

Abstract
The insulin-regulated glucose transporter-4 (GluT4) is critical for insulin- and contractile-mediated glucose uptake in skeletal muscle. GluT4 is also expressed in some hippocampal neurons, but its functional role in the brain is unclear. Several established molecular modulators of memory processing regulate hippocampal GluT4 trafficking and hippocampal memory formation is limited by both glucose metabolism and insulin signaling. Therefore, we hypothesized that hippocampal GluT4 might be involved in memory processes. Here, we show that, in male rats, hippocampal GluT4 translocates to the plasma membrane after memory training and that acute, selective intrahippocampal inhibition of GluT4-mediated glucose transport impaired memory acquisition, but not memory retrieval. Other studies have shown that prolonged systemic GluT4 blockade causes insulin resistance. Unexpectedly, we found that prolonged hippocampal blockade of glucose transport through GluT4-upregulated markers of hippocampal insulin signaling prevented task-associated depletion of hippocampal glucose and enhanced both working and short-term memory while also impairing long-term memory. These effects were accompanied by increased expression of hippocampal AMPA GluR1 subunits and the neuronal GluT3, but decreased expression of hippocampal brain-derived neurotrophic factor, consistent with impaired ability to form long-term memories. Our findings are the first to show the cognitive impact of brain GluT4 modulation. They identify GluT4 as a key regulator of hippocampal memory processing and also suggest differential regulation of GluT4 in the hippocampus from that in peripheral tissues.
SIGNIFICANCE STATEMENT The role of insulin-regulated glucose transporter-4 (GluT4) in the brain is unclear. In the current study, we demonstrate that GluT4 is a critical component of hippocampal memory processes. Memory training increased hippocampal GluT4 translocation and memory acquisition was impaired by GluT4 blockade. Unexpectedly, whereas long-term inhibition of GluT4 impaired long-term memory, short-term memory was enhanced. These data further our understanding of the molecular mechanisms of memory and have particular significance for type 2 diabetes (in which GluT4 activity in the periphery is impaired) and Alzheimer's disease (which is linked to impaired brain insulin signaling and for which type 2 diabetes is a key risk factor). Both diseases cause marked impairment of hippocampal memory linked to hippocampal hypometabolism, suggesting the possibility that brain GluT4 dysregulation may be one cause of cognitive impairment in these disease states.
Keywords: glucose, GluT4, hippocampus, memory
Introduction
Insulin resistance is associated with Alzheimer's disease (de la Monte, 2012a; Biessels and Reagan, 2015). Numerous data suggest that insulin resistance can occur, not only peripherally, but also in the brain (Duarte et al., 2012; de la Monte, 2012b; Biessels and Reagan, 2015). In the periphery, insulin resistance is associated with a loss-of-function in insulin-regulated glucose transporter-4 (GluT4) (Zisman et al., 2000; Wallberg-Henriksson and Zierath, 2001). However, it is unclear whether brain insulin resistance follows brain GluT4 dysfunction. In the brain, GluT4 is found in neurons of specific regions involved in motor and episodic memory such as the cerebellum, motor cortex, and especially the hippocampus (Vannucci et al., 1998; Alquier et al., 2006). However, the functional role of brain GluT4 is largely undetermined, although recent studies have confirmed a role for hypothalamic GluT4 in metabolic regulation (Ren et al., 2015; Ren et al., 2014). As in the periphery, brain GluT4 is regulated by insulin, which stimulates translocation of brain GluT4 (Bakirtzi et al., 2009; Grillo et al., 2009) via insulin receptors (Ren et al., 2014). Both insulin and glucose are potent enhancers of hippocampal memory processes and it has been suggested that insulin's cognitive effects may occur via upregulation in GluT4-mediated glucose uptake (McNay et al., 2010; McNay and Recknagel, 2011). However, neither the role of GluT4 in memory processing or hippocampal function nor the impact of brain GluT4 blockade has been studied.
Many of GluT4's upstream regulators are involved in memory processes, suggesting that GluT4 may contribute directly to memory. When inactive, Glut4 is tethered within the cytosol in GluT4 storage vesicles (GSVs). Translocation of GluT4 from GSVs to the cell surface occurs in response to insulin and to several molecules involved in hippocampal learning and memory processes, including estrogen, norepinephrine, and brain-derived neurotrophic factor (BDNF) (Kishi et al., 1996; Barros et al., 2006b; Suwa et al., 2010). This translocation is also regulated by protein kinase C-zeta, histone acetylating proteins, protein kinase A, and Ca2+/calmodulin kinase II, all of which can affect memory processing (Barros et al., 2006b; Smith et al., 2007; Li et al., 2008; Mukwevho et al., 2008; Smith et al., 2008; Suwa et al., 2010; Zhang et al., 2010; Dehvari et al., 2012; Ojuka et al., 2012).
Importantly, GSVs house, not only GluT4, but also additional molecules [e.g., Ras-GTPase-activating-like protein (IQGAP1), insulin-regulated aminopeptidase (IRAP), and insulin-like growth factor receptor-2 (IGF2R)] that are well documented regulators of memory (Schrick et al., 2007; Fernando et al., 2008; Jedrychowski et al., 2010; Chen et al., 2011; Gao et al., 2011; Kandror and Pilch, 2011). Changes in ionic concentration gradients that occur with synaptic events critical for memory, such as elevated extracellular [K+] and increased intracellular [Ca2+], also facilitate GSV translocation (Yu et al., 1999; Xu et al., 2004; Li et al., 2006; Wijesekara et al., 2006; Fernando et al., 2008). Therefore, to examine the role of GluT4 in memory processing, it is important to block glucose uptake selectively through GluT4 rather than simply blocking GSV translocation. For example, inhibition of IRAP can increase performance on memory tasks, potentially by increasing GluT4-mediated glucose uptake (Fernando et al., 2008). To block GluT4 in the hippocampus, we used direct brain delivery of indinavir, which targets the cytosolic glucose-binding domain of GluT4, thus preventing glucose transport (Hertel et al., 2004; Hruz et al., 2008; Vyas et al., 2010; Hresko and Hruz, 2011). This treatment allowed us to block GluT4-mediated transport of glucose without affecting the translocation of GSVs to the plasma membrane (PM) or the function of other molecules housed in the GSVs. Because many molecules and events associated with memory regulate GluT4 trafficking and neuronal glucose supply and metabolism is necessary for memory, we hypothesized that hippocampal GluT4-mediated glucose uptake would be critical for memory formation (McNay et al., 2000; Hoyer, 2003). To test this hypothesis, indinavir was used to block GluT4-mediated glucose uptake. Indinavir blocks the cytoplasmic glucose-binding domain of GluT4 and its selectivity for GluT4 is well validated (Nolte et al., 2001; Hertel et al., 2004; Hruz et al., 2008; Hresko and Hruz, 2011). Our study identifies novel contributions of GluT4 to memory processing and glucose regulation in the hippocampus.
Materials and Methods
Subjects
Juvenile male Sprague Dawley rats (Charles River Laboratories), ∼300 g at the time of arrival, were housed in pairs on a 12:12 h light:dark schedule with food and water available ad libitum. Rats were given at least 1 week to acclimate before any testing, during which time they were handled extensively. Each rat was used only once. All procedures were approved by the University at Albany Institutional Animal Care and Use Committee.
Surgeries and drug administration
Acute GluT4 studies
Rats given surgeries received either an intrahippocampal microinjection cannula (Plastics One) or an intrahippocampal microdialysis (mD) cannula (BASi) using standard sterile stereotaxic methods as detailed previously (McNay et al., 2010, 2013,Pearson-Leary and McNay, 2012). Rats were anesthetized with isoflurane and cannulae were implanted into the left hippocampus. The nose bar was set at 4.6 mm above the interaural line. Microinjection coordinates were 5.6 mm posterior to bregma, +4.6 lateral, and 3.3 ventral from dura; mD coordinates (to accommodate the 4 mm mD probe) were 5.6 mm posterior to bregma, +5.0 lateral, and 3.0 ventral from dura. Aseptic procedures were used during all surgeries and animals were monitored for any signs of infection after recovery. All rats received chewable Rimadyl (analgesia) and Baytril (antibiotic) tablets for 3 d after surgery.
Prolonged GluT4 blockade studies
Using similar methods, a minosmotic pump (Alzet) was placed subcutaneously and connected to a catheter and a low-profile cannula implanted into the lateral ventricle (0.8 mm posterior to bregma, +1.5 lateral, and 4.5 ventral from dura). Some rats also received a simultaneously implanted hippocampal mD cannula. Two days before surgery, pumps were incubated in sterile saline at 36°C for 2 d to allow pump priming. After implantation, the pumps delivered treatments for 2 weeks at a flow rate of 0.5 μL/h. All pumps contained artificial extracellular fluid (aECF) containing the following (in mm): 153.5 Na, 4.3 K, 0.41 Mg, 0.71 Ca, and 139.4 Cl, buffered at pH 7.4 (McNay et al., 2004) either alone or containing indinavir sulfate.
GluT4 blockade
Indinavir sulfate was used to block GluT4-mediated glucose uptake (Hertel et al., 2004; Hruz et al., 2008; Hresko and Hruz, 2011) and was purchased from Toronto Research Chemicals (catalog #I525000). Indinavir sulafte was solubilized in aECF and filtered with a 0.22 μm filter immediately before injection.
Behavioral testing
We measured memory for an aversive stimulus using an inhibitory avoidance protocol based on previous work (Brioni et al., 1989). In the acquisition phase, rats were placed a brightly lit compartment of a custom-made avoidance apparatus facing the door between chambers, as described previously (McNay et al., 2013). After 20 s in the light chamber, the door to a dark compartment was opened and latency to enter was recorded. When rats had entered the dark chamber completely (all four paws on floor), the door closed and they received a 0.5 mA shock for 2 s, after which the door opened immediately and animals were returned to their home cages. Rats were tested 48 h later for retention. In the retention phase (labeled “retrieval” to indicate memory retrieval), rats were again placed in the light compartment facing the door and, after 20 s, the door was opened. Latency to enter the dark chamber was measured and the experiment was terminated when the rat entered the dark chamber (all four paws on the floor) or after 1200 s; longer latencies to enter were interpreted as indicating superior memory for the foot shock. A nonparametric test (Mann–Whitney U) was used to determine significance of performance differences between groups.
Spontaneous alternation (SA) testing was used to assess spatial working memory (McNay and Gold, 1999; McNay et al., 2000, 2005, 2006b, 2010; Pearson-Leary and McNay, 2012). Individual rats were placed into a custom four-arm Plexiglas plus maze and allowed to explore freely for 20 min. During this task, rats will tend to visit the least recently visited arm, using spatial working memory to recall arm-visit history. Alternation scores are calculated by dividing the percentage of alternations (defined as a visit to each of the four arms within each span of five consecutive entries), with chance performance being 44% (McNay and Gold, 1998; McNay et al., 2000; Pearson-Leary and McNay, 2012).
Contextual fear memory testing was performed using a trough-style fear-conditioning chamber. Rats were acclimated to the lit chamber for 5 min the day before testing. On the acquisition testing day, rats were allowed 1 min in the lit chamber before training. Each training block consisted of a 10 s tone at 80 dB and 5000 HZ followed by a 10 s shock at 0.8 mA. Each rat received 3 training blocks with a 1 min intertrial interval. After training, rats were placed back in their home cages and tested for retention after an interval of either 30 min (to assess short-term memory; STM) or 72 h (to measure long-term memory; LTM); animals were only tested once. Retention trials consisted of 3 min spent in the context-cue environment with tone reminders given for 10 s each minute. Fear memory was assessed by measuring time spent freezing, defined as a complete absence of movement other than respiration. Context-cue fear memory is sensitive to hippocampal function (Curzon et al., 2009).
For novel object memory testing, rats were habituated to a cubical Plexiglas box (60 cm per side) in a dimly lit room for 2 d before testing and allowed to explore for 5 min during each session. During the training (memory acquisition) trial, rats were placed back into the chamber with two novel objects and allowed to explore each object. In the testing trial (memory retrieval), a novel object replaced one object; time spent exploring each object was recorded during both trials. Greater time spent with the novel object during the testing trial is interpreted as increased memory because rats tend to interact with novel objects more than familiar objects (Bevins and Besheer, 2006). Performance was calculated as the percentage of total test exploration time spent exploring the novel object. STM was tested after a 30 min intertrial interval and LTM was tested at 16 h. LTM time points were based on pilot studies to determine memory performance sensitivity to treatment. Each animal was tested at only one of these time points. Twenty-four hours after object recognition testing, rats were given a lethal dose of pentobarbital, decapitated, and brains were flash frozen on dry ice then stored at −80°C. Brains from this cohort were used for protein analyses.
Microinjections and in vivo mD
mD was performed as described previously (McNay and Gold, 1999, 2001; McNay et al., 2000, 2001; McNay and Sherwin, 2004; Pearson-Leary and McNay, 2012). Samples were collected before, during, and after behavioral testing. On the day of testing, a fresh 4 mm probe (MD-2264; BASi) was placed into the guide cannula and the animal was allowed to acclimate for at least 1 h while being perfused with aECF including 1.25 mm glucose at a rate of 1.5 μL/min (McNay and Sherwin, 2004). Perfusate was collected in 10 min sample bins. Two baseline samples were collected before SA testing. Glucose and lactate were measured in a CMA600 analyzer (CMA/Microdialysis, Sweden) and corrected for in vivo probe recovery using a zero-net-flux plot for ECF glucose measurements under the same experimental conditions. Microinjections were given at 1.25 μL/min for 4 min, with the injection needle left in place for a further 5 min before experimental testing.
Cell fractionation
PM fractions were collected using a commercially available kit (Biovision). Whole hippocampi were homogenized with a Polytron hand-held electric homogenizer in 3 volumes of (1 mg/3 μL) lysis buffer with lyophilized protease inhibitors (Biovision). After the initial homogenization, 30 μL of the total lysate was added to 200 μL of ice-cold RIPA buffer (50 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X) containing fresh protease and phosphatase inhibitors (Pierce) and stored at −80°C until protein quantification. For the remaining lysate, PM separation proceeded according to the kit's instructions.
Western blotting
Protein concentration was determined using a commercially available kit for the bicinchoninic assay (Pierce). Then, 10–20 μg of protein was placed in sample buffer in denaturing and reducing conditions. Samples tested for total protein content were heated at 95°C for 5 min before sample loading. Samples analyzed for plasma membrane associated proteins were not heated before gel loading. Samples were resolved on 8% or 12% SDS polyacrylamide mini-gels (Pierce or Bio-Rad) and transferred onto PVDF membranes (Bio-Rad), followed by detection with the appropriate primary antibodies: anti-rabbit directed against GluT4 (Millipore catalog #07–1404), GluT1 (Abcam catalog #ab652; this antibody recognizes both the 45 kDa astrocytic GluT1 and the 55 kDa endothelial GluT1), GluT3 (Abcam catalog #ab41525), phosphorylated Akt at serine 473 (Cell Signaling Technology catalog #4060L), total Akt (Cell Signaling Technology catalog #4691L), IRAP (Cell Signaling Technology catalog #6918), AMPA receptor GluR1 subunit (Cell Signaling Technology catalog #8850), NMDA receptor NR2B subunit (Cell Signaling Technology catalog #4212). β-actin (Sigma-Aldrich catalog #A2228) was used as a loading control for total protein samples and PM calcium pump ATPase (Abcam catalog #ab2825) was used as a PM loading control. After wet transfer at 350 mA at 4°C for 60 min, PVDF membranes were blocked for 1 h with 5% milk or 5% BSA in Tris-buffered saline containing 0.1% Tween 20. Membranes were incubated with primary antibodies in blocking solution overnight at 4°C in conditions optimized for immunodetection by each antibody. Secondary incubation in biotin-conjugated goat anti-mouse (Pierce) or goat anti-rabbit (Vector Laboratories) IgG was followed by incubation in HRP strepdavidin (Pierce) for enhanced immunoreactivity, both for 1 h at room temperature. Immunoblots were developed with chemiluminscent detection using the SuperSignal West Pico Chemiluminescent Kit (Pierce). Images were taken in a Bio-rad ChemiDoc XRS Image Analyzer. Membranes were then stripped with Restore Plus Stripping Buffer (Pierce) and reprobed for loading controls or total protein content. Band intensity was measured by densitometry in ImageQuant software and normalized to loading controls or total protein content. To control for between-gel variations in band intensity, vehicle control values were normalized to 100% and treatment groups were analyzed as percentage of control values.
ELISA
A total of 30 μg of total hippocampal lysate from each rat was analyzed for BDNF or amyloid-β x-40 levels. BDNF was measured using a commercially available kit from Millipore (catalog #CYT306). Amyloid-β x-40 was measured using a commercially available kit from Covance (catalog #SIG-38950). The procedures were performed according to the manufacturers' instructions.
Statistical analyses
For statistical comparisons of two groups, we used the Student's t test or Mann– Whitney U test and, for comparisons of more than two groups, we used an ANOVA followed by Bonferroni post hoc tests. An α level of 0.05 (two-tailed) was set for significance. All statistical analyses were made in SPSS version 17 or Prism 5.
Results
Hippocampal GluT4 is necessary for aversive memory acquisition but not retrieval
We first investigated whether hippocampal GluT4-mediated glucose uptake was necessary for aversive memory acquisition or retrieval. Because several conditions that occur during memory acquisition also trigger translocation of GSVs to the PM or alter the intrinsic activity of GluT4 already in the PM and because optimal neural glucose metabolism is critical for memory formation, we hypothesized that GluT4-mediated glucose uptake would be critical for memory acquisition (Mora et al., 1995; Thai et al., 1998; McNay et al., 2001; McNay and Gold, 2002; Barros et al., 2006a; Barros et al., 2006b; Smith et al., 2007; Li et al., 2008; Mukwevho et al., 2008; Smith et al., 2008; McNay and Cotero, 2010; Suwa et al., 2010; Zhang et al., 2010; Dehvari et al., 2012). Indinavir was used to block GluT4-mediated glucose uptake because this drug binds selectively to the glucose-binding domain of GluT4 and impairs insulin-mediated glucose uptake without affecting insulin signaling upstream of GluT4 or on different pathways (Nolte et al., 2001). The experimental outline is presented in Figure 1A.
Figure 1.

Inhibitory avoidance learning (0.5 mA shock, 2 s). A, Experimental outline for inhibitory avoidance procedures used to test the effects of intrahippocampal GluT4 inhibition on memory acquisition or retrieval (n = 8–10 per group). Graphs depict latencies for the retention trial. B, Intrahippocampal administration of the GluT4 inhibitor indinavir (200 ng dose) 10 min before the training trial (“Acquisition” group) decreased latencies to enter the dark chamber during the retention trial. C, Intrahippocampal administration of indinavir 10 min before day 2 testing (“Retrieval” group) had no effect on latencies to enter the dark chamber on retention trial testing (day 2 latencies are shown in the figure). Data are presented as the mean ± SEM; *p < 0.05.
The GluT4 blocker indinavir (180 ng) delivered intrahippocampally (unilateral, left hippocampus) 10 min before training decreased latencies to cross into the dark chamber during the retention trial 48 h later (“Acquisition” group; Mann–Whitney U = 19, p = 0.02; Fig. 1B). In contrast, when administered 10 min before the retention trial (“Retrieval” group), indinavir had no effect on latency to cross into the dark chamber (Mann–Whitney U = 24, p = n.s.; Fig. 1C). These data suggest that hippocampal GluT4-mediated glucose uptake is necessary for memory acquisition, but not retrieval.
PM expression of GluT4 was increased after memory acquisition training
In a separate experiment, rats were killed immediately after the training trial to determine whether inhibitory avoidance training affected GluT4 translocation in the hippocampus (the experimental outline is shown in Fig. 2A). The ratio of PM to total GluT4 increased 30 min after the inhibitory avoidance training trial (Student's t = 3.78, p = 0.001; Fig. 2B,C) relative to control rats that did not undergo training on IA. There were no significant effects of training on PM expression of other hippocampal glucose transporters examined. Because translocation of GluT4 after stimulation is necessary for GluT4 activation, these data suggest that memory acquisition training increases GluT4 activity (Robinson et al., 1992; Wallberg-Henriksson and Zierath, 2001; Thong et al., 2005; Rowland et al., 2011).
Figure 2.

Western blotting for glucose transporter expression in the hippocampus of rats killed immediately after the training trial of inhibitory avoidance. A, Experimental design outline. B, Western blot images from hippocampi of rats killed 30 min after the retention trial. For GluT1, the antibody used detected both endothelial (top band, 55 kDa) and astrocytic isoforms (bottom band, 45 kDa). C, There were no significant differences in PM glucose transporter expression between rats that were not tested for inhibitory avoidance (“No Training”). Rats that were killed 30 min after the training trial (“Training”) had increased PM GluT4 relative to PM GluT4 expression in untrained control rats. Inhibitory avoidance training did not lead to differences in expression of other hippocampal glucose transporters (n = 4–8 per group). Data are presented as the mean ± SEM; *p < 0.05.
Two weeks of brain GluT4 inhibition increased STM but impaired LTM
Based on our findings from acute brain delivery of indinavir on impairing memory acquisition, we measured object recognition memory after treatment with either 180 ng/h indinavir or vehicle control (aECF) for 2 weeks before testing via an intracerebroventricular Alzet minipump. This dose was chosen based on dose–response studies on metabolism and SA behavior presented below. We habituated rats to the testing box on day 21 of the experiment, with training 1 d later and a recognition test after a further 30 min (STM test) or 16 h (LTM test); the experimental outline shown in Figure 3A. A two-way ANOVA revealed a significant interaction effect (F(1,36) = 12.92, p = 0.001) and post hoc analyses showed that GluT4 inhibition impaired LTM, but led to an apparently paradoxical increase in STM (p < 0.05; Fig. 3B).
Figure 3.
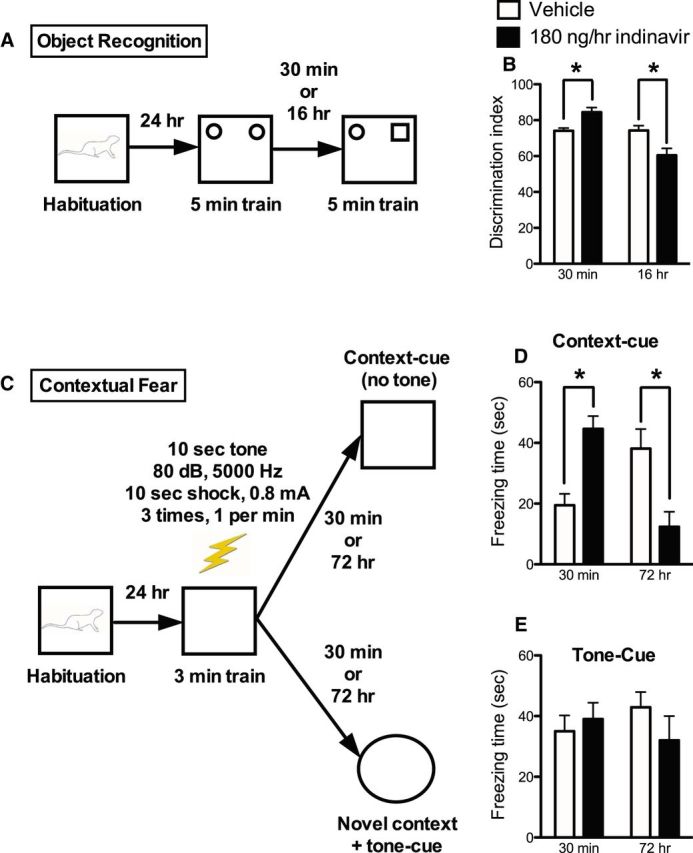
Increased STM but impaired LTM after 2-week treatment with 180 ng/h indinavir. A, Experimental outline for object recognition testing. Separate cohorts of rats were tested 30 min (STM) or 16 h (LTM) after the 5 min training session (n = 8–13 per group). B, The 2 × 2 interaction was significant, with post hoc t tests showing increased discrimination index scores at 30 min (suggesting increased STM) and decreased scores at 16 h (suggesting decreased LTM). C, Experimental outline for contextual fear testing. Separate cohorts of rats were tested 30 min (STM) or 72 h (LTM) after the 3 min training session (n = 5–8 per group). D, Indinavir-treated rats had increased freezing behavior 30 min after the 3 min training session and decreased freezing behavior 72 h after the training session. E, There were no differences in freezing time in response to the tone cue. Data were analyzed statistically using a two-way ANOVA followed by Bonferroni's post hoc test and are presented as the mean ± SEM; *p < 0.05.
Because there is some debate as to the extent of hippocampal involvement in object recognition (Broadbent et al., 2010; Barker and Warburton, 2011), we used the hippocampally dependent contextual fear task (Anagnostaras et al., 2001) to further examine the memory pathways affected by chronic central GluT4 inhibition. Additional groups of rats were treated identically to those tested on object recognition, then habituated and given contextual fear training (Fig. 3C), with measurement of contextual fear (as shown by freezing behavior) after either 30 min or 72 h. A significant interaction of treatment with time of testing was again found (F(1,24) = 26.09, p = 0.0005) and post hoc analyses showed that indinavir-treated rats had significantly increased freezing times when tested at 30 min, but significantly decreased freezing times 72 h later (p < 0.05; Fig. 3D). The hippocampus is critical for response to contextual cues (Anagnostaras et al., 2001). However, freezing in response to the tone cue alone (in a novel context) is amygdala dependent and hippocampus independent (Anagnostaras et al., 2001). Because there is a high concentration of GluT4 in the hippocampus, but little GluT4 is expressed in the amygdala, we investigated whether indinavir treatment would affect amygdala-dependent memory by assessing freezing in response to the tone cue (Fig. 3E). We found no significant difference in response to the tone cue (p = n.s.). Collectively, these data strongly support a specific role for GluT4 in mediating hippocampally dependent memory.
Two weeks inhibition of glucose transport through hippocampal GluT4 prevented clearance of extracellular glucose during spatial working memory testing
SA was used to assess spatial working memory. We used this task because it is hippocampally dependent and metabolically taxing: performance on this task causes a large dip in hippocampal extracellular glucose that limits performance (McNay et al., 2000, 2001; McNay and Cotero, 2010; Pearson-Leary and McNay, 2012), whereas exogenous glucose improves performance on this task and reverses the task-associated dip in ECF glucose (McNay et al., 2000, 2001; McNay and Cotero, 2010). Indinavir treatment enhanced SA performance: there was a significant main effect for dose (F(4,19) = 4.48, p = 0.01; Fig. 4A) and Bonferroni post hoc analyses showed that the highest dose of indinavir (180 ng/h) produced a significant enhancement in SA performance relative to vehicle (p < 0.05), although there were no significant post hoc differences between the indinavir groups. There was no significant difference in total arms entered (p = n.s.), suggesting that motor behaviors cannot account for behavioral differences (Fig. 4B).
Figure 4.

In vivo mD to measure markers of hippocampal metabolism while rats were engaged in a spatial working memory task. A, Two weeks of brain indinavir administration via intracerebroventricular Alzet minipump infusion led to a dose-dependent increase in spontaneous alternation behavior (n = 4–6 per group). B, Treatment did not affect total number of arms entered. C, Hippocampal glucose levels measured by in vivo mD showing no difference in hippocampal glucose levels after indinavir treatment. D, Indinavir prevented the task-associated dip in glucose that is well established to occur during spontaneous alternation testing (#p < 0.05 for comparison between vehicle and 180 ng/h indinavir at bin 3). E, Rats treated with vehicle or indinavir both showed significant increase in ECF lactate from baseline levels during SA testing (n = 9 per group for mD experiments). Data are presented as the mean ± SEM; *p < 0.05.
To determine whether indinavir's cognitive enhancing effects on SA performance affected the well established hippocampal extracellular glucose “dip” that's thought to represent utilization of glucose during SA, we measured hippocampal ECF glucose and lactate. Hippocampal ECF glucose levels showed a significant 12.4% dip in vehicle-treated rats during SA testing (Student's t = 3.02, p = 0.006; Fig. 4D), consistent with several previous reports (McNay et al., 2000; Newman et al., 2011; Pearson-Leary and McNay, 2012). Hippocampal ECF glucose levels did not change significantly during testing in indinavir-treated rats (p = n.s.). The difference between ECF glucose levels during SA testing was significantly different between vehicle- and indinavir-treated rats (Student's t = 3.05, p = 0.005). Because it is possible that long-term blockade of GluT4 caused hyperglycemia, as observed in insulin-resistant subjects, we examined ECF glucose levels at baseline between conditions. Such findings could indicate that the null effect for a dip in ECF glucose during maze testing was because there was enhanced glucose provision during SA testing; however, we found no significant difference in ECF glucose levels between vehicle- or indinavir-treated rats after correcting for probe recovery via zero net flux (p = n.s; Fig. 4C). Hippocampal ECF lactate levels increased during maze testing by ∼22% in both in both vehicle- and indinavir-treated rats relative to baseline hippocampal ECF lactate levels (vehicle: Student's t = 9.09, p = 0.0001; indinavir: Student's t = 9.19, p = 0.0001; Fig. 4E). These data are consistent with several other reports that ECF lactate levels rise dramatically in the hippocampus during behavioral testing (Newman et al., 2011; Suzuki et al., 2011). ECF lactate measured during memory testing likely reflects astrocytic export of lactate and the null effect of indinavir on lactate levels suggests a specific effect of treatment on neurons, which solely express GluT4 in the CNS (Suzuki et al., 2011).
Chronic brain blockade of GluT4 did not cause metabolic abnormalities or affect amyloid processing in the hippocampus
To determine whether the cognitive effects of indinavir were because of impaired peripheral metabolism or via induction of amyloid processing in the brain, we assessed changes in body composition, plasma glucose levels, and brain amyloidogenic processing of amyloid precursor protein (a marker of AD-like pathology; Huse and Doms, 2000). Prolonged inhibition of GluT4-mediated glucose uptake in peripheral tissues via indinavir or similar drugs is known to cause rapid insulin resistance (Ben-Romano et al., 2003; Hertel et al., 2004; Vyas et al., 2010; Hresko and Hruz, 2011). Because GluT4-mediated glucose uptake is a critical event in the canonical insulin signaling pathway linking insulin signaling to glucose metabolism (Robinson et al., 1992; Wallberg-Henriksson and Zierath, 2001; Thong et al., 2005; Rowland et al., 2011), we hypothesized that chronic blockade (i.e., >2 weeks) of brain GluT4 would increase amyloid processing in the hippocampus without inducing peripheral insulin resistance. Confirmation of this hypothesis would suggest that chronic blockade of brain GluT4 could be a novel model of brain insulin resistance.
No intracerebroventricular dose tested affected bodyweight gain or plasma glucose levels (p = n.s.; Fig. 5A,B). These data are in contrast to peripheral administration of indinivar, which induces insulin resistance and increases plasma glucose levels rapidly in rats (Hertel et al., 2004; Hruz et al., 2008; Vyas et al., 2010; Hresko and Hruz, 2011). Therefore, any observed effects of indinavir in this study were likely caused by direct action in the brain rather than being secondary consequences of peripheral insulin resistance.
Figure 5.

Measurements of peripheral metabolism and hippocampal AD-like pathology after central GluT4 inhibition. A, There was no change in bodyweight after 2 weeks of indinavir at any dose tested. B, There was no change in plasma glucose levels after 2 weeks of indinavir treatment (n = 5–7 per group). C, D, Representative Western blot images and graphs of total APP (C) and the intracellular C-terminal fragment of APP (D). There was no change in APP or C-terminal protein (n = 4–6 per group). E, ELISA data showing no change in Aβx-40 after indinavir treatment (n = 7–8 per group). Data are presented as the mean ± SEM; *p < 0.05.
To determine whether brain GluT4 blockade induces AD-like brain pathology, we measured hippocampal amyloid-β after indinavir administration. Along with tau phosphorylation, increased amyloid-β is a key measure of AD-like brain pathology (Hampel et al., 2010). Brain GluT4 blockade did not alter the levels of amyloid-β (x-40) in the hippocampus (Fig. 5E) or affect the levels of C-terminal fragment of amyloid precursor protein (APP) or total APP (p = n.s.; Fig. 5C,D). These data suggest that inhibition of GluT4-mediated glucose uptake in the hippocampus alone is insufficient to cause the abnormal increase in hippocampal amyloid-β observed in insulin resistance (Takeda et al., 2010; Biessels and Reagan, 2015).
Two weeks of brain GluT4 inhibition increased neuronal glucose transporter trafficking
Indinavir binds to the cytoplasmic glucose-binding domain of PM GluT4, thus impairing entry of glucose into cells through GluT4. In skeletal muscle, indinavir decreases GluT4 trafficking, causing a reduction in the ratio of PM to total GluT4 (Hertel et al., 2004; Hresko and Hruz, 2011). After 2 weeks of intracerebroventricular indinivar, we show an opposite response in the hippocampus: the ratio of PM to total GluT4 was increased (Student's t = 2.91, p = 0.01; Fig. 6A). This increase was accompanied by an increase in phosphorylation of Akt at serine-9 residues within hippocampal neurons (Student's t = 2.15 p = 0.05; Fig. 6C), which is critical for GluT4 trafficking (Summers and Birnbaum, 1997). These data suggest that intracerebroventricular indinavir treatment increased hippocampal trafficking of GluT4 to the PM via Akt-mediated signaling.
Figure 6.

GluT and PI3K activity in the hippocampus after 2 weeks of brain GluT4 inhibition (180 ng/h indinavir). A, Western blot demonstrating increased ratio of PM to total GluT4 in indinavir-treated rats. B, There was no effect of treatment on the GSV co-passenger IRAP. C, Ratio of phosphorylated Akt at serine 473 to total Akt was increased after indinavir treatment. D, Increase PM expression of GluT3 after indinavir treatment (n = 4–8 per group). Data are presented as the mean ± SEM; *p < 0.05.
We also examined the expression of IRAP, which colocalizes with GluT4 in GSVs, as a control to determine whether increased GluT4 was accompanied by increased net GSV activity and delivery of cargo components. As discussed earlier, GSVs transport several proteins involved in memory to the PM, such as IQGAP1, IGF2 receptors, and IRAP (Jedrychowski et al., 2010; Kandror and Pilch, 2011). Therefore, increased translocation of GSVs could potentially confound interpretation of our behavioral results if membrane localization of proteins other than GluT4 were altered. However, there was no effect of indinavir treatment on PM expression of IRAP (p = n.s.; Fig. 6A), so it is likely that the upregulation of GluT4 membrane expression observed here was specific to GluT4 delivery and not increased delivery of other GSV cargo proteins.
Finally, we examined the expression of the neuronal glucose transporter GluT3, which is expressed on neurons ubiquitously across the CNS and considered the canonical constitutive neuronal glucose transporter (McEwen and Reagan, 2004). Moreover, synaptic activity was shown to increase PM expression of GluT3 (Ferreira et al., 2011). Consistent with our memory data showing increased STM after indinavir treatment, which suggests increased synaptic activity, we also observed an increase in PM GluT3 expression (Student's t = 2.93, p = 0.03; Fig. 6D).
Two weeks of brain GluT4 inhibition increased hippocampal membrane localization of AMPA receptors
PM expression of AMPA GluR1 was increased significantly in the indinavir condition relative to vehicle (Student's t = 2.31, p = 0.05; Fig. 7A). There was no effect of treatment on PM expression of NMDA NR2B subunits (p = n.s.; Fig. 7A). These data suggest that indinavir treatment may affect memory processes in part via enhanced glutamatergic signaling at AMPA receptors in the hippocampus.
Figure 7.

Glutamate receptor expression in the hippocampus after 2 weeks of brain GluT4 inhibition. A, B, Ratio of PM to total AMPA GluR1 subunits were increased after indinavir treatment, but there was no change in the expression of NMDA NR2B subunits (n = 4–6 per group). Data are presented as the mean ± SEM; *p < 0.05.
BDNF levels were decreased after chronic GluT4-mediated glucose transport inhibition
BDNF increases GluT4 expression in skeletal muscle via PI3K activation (Suwa et al., 2010). Because BDNF is critical for LTM formation (Alonso et al., 2002a; Alonso et al., 2002b; Alonso et al., 2005) and the 2-week inhibition of GluT4 impaired such memory, we envisioned a potential role for glucose flux through GluT4 in the regulation of BDNF: inhibition of GluT4 might decrease BDNF expression and therefore impair LTM. Consistent with this hypothesis, there was decreased BDNF in the hippocampus of indinavir-treated rats (Student's t = 2.89 p = 0.018; Fig. 8), suggesting that decreased hippocampal BDNF might have mediated the deficits in LTM observed in these rats. Together with previous work showing that BDNF can regulate GluT4 expression (Suwa et al., 2010), our data suggest a bidirectional effect whereby GluT4 and BDNF may mutually regulate each other in the brain.
Figure 8.

BDNF expression in the hippocampus after 2 weeks of brain GluT4 inhibition. Indinivar treatment decreased the expression of hippocampal BDNF (n = 6 per group). Data are presented as the mean ± SEM; *p < 0.05.
Discussion
Because hippocampal memory processes are well established to be sensitive to glucose supply and several mechanisms involved in memory formation and consolidation increase GluT4 translocation and/or increase the intrinsic activity of GluT4 already present in the PM, we hypothesized that glucose flux through hippocampal GluT4 might modulate hippocampal memory processing. Both acute and chronic intracerebroventricular blockade of the glucose transporter GluT4 affected subsequent performance on hippocampally mediated (but not amygdala-mediated) memory tasks. These data are, we believe, the first to identify a role for GluT4 in hippocampally dependent memory. Based on the results of acute blockade of hippocampal GluT4, we conclude that GluT4 is critical for both acquisition and consolidation of memory.
This conclusion is consistent with our finding that testing on a hippocampally dependent memory task increased GluT4 translocation during acquisition and suggests that memory formation increases glucose flux through GluT4. It has been suggested that brain GluT4 acts as a mechanism to permit increased glucose flux when needed to support the metabolic demands of sustained neuronal activity during cognitively demanding tasks or memory formation. That is, hippocampal GluT4 might act as a “reserve” glucose supply channel in neurons, preventing neuroglycopenia during cognitive load (Kobayashi et al., 1996; Leloup et al., 1996; Vannucci et al., 1998). We showed previously that hippocampal memory tasks cause metabolic demands on the hippocampus that exceed the brain's ability to supply glucose under baseline conditions (McNay et al., 2000). Translocation of GluT4 may be a mechanism that permits increased metabolic support at times of high demand, such as during cognitive challenge, without causing constitutively increased hippocampal metabolism.
Unexpectedly, long-term blockade of brain GluT4 improved STM but impaired LTM. However, several postmortem protein measures are consistent with this pattern of behavioral change: we observed increases in both glutamate receptors and glucose transporters at the cell surface, which may support enhanced STM (Baddeley et al., 2000; Izquierdo and McGaugh, 2000; Barros et al., 2002; Bevilaqua et al., 2003; Sanderson et al., 2008; Schmidt et al., 2010) and we also found a decrease in hippocampal BDNF that has been identified as a key component of LTM (Alonso et al., 2002a; Alonso et al., 2002b). Further support for our results comes from consistent findings in the literature. Increased phosphorylation of AMPA GluR1 subunits and PM expression of GluR1 subunit containing AMPA receptors have been documented under stimulated conditions (Lee et al., 2010). Although we did not measure AMPA GluR1 phosphorylation, we did observe increased PM expression of AMPA GluR1 subunits, consistent with our suggestion of increased neural activity in indinavir-treated rats. AMPA GluR1 subunit deletion results in decreased STM, but increased LTM (Sanderson et al., 2009), a result that corresponds to the increased STM and impaired LTM seen here in the presence of upregulated AMPA GluR1 membrane expression. Therefore, chronic reduction in GluT4-mediated glucose uptake may have increased transient synaptic excitability by increasing membrane content of AMPA GluR1 subunits and GluT3 expression. This could feasibly allow increased information storage in the short term, but in the absence of appropriate signals for transition of information from STM to LTM (i.e., decreased BDNF), the memory is not transferred to LTM. Our data are consistent with a dual process model of memory in which hippocampal STM and LTM are mediated, at least in part, by separate processes (Izquierdo et al., 1999; Vianna et al., 2000).
An important finding is that long-term brain GluT4-mediated glucose transport inhibition did not appear to impair hippocampal insulin signaling or alter hippocampal amyloid-β levels. On the contrary, Western blot analyses revealed that inhibition of glucose transport through brain GluT4 increased expression of hippocampal GluT4 and GluT3 and increased phosphorylated Akt, suggesting increased insulin signaling or increased insulin-independent glucose utilization (Clodfelder-Miller et al., 2005; McNay et al., 2010). These findings stand in contrast to results obtained with systemic/peripheral GluT4 blockade, which causes insulin resistance rapidly (Kahn, 1996; Garvey et al., 1998; Brady and Saltiel, 1999; Zisman et al., 2000; Wallberg-Henriksson and Zierath, 2001). Intriguingly, physiologically relevant increases in brain glucose were shown previously to increase Akt phosphorylation (Clodfelder-Miller et al., 2005), which suggests that neuronal sensing of increased brain glucose (e.g., as we observed during spontaneous alternation testing) may activate Akt signaling to promote increased GluT4 trafficking. Collectively, these findings suggest that brain GluT4 is regulated distinctly from peripheral GluT4. One limitation to this work is that we did not measure other aspects of AD-like pathology such as tau phosphorylation, which has been shown to accompany, for example, insulin resistance.
In this study, intracerebroventricular blockade of GluT4-mediated glucose transport led to an increase in hippocampal membrane expression of neuronal glucose transporters GluT4 and GluT3 when brains were examined 24 h after memory testing. Other research has shown that knock-out of GluT3 in mice leads to a suggested adaptive increase in other glucose transporters, including GluT1 and GluT4 (Zhao et al., 2010). Mice with a brain GluT3 haploinsufficiency have no difference in brain glucose metabolism relative to their wild-type littermates, suggesting that counter-regulatory mechanisms compensate for loss of neuronal glucose transporter activity (Stuart et al., 2011). GluT4 was not examined, but the failure to find an effect on expression of other glucose transporters suggests the possibility that GluT4 might have been upregulated adaptively. Several polyphenols that can improve glucose tolerance can increase GluT expression (Cao et al., 2008; Manzano and Williamson, 2010); these results parallel the present data in that such polyphenols can block the glucose binding domain of glucose transporters, a mechanism similar to that of indinavir (Kwon et al., 2007; Manzano and Williamson, 2010). In the current study, memory testing led to an acute increase in hippocampal PM GluT4 expression. Therefore, a potential future study could investigate whether chronic indinavir treatment blocks this increase in PM GluT4 expression after memory testing. Together, these data suggest an adaptive capacity of the brain to upregulate glucose transporter levels in response to inhibition of glucose transport.
Our mD findings may suggest that inhibition of GluT4 can affect the uptake of glucose during cognitively demanding tasks, but does not affect lactate export or clearance. Others have used high-resolution [14C]-2DG imaging to show that increased glucose utilization during upregulated neuronal activity occurs primarily in synaptic areas (the neuropil) rather than the perikarya (Sokoloff, 1979, 1981; Sokoloff, 1984). Because GluT4 is expressed in the perikarya rather than in the neuropil (Leloup et al., 1996; Vannucci et al., 1998), which shows limited 2DG phosphorylation during hippocampally dependent memory acquisition, the increased neuronal glucose utilization seen during SA testing may not have been mediated entirely, or even primarily, through GluT4. Instead, it is likely that enhanced glucose utilization was at least in part because of increased GluT3 (Km = 1–2 mm), which has a higher affinity for glucose than GluT4 (Km = 3–5 mm) and is enriched in the neuropil (Wood and Trayhurn, 2003; Simpson et al., 2008). Moreover, GluT3 is not affected by indinavir (Hertel et al., 2004). Note that we have no reason to believe that indinavir prevents training-induced GluT4 membrane translocation; rather, this treatment blocks glucose uptake through GluT4, shown previously in vitro with regard to depolarization-induced glucose uptake in hippocampal slices (Fernando et al., 2008).
Our findings are consistent with prior studies that found enhanced memory as a consequence of prior reductions in glucose availability. Recurrent mild hypoglycemia caused improved hippocampal glucose supply together with increased spatial memory performance and induced increased paired-pulse inhibition and LTP, electrophysiological correlates of cellular memory processes (McNay et al., 2006a). Several glucose transport inhibitors can have similar, apparently paradoxical effects on STM. For instance, the flavonoid phlorizin can enhance memory even though it blocks glucose transport (Hall et al., 1992; Boccia et al., 1999).
Although associations among peripheral insulin resistance, hippocampal neurodegeneration, and memory loss are well established, the impact of insulin resistance specifically within the brain has been less studied. It is clear that organs affected by insulin resistance can include the brain, with consequences including decreased tyrosine phosphorylation of the insulin receptor and decreased Akt phosphorylation (Han and Li, 2010; Profenno et al., 2010; Correia et al., 2011; Duarte et al., 2012; de la Monte, 2012a, 2012b). Systemic insulin resistance causes impaired hippocampal glucose metabolism and memory deficits (McNay et al., 2010). Study of brain insulin resistance has acquired increased importance with the finding that Alzheimer's disease often correlates with impaired brain insulin signaling (Steen et al., 2005; de la Monte and Wands, 2008) and improved models of brain insulin resistance are still required. A common approach to inducing brain insulin resistance is to inject the GluT2 toxin streptozotocin, a drug that reliably induces insulin-dependent diabetes in the periphery, into the brain. However, because the brain has little to no GluT2 expression, the efficacy of this approach is likely limited (Leloup et al., 1994; Rayner et al., 1994; Roncero et al., 2004; Stranahan, 2015). Because GluT4 blockade is a reliable method of producing insulin resistance in the periphery, we hypothesized that intracerebroventricular GluT4 blockade would be an effective means of producing brain insulin resistance. Our data did not support this hypothesis; instead, they suggest that the interaction between GluT4-mediated glucose flux and insulin signaling within the brain may differ from that in the periphery. We speculate that this might be in part because insulin has several effects in the brain other than regulation of glucose transport, such as regulation of synaptic plasticity and neurotransmitter release (McNay and Recknagel, 2011). Moreover, as discussed, brain GluT4 is regulated by multiple molecules in addition to insulin (Kobayashi et al., 1996).
In summary, this study demonstrates the behavioral and molecular effects of central GluT4 blockade. Further research examining, for example, the impact of upregulated GluT4 function may further our understanding of the role that GluT4 plays in cognitive processes.
Footnotes
This work was supported by the National Institutes of Health (Grant R01 DK 077106 to E.C.M.) and the American Diabetes Association (Grant 7-12-BS-126 to E.C.M.). We thank Alvin George, Jessica Sage, Rachel Tobin, and Dennis Fitzgerald for assistance with data collection and Paul Hruz and Monique Heitmeier for HPLC analyses of indinavir samples.
The authors declare no competing financial interests.
References
- Alonso M, Vianna MR, Izquierdo I, Medina JH. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol. 2002a;22:663–674. doi: 10.1023/A:1021848706159. [DOI] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus. 2002b;12:551–560. doi: 10.1002/hipo.10035. [DOI] [PubMed] [Google Scholar]
- Alonso M, Bekinschtein P, Cammarota M, Vianna MR, Izquierdo I, Medina JH. Endogenous BDNF is required for long-term memory formation in the rat parietal cortex. Learn Mem. 2005;12:504–510. doi: 10.1101/lm.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alquier T, Leloup C, Lorsignol A, Pénicaud L. Translocable glucose transporters in the brain. Diabetes. 2006;55:S131–S138. doi: 10.2337/db06-S021. [DOI] [Google Scholar]
- Anagnostaras SG, Gale GD, Fanselow MS. Hippocampus and contextual fear conditioning: recent controversies and advances. Hippocampus. 2001;11:8–17. doi: 10.1002/1098-1063(2001)11:1<8::AID-HIPO1015>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Baddeley A, Bueno O, Cahill L, Fuster JM, Izquierdo I, McGaugh JL, Morris RG, Nadel L, Routtenberg A, Xavier G, Da Cunha C. The brain decade in debate: I. Neurobiology of learning and memory. Braz J Med Biol Res. 2000;33:993–1002. doi: 10.1590/S0100-879X2000000900002. [DOI] [PubMed] [Google Scholar]
- Bakirtzi K, Belfort G, Lopez-Coviella I, Kuruppu D, Cao L, Abel ED, Brownell AL, Kandror KV. Cerebellar neurons possess a vesicular compartment structurally and functionally similar to Glut4-storage vesicles from peripheral insulin-sensitive tissues. J Neurosci. 2009;29:5193–5201. doi: 10.1523/JNEUROSCI.0858-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Warburton EC. When is the hippocampus involved in recognition memory? J Neurosci. 2011;31:10721–10731. doi: 10.1523/JNEUROSCI.6413-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros DM, Pereira P, Medina JH, Izquierdo I. Modulation of working memory and of long- but not short-term memory by cholinergic mechanisms in the basolateral amygdala. Behav Pharmacol. 2002;13:163–167. doi: 10.1097/00008877-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Barros RP, Machado UF, Gustafsson JA. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med. 2006a;12:425–431. doi: 10.1016/j.molmed.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Barros RP, Machado UF, Warner M, Gustafsson JA. Muscle GLUT4 regulation by estrogen receptors ERbeta and ERalpha. Proc Natl Acad Sci U S A. 2006b;103:1605–1608. doi: 10.1073/pnas.0510391103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Romano R, Rudich A, Török D, Vanounou S, Riesenberg K, Schlaeffer F, Klip A, Bashan N. Agent and cell-type specificity in the induction of insulin resistance by HIV protease inhibitors. AIDS. 2003;17:23–32. doi: 10.1097/00002030-200301030-00005. [DOI] [PubMed] [Google Scholar]
- Bevilaqua LR, Kerr DS, Medina JH, Izquierdo I, Cammarota M. Inhibition of hippocampal Jun N-terminal kinase enhances short-term memory but blocks long-term memory formation and retrieval of an inhibitory avoidance task. Eur J Neurosci. 2003;17:897–902. doi: 10.1046/j.1460-9568.2003.02524.x. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Reagan LP. Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci. 2015;16:660–671. doi: 10.1038/nrn4019. [DOI] [PubMed] [Google Scholar]
- Boccia MM, Kopf SR, Baratti CM. Phlorizin, a competitive inhibitor of glucose transport, facilitates memory storage in mice. Neurobiol Learn Mem. 1999;71:104–112. doi: 10.1006/nlme.1998.3856. [DOI] [PubMed] [Google Scholar]
- Brady MJ, Saltiel AR. Closing in on the cause of insulin resistance and type 2 diabetes. J Clin Invest. 1999;104:675–676. doi: 10.1172/JCI8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brioni JD, Nagahara AH, McGaugh JL. Involvement of the amygdala GABAergic system in the modulation of memory storage. Brain Res. 1989;487:105–112. doi: 10.1016/0006-8993(89)90945-1. [DOI] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem. 2010;17:5–11. doi: 10.1101/lm.1650110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Urban JF, Jr, Anderson RA. Cinnamon polyphenol extract affects immune responses by regulating anti- and proinflammatory and glucose transporter gene expression in mouse macrophages. J Nutr. 2008;138:833–840. doi: 10.1093/jn/138.5.833. [DOI] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clodfelder-Miller B, De Sarno P, Zmijewska AA, Song L, Jope RS. Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J Biol Chem. 2005;280:39723–39731. doi: 10.1074/jbc.M508824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Insulin-resistant brain state: the culprit in sporadic Alzheimer's disease? Ageing Res Rev. 2011;10:264–273. doi: 10.1016/j.arr.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curzon P, Rustay NR, Browman KE. Cued and contextual fear conditioning for rodents. In: Buccafusco JJ, editor. Methods of behavior analysis in neuroscience. Ed 2. Boca Raton, FL: CRC; 2009. [PubMed] [Google Scholar]
- de la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res. 2012a;9:35–66. doi: 10.2174/156720512799015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer's disease. Drugs. 2012b;72:49–66. doi: 10.2165/11597760-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehvari N, Hutchinson DS, Nevzorova J, Dallner OS, Sato M, Kocan M, Merlin J, Evans BA, Summers RJ, Bengtsson T. beta(2)-Adrenoceptors increase translocation of GLUT4 via GPCR kinase sites in the receptor C-terminal tail. Br J Pharmacol. 2012;165:1442–1456. doi: 10.1111/j.1476-5381.2011.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. J Aging Res. 2012;2012:384017. doi: 10.1155/2012/384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando RN, Albiston AL, Chai SY. The insulin-regulated aminopeptidase IRAP is colocalised with GLUT4 in the mouse hippocampus–potential role in modulation of glucose uptake in neurones? Eur J Neurosci. 2008;28:588–598. doi: 10.1111/j.1460-9568.2008.06347.x. [DOI] [PubMed] [Google Scholar]
- Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31:1991–1999. doi: 10.1523/JNEUROSCI.1850-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Frausto SF, Guedea AL, Tronson NC, Jovasevic V, Leaderbrand K, Corcoran KA, Guzmán YF, Swanson GT, Radulovic J. IQGAP1 regulates NR2A signaling, spine density, and cognitive processes. J Neurosci. 2011;31:8533–8542. doi: 10.1523/JNEUROSCI.1300-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009;1296:35–45. doi: 10.1016/j.brainres.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JL, Reilly RT, Cottrill KL, Stone WS, Gold PE. Phlorizin enhancement of memory in rats and mice. Pharmacol Biochem Behav. 1992;41:295–299. doi: 10.1016/0091-3057(92)90101-K. [DOI] [PubMed] [Google Scholar]
- Hampel H, Blennow K, Shaw LM, Hoessler YC, Zetterberg H, Trojanowski JQ. Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol. 2010;45:30–40. doi: 10.1016/j.exger.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Li C. Linking type 2 diabetes and Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:6557–6558. doi: 10.1073/pnas.1002555107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, Struthers H, Horj CB, Hruz PW. A structural basis for the acute effects of HIV protease inhibitors on GLUT4 intrinsic activity. J Biol Chem. 2004;279:55147–55152. doi: 10.1074/jbc.M410826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer S. Memory function and brain glucose metabolism. Pharmacopsychiatry. 2003;36:S62–S67. doi: 10.1055/s-2003-40452. [DOI] [PubMed] [Google Scholar]
- Hresko RC, Hruz PW. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS One. 2011;6:e25237. doi: 10.1371/journal.pone.0025237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruz PW, Yan Q, Struthers H, Jay PY. HIV protease inhibitors that block GLUT4 precipitate acute, decompensated heart failure in a mouse model of dilated cardiomyopathy. FASEB J. 2008;22:2161–2167. doi: 10.1096/fj.07-102269. [DOI] [PubMed] [Google Scholar]
- Huse JT, Doms RW. Closing in on the amyloid cascade: recent insights into the cell biology of Alzheimer's disease. Mol Neurobiol. 2000;22:81–98. doi: 10.1385/MN:22:1-3:081. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, McGaugh JL. Behavioural pharmacology and its contribution to the molecular basis of memory consolidation. Behav Pharmacol. 2000;11:517–534. doi: 10.1097/00008877-200011000-00001. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Medina JH, Vianna MR, Izquierdo LA, Barros DM. Separate mechanisms for short- and long-term memory. Behav Brain Res. 1999;103:1–11. doi: 10.1016/S0166-4328(99)00036-4. [DOI] [PubMed] [Google Scholar]
- Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, Pilch PF. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285:104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB. Lilly lecture 1995. Glucose transport: pivotal step in insulin action. Diabetes. 1996;45:1644–1654. doi: 10.2337/diab.45.11.1644. [DOI] [PubMed] [Google Scholar]
- Kandror KV, Pilch PF. The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic. 2011;12:665–671. doi: 10.1111/j.1600-0854.2011.01175.x. [DOI] [PubMed] [Google Scholar]
- Kishi K, Hayashi H, Wang L, Kamohara S, Tamaoka K, Shimizu T, Ushikubi F, Narumiya S, Ebina Y. Gq-coupled receptors transmit the signal for GLUT4 translocation via an insulin-independent pathway. J Biol Chem. 1996;271:26561–26568. doi: 10.1074/jbc.271.43.26561. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Nikami H, Morimatsu M, Saito M. Expression and localization of insulin-regulatable glucose transporter (GLUT4) in rat brain. Neurosci Lett. 1996;213:103–106. doi: 10.1016/0304-3940(96)12845-7. [DOI] [PubMed] [Google Scholar]
- Kwon O, Eck P, Chen S, Corpe CP, Lee JH, Kruhlak M, Levine M. Inhibition of the intestinal glucose transporter GLUT2 by flavonoids. FASEB J. 2007;21:366–377. doi: 10.1096/fj.06-6620com. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup C, Arluison M, Lepetit N, Cartier N, Marfaing-Jallat P, Ferré P, Pénicaud L. Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res. 1994;638:221–226. doi: 10.1016/0006-8993(94)90653-X. [DOI] [PubMed] [Google Scholar]
- Leloup C, Arluison M, Kassis N, Lepetit N, Cartier N, Ferré P, Pénicaud L. Discrete brain areas express the insulin-responsive glucose transporter GLUT4. Brain Res Mol Brain Res. 1996;38:45–53. doi: 10.1016/0169-328X(95)00306-D. [DOI] [PubMed] [Google Scholar]
- Li P, Koike T, Qin B, Kubota M, Kawata Y, Jia YJ, Oshida Y. A high-fructose diet impairs Akt and PKCzeta phosphorylation and GLUT4 translocation in rat skeletal muscle. Horm Metab Res. 2008;40:528–532. doi: 10.1055/s-2008-1073162. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang P, Xu J, Desir GV. Voltage-gated potassium channel Kv1.3 regulates GLUT4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am J Physiol Cell Physiol. 2006;290:C345–C351. doi: 10.1152/ajpcell.00091.2005. [DOI] [PubMed] [Google Scholar]
- Manzano S, Williamson G. Polyphenols and phenolic acids from strawberry and apple decrease glucose uptake and transport by human intestinal Caco-2 cells. Mol Nutr Food Res. 2010;54:1773–1780. doi: 10.1002/mnfr.201000019. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol. 2004;490:13–24. doi: 10.1016/j.ejphar.2004.02.041. [DOI] [PubMed] [Google Scholar]
- McNay EC. The impact of recurrent hypoglycemia on cognitive function in aging. Neurobiol Aging. 2005;26:76–79. doi: 10.1016/j.neurobiolaging.2005.08.014. [DOI] [PubMed] [Google Scholar]
- McNay EC, Cotero VE. Mini-review: impact of recurrent hypoglycemia on cognitive and brain function. Physiol Behav. 2010;100:234–238. doi: 10.1016/j.physbeh.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Memory modulation across neural systems: intra-amygdala glucose reverses deficits caused by intraseptal morphine on a spatial task but not on an aversive task. J Neurosci. 1998;18:3853–3858. doi: 10.1523/JNEUROSCI.18-10-03853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem. 1999;72:785–790. doi: 10.1046/j.1471-4159.1999.720785.x. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Age-related differences in hippocampal extracellular fluid glucose concentration during behavioral testing and following systemic glucose administration. J Gerontol A Biol Sci Med Sci. 2001;56:B66–B71. doi: 10.1093/gerona/56.2.B66. [DOI] [PubMed] [Google Scholar]
- McNay EC, Gold PE. Food for thought: fluctuations in brain extracellular glucose provide insight into the mechanisms of memory modulation. Behav Cogn Neurosci Rev. 2002;1:264–280. doi: 10.1177/1534582302238337. [DOI] [PubMed] [Google Scholar]
- McNay EC, Recknagel AK. Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem. 2011;96:432–442. doi: 10.1016/j.nlm.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Sherwin RS. From artificial cerebro-spinal fluid (aCSF) to artificial extracellular fluid (aECF): microdialysis perfusate composition effects on in vivo brain ECF glucose measurements. J Neurosci Methods. 2004;132:35–43. doi: 10.1016/j.jneumeth.2003.08.014. [DOI] [PubMed] [Google Scholar]
- McNay EC, Fries TM, Gold PE. Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci U S A. 2000;97:2881–2885. doi: 10.1073/pnas.050583697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, McCarty RC, Gold PE. Fluctuations in brain glucose concentration during behavioral testing: dissociations between brain areas and between brain and blood. Neurobiol Learn Mem. 2001;75:325–337. doi: 10.1006/nlme.2000.3976. [DOI] [PubMed] [Google Scholar]
- McNay EC, Williamson A, McCrimmon RJ, Sherwin RS. Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes. 2006a;55:1088–1095. doi: 10.2337/diabetes.55.04.06.db05-1314. [DOI] [PubMed] [Google Scholar]
- McNay EC, Canal CE, Sherwin RS, Gold PE. Modulation of memory with septal injections of morphine and glucose: effects on extracellular glucose levels in the hippocampus. Physiol Behav. 2006b;87:298–303. doi: 10.1016/j.physbeh.2005.10.016. [DOI] [PubMed] [Google Scholar]
- McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. 2010;93:546–553. doi: 10.1016/j.nlm.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay EC, Sandusky LA, Pearson-Leary J. Hippocampal insulin microinjection and in vivo microdialysis during spatial memory testing. J Vis Exp. 2013;71:e4451. doi: 10.3791/4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora S, Kaliman P, Chillarón J, Testar X, Palacín M, Zorzano A. Insulin and insulin-like growth factor I (IGF-I) stimulate GLUT4 glucose transporter translocation in Xenopus oocytes. Biochem J. 1995;311:59–65. doi: 10.1042/bj3110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukwevho E, Kohn TA, Lang D, Nyatia E, Smith J, Ojuka EO. Caffeine induces hyperacetylation of histones at the MEF2 site on the Glut4 promoter and increases MEF2A binding to the site via a CaMK-dependent mechanism. Am J Physiol Endocrinol Metab. 2008;294:E582–E588. doi: 10.1152/ajpendo.00312.2007. [DOI] [PubMed] [Google Scholar]
- Newman LA, Korol DL, Gold PE. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One. 2011;6:e28427. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte LA, Yarasheski KE, Kawanaka K, Fisher J, Le N, Holloszy JO. The HIV protease inhibitor indinavir decreases insulin- and contraction-stimulated glucose transport in skeletal muscle. Diabetes. 2001;50:1397–1401. doi: 10.2337/diabetes.50.6.1397. [DOI] [PubMed] [Google Scholar]
- Ojuka EO, Goyaram V, Smith JA. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am J Physiol Endocrinol Metab. 2012;303:E322–E331. doi: 10.1152/ajpendo.00091.2012. [DOI] [PubMed] [Google Scholar]
- Pearson-Leary J, McNay EC. Intrahippocampal administration of amyloid-beta(1–42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J Alzheimers Dis. 2012;30:413–422. doi: 10.3233/JAD-2012-112192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Rayner DV, Thomas ME, Trayhurn P. Glucose transporters (GLUTs 1–4) and their mRNAs in regions of the rat brain: insulin-sensitive transporter expression in the cerebellum. Can J Physiol Pharmacol. 1994;72:476–479. doi: 10.1139/y94-069. [DOI] [PubMed] [Google Scholar]
- Ren H, Lu TY, McGraw TE, Accili D. Anorexia and impaired glucose metabolism in mice with hypothalamic ablation of Glut4 neurons. Diabetes. 2015;64:405–417. doi: 10.2337/db14-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Yan S, Zhang B, Lu TY, Arancio O, Accili D. Glut4 expression defines an insulin-sensitive hypothalamic neuronal population. Mol Metab. 2014;3:452–459. doi: 10.1016/j.molmet.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LJ, Pang S, Harris DS, Heuser J, James DE. Translocation of the glucose transporter (GLUT4) to the cell surface in permeabilized 3T3–L1 adipocytes: effects of ATP insulin, and GTP gamma S and localization of GLUT4 to clathrin lattices. J Cell Biol. 1992;117:1181–1196. doi: 10.1083/jcb.117.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncero I, Alvarez E, Chowen JA, Sanz C, Rábano A, Vázquez P, Blázquez E. Expression of glucose transporter isoform GLUT-2 and glucokinase genes in human brain. J Neurochem. 2004;88:1203–1210. doi: 10.1046/j.1471-4159.2003.02269.x. [DOI] [PubMed] [Google Scholar]
- Rowland AF, Fazakerley DJ, James DE. Mapping insulin/GLUT4 circuitry. Traffic. 2011;12:672–681. doi: 10.1111/j.1600-0854.2011.01178.x. [DOI] [PubMed] [Google Scholar]
- Sanderson DJ, Good MA, Seeburg PH, Sprengel R, Rawlins JN, Bannerman DM. The role of the GluR-A (GluR1) AMPA receptor subunit in learning and memory. Prog Brain Res. 2008;169:159–178. doi: 10.1016/S0079-6123(07)00009-X. [DOI] [PubMed] [Google Scholar]
- Sanderson DJ, Good MA, Skelton K, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM. Enhanced long-term and impaired short-term spatial memory in GluA1 AMPA receptor subunit knockout mice: evidence for a dual-process memory model. Learn Mem. 2009;16:379–386. doi: 10.1101/lm.1339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Trümbach D, Weber P, Wagner K, Scharf SH, Liebl C, Datson N, Namendorf C, Gerlach T, Kühne C, Uhr M, Deussing JM, Wurst W, Binder EB, Holsboer F, Müller MB. Individual stress vulnerability is predicted by short-term memory and AMPA receptor subunit ratio in the hippocampus. J Neurosci. 2010;30:16949–16958. doi: 10.1523/JNEUROSCI.4668-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron. 2007;55:786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab. 2008;295:E242–E253. doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Collins M, Grobler LA, Magee CJ, Ojuka EO. Exercise and CaMK activation both increase the binding of MEF2A to the Glut4 promoter in skeletal muscle in vivo. Am J Physiol Endocrinol Metab. 2007;292:E413–E420. doi: 10.1152/ajpendo.00142.2006. [DOI] [PubMed] [Google Scholar]
- Smith JA, Kohn TA, Chetty AK, Ojuka EO. CaMK activation during exercise is required for histone hyperacetylation and MEF2A binding at the MEF2 site on the Glut4 gene. Am J Physiol Endocrinol Metab. 2008;295:E698–E704. doi: 10.1152/ajpendo.00747.2007. [DOI] [PubMed] [Google Scholar]
- Sokoloff L. Mapping of local cerebral funcitonal activity by measurement of of local cerebral glucose utilization with (14) deoxyglucose. Brain. 1979;102:653–668. doi: 10.1093/brain/102.4.653. [DOI] [PubMed] [Google Scholar]
- Sokoloff L. Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. J Cereb Blood Flow Metab. 1981;1:7–36. doi: 10.1038/jcbfm.1981.4. [DOI] [PubMed] [Google Scholar]
- Sokoloff L. Modeling metabolic processes in the brain in vivo. Ann Neurol. 1984;15:S1–S11. doi: 10.1002/ana.410150703. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease: is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Stranahan AM. Models and mechanisms for hippocampal dysfunction in obesity and diabetes. Neuroscience. 2015;309:125–139. doi: 10.1016/j.neuroscience.2015.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart CA, Ross IR, Howell ME, McCurry MP, Wood TG, Ceci JD, Kennel SJ, Wall J. Brain glucose transporter (Glut3) haploinsufficiency does not impair mouse brain glucose uptake. Brain Res. 2011;1384:15–22. doi: 10.1016/j.brainres.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA, Birnbaum MJ. A role for the serine/threonine kinase, Akt, in insulin-stimulated glucose uptake. Biochem Soc Trans. 1997;25:981–988. doi: 10.1042/bst0250981. [DOI] [PubMed] [Google Scholar]
- Suwa M, Yamamoto KI, Nakano H, Sasaki H, Radak Z, Kumagai S. Brain-derived neurotrophic factor treatment increases the skeletal muscle glucose transporter 4 protein expression in mice. Physiol Res. 2010;59:619–623. doi: 10.33549/physiolres.931784. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai MV, Guruswamy S, Cao KT, Pessin JE, Olson AL. Myocyte enhancer factor 2 (MEF2)-binding site is required for GLUT4 gene expression in transgenic mice: regulation of MEF2 DNA binding activity in insulin-deficient diabetes. J Biol Chem. 1998;273:14285–14292. doi: 10.1074/jbc.273.23.14285. [DOI] [PubMed] [Google Scholar]
- Thong FS, Dugani CB, Klip A. Turning signals on and off: GLUT4 traffic in the insulin-signaling highway. Physiology (Bethesda) 2005;20:271–284. doi: 10.1152/physiol.00017.2005. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res. 1998;797:1–11. doi: 10.1016/S0006-8993(98)00103-6. [DOI] [PubMed] [Google Scholar]
- Vianna MR, Izquierdo LA, Barros DM, Walz R, Medina JH, Izquierdo I. Short- and long-term memory: differential involvement of neurotransmitter systems and signal transduction cascades. Anais da Academia Brasileira de Ciencias. 2000;72:353–364. doi: 10.1590/S0001-37652000000300009. [DOI] [PubMed] [Google Scholar]
- Vyas AK, Koster JC, Tzekov A, Hruz PW. Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J Biol Chem. 2010;285:36395–36400. doi: 10.1074/jbc.M110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg-Henriksson H, Zierath JR. GLUT4: a key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review) Mol Membr Biol. 2001;18:205–211. doi: 10.1080/09687680110072131. [DOI] [PubMed] [Google Scholar]
- Wijesekara N, Tung A, Thong F, Klip A. Muscle cell depolarization induces a gain in surface GLUT4 via reduced endocytosis independently of AMPK. Am J Physiol Endocrinol Metab. 2006;290:E1276–E1286. doi: 10.1152/ajpendo.00573.2005. [DOI] [PubMed] [Google Scholar]
- Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89:3–9. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci U S A. 2004;101:3112–3117. doi: 10.1073/pnas.0308450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Poirier LA, Nagy LE. Mobilization of GLUT-4 from intracellular vesicles by insulin and K(+) depolarization in cultured H9c2 myotubes. Am J Physiol. 1999;277:E259–E267. doi: 10.1152/ajpendo.1999.277.2.E259. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Yang JP, Wang GH, Xia Z, Duan SZ, Wu Y. Role of PKCzeta translocation in the development of type 2 diabetes in rats following continuous glucose infusion. Diabetes Metab Res Rev. 2010;26:59–70. doi: 10.1002/dmrr.1056. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Fung C, Shin D, Shin BC, Thamotharan S, Sankar R, Ehninger D, Silva A, Devaskar SU. Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol Psychiatry. 2010;15:286–299. doi: 10.1038/mp.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]