al., 2010).
Animals.
The PS1-M146V knock-in mice (PS1KI) (Guo et al., 1999) were kindly provided by Hui Zheng (Baylor University). APPKI mice were kindly provided by Takaomi Saido (Riken, Japan) (Saito et al., 2014). Wild-type (WT) mice of the same strain (C57BL/6) were used in control experiments. PS1KIGFP and APPKIGFP mice were generated by crossing PS1KI or APPKI mice with Line M GFP mice (C57BL/6 strain) (Feng et al., 2000). All mice colonies were established and housed in a vivarium (four per cage) with 12 h light/dark cycle at the University of Texas (UT) Southwestern Medical Center barrier facility. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of the UT Southwestern Medical Center at Dalla, in accordance with the National Institutes of Health's Guidelines for the Care and Use of Experimental Animals.
Antibodies.
Anti-TRPC6 pAb (1:500, SAB4300572; Sigma-Aldrich), anti-Orai2 pAb (1:200, sc-292103; Santa Cruz Biotechnology), anti-TRPC1 mAb (1:200, sc-133076; Santa Cruz Biotechnology), anti-TRPC4 pAb (1:400, AB5812; Chemicon), anti-Orai1 pAb (1:200, sc-68895; Santa Cruz Biotechnology), anti-STIM2 goat antibody (used for immunoprecipitation) (sc-79110; Santa Cruz Biotechnology), anti-GFP mAb (1:2000, MA5-15256; Pierce), anti-FLAG (1:1000, F3165; Sigma-Aldrich), anti-HA (1:3000, MMS-101R; Covance), anti-STIM2 pAb (used for Western blotting) (1:500, 4917s; Cell Signaling Technology), anti-phospho-CaMKII (1:1000, 3361s; Cell Signaling Technology), anti-CaMKII (1:1000, MAB8699; Millipore), anti-PSD95 (1:1000, 3450s; Cell Signaling Technology), anti-GAPDH (1:1000, Millipore, MAB374), and Anti-Aβ 6E10 mAb (1:1000, SIG-39300; Covance) were used. HRP-conjugated anti-rabbit and anti-mouse secondary antibodies were from Jackson ImmunoReseach ((115-035-146 and 111-035-144, respectively).
Quantitative reverse transcription PCR (qRT-PCR).
For mouse gene expression profiling, different brain region tissue were obtained from 7- to 8-week-old male C57BL/6 mice (n = 6). RNA was extracted using RNAStat60 (TelTest) according to the manufacturer's directions. Total RNA was pooled in equal quantities for each tissue (n = 6). Genomic DNA contamination was eliminated by DNase I (Roche). cDNA for qRT-PCR assays was prepared using the High Capacity cDNA Reverse Transcription kit (Life Technologies). Gene expression levels were measured on an Applied Biosystems 7900HT with SYBR green chemistry using the primers shown in Table 1. Normalized mRNA levels are expressed as arbitrary units and were obtained by dividing the averaged, efficiency-corrected values for mRNA expression by that for 18s rRNA (mouse 18s rRNA forward: accgcagctaggaataatgga and mouse 18s rRNA reverse: gcctcagttccgaaaacca). The resulting values were multiplied by 105 for graphical representation. Average technical variance was the SDs of the measured values in n = 3 technical replicates.
Table 1.
Genes and primers used in the present study
| Gene | Forward | Reverse |
|---|---|---|
| Trpc1 | tgaacttagtgctgacttaaaggaac | cgggctagctcttcataatca |
| Trpc2 | acgaaaggagcctgagtttaag | ccagcaactcgaagccatag |
| Trpc3 | ttaattatggtctgggttcttgg | tccacaactgcacgatgtact |
| Trpc4 | aaggaagccagaaagcttcg | ccaggttcctcatcacctct |
| Trpc5 | gcctgatacaaaatcaacattatca | gcccctcatttgttttgga |
| Trpc6 | gcagctgttcaggatgaaaac | ttcagcccatatcatgccta |
| Trpc7 | cctgcgtattctactctcgatg | cgttgaacatgtaggcagga |
| Orai1 | tacttaagccgcgccaag | acttccaccatcgctacca |
| Orai2 | gggaggagaagatgacctctg | gccttgaacccctgatcc |
| Orai3 | cacatctgctctgctgtcg | ggtgggtattcatgatcgttct |
Hippocampal synaptosome fraction and coimmunoprecipitation.
Hippocampal regions were extracted from 1-month-old mice, homogenized in 0.32 m sucrose and 25 mm HEPES, pH 7.2, and centrifuged for 10 min at 800 × g to remove the nuclei. The low-speed supernatant was then centrifuged for 20 min at 12,000 × g to separate the synaptosomal supernatant and synaptosomal membrane fractions (P2 pellet). The P2 pellet were solubilized in lysis buffer containing 1% CHAPS plus the following (in mm): 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.4 KH2PO4, pH 7.2, 5 EDTA, and 5 EGTA and protease inhibitors for 2 h at 4°C. Insoluble material was removed by centrifugation of samples for 20 min at 16,300 × g. Protein concentration in the synaptosome fraction was measured with a Nanodrop OD280. For each coimmunoprecipitation reaction, 500 μg of total protein lysates were first precleaned with normal rabbit, mouse, or goat IgG and protein A/G beads at 4°C for 1 h, incubated with 2 μg of primary antibody at 4°C for 1 h, and incubated with 20 μl of protein A/G agarose beads at 4°C overnight on a rocking platform. Precipitated samples were then washed three times with lysis buffer, the final beads pellet resuspended in 1× SDS loading buffer, and analyzed by SDS-PAGE and Western blot.
GST pull-down assays.
GST-fusion proteins were expressed in the BL21 E. coli strain and purified as described previously (Zhang et al., 2005). YFP-TRPC6 or HA-Orai2 proteins were expressed in HEK293 cells and extracted in lysis buffer containing 1% CHAPS plus the following (in mm): 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.4 KH2PO4, pH 7.2, 5 EDTA, 5 EGTA, and protease inhibitors for 1 h at 4°C. Extracts were clarified by centrifugation and incubated for 1 h at 4°C with the corresponding GST fusion protein. Beads were washed four times with the extraction buffer and attached proteins were separated by SDS-PAGE and probed with the anti-GFP or anti-HA antibody.
Fura-2 Ca2+-imaging experiments.
HEK293 cells were transfected with EGFP plasmid or a mixture of EGFP and TRPC6 plasmids (at 1:5 ratio), cultured for 40–48 h, and loaded with Fura2-AM. Fura-2 340/380 ratio images were collected using a DeltaRAM-X illuminator, Evolve camera, and IMAGEMASTER PRO software (all from Photon Technology International) from GFP-positive cells. To test the direct effect of Hyp or NSN, the cells were loaded with Fura-2 and incubated in artificial CSF (aCSF) containing the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.3. After basal recordings for 30 s, 1 μm Hyp or 1 μm NSN were applied and Ca2+ responses measured. To test the effect of NSN on OAG response, cells were moved to aCSF medium containing 0.1 mm Ca2+ for 2 min and then returned to aCSF medium containing 2 mm Ca2+ and 100 μm OAG. These experiments were performed in the presence or absence of 1 μm NSN. The maximal amplitude (peak) of Ca2+ influx was determined from the Fura-2 340 nm/380 nm ratio. All Ca2+-imaging experiments were performed at room temperature.
Dendritic spine analysis in primary hippocampal neuronal cultures.
The hippocampal cultures of PS1KI, APPKI, and WT mice were established from postnatal day 0–1 pups and maintained in culture as we described previously (Zhang et al., 2010). For the assessment of synapse morphology, hippocampal cultures were transfected with TD-Tomato plasmid at d in vitro 7 (DIV7) using the calcium phosphate method and fixed (4% formaldehyde, 4% sucrose in PBS, pH 7.4) at DIV16–17. A Z-stack of optical section was captured using 100× objective with a confocal microscope (Carl Zeiss Axiovert 100M with LSM510). A total of 18–20 cultured neurons from three batches of cultures were used for quantitative analysis per genotype. Quantitative analysis for dendritic spines was performed with the NeuronStudio software package (Rodriguez et al., 2008). To classify the shape of neuronal spines in culture, we adapted an algorithm from a previously published method (Rodriguez et al., 2008). In classification of spine shapes, we used the following cutoff values: aspect ratio for thin spines (AR_thin(crit)) = 2.5, head to neck ratio (HNR(crit)) = 1.4, and head diameter (HD(crit)) = 0.5 μm. These values were defined and calculated exactly as described previously (Rodriguez et al., 2008).
GCamp5.3 Ca2+-imaging experiments.
GCamp5.3-imaging experiments were performed as we described previously (Zhang et al., 2015b). Briefly, cultured hippocampal neurons were transfected with GCamp5.3 expression plasmid using the calcium phosphate transfection method at DIV7. GCamp5.3 fluorescent images were collected using Olympus IX70 inverted epifluorescence microscope equipped with a 60× lens, Cascade 650 digital camera (Roper Scientific), and Prior Lumen 200 illuminator. The experiments were controlled by the MetaFluor image acquisition software package (Universal Imaging). To measure spine nSOC, the neurons were moved from aCSF to calcium-free medium with 0.4 mm EGTA and 1 μm thapsigargin for 30 min after recording 30 s basal, 100 μm DHPG in calcium-free aCSF was added, and then 50 s later, neurons were returned to aCSF with addition of a Ca2+ channel inhibitor mixture (1 μm TTX, 50 μm AP5, 10 μm CNQX, and 50 μm nifedipine). Analysis of the data was performed using ImageJ software. The region of interest used in the image analysis was chosen to correspond to spines. All Ca2+ -imaging experiments were done at room temperature.
Hippocampal slice field recordings.
Hippocampal slice field recordings were performed as described previously (Zhang et al., 2015a). Briefly, hippocampal slices (400 μm) were prepared from 6-month-old animals of either sex. Mice were anesthetized and transcardially perfused with dissection buffer before decapitation. The brain was removed, dissected, and sliced in ice-cold dissection buffer containing the following (in mm): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose, and 10 dextrose, using a vibratome (VT 1000S; Leica). CA3 were cut off to avoid epileptogenic activity. The slices were transferred into a reservoir chamber filled with aCSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2, and 10 dextrose. Slices were allowed to recover for 2–5 h at 30°C. aCSF and dissection buffer were equilibrated with 95% O2/5% CO2. For recording, slices were transferred to a submerged recording chamber, maintained at 30°C, and perfused continuously with aCSF at a rate of 2–3 ml/min. Field potentials (FPs) were recorded with extracellular recording electrodes (1 MΩ) filled with aCSF and placed in stratum radiatum of area CA1. FPs were evoked by monophasic stimulation (100 μs duration) of Schaffer collateral/commissural afferents with a concentric bipolar tungsten stimulating electrode (FHC). Stable baseline responses were collected every 30 s using a stimulation intensity (15–30 μA) yielding 50% of the maximal response. The initial slope of the FPs was used to measure stability of synaptic responses and to quantify the magnitude of LTP. The LTP was induced by two trains of 100 Hz frequency stimulation for 1 s, with each train separated by a 20 s interval. For NSN treatment experiments, hippocampal slices were preincubated with 300 nm NSN for 2–3 h before initiation of recordings in aCSF.
Dendritic spine analysis in hippocampal slices.
To analyze the shape of the spines in hippocampus slices, we used WTGFP, PS1KIGFP, and APPKIGFP mice. Hippocampal slices were prepared as above and allowed to recover for 1 h at 30°C. Then, half of the slices were treated with 300 nm NSN for 3.5 h at 30°C and the other half were kept in the aCSF as control. After incubation, slices were fixed in 4% formaldehyde, 0.125% glutaraldehyde in PBS. GFP images were acquired by two-photon imaging (LSM780; Zeiss) with a 40× lens and 5× zoom. The Z interval was 0.5 μm. The secondary apical dendrites of hippocampal CA1 pyramidal neurons were selected for taking images. Approximately 25 neurons from five mice were analyzed for each genotype. To classify the shape of neuronal spines in slices, we also used the NeuronStudio software package and an algorithm from Rodriguez et al. (2008) with the following cutoff values: AR_thin(crit) = 2.5, HNR(crit) = 1.4, and HD(crit) = 0.5 μm.
Hoechst 33342 and propidium iodide (PI) live/dead cell staining.
DIV15 hippocampal neuronal cultures were treated with 30 nm, 300 nm, or 1 μm NSN or Hyp for 18 h and stained with 5 μg/ml Hoechst 33342 and 1 μg/ml PI in aCSF solution for 5 min at 37°C. After staining, the live cells were transferred to aCSF medium and confocal imaging was performed. The ratio of PI-positive (dead) cells to Hoechst 33342-stained cells (total) was determined for each field of view by a blinded observer.
Dendritic spine analysis in mouse hippocampus.
To analyze the shape of the spines in hippocampus in vivo, we used the GFP-M mouse line (Feng et al., 2000) (WTGFP). To simplify the analysis, we crossed Line M GFP mice with PS1KI and APPKI mice to yield PS1KIGFP and APPKIGFP mice. Five female mice for each group (WTGFP, PS1KIGFP and APPKIGFP) were injected intraperitoneally 3 times/week with 10 mg/kg NSN starting at 4 months of age. Control groups of mice were injected with the same solvent solution. After 6 weeks, the injection routine was changed to 2 times per week. After 10 weeks, all mice were killed for in vivo spine analysis. Mice were intracardially perfused with ice-cold 4% paraformaldehyde (PFA) solution in phosphate buffer, pH 7.4, 30 ml in 3 min. The brains were extracted and postfixed in 4% PFA solutions for 16 h before cutting. Then, 50 μm hippocampal sections from the fixed brains were obtained using vibratome (Leica 1200S). A Z-stack of optical sections was captured using a 100× objective with a confocal microscope (Carl Zeiss Axiovert 100M with LSM510). The Z interval was 0.5 μm. The apical dendrites of hippocampal CA1 pyramidal neurons were selected for taking images. Approximately 25 neurons from five mice were analyzed for each group of mice. To classify the shape of neuronal spines in slices, we also used the NeuronStudio software package and an algorithm from Rodriguez et al. (2008) with the following cutoff values: AR_thin(crit) = 2.5, HNR(crit) = 1.4, and HD(crit) = 0.5 μm.
Statistical analyses.
The results are presented as mean ± SEM (n = number of neurons, slices, or mice as indicated). Statistical comparisons of results obtained in experiments were performed by Student's t test for two-group comparisons and one-way or two-way ANOVA followed by Tukey's test for multiple comparisons among more than two groups. The p-values are indicated in the text and figure legends as appropriate.
Results
TRPC6 and Orai2 support STIM2-gated nSOC in hippocampal mushroom spines
To identify molecular components of nSOC channels in the spines, we took a candidate approach. Previous studies suggested that the two major families of proteins, the TRPC and Orai channels, play a key role in supporting SOC in a variety of cells (Sun et al., 2014b; Majewski and Kuznicki, 2015). There are six TRPC proteins in humans (TRPC1 and TRPC3–TRPC7), which have been divided into two subfamilies, TRPC1/TRPC4/TRPC5 and TRPC3/TRPC6/TRPC7, based on biochemical and functional similarities. The remaining member, TRPC2, is a pseudogene in humans but is expressed in other species in a restricted expression pattern (Cheng et al., 2013). There are three Orai channels (Orai1–Orai3) but, so far, most studies have been focused on Orai1. We reasoned that the members of TRPC and/or Orai channel families are the most likely candidates to encode nSOC channels in the spines.
In previous studies, we demonstrated that overexpression of STIM2 rescues mushroom spine deficit in PS1KI neurons (Sun et al., 2014a). We reasoned that overexpression of a channel subunit supporting spine nSOC should exert a similar rescue effect. To identify the TRPC isoform that supports spine nSOC, we cotransfected TD-Tomato plasmid together with various TRPC expression constructs to PS1KI hippocampal neurons, fixed the cells, and evaluated spine morphology by confocal imaging (Fig. 1A,B). The fraction of mushroom spines in each culture was determined by automated analysis of obtained confocal images (Fig. 1C). Consistent with previous studies (Sun et al., 2014a), the fraction of mushroom spines in WT cultures was 30 ± 2% (n = 20), but the fraction of mushroom spines in PS1KI cultures was only ∼17 ± 1% (n = 20) (Fig. 1B,C). In our analysis, we determined that overexpression of TRPC1 did not have any rescue effect on spine morphology or mushroom spine density (Fig. 1B,C). Overexpression of TRPC3, TRPC5, or TRPC7 constructs resulted in enlargement of mushroom head in a few spines (Fig. 1B), but had no significant effect on mushroom spine density (Fig. 1C). Overexpression of TRPC4 resulted in an increase in a fraction of mushroom spines to 21 ± 3% (n = 20), but this effect was not statistically significant (p = 0.14). In contrast to all other isoforms, overexpression of TRPC6 rescued the fraction of mushroom spines in PS1KI cultures to 30 ± 1% (n = 20) (Fig. 1B,C), which was significantly (p < 0.001) higher than in control PS1KI cultures and the same as WT control cultures (Fig. 1B,C). From these results, we concluded that TRPC6 is a candidate channel for mediating spine nSOC in hippocampal mushroom synaptic spines.
Figure 1.
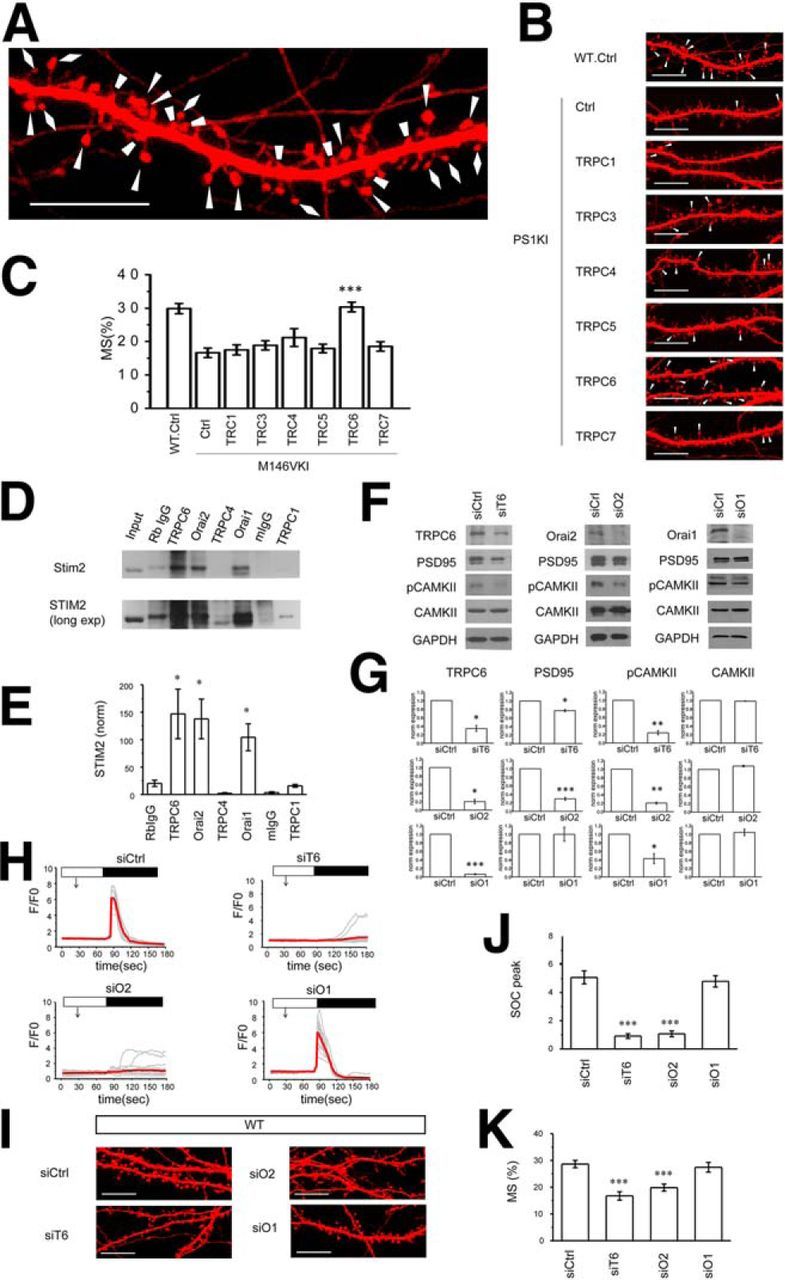
TRPC6 and Orai2 support spine nSOC in hippocampal neurons. A, Spine shape of primary hippocampal neurons from WT mice was visualized with TD-Tomato (same picture as control WT image on B is shown at higher magnification). Mushroom spines are marked by triangles; thin spines are marked by diamonds; stubby spines are marked by trapezoids. B, Confocal images of WT and PS1KI hippocampal neurons transfected with TD-Tomato and TRPC expression constructs at DIV7 and fixed at DIV16–17. Control neurons (Ctrl) transfected with TD-Tomato alone. Scale bar, 10 μm. C, Average fraction of mushroom spines for each group of cells shown in B is presented as mean ± SE (n = 20 neurons). ***p < 0.001 compared with the PS1KI control group. D, Immunoprecipitation experiments were performed with hippocampal synapsomal lysates using anti-TRPC6, and-Orai2, anti-TRPC4, anti-Orai1, and anti-TRPC1 antibodies as indicated. Control immunoprecipitations were performed with rabbit IgG (Rb IgG) or mouse IgG (m IgG). Precipitated samples were blotted with anti-STIM2 rabbit antibodies. Input is 1/10 of lysate used for immunoprecipitation. Longer exposure of the same blot is shown below. E, Quantification of STIM2 band intensity (normalized to input) for each lane in D. Data are shown as mean ± SE (n = 2–4 repeated experiments). *p < 0.05 when compared with Rb IgG sample. F, Western blot analysis of lysates from WT hippocampal neurons cultures infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), RNAi against Orai2 (siO2), or RNAi against Orai1 (siO1). The lysates were blotted with antibodies against TRPC6, Orai2, Orai1 PSD95, pCaMKII, and CaMKII as indicated. GAPDH was used as a loading control. Representative results from two to three independent cultures are shown. Quantification is shown in G. H, Time course of GCaMP5.3 fluorescence signal changes in the spines of WT hippocampal neurons infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), RNAi against Orai2 (siO2), or RNAi against Orai1 (siO1). The time of extracellular Ca2+ readdition is indicated by a black bar above the traces. The time of 100 μm DHPG addition is indicated by an arrow. For each experimental group, individual spine (gray) and average (red) fluorescence traces are shown. I, Confocal images of WT hippocampal neurons transfected with TD-Tomato at DIV7 and infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), RNAi against Orai2 (siO2), or RNAi against Orai1 (siO1). The neurons were fixed at DIV16–17. Scale bar, 10 μm. J, Average nSOC spine peak amplitude is shown for each group of cells. H, Mean ΔF/F0 signals for each group and presented as mean ± SE (n ≥ 105 spines). ***p < 0.001 compared with the siCtrl group. K, Average fraction of mushroom spines for each group of cells shown in I is presented as mean ± SE (n = 20 neurons). ***p < 0.001 compared with the siCtrl group.
Additional support to this hypothesis was provided by analysis of the SOC components' brain expression pattern. The expression of STIM2 is highly enriched in hippocampus (Skibinska-Kijek et al., 2009; Sun et al., 2014a) (Fig. 2). We reasoned that other components of STIM2-gated nSOC channels should have a similar expression pattern. Analysis of data from the Allen Brain Atlas revealed that the expression of TRPC6 is also highly enriched in the hippocampus compared with all other TRPC isoforms (Fig. 2A). To confirm these results, we performed a series of qRT-PCR experiments with cDNA samples prepared from different brain regions. Analysis of these data revealed that TRPC6 is highly enriched in hippocampus and hippocampal expression was also observed for TRPC1 and TRPC4 (Fig. 2B). Expression data also revealed that the Orai2 subunit is highly enriched in the hippocampus, but the Orai1 subunit is uniformly expressed in the brain at low levels (Fig. 2).
Figure 2.
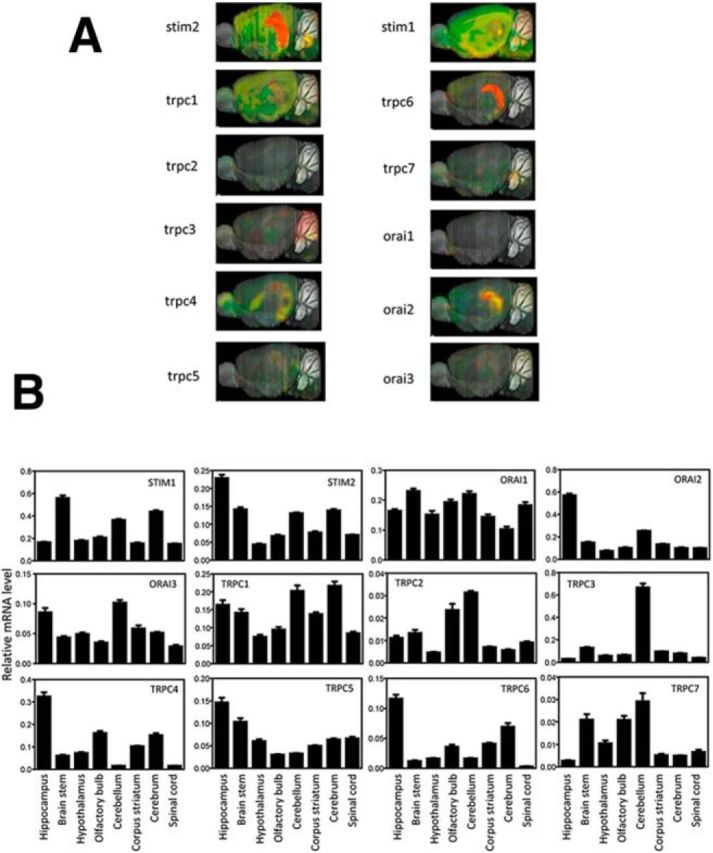
STIM, TRPC, and Orai expression in mouse brain. A, In situ hybridization images from the Allen Brain Atlas show expression of STIMs, TRPCs, and Orai channels in mouse brain. B, Gene expression profile of STIMs, TRPC, and Orai channels in different mouse brain regions. qRT-PCR results are presented as mean ± SD (triplicate technical measurements) for each gene transcript and brain region as indicated.
To further identify components of the STIM2-gated channel complex, we prepared hippocampal synaptosomes and performed a series of immunoprecipitation experiments. We discovered that antibodies against TRPC6, Orai2, or Orai1 can pull down STIM2 from synaptosomal lysates (Fig. 1D,E). The apparent molecular weight of immunoprecipitated STIM2 was higher than the molecular weight of STIM2 in the input lane (Fig. 1D). It is likely that TRPC6 and Orai form a complex with STIM2 that has undergone posttranslational modification such as phosphorylation (Smyth et al., 2012). Consistent with this observation, STIM2 coimmunoprecipitated with Orai1 from cortical lysates also displayed higher molecular weight on the gel (Gruszczynska-Biegala and Kuznicki, 2013). In contrast to TRPC6 immunoprecipitation, a very weak STIM2 signal was observed in TRPC1 immunoprecipitation experiments and no signal was observed in immunoprecipitation experiments with TRPC4 antibodies (Fig. 1D,E). These results are consistent with the inability of TRPC1 and TRPC4 to rescue mushroom spines in PS1KI neurons (Fig. 1B,C). A similar STIM2 interaction pattern was obtained in reverse immunoprecipitation experiments performed with hippocampal synaptosomal lysates except that the Orai1 interaction with STIM2 was very weak (data not shown).
To further determine whether TRPC6, Orai1, and Orai2 may indeed act as components of STIM2-gated nSOC channels in the spines, we performed knock-down of TRPC6, Orai1, and Orai2 proteins in WT mouse hippocampal neuronal cultures by using lentiviral-mediated shRNAi delivery. We confirmed efficient knock-down of target proteins by Western blotting of hippocampal culture lysates (Fig. 1F,G). In previous studies, we demonstrated that the activity of synaptic CaMKII is regulated by the nSOC pathway and that the levels of autophosphorylated pCaMKII can be used as biochemical readout for steady-state CaMKII activity in the spines (Sun et al., 2014a). We also demonstrated previously that inhibition of nSOC results in a loss of PSD95 expression in the spines (Sun et al., 2014a). In our experiments, we discovered that RNAi-mediated knock-down of TRPC6 or Orai2 resulted in a reduction in PSD95 expression and reduced levels of pCaMKII (Fig. 1E,F). Total levels of CaMKII remained unaffected (Fig. 1F,G). The reduction of pCaMKII and PSD95 levels after TRPC6 or Orai2 knock-down is consistent with the changes induced by STIM2 reduction or application of nSOC inhibitors in our previous studies (Sun et al., 2014a). Knock-down of Orai1 had no effect on the expression levels of PSD95 but resulted in some reduction in pCaMKII levels, although this was less significant than knock-down of Orai2 or TRPC6 (Fig. 1F,G).
To evaluate nSOC activity more directly, we performed a series of Ca2+-imaging experiments. To perform Ca2+ imaging in the spines, we transfected WT hippocampal neurons with GCamp5.3 plasmid to enable us to visualize the dendritic spines and measure local Ca2+ signals simultaneously (Sun et al., 2014a). In these experiments, we discovered that knock-down of TRPC6 or Orai2 resulted in a drastic reduction in spine nSOC (Fig. 1H,J). In contrast, knock-down of Orai1 had no effect on spine nSOC (Fig. 1H,J). To evaluate the morphology of synaptic spines after knock-down of TRPC6, Orai2, or Orai1, we transfected WT hippocampal cultures with TD-Tomato plasmid, fixed the cells, and performed confocal imaging experiments for each experimental group (Fig. 1I). Automated analysis of spine shapes revealed that the fraction of mushroom spines was reduced significantly after knock-down of TRPC6 or Orai2 proteins, but not after knock-down of Orai1 protein (Fig. 1I,K). Based on these results, we ruled out Orai1 as important component of spine nSOC in mature hippocampal neurons. This conclusion is consistent with low levels of Orai1 expression in the hippocampus (Fig. 2) and our inability to confirm STIM2-Orai1 association in reverse immunoprecipitation experiments with STIM2 antibodies (data not shown). Notably, a recently published study suggested that Orai1 may have an important role in the formation of new spines in immature hippocampal neurons (Korkotian et al., 2016). Our functional experiments (Fig. 1B,C,F–K) suggested that the TRPC6 and Orai2 proteins are the most likely candidates to support STIM2-gated spine nSOC in mature hippocampal neurons. This conclusion is also supported by analysis of brain expression pattern of TRPC and Orai isoforms (Fig. 2) and by STIM2 immunoprecipitation experiments with hippocampal synaptosomal lysates (Fig. 1D,E).
In the previous studies, we demonstrated that overexpression of STIM2 rescues nSOC and mushroom spine deficit in PS1KI neurons (Sun et al., 2014a). To further validate TRPC6 and Orai2 as STIM2-gated nSOC channels, we used RNAi to knock down TRPC6 or Orai2 in PS1KI neurons and performed STIM2 overexpression experiments. Consistent with earlier findings (Sun et al., 2014a), we demonstrated that overexpression of STIM2 can rescue spine nSOC in PS1KI hippocampal neurons infected with control RNAi construct (Fig. 3A,B). However, overexpression of STIM2 failed to rescue nSOC in spines of PS1KI neurons after RNAi-mediated knock-down of TRPC6 or Orai2 (Fig. 3A,B). In mushroom spine analysis, we found that knock-down of TRPC6 or Orai2 could cause an additional reduction of the mushroom spine fraction in PS1KI neurons (Fig. 3C,D). More importantly, knock-down of TRPC6 or Orai2 completely abolished the rescue effect of STIM2 overexpression in PS1KI neurons (Fig. 3C,D). These results further indicate that both the TRPC6 and Orai2 subunits are necessary for the activity of STIM2-gated nSOC channels in hippocampal mushroom spines.
Figure 3.

TRPC6 and Orai2 are necessary for STIM2-mediated rescue of PS1KI mushroom spines. A, Time course of GCaMP5.3 fluorescence signal changes in the spines of PS1KI hippocampal neurons. The time of 100 μm DHPG addition is indicated by an arrow. The time of extracellular Ca2+ readdition is indicated by a black bar above the traces. The neurons were infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), or RNAi against Orai2 (siO2). The results for control neurons (Con) and neurons cotransfected with STIM2 expression plasmid (+STIM2) are shown. For each experimental group, individual spine (gray) and average (red) fluorescence traces are shown. B, Average nSOC spine peak amplitude is shown for each group of cells shown in A. Mean ΔF/F0 signals for each group are presented as mean ± SE (n ≥ 45 spines). ***p < 0.001. C, Confocal images of PS1KI hippocampal neurons transfected with TD-Tomato at DIV7 and fixed at DIV16–17. The neurons were infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), or RNAi against Orai2 (siO2). The results for control neurons (Con) and neurons cotransfected with STIM2 expression plasmid (+STIM2) are shown. Scale bar, 10 μm. D, Average fraction of mushroom spines for each group of cells shown in C is presented as mean ± SE (n = 20 neurons). ***p < 0.001, *p < 0.05.
TRPC6 and Orai2 form a complex with STIM2 in the hippocampal synapse
Does STIM2 bind directly to TRPC6 and/or Orai2? STIM1 and STIM2 proteins share a similar domain structure (Fig. 4A) and 76% sequence similarity (Collins and Meyer, 2011; Stathopulos and Ikura, 2013). The STIM2 protein has not been studied extensively, but structure–functional analysis of STIM1 protein has been performed previously by several groups of investigators. The domain structure of STIM proteins consists of the Ca2+-sensing EF-SAM domain in the ER lumen, an extended coiled-coiled cytosolic domain, an Orai-associated SOAR/CAD domain, and a carboxy-terminal polybasic motif that is associated with negatively charged lipids on the plasma membrane inner surface (Fig. 4A) (Muik et al., 2009; Park et al., 2009; Yuan et al., 2009; Covington et al., 2010; Collins and Meyer, 2011; Stathopulos and Ikura, 2013). A double mutation in the STIM1 SOAR domain sequence (L373S, A376S) disrupted association between STIM1 and Orai and rendered STIM1 inactive (Frischauf et al., 2009). Guided by sequence homology between STIM1 and STIM2, we generated a GST-fusion construct of the WT STIM2-SOAR domain (S2-SOAR) and the corresponding mutant (L377S, A380S) in the STIM2-SOAR sequence (S2-LASS) (Fig. 4A). We used these constructs in pull-down experiments with lysates from HEK293 cells transfected with HA-tagged Orai2 or YFP-tagged TRPC6. As expected, we discovered that the STIM2-SOAR domain associated strongly with the Orai2 protein and that this association was disrupted by the LASS mutation (Fig. 4B). In contrast, association of the STIM2-SOAR domain with TRPC6 was weak and was not affected by the LASS mutation (Fig. 4B). These results suggested that STIM2 is associated strongly and directly with Orai2 via the SOAR domain, but is only associated weakly with TRPC6. This is consistent with previous analysis of STIM1, which has been shown to interact with Orai1 via the SOAR domain and with TRPC1/2/4, but not with TRPC3/6/7, via the ERM domain (Huang et al., 2006). To explain the ability of TRPC6 antibodies to precipitate STIM2 from synaptosomal lysates (Fig. 1C), we reasoned that Orai2 and TRPC6 may form a complex in the membrane. A similar complex has been proposed previously for TRPC6/3 and Orai1 in nonexcitable cells (Liao et al., 2007; Jardin et al., 2009). To test this hypothesis, we cotransfected FLAG-tagged TRPC6 and HA-tagged Orai2 to HEK293 cells and confirmed the formation of the TRPC6/Orai2 complex in coimmunoprecipitation experiments (Fig. 4C).
Figure 4.
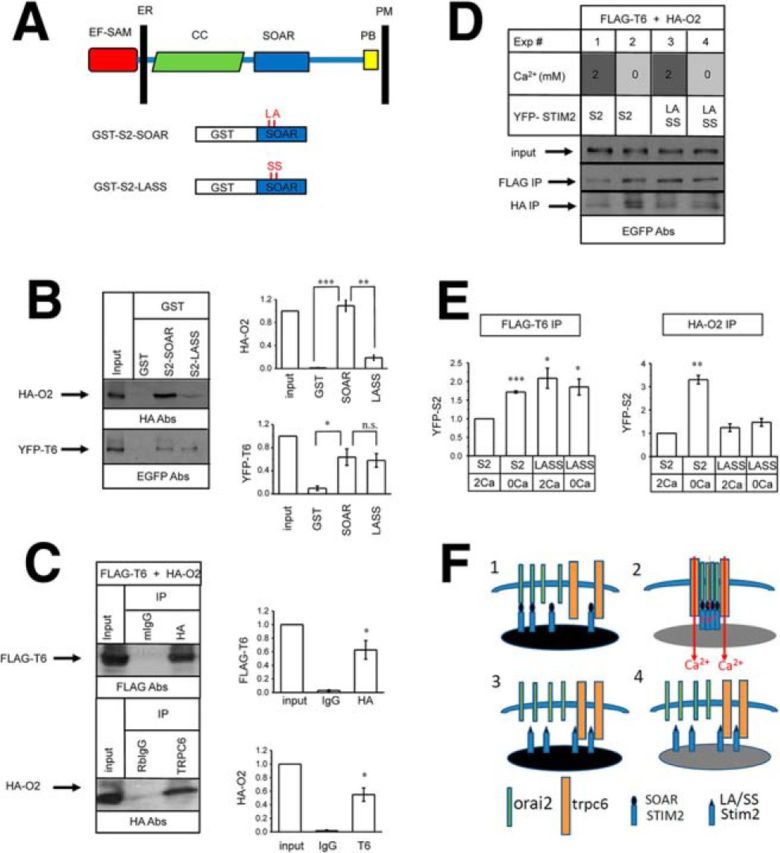
TRPC6 and Orai2 form a STIM2-regulated channel complex in hippocampus. A, Domain structure of STIM proteins. (Adapted from Collins and Meyer, 2011). The ER membrane and plasma membrane (PM) are indicated. Also indicated are the main functional domains of STIM proteins: EF-SAM ER Ca2+ sensor domain, coiled-coiled region (CC), CAD/SOAR domain, and polybasic motif (PB). The GST-STIM2-SOAR construct and GST-STIM2-SOAR-LASS mutant (L377S, A380S) constructs are also shown. B, GST, GST-S2-SOAR, and GST-S2-LASS recombinant proteins were used in pull-down experiments with lysates from HEK293 cells transfected with HA-Orai2 or YFP-TRPC6 expression constructs. The input is 1/50 of lysate used for pull-down experiments. Quantification is shown in the right. For each lane, the signal was normalized to the input lane. Normalized and averaged data are shown as mean ± SE (n = 3 experiments). ***p < 0.001, *p < 0.05 compared with GST sample. C, Anti-HA mouse monoclonal antibodies or anti-TRPC6 rabbit polyclonal antibodies were used in immunoprecipitation experiments with lysates from HEK293 cells cotransfected with YFP-TRPC6 and HA-Orai2 expression plasmids. The input lane is 1/20 of lysate used for immunoprecipitation. Quantification is shown on the right. For each lane, the signal was normalized to the input lane. Normalized and averaged data are shown as mean ± SE (n = 2 experiments). *p < 0.05 compared with control IgG sample. D, Anti-FLAG or anti-HA mouse monoclonal antibodies were used in immunoprecipitation experiments with lysates from HEK293 cells cotransfected with HA-Orai2, FLAG-TRPC6, and YFP-STIM2 or YFP-STIM2-LASS constructs. Experiments 1 and 3 were performed with lysates prepared from cells incubated in normal aCSF (2 mm Ca2+) for 10 min before lysis. Experiments 2 and 4 were were performed with lysates prepared from cells incubated in Ca2+-free aCSF (with addition of 400 μm EGTA) for 10 min before lysis. The input lanes for Experiments 1–4 contain 1/50 of total lysate used for immunoprecipitation. E, Quantification of anti-FLAG-TRPC6 and anti-HA-Orai2 immunoprecipitation experiments. For each lane, the YFP-STIM2 signal was normalilzed to the input lane. Normalized and averaged data are shown as mean ± SE (n = 2–3 experiments). *p < 0.05, **p < 0.01, ***p < 0.001 compared with the signal observed in Experiment 1 (YFP-STIM2 and 2 mm Ca2+). F, Model explaining results shown in D (Experiments 1–4). STIM2 binds to Orai2 directly and strongly via the SOAR domain and weakly to TRPC6 via different region (Experiment 1). Depletion of Ca2+ stores promotes assemembly of functional TRPC6/Orai2-STIM2 complex (Experiment 2). The STIM2-LASS mutant does not bind to Orai2 and instead is recruited to nonproductive complexes with TRPC6 (Experiment 3). Association of the STIM2-LASS mutant with TRPC6 is not affected by ER store depletion (Experiment 4).
To further investigate the function of the STIM2-Orai2-TRPC6 complex, we cotransfected HEK293 cells with FLAG-tagged TRPC6, HA-tagged Orai2, and YFP-tagged STIM2 or STIM2-LASS mutant constructs. Previous results suggest that association among the STIM1, Orai, and TRPC channels can be modulated by the depletion state of ER Ca2+ stores (Liao et al., 2007; Ong et al., 2007; Cheng et al., 2008; Liao et al., 2008; Zeng et al., 2008; Jardin et al., 2009; Liao et al., 2009; Cheng et al., 2011; Cheng et al., 2013). To account for this possibility, we prepared lysates from transfected HEK239 cells in standard culture conditions (2 mm extracellular Ca2+) or after incubation in Ca2+-free medium to cause store depletion. The lysates were precipitated with anti-FLAG or anti-HA antibodies and the presence of YFP-STIM2 was analyzed by Western blotting with anti-EGFP antibodies. In these experiments, we found that, under normal Ca2+ conditions (2 mm Ca2+), STIM2 associated with TRPC6 and Orai2 weakly (Fig. 4D, lane 1, E), but this association was facilitated by store depletion (Fig. 4D, lane 2, E). As expected, the LASS mutation in the STIM2-SOAR domain sequence disrupted the STIM2 association with Orai2 (Fig. 4D, lanes 3 and 4, E). In contrast, the LASS mutation in the STIM2-SOAR domain sequence did not disrupt association of STIM2 with TRPC6, but this interaction was no longer modulated by ER Ca2+ levels (Fig. 4D, lanes 3 and 4, E).
To explain the results of our functional and biochemical experiments (Figs. 1, 3, 4), we proposed a model depicted in Figure 4F. We propose that, in conditions of filled ER Ca2+ stores, there are some STIM2 proteins that interact strongly with Orai2 via the SOAR domain and some that interact weakly with TRPC6 via a different region (Fig. 4F, panel 1). After ER store depletion and oligomerization of STIM2, more Orai2 and TRPC6 proteins are recruited and a functional complex of TRPC6, Orai2, and STIM2 is assembled (Fig. 4F, panel 2). In this complex, TRPC6 serves as a Ca2+-conducting channel and Orai2 is involved in sensing ER Ca2+ levels by means of association with STIM2. Similar ideas have been proposed before to explain the function of the STIM1-TRPC3/6-Orai1 complex (Liao et al., 2007; Jardin et al., 2009). We further argue that the LASS mutation in the STIM2-SOAR domain disrupts its association with Orai2 and results in enhanced nonproductive association with TRPC6 due to loss of competition with Orai2 (Fig. 4F, panel 3). Because of its inability to bind Orai2, the STIM2-LASS association with TRPC6 is no longer regulated by ER Ca2+ store depletion (Fig. 4F, panel 4). In the remainder of this manuscript, we will be guided by this model (Fig. 4F) to evaluate the function of the TRPC6/Orai2-STIM2 complex in hippocampal spines.
Distinct functional roles of TRPC6 and Orai2 as components of hippocampal spine nSOC
In previous studies, we demonstrated that STIM2 overexpression rescues nSOC and mushroom spine defects in PS1KI and APPKI mouse models of familial AD (Sun et al., 2014a; Zhang et al., 2015b). Our initial results suggested that overexpression of TRPC6 rescues mushroom spine deficit in PS1KI hippocampal neurons (Fig. 1B,C). In agreement with these findings, we discovered that overexpression of TRPC6 rescues the nSOC defect in spines of PS1KI and APPKI hippocampal neurons (Fig. 5A,B) and also rescues mushroom spine deficit in PS1KI and APPKI hippocampal neurons (Fig. 5C,D). In contrast, overexpression of Orai2 fails to rescue spine nSOC in PS1KI and APPKI hippocampal neurons (Fig. 5A,B). In fact, overexpression of Orai2 significantly impairs spine nSOC responses, even in WT neurons (Fig. 5A,B). Consistent with our findings, overexpression of Orai2 has been reported to inhibit SOCE in nonexcitable cells (Mercer et al., 2006; Hoover and Lewis, 2011; Inayama et al., 2015). Approximately 35% of PS1KI neurons transfected with Orai2 displayed abnormal morphology and dendritic spines could not be identified clearly in these cells (Fig 5E). Similar results were obtained when WT or APPKI neurons were transfected with Orai2 plasmid (data not shown). From these results, we concluded that overexpressed Orai2 binds STIM2 with high affinity, but does not yield functional Ca2+ influx channels in the absence of stoichiometric amounts of TRPC6 and/or STIM2. To test this hypothesis, we cotransfected PS1KI hippocampal neurons with the Orai2 construct together with the STIM2 construct, the TRPC6 construct, or the STIM2 and TRPC6 construct combination. We discovered that the abnormal neuronal morphology was recovered partially in the presence of STIM2 or TRPC6 and recovered fully in the presence of STIM2 and TRPC6 (Fig. 5E). We also analyzed the PS1KI neurons, which display normal morphology after overexpressing Orai2, and detected a small elevation in the fraction of mushroom spines in these cells (Fig. 5F). Cotransfection of Orai2 together with TRPC6 or the TRPC6 and STIM2 combination resulted in complete rescue of the mushroom spine fraction in PS1KI neurons (Fig. 5F). These results support the hypothesis that the stochiometric complex of Orai2, TRPC6, and STIM2 is necessary to support function of nSOC in the spines.
Figure 5.
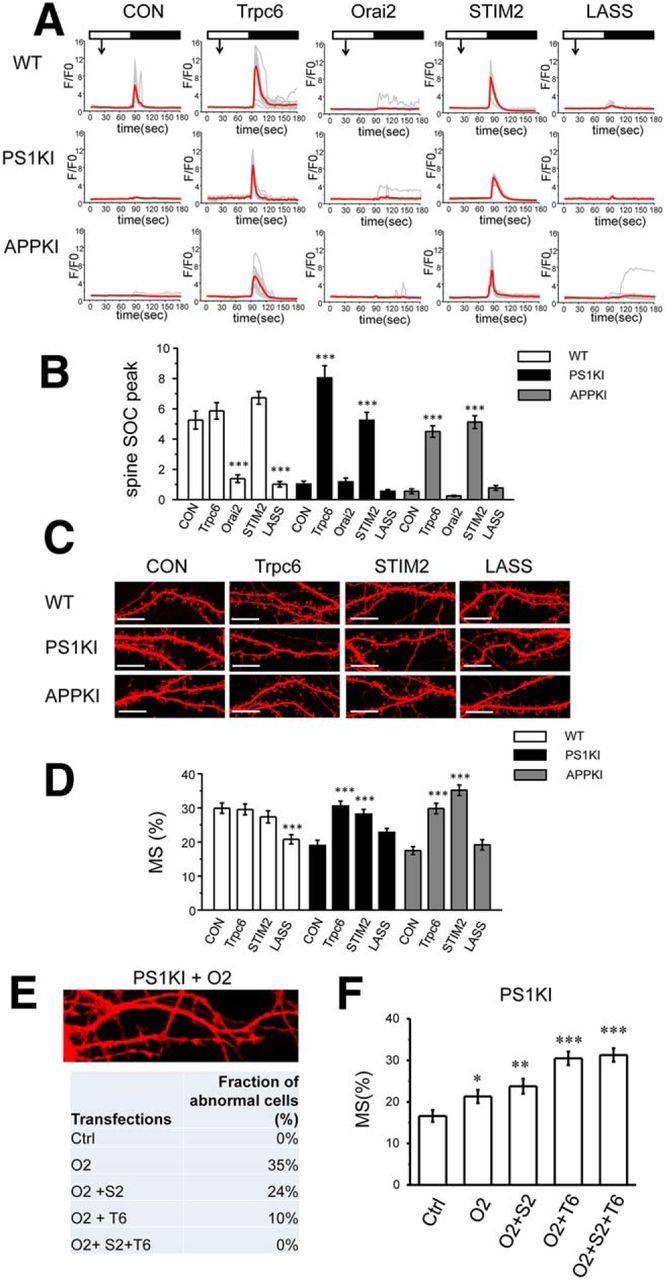
Functional roles of TRPC6 and Orai2 in supporting spine nSOC and hippocampal mushroom spines. A, Time course of GCaMP5.3 fluorescence signal changes in the spines of WT, PS1KI and APPKI hippocampal neurons. The time of 100 μm DHPG addition is indicated by an arrow. The time of extracellular Ca2+ readdition is indicated by a black bar above the traces. The results are shown for control (Con) neurons and for neurons cotransfected with TRPC6, Orai2, STIM2, or STIM2-LASS plasmids as indicated. For each experimental group, individual spine (gray) and average (red) fluorescence traces are shown. B, Average spine nSOC peak amplitude is shown for each group of cells shown in. A, Mean ΔF/F0 signals for each group are presented as mean ± SE (n ≥ 73 spines). ***p < 0.001 compared with control group of the same genotype. C, Confocal images of WT, PS1KI, and APPKI hippocampal neurons transfected with TD-Tomato at DIV7 and fixed at DIV16–17. The images are shown for control neurons (Con) and for neurons cotransfected with TRPC6, STIM2, or STIM2-LASS plasmids. Scale bar, 10 μm. D, Average fraction of mushroom spines for each group of cells shown in C is presented as mean ± SE (n ≥ 19 neurons). ***p < 0.001 compared with control group of the same genotype. E, Confocal image of typical abnormal neuronal morphology in PS1KI hippocampal neurons cotransfected with TD-Tomato and Orai2 expression plasmids. The fraction of neuronal cells with abnormal morphology in DIV16–17PS1KI hippocampal cultures was analyzed. Ctrl cells were transfected with TD-Tomato plasmid. Other groups of cells were cotransfected with DT-Tomato plasmid and Orai2 (O2), Orai2 and STIM2 (O2 + S2), Orai2 and TRPC6 (O2 + T6), or Orai2, Stim2, and TRPC6 (O2 + S2 + T6). The fraction of cells with abnormal morphology was estimated by visual inspection of confocal images for each group of cells. F, Average fraction of mushroom spines for PS1KI cells with normal morphology. Ctrl cells were transfected with TD-Tomato plasmid. Other groups of cells were cotransfected with DT-Tomato plasmid and Orai2 (O2), Orai2 and STIM2 (O2 + S2), Orai2 and TRPC6 (O2 + T6), or Orai2, Stim2, and TRPC6 (O2 + S2 + T6). The fraction of mushroom spines was calculated by automated analysis of confocal images and is presented as mean ± SE (n ≥ 13 neurons). *p < 0.05, **p < 0.01, ***p < 0.001 compared with control group PS1KI cells.
To further test this hypothesis, we compared the effects of STIM2 and STIM2-LASS mutant overexpression. In agreement with the previous studies (Sun et al., 2014a; Zhang et al., 2015b), the expression of STIM2 rescued spine nSOC (Fig. 5A,B) and mushroom spine loss (Fig. 5C,D) in PS1KI and APPKI hippocampal neurons. In contrast, the expression of the STIM2-LASS mutant failed to rescue spine nSOC (Fig. 5A,B) and mushroom spine loss (Fig. 5C,D) in PS1KI and APPKI hippocampal neurons. In fact, expression of STIM2-LASS mutant exerted a dominant-negative effect on spine nSOC in WT neurons (Fig. 5A,B) and resulted in mushroom spine loss in these neurons (Fig. 5C,D). To explain these results, we reasoned that the STIM2-LASS mutant does not bind to Orai2 and instead traps TRPC6 in nonfunctional complex (Fig. 4F, panel 4). From these results, we concluded that TRPC6 is a major Ca2+ influx channel in dendritic spines and Orai2 is a regulatory subunit of the complex that is gated by STIM2 in a store-depletion-dependent manner. A similar model has been proposed previously for the STIM1-TRPC3/6-Orai1 complex in nonexcitable cells (Liao et al., 2007; Jardin et al., 2009).
NSN and Hyp activate spine nSOC channels
Our genetic rescue experiments (Sun et al., 2014a; Zhang et al., 2015b) (Fig. 5) suggested that pharmacological activators of TRCP6/Orai2 nSOC channels in spines may help to prevent mushroom spine loss and have a therapeutic potential for AD. Hyp is a known activator of TRPC6 (Leuner et al., 2007) (Fig. 6A). We recently identified a novel nSOC activator, NSN (molecular weight 322) (Fig. 6A). This compound was serendipitously discovered in the process of analyzing novel nSOC inhibitors in our previous study (Wu et al., 2011). In Ca2+-imaging experiments, we discovered that application of 300 nm Hyp or NSN before Ca2+ readdition rescued spine nSOC in PS1KI and APPKI hippocampal neurons (Fig. 6B,C). Interestingly, neither compound had any significant effect on nSOC in WT spines (Fig. 6B,C), suggesting that the spine nSOC pathway is already maximally activated in normal conditions. In further experiments, we incubated TD Tomato-transfected hippocampal neuronal cultures with 30 nm concentrations of Hyp or NSN for 16 h and performed analysis of spine shapes by confocal imaging (Fig. 6D). We discovered that incubation with Hyp or NSN resulted in complete rescue of mushroom spines in both PS1KI and APPKI neurons (Fig. 6D,E). Neither compound had any significant effect on the fraction of the mushroom spines in WT neurons (Fig. 6D,E). In additional experiments, we demonstrated that 4 h of treatment with 300 nm Hyp or NSN exerted similar rescue effect on mushroom spines in PS1KI and APPKI hippocampal neurons (data not shown).
Figure 6.

NSN and Hyp rescue spine nSOC and mushroom spine loss in AD hippocampal neurons. A, Chemical structure of NSN and Hyp. B, Time course of GCaMP5.3 fluorescence signal changes in the spines of WT, PS1KI, and APPKI hippocampal neurons. The time of 100 μm DHPG addition is indicated by an arrow. The time of extracellular Ca2+ readdition is indicated by a black bar above the traces. The results are shown for control (Con) neurons and for neurons pretreated with 300 nm NSN (+NSN) or 300 nm Hyp (+Hyp) for 30 min as indicated. For each experimental group, individual spine (gray) and average (red) fluorescence traces are shown. C, Average spine nSOC peak amplitude is shown for each group of cells shown in B. Mean ΔF/F0 signals for each group are presented as mean ± SE (n ≥ 45 spines). ***p < 0.001 compared with control group of the same genotype. D, Confocal images of WT, PS1KI, and APPKI hippocampal neurons transfected with TD-Tomato and fixed at DIV16–17. The images are shown for control neurons (Con) and for neurons treated with 30 nm NSN (+NSN) or 30 nm Hyp (+Hyp) for 16 h before fixation. Scale bar, 10 μm. E, Average fraction of mushroom spines for each group of cells shown in D is presented as mean ± SE (n ≥ 18 neurons). ***p < 0.001 compared with control group of the same genotype.
To confirm the target for Hyp and NSN compounds, we overexpressed TRPC6 in HEK293 cells and performed a series of Fura-2 Ca2+-imaging experiments. Consistent with the published reports (Leuner et al., 2007), application of 1 μm Hyp activated Ca2+ influx in TRPC6-transfected HEK293 cells, but not in control cells transfected with EGFP plasmid (Fig. 7A,C). In contrast to Hyp, application of 1 μm NSN did not trigger Ca2+ influx in TRPC6-transfected cells (Fig. 7A,C). In additional experiments, we evaluated effects of a 1 μm NSN compound in experiments with HEK293 cells transfected with other TRPCs (TRPC1-7), Orais (Orai1-3), or a combination of TRPC6 and Orai2. The NSN compound failed to induce Ca2+ influx in any of these experiments (data not shown). From these experiments, we concluded that Hyp acts as a direct activator of TRPC6 channels, but actions of the NSN compound are more complex. In further experiments, we measured SOC in conditions of store depletion. To achieve this, HEK239 cells were preincubated in Ca2+-free medium containing 1 μm Tg. In these experiments, we observed an endogenous SOC response in EGFP-transfected cells, which was further enhanced in TRPC6-transfected cells. However, application of the NSN compound had no additional effect on SOC in control or TRPC6 cells in these conditions (data not shown). TRPC6 channels are known to be activated by diacyl glycerol (DAG) (Estacion et al., 2004). In our spine nSOC measurements, the addition of 100 μm DHPG was required to generate robust Ca2+ responses. Therefore, in the next series of experiments, we evaluated effects of 1-Oleoyl-2-acetyl-glycerol (OAG), a synthetic and stable analog of DAG. In standard recording conditions, application of 50 μm OAG to TRPC6-transfected cells resulted in highly variable responses, with some batched of cells displaying Ca2+ influx and some batches nonresponsive (data not shown). However, we discovered that 50 μm OAG could produce more consistent responses when stores were partially depleted by preincubation of cells in extracellular medium containing 0.1 mm Ca2+ (Fig. 7B,C). The effect of OAG was observed in TRPC6-transfected cells, but not in control EGFP-transfected cells (Fig. 7B,C). Interestingly, preincubation with 1 μm NSN resulted in significant potentiation of OAG-induced responses in these conditions in TRPC6-transfected cells, but not in control cells (Fig. 7B,C). Additional control experiments demonstrated that, even after a partial depletion protocol, the NSN compound was not able to activate Ca2+ influx in TRPC6-transfected HEK293 cells in the absence of OAG (data not shown). From these results, we concluded that NSN compound most likely acts by potentiating the effects of endogenous DAG on TRPC6 channels in neurons.
Figure 7.
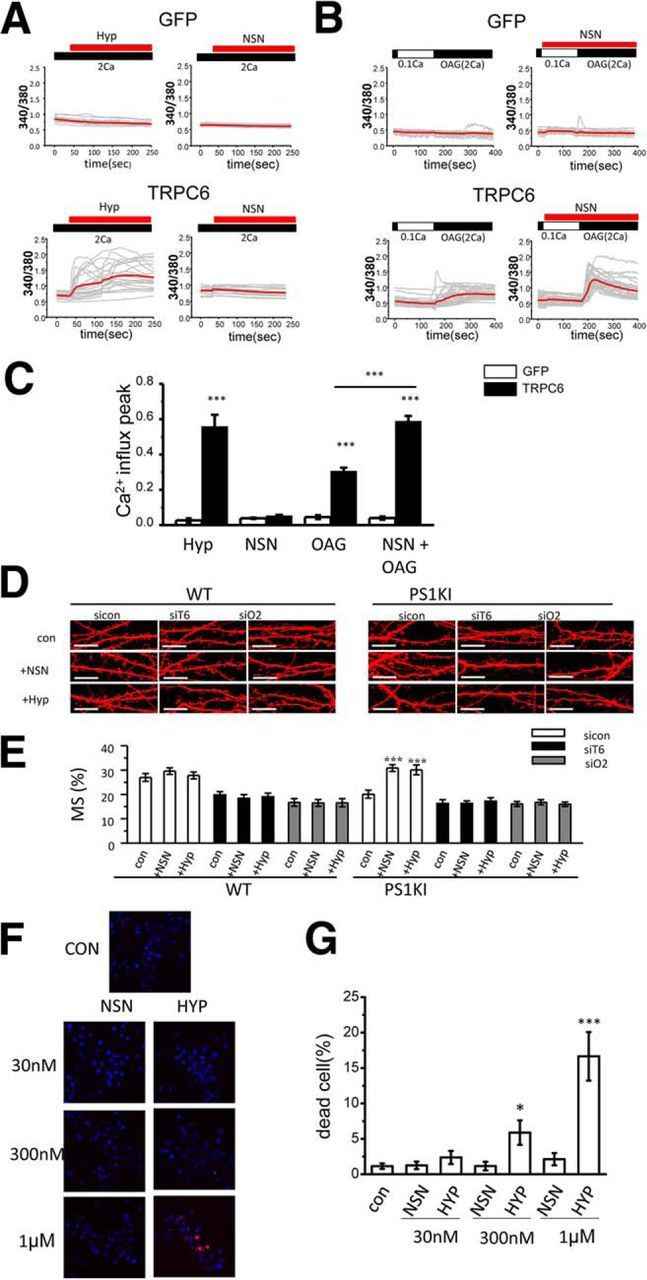
TRPC6 is a molecular target for NSN and Hyp. A, B, Time course of Fura-2 Ca2+ signals is shown for HEK293 cells transfected with EGFP plasmid (GFP) or combination of EGFP and TRPC6 plasmids (TRPC6). Cells were incubated in aCSF medium containing 2 mm Ca2+. In experiments shown in B, cells were moved to modified aCSF medium containing 0.1 mm Ca2+ for 2 min and then returned to the medium containing 2 mm Ca2+ with the addition of 50 μm OAG. The time of addition of 1 μm Hyp or NSN is indicated by red bars above the Fura-2 traces. For each experimental group, individual cell (gray) and average (red) ratio traces are shown. C, Average Ca2+ influx peak is shown for experiments presented in A and B as mean ± SE (n ≥ 81cells). ***p < 0.001. D, Confocal images of WT and PS1KI hippocampal neurons transfected with TD-Tomato and fixed at DIV16–17. The neurons were infected with lentiviruses encoding control RNAi (siCtrl), RNAi against TRPC6 (siT6), or RNAi against Orai2 (siO2). The images are shown for no drug treated neurons (Con) and for neurons treated with 30 nm NSN (+NSN) or 30 nm Hyp (+Hyp) for 16 h before fixation. Scale bar, 10 μm. Control group of WT and PS1KI neurons data are from the same experiments as shown in Figure 1B and Figure 3C. E, Average fraction of mushroom spines for each group of cells shown in D is presented as mean ± SE (n ≥ 19 neurons). ***p < 0.001. F, Confocal images of DIV16 live WT hippocampal neuronal cultures stained with Hoechst 33342 (blue) and PI (red). Control cells (CON) were untreated. Other cells were treated with 30 nm, 300 nm, or 1 μm NSN or Hyp for 18 h as indicated. G, Ratio of PI-positive (dead) cells to Hoechst 33342-stained cells (total) was determined for each group of cells shown in F. Average fraction of dead cell data are presented as mean ± SE (n = 10 random fields of view from two independent experiments). *p < 0.05, ***p < 0.001 compared with control (untreated) group of cells.
To validate TRPC6 as the target for Hyp and NSN compounds in the spines, we performed experiments with WT and PS1KI hippocampal neurons infected with Lenti-RNAi against TRPC6. These cultures were transfected with TD Tomato, incubated with 30 nm Hyp or NSN for 16 h, and analyzed by confocal microscopy (Fig. 7D). Knock-down of TRPC6 resulted in loss of mushroom spines in WT neurons in these experiments (Fig. 7D,E). Incubation with 30 nm Hyp or NSN failed to rescue this phenotype (Fig. 7D,E), rescued mushroom spine loss in PS1KI cultures infected with control RNAi lentiviruses, but failed to rescue mushroom spine loss in PS1KI hippocampal neurons after knock-down of TRPC6 (Fig. 7D,E). To further confirm that spine nSOC channel is composed of TRPC6 and Orai2, we infected WT and PS1KI neurons with Lenti-RNAi against Orai2. We discovered that 30 nm Hyp or NSN failed to rescue mushroom spine loss in WT and PS1KI hippocampal neurons after knock-down of Orai2 (Fig. 7D,E). These results are consistent with the hypothesis that Hyp and NSN rescue spine nSOC and mushroom spines in AD neurons by activating the TRPC6/Orai2 channel complex in the spines.
To determine whether difference in mechanisms of action affect toxicity of Hyp and NSN compounds, we incubated primary hippocampal neuronal cultures in the presence of increasing concentrations of Hyp or NSN. After 18 h of treatment, the cells were stained with Hoechst 33342 and PI and the fraction of PI-positive neuronal nuclei (dead cells) was calculated. In these experiments, we discovered that NSN compound did not induce cell death of hippocampal neurons when tested in concentrations as high as 1 μm (Fig. 7F,G). Hyp started to induce cell death at a 300 nm concentration and resulted in very significant toxicity at 1 μm concentration after overnight incubation (Fig. 7F,G). Therefore, we reasoned that the NSN compound may have a potentially wider therapeutic window than Hyp due to reduced toxicity. Effects of Hyp and its derivatives in AD mouse models have been described previously (see Discussion) and we focused on the analysis of the NSN compound for the remainder of the study.
NSN rescues mushroom spine and plasticity defects in hippocampal slices from AD mouse models
To further evaluate synaptic effects of NSN compound, we performed a series of experiments with hippocampal slices. To simplify the analysis, we crossed Line M GFP mice (Feng et al., 2000) with PS1KI and APPKI mice to yield PS1KIGFP and APPKIGFP mice. Hippocampal slices were prepared from 6-month-old Line M GFP mice (WTGFP), PS1KIGFP and APPKIGFP mice. The slices were treated with 300 nm NSN for 3.5 h, fixed, and analyzed by confocal imaging (Fig. 8A). Consistent with our previous studies (Sun et al., 2014a; Zhang et al., 2015b), analysis of spine shapes revealed a significant loss of mushroom spines in 6-month-old PS1KIGFP and APPKIGFP mice compared with WTGFP mice (Fig. 8A,B). Treatment with 300 nm NSN had no effect on mushroom spines in WTGFP mice, but resulted in complete rescue of mushroom spines in PS1KIGFP and APPKIGFP hippocampal slices (Fig. 8A,B).
Figure 8.

NSN rescues mushroom spine and synaptic plasticity defects in AD hippocampal slices. A, Confocal images of CA1 hippocampal slices from 6-month-old WTGFP, PS1KIGFP, and APPKIGFP mice. The images are shown for untreated slices (CON) and for slices treated with 300 nm NSN (+NSN) for 3.5 h before fixation. Scale bar, 10 μm. B, Fraction of mushroom spines in hippocampal CA1 neurons from 6-month-old WTGFP, PS1KIGFP, and APPKIGFP mice. The results are shown for untreated slices (CON) and for slices treated with 300 nm NSN (+NSN). The average fraction of mushroom spines for each group of cells is shown as mean ± SE (n = 5 mice). ***p < 0.001. C, Sample fEPSP traces are shown for 6-month-old WT and APPKI hippocampal slices before stimulation (basal), immediately after tetanus stimulation (induction), and 1 h after tetanus stimulation (after 1 h). The results are shown for untreated slices and for slices pretreated with 300 nm NSN (+NSN) for 2–3 h before tetanus stimulation. D, Normalized and averaged fEPSP slope is shown as a function of time in the experiments with 6-month-old WT and APPKI slices with (+NSN) or without 300 nm NSN pretreatment. At each time point, the average normalized fEPSP slope is shown as mean +S.E (n ≥ 6 mice). E, Average normalized fEPSP slope 1 h after tetanus stimulation is shown for 6-month-old WT and APPKI slices. The results are shown for untreated slices (CON) and for slices pretreated 300 nm NSN (+NSN) as mean ± SE (n ≥ 6 mice in each group). *p < 0.05.
Next, we evaluated effects of NSN compound on synaptic plasticity phenotypes in electrophysiological experiments. PS1KI mice do not display E-LTP defects (Oddo et al., 2003; Chakroborty et al., 2009) and only the L-LTP phenotype was reported for these mice (Auffret et al., 2010; Zhang et al., 2015a). APPKI mice have been generated recently (Saito et al., 2014) and no LTP studies have been performed with these mice so far. In our studies, we discovered that two trains of high-frequency stimulation could induce similar synaptic potentiation in 6-month-old WT and APPKI hippocampal slices (Fig. 8C,D). However, this potentiation did not last in APPKI hippocampal slices (Fig. 8C,D). On average, for APPKI slices, the slope of fEPSP dropped back to prestimulation levels within 60 min (Fig. 8C–E). In contrast, the slope of fEPSP stayed elevated for WT slices, with an average increase of 175% at the 60 min time point (Fig. 8C–E). These results suggested that APPKI mice display a robust LTP defect at 6 months of age, which could be expected from the previously described Aβ42 effect on hippocampal LTP (Chapman et al., 1999; Walsh et al., 2002; Shankar et al., 2007; Shankar et al., 2008). Pretreatment of hippocampal slices with a 300 nm NSN compound for 2–3 h had no significant effect on LTP in WT slices, but completely rescued the LTP defect in APPKI slices (Fig. 8C–E). These results are consistent with the earlier studies demonstrating the important role of nSOC in supporting hippocampal LTP (Baba et al., 2003). From these experiments, we concluded that activation of spine nSOC pathway by the NSN compound can rescue synaptic plasticity defects in APPKI hippocampal neurons.
In vivo effects of NSN in AD mouse models
To determine whether the NSN compound can exert beneficial effects in vivo, we performed pilot metabolic stability studies of this compound. We discovered that the NSN compound is stable in commercial liver S9 fractions in the presence of phase I cofactors, which comprise an NADPH-regenerating system, and is stable in commercial CD-1 mouse plasma (data not shown). After intraperitoneal injection of NSN at 10 mg/kg, the compound reached modest levels in plasma and brain penetration of this compound was low, with peak concentration of NSN in brain ∼20 ng/ml (60 nm) (data not shown). Because the NSN compound was effective at a 30 nm concentration in spine rescue experiments (Figs. 6, 7), we initiated whole animal studies. In these experiments, NSN compound was injected intraperitoneally 3 times per week at a 10 mg/kg concentration in WTGFP, PS1KIGFP, and APPKIGFP mice starting from 4 months of age. We did not observe any obvious toxicity in injected mice, but there was some weight loss in NSN-injected mice after 10 weeks of treatment (Fig. 9A). This weight loss may result from activation of TRPC6 channels in the gut smooth muscles, which can accelerate intestinal motility (Tsvilovskyy et al., 2009). The mice were killed at 6.5 months of age and analysis of spine shapes was performed by confocal imaging of hippocampal sections (Fig. 9B). Consistent with our previous studies (Sun et al., 2014a; Zhang et al., 2015b), we observed a significant loss of mushroom spines in the control group of PS1KIGFP and APPKIGFP mice compared with WTGFP mice (Fig. 9B,C). Injections of the NSN compound had no effect on mushroom spines in WTGFP mice, but resulted in rescue of mushroom spine deficiency in PS1KIGFP and APPKIGFP mice (Fig. 9B,C). These results are comparable to in vivo rescue of mushroom spines in PS1KI and APPKI mice after hippocampal injection of AAV1-STIM2 virus (Sun et al., 2014a; Zhang et al., 2015b).
Figure 9.

NSN rescues phenotypes of AD mice in vivo. A, Body weight of 6.5-month-old WTGFP, PS1KIGFP, and APPKIGFP mice injected intraperitoneally with vehicle solution (CON) and injected with 10 mg/kg NSN (+NSN) for 10 weeks. B, Confocal images of CA1 hippocampal slices from 6.5-month-old WTGFP, PS1KIGFP, and APPKIGFP mice. The images are shown for mice injected intraperitoneally with vehicle solution (CON) and for mice injected with 10 mg/kg NSN (+NSN) for 10 weeks. Scale bar, 10 μm. C, Fraction of mushroom spines and mushroom spine density in hippocampal CA1 neurons from 6.5-month-old WTGFP, PS1KIGFP, and APPKIGFP mice. The results are shown for mice injected intraperitoneally with vehicle solution (CON) and for mice injected with 10 mg/kg NSN (+NSN) for 10 weeks. The average fraction of mushroom spines and mushroom spine density in each group is shown as mean ± SE (n = 5 mice). ***p < 0.001. D, Model of STIM2-gated TRPC6/Orai2 nSOC channels and spine maintenance. STIM2-gated TRPC6/Orai2 nSOC channels play a critical role in maintenance of mushroom spines. Extracellular glutamate activates mGluR receptors in the spines, leading to activation of PLC, hydrolysis of PIP2, and generation of InsP3 and DAG. InsP3 causes activation of InsP3R1 in the ER Ca2+ stores in the spines, leading release of Ca2+ and depletion of the stores. Depletion of the stores causes oligomerization of STIM2 and activation of TRPC6/Orai2 Ca2+ influx channels. DAG generated after PIP2 hydrolysis acts as a cofactor in activating TRPC6/Orai2 channels. Resulting Ca2+ influx supports activity of CaMKII in the spines, which is necessary for long-term mushroom spine maintenance. The NSN compound acts as a positive modulator of TRPC6/Orai2 channel activity, promoting STIM2-gated and DAG-activated nSOC Ca2+ influx in the spines and leading to rescue of mushroom spines and LTP in AD mouse models.
Discussion
TRPC6 and Orai2 form STIM2-regulated nSOC channel in hippocampal mushroom spines
In previous studies, we demonstrated that STIM2-mediated nSOC in mushroom spines is important for stability of these spines (Sun et al., 2014a). We further concluded that nSOC-mediated Ca2+ influx causes constitutive activation of synaptic CaMKII, which is necessary for stability of mushroom spines (Sun et al., 2014a). Importantly, we demonstrated that STIM2-nSOC-CaMKII pathway is compromised in PS1KI neurons, APPKI neurons, aging neurons, and sporadic AD brains due to downregulation of the STIM2 protein (Sun et al., 2014a; Zhang et al., 2015b). In the present study, we determined the molecular identity of STIM2-gated nSOC channels in hippocampal spines. Starting with the candidate approach, we identified TRPC6 and Orai2 channels as key components of STIM2-gated nSOC. We demonstrate that overexpression of TRPC6, but not any other member of TRPC family, is able to rescue the mushroom spine defect in PS1KI hippocampal neurons (Fig. 1B,C). We further demonstrated that knock-down of TRPC6 or Orai2, but not knock-down of Orai1, causes inhibition of spine nSOC and loss of mushroom spines in WT neurons (Fig. 1H–K). We confirmed association of TRPC6 and Orai2 with STIM2 in biochemical experiments (Figs. 1D,E, 4) and determined that STIM2, TRPC6, and Orai2 are highly enriched in the hippocampus (Fig. 2). We further demonstrated that overexpression of STIM2 is not able to rescue spine nSOC defects or mushroom spine loss in PS1KI neurons if expression of TRPC6 or Orai2 is suppressed by RNAi (Fig. 3). Our results are consistent with the hypothesis that TRPC6/Orai2 form STIM2-gated nSOC channel in the mushroom synaptic spines (Fig. 9D).
Similar to nonexcitable cells, previous studies of nSOC has been focused primarily on Orai1 (Klejman et al., 2009; Mitchell et al., 2012). A recently published study suggested that Orai1 may have an important role in the formation of new spines in immature hippocampal neurons (Korkotian et al., 2016). Based on the neuronal expression pattern, it has been postulated that Orai2 may play an important role in supporting nSOC (Hoth and Niemeyer, 2013; Majewski and Kuznicki, 2015), but no direct experimental evidence to support this claim has been obtained until our results. TRPC6 has been suggested previously to be critical for spine morphology and neurite growth (Tai et al., 2008; Zhou et al., 2008; Heiser et al., 2013). TRPC6 transgenic mice showed enhancement in spine formation and spatial learning and memory in the Morris water maze (Zhou et al., 2008). TRPC6 gene disruption in human subjects was recently linked with autism spectrum disorders (Griesi-Oliveira et al., 2015). Although TRPC6 has been implicated in SOC in some studies, this channel is largely believed to be a receptor-operated channel (ROC), that can be activated directly by DAG (Cheng et al., 2013; Sun et al., 2014b). In our experiments, we discovered that robust spine nSOC measurements require the application of 100 μm DHPG before Ca2+ add back. Interestingly, the effects of DHPG were not mimicked by the synthetic DAG analog OAG and direct application of 100 μm OAG to hippocampal neuronal cultures in 2 mm Ca2+ aCSF induced Ca2+ responses in just a few spines (data not shown). From these results, we concluded that the activation of TRPC6 channels in the spines requires depletion of the local Ca2+ stores and could not be achieved by OAG alone. A similar conclusion is reached based on the experiments with STIM2-LASS mutant. Expression of this mutant, which is not able to interact with Orai2, exerted a dominant-negative effect on spine nSOC in WT neurons (Fig. 5A,B). During neuronal synaptic activity, activation of mGluR receptors in the spines is coupled to activation of PLC, breakdown of PIP2, generation of DAG and InsP3, and InsP3R1-mediated Ca2+ release from ER stores in the spines. Our results suggest that activation of TRPC6/Orai2 channels complex in the spines occurs primarily as a result of local ER Ca2+ stores depletion and is mediated by STIM2 (Fig. 9D). Local generation of DAG is likely to contribute to activation of TRPC6/Orai2 channels in the spines, and our results (Fig. 7B,C) suggest that the NSN compound may target this step in the activation of spine nSOC. Based on the results obtained, we concluded that the TRPC6 channel mediates Ca2+ influx in the spines and that Orai2 confers ER Ca2+ sensitivity by means of direct interaction with STIM2-SOAR (Fig. 9D). Therefore, both TRPC6 and Orai2 are necessary for store-depletion-mediated activation of nSOC in spines. A similar model has been proposed previously for the STIM1-TRPC3/6-Orai1 complex in nonexcitable cells (Liao et al., 2007; Jardin et al., 2009).
TRPC6/Orai2 nSOC channel complex as a novel therapeutic target for AD
Our results further indicate that the STIM2-gated TRPC6/Orai2 nSOC channel in the spines is a promising therapeutic target for AD and age-related memory loss. In previous studies, we demonstrated that STIM2 overexpression rescues nSOC and mushroom spine defects in PS1KI and APPKI mouse models of familial AD (Sun et al., 2014a; Zhang et al., 2015b) and protects mushroom spines from synaptotoxic effects of Aβ42 oligomers (Popugaeva et al., 2015). In the present study, we demonstrate that overexpression of TRPC6 also rescued nSOC and mushroom spine defects in PS1KI and APPKI mouse models (Fig. 5). Moreover, we demonstrated that a known TRPC6 activator, Hyp, and a novel nSOC-positive modulator, NSN, also rescued nSOC and mushroom spine defects in PS1KI and APPKI mouse models (Fig. 6). It has been demonstrated in previous studies that Hyp and its derivatives were able to prevent β-amyloid neurotoxicity and spatial memory impairments in AβPPSwe/PSEN1ΔE9 (AβPP/PS1) transgenic mice (Dinamarca et al., 2006; Cerpa et al., 2010; Inestrosa et al., 2011). However, the mechanism of Hyp action in these experiments was not clarified. It has been suggested that Hyp exerts its beneficial effects in these experiments by affecting acetylcholinesterase activity, reducing Aβ deposits, or promoting mitochondrial function and neurogenesis (Dinamarca et al., 2006; Cerpa et al., 2010; Inestrosa et al., 2011; Abbott et al., 2013; Carvajal et al., 2013; Zolezzi et al., 2013). Tetrahydrohyperforin (IDN5706) was also recently reported to enhance autophagic clearance of APP (Cavieres et al., 2015). In a recent study, it was suggested that tetrahydrohyperforin rescued Aβ-induced synaptic plasticity defects by activating TRPC3/6/7 channels in neurons (Montecinos-Oliva et al., 2014). It was also reported recently that Hyp modulates dendritic spine morphology in hippocampal slice cultures through activation of TRPC6 channels (Leuner et al., 2013). In hippocampal neurons, activation of TRPC6 has been reported previously to lead to activation of the RAS/MEK/ERK, PI3K, and CAMKIV pathways (Tai et al., 2008; Heiser et al., 2013). Our results with Hyp (Fig. 6) are consistent with the conclusion that Hyp and its derivatives exert beneficial effects in AD models by stimulation of TRPC6-mediated nSOC in mushroom spines. Interestingly, a recent study suggested that TRPC6 may also affect APP processing by γ-secretase via a protein–protein interaction mechanism independently from its ion channel activity (Wang et al., 2015).
We established that both Hyp and the NSN compound act on the Trpc6/Orai2 channel complex because knock-down of either TRPC6 or Orai2 made these compounds ineffective in a spine rescue assay (Fig. 6). However, the mechanisms of action of these two compounds differ from each other. Hyp was able to activate TRPC6 channels expressed in HEK293 cells directly in standard recording conditions (Fig. 7). In contrast, NSN compound was not effective in these experiments, but was able to facilitate OAG-induced Ca2+ influx through TRPC6 channels in conditions of partially depleted intracellular stores (Fig. 7). These results suggested that Hyp acts as direct activator of TRPC6, but that the NSN compound acts as a positive modulator of TRPC6 (Figs. 7, 9D). The exact mechanism of action for the NSN compound requires further investigation, but the ability of this compound to act as a positive modulator of endogenous spine nSOC channels in physiological conditions may offer additional benefits for therapeutic applications in AD. Indeed, in primary hippocampal culture experiments, we discovered that the NSN compound causes significantly less toxicity than Hyp (Fig. 7F,G). Upregulation of TRPC6 channel expression and activity has been implicated in human intestinal fibrotic stenosis and excessive Crohn's disease fibrosis (Kurahara et al., 2015). Upregulation of TRPC6 has been also been implicated recently in carcinogenesis and enhanced aggressive phenotype and tumor invasiveness (Ding et al., 2010a; Ding et al., 2010b; Zhang et al., 2013). Mutations causing overactivation of TRPC6 have been linked with kidney disease (Reiser et al., 2005). These results suggest that strong activators of TRPC6 may result in undesirable side effects. Conversely, Hyp is one of the active components of Hypericum perforatum (St. John's wort), the extracts of which have a long history of safe usage in humans as anti-depressants with minimal side effects (Linde et al., 2008; Kasper et al., 2010). Common adverse side effects of using St. John's wort include gastrointestinal symptoms, consistent with high expression levels of TRPC6 channels in the gut smooth muscles (Tsvilovskyy et al., 2009). Our results (Fig. 7) suggest that the NSN compound may have a wider therapeutic window and result in fewer side effects than direct activators of TRPC6 such as Hyp and its derivatives.
In the present study, we performed preclinical evaluation of the NSN compound in genetic mouse models of AD. We demonstrated that this compound was able to rescue mushroom spine loss in hippocampal cultures and slices from PS1KI and APPKI mouse models (Figs. 6D,E, 8A,B) and rescue hippocampal LTP defects in APPKI mice (Fig. 8C–E). Moreover, NSN rescued mushroom spine loss in PS1KI and APPKI mice when delivered by intraperitoneal injections (Fig. 9B,C). Based on the obtained results (Figs. 6, 7, 8, 9), we concluded that NSN is a potential candidate molecule for therapeutic intervention in brain aging and AD.
Footnotes
This work was supported by the National Institutes of Health (Grant R01NS080152 to I.B.; Figs. 1, 2, 3, 4, 5, 6, 7, and 8A,B) and the Russian Science Foundation (Grant 14-25-00024 to I.B.; Fig. 8C–E). The Preclinical Pharmacology Core receives institutional support from the Institute for Innovations in Medical Technology. I.B. is a holder of the Carl. J, and Hortense M. Thomsen Chair in Alzheimer's Disease Research. Based on the results described in this manuscript, International Patent Application PCT/US2016/030704 “Activation of Neuronal Store-Operated Calcium Entry Pathway for the Treatment of Alzheimer's Disease” was filed by I.B. and H.Z. We thank Dr. Hui Zheng for providing PS1-M146V KI mice; Dr. Takaomi Saido for providing APPKI mice; Dr Ryan Potts for generous help with qRT-PCR experiments; Drs. Craig Montell, Joseph Yuan, and Jen Liou for providing expression constructs; Dr. Noelle Williams in the Preclinical Pharmacology Core for assistance with metabolic stability and PK analysis; members of Ilya Bezprozvanny laboratory for advice and suggestions; and Leah Taylor and Polina Plotnikova for administrative assistance.
The authors declare no competing financial interests.
References
- Abbott AC, Calderon Toledo C, Aranguiz FC, Inestrosa NC, Varela-Nallar L. Tetrahydrohyperforin increases adult hippocampal neurogenesis in wild-type and APPswe/PS1DeltaE9 mice. J Alzheimers Dis. 2013;34:873–885. doi: 10.3233/JAD-121714. [DOI] [PubMed] [Google Scholar]
- Auffret A, Gautheron V, Mattson MP, Mariani J, Rovira C. Progressive age-related impairment of the late long-term potentiation in Alzheimer's disease presenilin-1 mutant knock-in mice. J Alzheimers Dis. 2010;19:1021–1033. doi: 10.3233/JAD-2010-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hiesinger PR. The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol Neurodegener. 2013;8:23. doi: 10.1186/1750-1326-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal FJ, Zolezzi JM, Tapia-Rojas C, Godoy JA, Inestrosa NC. Tetrahydrohyperforin decreases cholinergic markers associated with amyloid-beta plaques, 4-hydroxynonenal formation, and caspase-3 activation in AbetaPP/PS1 mice. J Alzheimers Dis. 2013;36:99–118. doi: 10.3233/JAD-130230. [DOI] [PubMed] [Google Scholar]
- Cavieres VA, González A, Muñoz VC, Yefi CP, Bustamante HA, Barraza RR, Tapia-Rojas C, Otth C, Barrera MJ, González C, Mardones GA, Inestrosa NC, Burgos PV. Tetrahydrohyperforin inhibits the proteolytic processing of amyloid precursor protein and enhances its degradation by Atg5-dependent autophagy. PLoS One. 2015;10:e0136313. doi: 10.1371/journal.pone.0136313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W, Hancke JL, Morazzoni P, Bombardelli E, Riva A, Marin PP, Inestrosa NC. The hyperforin derivative IDN5706 occludes spatial memory impairments and neuropathological changes in a double transgenic Alzheimer's mouse model. Curr Alzheimer Res. 2010;7:126–133. doi: 10.2174/156720510790691218. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008;283:12935–12940. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Ong HL, Liu X, Ambudkar IS. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr Top Membr. 2013;71:149–179. doi: 10.1016/B978-0-12-407870-3.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Meyer T. Evolutionary origins of STIM1 and STIM2 within ancient Ca(2+) signaling systems. Trends Cell Biol. 2011;21:202–211. doi: 10.1016/j.tcb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21:1897–1907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinamarca MC, Cerpa W, Garrido J, Hancke JL, Inestrosa NC. Hyperforin prevents beta-amyloid neurotoxicity and spatial memory impairments by disaggregation of Alzheimer's amyloid-beta-deposits. Mol Psychiatry. 2006;11:1032–1048. doi: 10.1038/sj.mp.4001866. [DOI] [PubMed] [Google Scholar]
- Ding X, He Z, Shi Y, Wang Q, Wang Y. Targeting TRPC6 channels in oesophageal carcinoma growth. Expert Opin Ther Targets. 2010a;14:513–527. doi: 10.1517/14728221003733602. [DOI] [PubMed] [Google Scholar]
- Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D, Cai R, Jin Y, Dong B, Xu Y, Wang Y. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J Natl Cancer Inst. 2010b;102:1052–1068. doi: 10.1093/jnci/djq217. [DOI] [PubMed] [Google Scholar]
- Estacion M, Li S, Sinkins WG, Gosling M, Bahra P, Poll C, Westwick J, Schilling WP. Activation of human TRPC6 channels by receptor stimulation. J Biol Chem. 2004;279:22047–22056. doi: 10.1074/jbc.M402320200. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K, Romanin C. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284:21696–21706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesi-Oliveira K, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2015;20:1350–1365. doi: 10.1038/mp.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J, Kuznicki J. Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J Neurochem. 2013;126:727–738. doi: 10.1111/jnc.12320. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Heiser JH, Schuwald AM, Sillani G, Ye L, Müller WE, Leuner K. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J Neurochem. 2013;127:303–313. doi: 10.1111/jnc.12376. [DOI] [PubMed] [Google Scholar]
- Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci U S A. 2011;108:13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Niemeyer BA. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr Top Membr. 2013;71:237–271. doi: 10.1016/B978-0-12-407870-3.00010-X. [DOI] [PubMed] [Google Scholar]
- Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- Inayama M, Suzuki Y, Yamada S, Kurita T, Yamamura H, Ohya S, Giles WR, Imaizumi Y. Orai1-Orai2 complex is involved in store-operated calcium entry in chondrocyte cell lines. Cell Calcium. 2015;57:337–347. doi: 10.1016/j.ceca.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Tapia-Rojas C, Griffith TN, Carvajal FJ, Benito MJ, Rivera-Dictter A, Alvarez AR, Serrano FG, Hancke JL, Burgos PV, Parodi J, Varela-Nallar L. Tetrahydrohyperforin prevents cognitive deficit, Abeta deposition, tau phosphorylation and synaptotoxicity in the APPswe/PSEN1DeltaE9 model of Alzheimer's disease: a possible effect on APP processing. Transl Psychiatry. 2011;1:e20. doi: 10.1038/tp.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardin I, Gómez LJ, Salido GM, Rosado JA. Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca(2+) entry pathways. Biochem J. 2009;420:267–276. doi: 10.1042/BJ20082179. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kasper S, Gastpar M, Möller HJ, Müller WE, Volz HP, Dienel A, Kieser M. Better tolerability of St. John's wort extract WS 5570 compared to treatment with SSRIs: a reanalysis of data from controlled clinical trials in acute major depression. Int Clin Psychopharmacol. 2010;25:204–213. doi: 10.1097/YIC.0b013e328335dc1a. [DOI] [PubMed] [Google Scholar]
- Klejman ME, Gruszczynska-Biegala J, Skibinska-Kijek A, Wisniewska MB, Misztal K, Blazejczyk M, Bojarski L, Kuznicki J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem Int. 2009;54:49–55. doi: 10.1016/j.neuint.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E, Oni-Biton E, Segal M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J Physiol. 2016 doi: 10.1113/JP272645. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahara LH, Sumiyoshi M, Aoyagi K, Hiraishi K, Nakajima K, Nakagawa M, Hu Y, Inoue R. Intestinal myofibroblast TRPC6 channel may contribute to stenotic fibrosis in Crohn's disease. Inflamm Bowel Dis. 2015;21:496–506. doi: 10.1097/MIB.0000000000000295. [DOI] [PubMed] [Google Scholar]
- Leuner K, Kazanski V, Müller M, Essin K, Henke B, Gollasch M, Harteneck C, Müller WE. Hyperforin-a key constituent of St. John's wort specifically activates TRPC6 channels. FASEB J. 2007;21:4101–4111. doi: 10.1096/fj.07-8110com. [DOI] [PubMed] [Google Scholar]
- Leuner K, Li W, Amaral MD, Rudolph S, Calfa G, Schuwald AM, Harteneck C, Inoue T, Pozzo-Miller L. Hyperforin modulates dendritic spine morphology in hippocampal pyramidal neurons by activating Ca(2+) -permeable TRPC6 channels. Hippocampus. 2013;23:40–52. doi: 10.1002/hipo.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci U S A. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde K, Berner MM, Kriston L. St John's wort for major depression. Cochrane Database Syst Rev. 2008;4:CD000448. doi: 10.1002/14651858.CD000448.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski L, Kuznicki J. SOCE in neurons: signaling or just refilling? Biochim Biophys Acta. 2015;1853:1940–1952. doi: 10.1016/j.bbamcr.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell CB, Gasperini RJ, Small DH, Foa L. STIM1 is necessary for store-operated calcium entry in turning growth cones. J Neurochem. 2012;122:1155–1166. doi: 10.1111/j.1471-4159.2012.07840.x. [DOI] [PubMed] [Google Scholar]
- Montecinos-Oliva C, Schuller A, Parodi J, Melo F, Inestrosa NC. Effects of tetrahydrohyperforin in mouse hippocampal slices: neuroprotection, long-term potentiation and TRPC channels. Curr Med Chem. 2014;21:3494–3506. doi: 10.2174/0929867321666140716091229. [DOI] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Derler I, Schindl R, Bergsmann J, Frischauf I, Groschner K, Romanin C. A Cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–8426. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Gill D, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Bezprozvanny I. Role of endoplasmic reticulum Ca2+ signaling in the pathogenesis of Alzheimer disease. Front Mol Neurosci. 2013;6:29. doi: 10.3389/fnmol.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popugaeva E, Supnet C., Bezprozvanny I. Presenilins, deranged calcium homeostasis, synaptic loss and dysfunction in Alzheimer's disease. Messenger. 2012;1:53–62. doi: 10.1166/msr.2012.1002. [DOI] [Google Scholar]
- Popugaeva E, Pchitskaya E, Speshilova A, Alexandrov S, Zhang H, Vlasova O, Bezprozvanny I. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol Neurodegener. 2015;10:37. doi: 10.1186/s13024-015-0034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinska-Kijek A, Wisniewska M.B., Gruszczynska-Biegala J., Methner A., Kuznicki J. Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol Exp (Wars) 2009;69:413–428. doi: 10.55782/ane-2009-1753. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Beg AM, Wu S, Putney JW, Jr, Rusan NM. Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr Biol. 2012;22:1487–1493. doi: 10.1016/j.cub.2012.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Ikura M. Structure and function of endoplasmic reticulum STIM calcium sensors. Curr Top Membr. 2013;71:59–93. doi: 10.1016/B978-0-12-407870-3.00003-2. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL, 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014a;82:79–93. doi: 10.1016/j.neuron.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Sukumaran P, Bandyopadhyay BC, Singh BB. Physiological function and characterization of TRPCs in neurons. Cells. 2014b;3:455–475. doi: 10.3390/cells3020455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Ghori A, Brandt R. Thin, stubby or mushroom: spine pathology in Alzheimer's disease. Curr Alzheimer Res. 2009;6:261–268. doi: 10.2174/156720509788486554. [DOI] [PubMed] [Google Scholar]
- Tai Y, Feng S, Ge R, Du W, Zhang X, He Z, Wang Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J Cell Sci. 2008;121:2301–2307. doi: 10.1242/jcs.026906. [DOI] [PubMed] [Google Scholar]
- Tsvilovskyy VV, Zholos AV, Aberle T, Philipp SE, Dietrich A, Zhu MX, Birnbaumer L, Freichel M, Flockerzi V. Deletion of TRPC4 and TRPC6 in mice impairs smooth muscle contraction and intestinal motility in vivo. Gastroenterology. 2009;137:1415–1424. doi: 10.1053/j.gastro.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener. 2014;9:48. doi: 10.1186/1750-1326-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang J, Lu R, Yang J, Li H, He Z, Jing N, Wang X, Wang Y. TRPC6 specifically interacts with APP to inhibit its cleavage by gamma-secretase and reduce Abeta production. Nat Commun. 2015;6:8876. doi: 10.1038/ncomms9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Shih HP, Vigont V, Hrdlicka L, Diggins L, Singh C, Mahoney M, Chesworth R, Shapiro G, Zimina O, Chen X, Wu Q, Glushankova L, Ahlijanian M, Koenig G, Mozhayeva GN, Kaznacheyeva E, Bezprozvanny I. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem Biol. 2011;18:777–793. doi: 10.1016/j.chembiol.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010;30:8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu J, Sun S, Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling, excitability, and synaptic plasticity defects in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015a;45:561–580. doi: 10.3233/JAD-142427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I. Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer's disease. J Neurosci. 2015b;35:13275–13286. doi: 10.1523/JNEUROSCI.1034-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SS, Wen J, Yang F, Cai XL, Yang H, Luo KJ, Liu QW, Hu RG, Xie X, Huang QY, Chen JY, Fu JH, Hu Y. High expression of transient potential receptor C6 correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Med Oncol. 2013;30:607. doi: 10.1007/s12032-013-0607-7. [DOI] [PubMed] [Google Scholar]
- Zhou J, Du W, Zhou K, Tai Y, Yao H, Jia Y, Ding Y, Wang Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat Neurosci. 2008;11:741–743. doi: 10.1038/nn.2127. [DOI] [PubMed] [Google Scholar]
- Zolezzi JM, Carvajal FJ, Ríos JA, Ordenes D, Silva-Alvarez C, Godoy JA, Inestrosa NC. Tetrahydrohyperforin induces mitochondrial dynamics and prevents mitochondrial Ca2+ overload after Abeta and Abeta-AChE complex challenge in rat hippocampal neurons. J Alzheimers Dis. 2013;37:735–746. doi: 10.3233/JAD-130173. [DOI] [PubMed] [Google Scholar]