

Abstract
Rett syndrome (RTT) is caused by mutations in the gene encoding methyl-CpG binding protein 2 (MECP2), an epigenetic regulator of mRNA transcription. Here, we report a test of the hypothesis of shared pathophysiology of RTT and fragile X, another monogenic cause of autism and intellectual disability. In fragile X, the loss of the mRNA translational repressor FMRP leads to exaggerated protein synthesis downstream of metabotropic glutamate receptor 5 (mGluR5). We found that mGluR5- and protein-synthesis-dependent synaptic plasticity were similarly altered in area CA1 of Mecp2 KO mice. CA1 pyramidal cell-type-specific, genome-wide profiling of ribosome-bound mRNAs was performed in wild-type and Mecp2 KO hippocampal CA1 neurons to reveal the MeCP2-regulated “translatome.” We found significant overlap between ribosome-bound transcripts overexpressed in the Mecp2 KO and FMRP mRNA targets. These tended to encode long genes that were functionally related to either cytoskeleton organization or the development of neuronal connectivity. In the Fmr1 KO mouse, chronic treatment with mGluR5-negative allosteric modulators (NAMs) has been shown to ameliorate many mutant phenotypes by correcting excessive protein synthesis. In Mecp2 KO mice, we found that mGluR5 NAM treatment significantly reduced the level of overexpressed ribosome-associated transcripts, particularly those that were also FMRP targets. Some Rett phenotypes were also ameliorated by treatment, most notably hippocampal cell size and lifespan. Together, these results suggest a potential mechanistic link between MeCP2-mediated transcription regulation and mGluR5/FMRP-mediated protein translation regulation through coregulation of a subset of genes relevant to synaptic functions.
SIGNIFICANCE STATEMENT Altered regulation of synaptic protein synthesis has been hypothesized to contribute to the pathophysiology that underlies multiple forms of intellectual disability and autism spectrum disorder. Here, we show in a mouse model of Rett syndrome (Mecp2 KO) that metabotropic glutamate receptor 5 (mGluR5)- and protein-synthesis-dependent synaptic plasticity are abnormal in the hippocampus. We found that a subset of ribosome-bound mRNAs was aberrantly upregulated in hippocampal CA1 neurons of Mecp2 KO mice, that these significantly overlapped with FMRP direct targets and/or SFARI human autism genes, and that chronic treatment of Mecp2 KO mice with an mGluR5-negative allosteric modulator tunes down upregulated ribosome-bound mRNAs and partially improves mutant mice phenotypes.
Keywords: autism, fragile X, intellectual disability, metabotropic glutamate receptor, Rett syndrome, synaptic protein synthesis
Introduction
Accumulating evidence suggests that aberrant synaptic protein synthesis may contribute to the pathophysiology underlying autism spectrum disorder (ASD) and intellectual disability (ID) (Kelleher and Bear, 2008). Synaptic control of local protein synthesis can be assayed in animal models by the study of synaptic long-term depression (LTD) induced by activation of metabotropic glutamate receptor 5 (mGluR5) (Huber et al., 2000, Huber et al., 2001). Studies over the past 15 years have shown that protein-synthesis-dependent mGluR-LTD is altered in multiple mouse models of ASD and ID, including those for fragile X, tuberous sclerosis, CYFIP1 and SYNGAP1 haplo-insufficiency, and chromosome 16p11.2 microdeletion (Huber et al., 2002, Auerbach et al., 2011, Bozdagi et al., 2012, Barnes et al., 2015, Tian et al., 2015). These results support the notion that aberrant regulation of synaptic protein synthesis represents a point of convergence for many varied gene mutations that manifest behaviorally as ASD and ID.
Rett syndrome (RTT), affecting 1 in 10,000 females, is a severe developmental disorder caused by mutations in an X-chromosome gene encoding methyl-CpG binding protein 2 (MECP2). RTT patients have seemingly normal development for the first 6–18 months before entering the stage of regression. Major symptoms include ASD, deceleration of head growth, loss of speech and purposeful hand movements, gait abnormalities, and irregular breathing patterns. The disease is rare in males because hemizygous mutation of MECP2 leads to early lethality. Postmortem studies show that RTT is not a neurodegenerative disorder, but neurons in patients' brains are smaller in size and more densely packed (for review, see Chahrour and Zoghbi, 2007). Heterozygous female mice with Mecp2 mutation exhibit relatively subtle symptoms until 20 weeks or older, with varied symptom onset age (Samaco et al., 2013). In contrast, hemizygous Mecp2 KO male (Mecp2−/y) mice recapitulate many clinically relevant symptomatic features at an earlier age, including locomotor deficits, irregular breathing with apneas, reduced neuronal soma size, premature death, and severe learning and memory impairment. Therefore, male Mecp2 KO mice are often used as a functionally relevant disease model for revealing the underlying pathophysiology of RTT (Chen et al., 2001, Guy et al., 2001).
MeCP2 selectively recognizes and binds to methylated cytosines in the genome and acts as a major effector protein for this epigenetic DNA modification (Lewis et al., 1992, Skene et al., 2010, Guo et al., 2014, Gabel et al., 2015). Although overall gene expression appears to be only modestly dysregulated in the absence of MeCP2 (Tudor et al., 2002, Jordan et al., 2007, Chahrour et al., 2008, Ben-Shachar et al., 2009, Gabel et al., 2015), MeCP2 was found recently to preferentially repress the transcription of many long genes associated with relatively high levels of intragenic non-CpG methylation (Sugino et al., 2014, Chen et al., 2015, Gabel et al., 2015). Moreover, MeCP2 may also regulate microRNA expression and thereby exert an additional layer of transcription/translation regulation (Szulwach et al., 2010, Wu et al., 2010).
Because transcription and translation are in general tightly coupled to allocate cellular resources efficiently (Li et al., 2014), we set out to test the hypothesis that loss of MeCP2 as a transcription repressor may lead to downstream translational dysregulation of a cohort of mRNAs that are under tight translational control by an mGluR5/FMRP pathway. In the current study, using integrated approaches of electrophysiology, biochemistry, genomics, and behavioral analyses, we find that in early symptomatic Mecp2 KO mice, hippocampal mGluR-LTD is protein synthesis independent and protein translation is abnormal, with an increase in ribosome-associated mRNAs for long genes. Furthermore, we provide evidence that many MeCP2-repressed and FMRP-bound mRNAs that are aberrantly upregulated in Mecp2 KO may be tuned down by treatment with a negative allosteric modulator of mGluR5, which suggests a mechanistic link between MeCP2-mediated transcription repression and FMRP-mediated translation regulation. Finally, we demonstrate that pharmacological modulation of the mGluR5 signaling pathway partially ameliorates a subset of RTT phenotypes.
Materials and Methods
Animals.
Mecp2 KO mice (Mecp2tm1.1bird, 003890; The Jackson Laboratory) and WT littermates were maintained on a C57BL/6 background by breeding Mecp2−/+ females with WT males from Charles River Laboratories (strain code: 027). The Cck-bacTRAP (GM39) line was provided by Dr. Nathaniel Heintz of The Rockefeller University. In this mouse, EGFP fused to the ribosomal protein L10a was inserted in the bacterial artificial chromosome (BAC) in such a way that expression was driven by the cholecystokinin (Cck) promoter. All animal experimentation was done in accordance with National Institutes of Health and Massachusetts Institute of Technology guidelines. All experiments were performed on littermate WT and Mecp2 KO mice with the experimenter blinded to genotype.
Hippocampal slice physiology.
Experiments were done as described previously (Auerbach et al., 2011). Acute hippocampal slices (350 μm) were prepared using a vibroslicer from postnatal day 30 (P30) to P35 mice in ice-cold dissection buffer containing the following (in mm): NaCl 87, sucrose 75, KCl 2.5, NaH2PO4 1.25, NaHCO3 25, CaCl2 0.5, MgSO4 7, ascorbic acid 1.3, and d-glucose 10 (saturated with 95% O2/5% CO2). Immediately after slicing, the CA3 region was removed. Slices were recovered in artificial CSF (ACSF) containing the following (in mm): NaCl 124, KCl 5, NaH2PO4 1.23, NaHCO3 26, CaCl2 2, MgCl2 1, and d-glucose 10 (saturated with 95% O2/5% CO2) at 32.5°C for at least 3 h before recording. Field EPSPs (fEPSPs) were evoked by stimulation of the Schaffer collaterals and recorded from CA1 stratum radiatum. Chemical LTD was induced by applying S-3,5-dihydroxyphenylglycine (DHPG, 50 μm; Sigma-Aldrich) for 5 min (Huber et al., 2001). For experiments with cycloheximide treatment, slices were incubated in ACSF containing 60 μm cycloheximide (Sigma-Aldrich) throughout the recording. The initial slope of the fEPSP was used to assess synaptic strength. Data were normalized to the baseline response and are presented as mean ± SEM. LTD was calculated by comparing the average response 55–60 min after DHPG application with the average of the last 5 min of baseline. Waveforms shown are from all traces averaged from all animals at the time points indicated. Genotype and drug effect were analyzed using two-way ANOVA.
Immunoblotting.
Hippocampal slices were lysed in Laemmli buffer and equal amounts of protein were loaded and resolved in SDS-PAGE and transferred onto nitrocellulose membrane. Total protein levels were quantified by MemCode reversible protein stain (Pierce). The following antibodies were used: phospho-ERK (p-ERK1/2; Thr202/Tyr204), ERK1/2, phospho-Akt (p-AKT; Ser473), phosphorylated mammalian target of rapamycin (p-mTOR; Ser2448), mTOR, p-S6 (Ser235/236), S6 (all from Cell Signaling Technology), mGluR5 (Abcam), mGluR1 (BD Biosciences), and β-actin (Abcam). Bands were detected by chemiluminescence (ECL; GE Healthcare) or IR fluorescence (LI-COR). Quantifications were done with Quantity-One (Bio-Rad). The relative expression levels of each protein were determined by the intensity of target immunoblot bands normalized to the total protein level of each lane (MemCode intensity). A paired two-tailed t test was used to determine whether differences between genotypes were statistically significant at p < 0.05.
Metabolic labeling.
Protein synthesis measurements were done as described previously with paired WT and Mecp2 KO littermates (Osterweil et al., 2010). Briefly, 500 μm hippocampal slices were prepared with a Stoelting tissue slicer and recovered for 3 h in 32.5°C ACSF, incubated for 30 min with 25 μm actinomycin D (EMD Biosciences), and then transferred to ACSF with 10 μCi/ml 35SMet/Cys (PerkinElmer) for 30 min. Labeled slices were homogenized in protein lysis buffer (10 mm HEPES, pH 7.4, 2 mm EDTA, 2 mm EGTA, 1% TX100, and protease inhibitor; Roche) and unincorporated amino acids were removed by precipitating proteins in the homogenate with trichloroacetic acid. Isotope incorporation was measured with a scintillation counter (Beckman). Protein concentration was measured by DC Protein assay (Bio-Rad). Protein synthesis rate was calculated as counts per minute per microgram of protein and the reading was normalized to the ACSF 35SMet/Cys counts. A paired two-tailed t test was used to determine whether differences between genotypes were statistically significant at p < 0.05.
TRAP-seq and differential gene expression analysis.
Translating mRNAs were immunoprecipitated from acutely dissected hippocampal tissue and purified as described previously (Heiman et al., 2008, Heiman et al., 2014). RNA samples were analyzed with a Bioanalyzer (Agilent) to confirm RNA integrity (RIN >7). RNA concentrations were determined with the Quant-it Ribogreen RNA assay (Life Technologies). Purified RNA (1∼1.5 ng) was converted to cDNAs with Ovation RNA-seq V2 system (NuGEN). A total of 500 ng of cDNAs from each sample were fragmented to ∼200 bp using a Covaris S2 ultrasonicator. Fragmented cDNA (100 ng) was end-repaired, dA-tailed, and ligated to adaptors using the NEBNext Ultra DNA library prep kit (New England Biolabs). Sequencing libraries were prepared using the KAPA HiFi Hotstart Ready Mix PCR Kit (Kapa Biosystems) and NEBNext Multiplex Oligos for Illumina (Index Primers Set1 and 2). Library quality was examined with the Qubit dsDNA HS Assay Kit (Life Technologies) and Bioanalyzer using a High Sensitivity DNA kit (Agilent). Final concentrations of sequencing libraries were quantified by qPCR using KAPA Library Quantification Kit (Kapa Biosystems). TRAP-seq libraries were sequenced on a HiSeq 2500 or a NextSeq 500 Sequencer (Illumina). Sequencing reads (50 bp, single-end) were first mapped to the Mus musculus primary assembly reference genome (Gencode GRCm38/release M6) using STAR-2.4.2a. Uniquely mapped reads that overlapped with exons of known protein coding genes were counted using htseq-count python script. Differential expression was calculated using DESeq2 (Love et al., 2014) and p-values were adjusted for multiple testing using the Benjamini–Hochberg procedure (GEO accession number: GSE82068).
Gene set enrichment analysis (GSEA).
GSEA analysis was performed as described previously (Subramanian et al., 2005) using a modified GSEA.1.0 R script (implemented in R-3.1.3). Normalized enrichment score and statistical significance were estimated by comparison with a null model generated by permuting the gene labels randomly 1000 times. FMRP direct targets used in GSEA analysis were downloaded from the FMRP HITS-CLIP dataset (Darnell et al., 2011).
Gene ontology (GO) analysis.
GO analysis (see Fig. 3d) was performed using the DAVID version 6.7 bioinformatics resource (http://david.abcc.ncifcrf.gov/).
Figure 3.
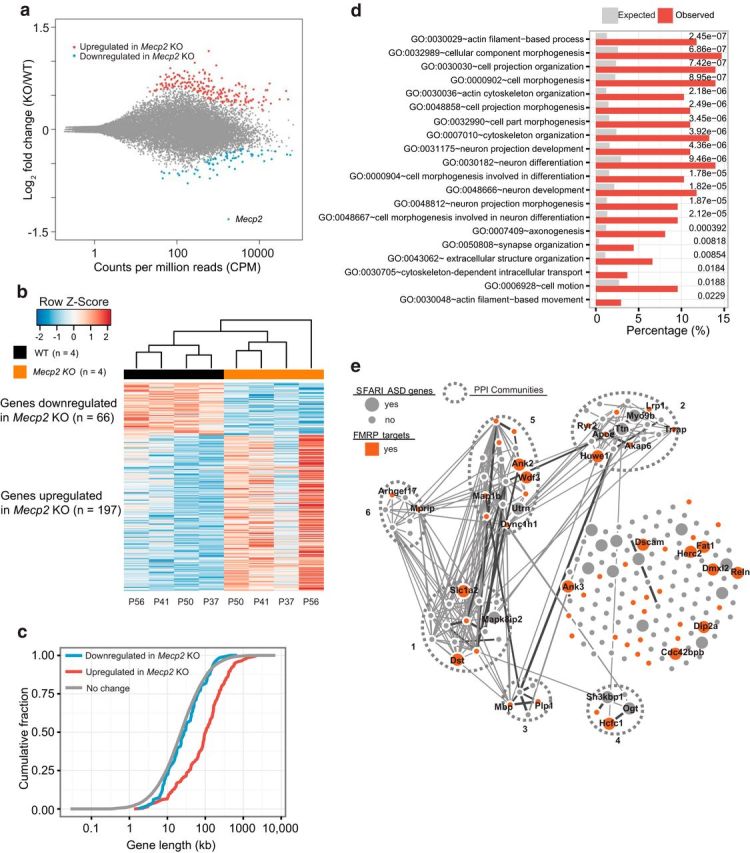
Analysis of MeCP2-regulated ribosome-associated mRNAs in hippocampal CA1 neurons. a, MA plot (DESeq2) showing log2-fold changes between Mecp2 KO/WT over the mean of normalized transcript counts. TRAP-seq analyzed translating mRNAs isolated from hippocampal CA1 neurons in WT Cck bacTRAP mice (n = 4) and Mecp2 KO Cck-bacTRAP mice (n = 4). Red dots represent transcripts that were significantly upregulated in Mecp2 KO (n = 197, false discovery rate [FDR] = 0.1) and blue dots represent transcripts that were significantly downregulated in Mecp2 KO (n = 66, FDR = 0.1). Gray dots represent transcripts not significantly altered. The data point corresponding to the Mecp2 mRNA is indicated. b, Heat map of transcripts that are significantly differentially expressed between WT and Mecp2 KO. Each column represents one biological replicate (animal) (FDR < 0.1) with age marked at the bottom. c, Cumulative distribution of gene lengths for genes upregulated in Mecp2 KO (red), genes downregulated in Mecp2 KO (blue), and all the unaltered genes detected in hippocampal CA1 neurons (gray). Two-sample KS test: upregulated versus no change, p = 4.6 × 10−34; downregulated versus no change, p = 0.21. d, Functional annotation analysis of genes upregulated in Mecp2 KO using DAVID Bioinformatics Resources 6.7 (National Institute of Allergy and Infectious Diseases–National Institutes of Health). The top 20 enriched GO terms with p < 0.05 (corrected for multiple comparisons by Benjamin adjustment) are shown with expected (gray bars) and observed (red bars) percentages of genome. e, PPI network analysis of 197 genes upregulated in Mecp2 KO generated by the GeNets algorithm using the InWeb PPI database (https://www.broadinstitute.org/genets). Curated and computationally derived PPIs are shown as lines. Line thickness reflects the level of confidence in the interaction. Sets of genes more connected with each other than to other groups of genes are defined as PPI communities and are enclosed by the ovals bounded by dashed lines. Genes without known PPI are arrayed to the right side. This analysis identified six distinct PPI communities within 197 MeCP2-repressed genes. Each community gene list is submitted to DAVID functional annotation tool. Community 1 is enriched for actin cytoskeleton and neuron projection. Community 5 is enriched for actin binding, cell junction, and synapse. In addition, genes that are identified as SFARI ASD genes (https://gene.sfari.org/autdb/HG_Home.do) are highlighted as larger nodes, whereas those overlapping with FMRP-bound targets are highlighted in red. Gene names are indicated for those that are identified as SFARI ASD genes and/ or FMRP targets.
Protein–protein network analyses.
Protein–protein interaction (PPI) network analysis was performed using the GeNets program (https://www.broadinstitute.org/genets) and the InWeb PPI database. A set of genes (nodes in the network) was grouped as a PPI community if they were more connected to one another than they were to other groups of genes. Curated and computationally derived PPIs were shown as lines. Each PPI interaction was assigned a credibility score between 0 and 1, where 1 s are “gold standard interactions,” indicated by thick black lines. The scores other than 1 were defined based on the number of citations and types of experiments that lend evidence toward the interaction. The connectivity p-value indicates whether the observed PPI network is significantly more connected than expected for a random gene set of same size. GSEA was performed using a Bonferroni-adjusted hypergeometric test and only the C2 set (4722 curated gene sets from BioCarta, KEGG, Reactome, PID, etc.) from the Molecular Signatures Database (MSigDB) was considered during this analysis. SFARI genes were downloaded from the SFARI human gene collection (https://gene.sfari.org/autdb/Welcome.do).
CTEP treatment.
Chronic CTEP treatment (0.4 mg/kg) was administered via subcutaneous injection (for mice younger than P30) or oral gavage (for mice older than P30) once every 48 h. This dose was chosen after a pilot study revealed that the dose of 2 mg/kg used in previous studies in fragile X (Michalon et al., 2012) and 16p11.2 microdeletion (Tian et al., 2015) mouse models caused significant weight loss in the Mecp2 mice.
Whole-body plethysmography.
Breathing was recorded in awake and unrestrained mice using whole-body plethysmography (EMKA Technologies) with a constant inflow of fresh air (0.8 L/min). Mice were habituated to the chamber for 20 min 1 d before being recording for up to 1.5 h. Raw data were exported and analyzed in Matlab (The MathWorks).
Immunohistochemistry and soma size analyses.
Mice were perfused with PBS, followed by 4% paraformaldehyde (PFA), and brains were sliced into 50-μm-thick sections in PBS with a vibratome (Leica). Brain sections were blocked and permeabilized for 1 h at room temperature in a blocking buffer of 1×PBS with 5% normal donkey serum and 0.16% Triton X-100. For immunostaining, sections were incubated with primary antibody overnight, washed 3 times with PBS, incubated with secondary antibody for 1.5 h, washed 3 times with PBS, and then mounted onto slides and coverslipped. The following antibodies were used for immunostaining: GFP (Abcam, rabbit polyclonal, 1/1500), NeuN (EMD Millipore, mouse monoclonal, 1/500). For Nissl staining, permeabilized brain sections were incubated with Neurotrace (Life Sciences, 1/100) for 30 min at room temperature, washed 3 times with PBS, and then mounted onto slides and coverslipped. Images were taken with a confocal microscope. Only neurons with clear nuclei were quantified and soma sizes were analyzed with Fiji (ImageJ) software by an experimenter blinded to genotype and treatment. Two sample Kolmogorov–Smirnov (KS) tests were used to determine whether the differences between each genotype/treatment (based on cell number) were statistically significant. Two-way ANOVA was used to determine whether the differences between genotype/treatment were statistically significant (based on averaged soma size per animal).
Open-field test.
Locomotor activity was measured with an open-field test as described previously (Crittenden et al., 2014). Mice were habituated to a test room with dim light for 15 min before being placed into 25 cm (W) × 25 cm (L) × 40 cm (H) arenas surrounded by Plexiglas walls with an activity monitoring system (TrueScan; Coulbourn Instruments). The sensor ring with infrared beams measured animal position every 100 ms and the software calculated the distance traveled in 5 min bins for a total time of 20 min per day for 2 consecutive days. Two-way ANOVA was used to determine whether the differences between genotype/treatment were statistically significant.
Inhibitory avoidance test.
Inhibitory avoidance test was performed as described previously (Dölen et al., 2007, Michalon et al., 2012). Mice were placed in the lit compartment of a two-chamber box for 90 s before the shutter separating the two compartments opened. At 0 h training time, once the animal entered the dark compartment, it received a single foot shock of 0.5 mA for 2 s. Mice then stayed in the dark compartment for 120 s before being returned to their home cages. Mice were placed back in the lit chamber at 24 and 48 h after conditioning to measure the latency to enter the dark compartment. They were enclosed in the dark compartment for 120 s without receiving any additional foot shock at 24 and 48 h time points. Two-way repeated-measures (RM) ANOVA was used to determine whether the differences between genotype/treatment were statistically significant.
Contextual fear conditioning.
Mice were fear conditioned to the training context with a single foot shock of 0.8 mA for 2 s as described previously (Auerbach et al., 2011). They were returned 24 h later either to the same context or to a novel context (varying distal cues, odor, lighting, and floor material). Conditioned fear response was assessed by measuring the percentage of time spent freezing during a test period of 3 min. Two-way ANOVA was used to determine whether the differences between genotype/treatment were statistically significant.
Elevated plus maze.
The elevated plus maze consisted of two open arms and two closed arms with an elevation of 30 cm from the ground. Each arm was 10 cm × 40 cm. Mice were placed in the center of an elevated plus maze facing an open arm and videotaped for 15 min. The total time spent on the open arms versus the close arms were recorded and calculated. Two-way ANOVA was used to determine whether the differences between genotype/treatment were statistically significant.
Marble-burying test.
The marble-burying test was performed as described previously (Deacon, 2006). Mice were placed and left for 30 min in a cage with ∼5 cm deep of bedding and 2 rows of marbles with regular spacing in between. The marbles that were buried (to 2/3 of their depth) or displaced from their original positions were counted. Two-way ANOVA was used to determine whether the differences between genotype/treatment were statistically significant.
Results
We first measured chemically induced mGluR-LTD at Schaffer collateral–CA1 synapses using hippocampal slices from P30–P35 mice as described previously (Huber et al., 2001). At this age, Mecp2 KO mice in our colony exhibited no hindlimb clasping, few tremor and mobility problems, and were considered to be “early symptomatic.” mGluR-LTD can be induced by brief application of the selective mGluR1/5 agonist DHPG or by repetitively stimulating Schaffer collateral axons using paired pulses (50 ms interval) at 1 Hz (PP-LFS). A previous study using a loss-of-function RTT model (Mecp2308/y mice) showed that DHPG-LTD magnitude was not altered, whereas PP-LFS-LTD was impaired (Moretti et al., 2006). The discrepancy is likely due to the fact that the PP-LFS protocol is dependent on presynaptic glutamate release to induce LTD and both Mecp2308/y mice and Mecp2 KO mice have presynaptic neurotransmission abnormalities (Moretti et al., 2006, Calfa et al., 2011; J.T. and M.F.B., unpublished observations). Because our interest was in postsynaptic regulation of protein synthesis, we reexamined the effects of DHPG in the presence and absence of cycloheximide (CHX), an mRNA translation inhibitor. We confirmed the finding that application of DHPG induces a similar magnitude of LTD in both WT and Mecp2 KO littermates (Fig. 1a). However, whereas mGluR-LTD was diminished in WT mice by application of CHX, in Mecp2 KO mice, mGluR-LTD was insensitive to CHX treatment (Fig. 1b). This phenotype is reminiscent of mGluR-LTD defects reported in mouse models of fragile X syndrome (Fmr1−/y), 16p11.2 microdeletion (16p11.2 df/+), and Syngap1 haploinsufficiency (Huber et al., 2002, Auerbach et al., 2011, Barnes et al., 2015, Tian et al., 2015), which has been interpreted as arising from increased basal synthesis of proteins that are normally rate limiting for LTD.
Figure 1.

Mecp2 KO mice exhibited dysregulated protein-synthesis-dependent mGluR-LTD in hippocampus. a, DHPG (50 μm; 5 min) induced comparable magnitudes of mGluR-LTD in hippocampal slices of P30∼P35 littermate WT (22.1 ± 1.6%, n = 10 mice) and Mecp2 KO (22.7 ± 1.4%, n = 11). b, The protein synthesis inhibitor CHX significantly reduced mGluR-LTD in WT (11.0 ± 4%, n = 10 mice), but did not alter mGluR-LTD in Mecp2 KO (20.9 ± 1.6%, n = 11). Statistics: 2-way ANOVA, interaction p = 0.031, genotype p = 0.021, treatment p = 0.006. Tukey's multiple-comparisons test WT vs WT+CHX **, KO vs KO+CHX ns (in this and subsequent figures, *p < 0.05; **p < 0.01; ***p < 0.001). c, Metabolic labeling of P30∼P35 hippocampal slices revealed a significant increase in basal protein synthesis in Mecp2 KO mice (KO: 113 ± 10%, n = 22 mice) compared with WT littermate (WT: 100 ± 8.4%, n = 22) (paired t test, p = 0.03). d, ERK1/2 phosphorylation were comparable between P30∼P35 WT and Mecp2 KO hippocampal slices stimulated with DHPG (50 μm, 5 min) (WT: 100 ± 4.6%, WT + DHPG: 122 ± 3.9%, KO: 95.7 ± 4.9%, KO + DHPG: 123.5 ± 4.2%, n = 17 animals for both WT and KO) (2-way ANOVA, treatment p < 0.0001, interaction and genotype n.s, Sidak's multiple-comparisons test WT vs WT + DHPG, p < 0.001, KO vs KO + DHPG, p < 0.0005). e, AKT phosphorylation was comparable between P30∼P35 WT and Mecp2 KO hippocampal slices stimulated with insulin (1 μm, 10 min) (WT: 100 ± 9.6%, WT + insulin: 200 ± 21%, KO: 108 ± 11.7%, KO + insulin: 209 ± 17.6%, n = 4 animals for both WT and KO) (2-way ANOVA, treatment p = 0.0009, interaction and genotype n.s., Sidak's multiple-comparisons test WT versus WT+DHPG, p < 0.05, KO vs KO+DHPG, p < 0.05). f, Normalized ratio of mGluR5 expression level to total protein amount between P30∼P35 WT and KO littermates was not significantly altered (WT: 1 ± 3.7%, n = 9; KO: 95 ± 4.0%, n = 9, 2-tailed paired t test p = 0.459). g, Normalized ratio of mGluR1 expression level to total protein amount between P30∼P35 WT and KO littermates was not significantly altered (WT: 100 ± 9.8%, n = 9; KO: 115.6 ± 15.6%, n = 9, 2-tailed paired t test p = 0.328).
To examine global protein translation directly, we measured bulk protein synthesis of hippocampal slices in the presence of the transcription inhibitor actinomycin D (Osterweil et al., 2010). Compared with yoked WT littermate controls, Mecp2 KO mice showed a small but significant increase in 35S-methionine/cysteine incorporation indicative of enhanced basal protein translation (Fig. 1c). However, we observed no difference in basal or evoked levels of p-ERK (stimulated by DHPG), and p-AKT (stimulated by insulin) in the mutant slices compared with those from WT littermates (Fig. 1d,e) and no differences in mGluR5 or mGluR1 in hippocampal tissue lysates (Fig. 1f,g). Collectively, these results indicate that the mGluR-ERK and AKT signaling pathways are unaffected in early symptomatic stage Mecp2 KO hippocampal slices and may not contribute to the elevated basal protein synthesis. However, we cannot exclude the possibility that signaling pathways may be altered in a cell-type-specific manner. Western blotting with hippocampal slices provides limited information toward that end.
Bulk protein synthesis measurements and candidate protein expression analysis (e.g., Western blotting of individual proteins) from whole hippocampus slices lack cell-type specificity and cannot determine the identities of dysregulated mRNAs without prior knowledge. Because the pyramidal neurons in CA1 are those displaying the mGluR-LTD phenotype, it is highly desirable to identify translational dysregulation specifically in these neurons in an unbiased manner. To this end, we used translating ribosome affinity purification followed by RNA-sequencing (TRAP-seq) to investigate the translatome in hippocampal CA1 neurons. Compared with standard RNA-seq analysis of specific cell populations isolated by fluorescence-activated cell sorting or laser dissection, TRAP-seq provides the unique advantage of cell-type specific profiling of mRNAs from intact undisturbed brain tissues (Doyle et al., 2008, Heiman et al., 2008, Sanz et al., 2009) and measures the levels of mRNAs that are associated with ribosomes. To analyze hippocampal CA1 neurons, we used a BAC transgenic mouse line with EGFP-L10a expression driven by a cholecystokinin (Cck) promoter. Cck is a neuropeptide that in hippocampus is widely expressed by pyramidal neurons in fields CA1–3 and by a specific class of inhibitory interneuron. However, in the Cck-bacTRAP line, hippocampal expression of L10a-EGFP is limited to pyramidal neurons in area CA1 (Fig. 2a; Doyle et al., 2008, http://genstat.org). We dissected the hippocampus from Cck-bacTRAP mice and affinity purified EGFP-L10a tagged ribosome-associated mRNAs by immunoprecipitation (IP). Compared with whole hippocampal lysate samples (input), IP samples showed a very distinctive enrichment of a subset of genes (e.g., Cck) and depletion of genes associated with glia (e.g., Gfap) and GABAergic interneurons (e.g., Gad2) (Fig. 2b). We further compared our results with the Cck-bacTRAP line with published datasets from TRAP lines that specifically label cerebellar granule neurons (Nd1), Bergmann glia (Sept4), and Purkinje neuron (Pcp2) (Mellén et al., 2012). Immunoprecipitated mRNAs from the Cck line expressed high levels of Slc17a7 (vesicular glutamate transporter, expressed only in glutamatergic neurons), but very low levels of Gad1 (glutamic acid decarboxylase, enriched in Purkinje cells) and Aldh1l1 (aldehyde dehydrogenase, a glial marker) (Fig. 2c). The robust enrichment of glutamatergic neuronal markers compared with non-neuronal and inhibitory neuronal markers demonstrates that the Cck-bacTRAP line is suitable to study translating mRNA dysregulation in hippocampal CA1 excitatory neurons.
Figure 2.

Characterization of Cck-bacTRAP line and immunoprecipitation of ribosome associated mRNAs from hippocampal CA1 neurons. a, Coronal section of a Cck-bacTRAP mouse brain stained for GFP and Nissl. Inset picture is an immunohistochemistry image of hippocampus with antibodies against GFP (cyan) and NeuN (magenta) showing colocalization in CA1. Scale bar, 100 μm. b, Volcano plot analysis revealed transcripts specifically enriched in Cck-bacTRAP IP samples versus input samples (whole hippocampus lysates). Red dots represent transcripts that were significantly enriched in IP samples (e.g., Cck) and blue dots represent transcripts that were significantly enriched in input samples (e.g., Gfap, Gad2). c, Quantification of expression levels of Slc17a7 (Vglut1, excitatory neuron marker), Gad1 (inhibitory neuron marker), and Aldh1l1 (glial marker) across different bacTRAP lines expressing in different cell types: Cck (hippocampal CA1, from current study), Nd1 (cerebellar granule neuron), Sept4 (cerebellar Bergmann glia), Pcp2 (cerebellar Purkinje cell); the latter three are all published datasets from Dr. Heintz's group (Mellén et al., 2012). FPKM, Fragments per kilobase of transcript per million mapped reads.
Next, we crossed the Mecp2+/− line with the Cck-bacTRAP line to obtain Mecp2+/y;Cck (WT) and Mecp2−/y;Cck (KO) littermates. Mecp2−/y;Cck mice exhibited the same behavioral phenotypes and premature death as Mecp2−/y mice. We profiled ribosome-bound mRNAs from four pairs of littermates (from P37–P60) and identified transcripts that showed consistent significant differential expression between Mecp2 KO mice and their littermate WT controls (Fig. 3a,b). Loss of MeCP2 caused dysregulation of a cohort of mRNAs (n = 263, false discovery rate < 0.1) across multiple biological replicates (Fig. 3b). The majority of the dysregulated transcripts were increased in the Mecp2 KO (197 genes, 74.9%); the remaining transcripts were downregulated (66 genes, 25.1%). This result indicates that, in hippocampal CA1 neurons, MeCP2 acts predominantly as a repressor of gene expression and suggests that the previously observed increased bulk protein synthesis might be explained by increased levels of mRNAs associated with translating ribosomes. Interestingly, aberrantly upregulated genes (n = 197, p = 4.6 × 10−34, KS test), but not downregulated genes (n = 66, p = 0.21, KS test), were significantly longer compared with unaltered genes detected in hippocampal CA1 neurons (Fig. 3c). Therefore, our hippocampal CA1 specific profiling data support the recent observations that MeCP2 represses long genes preferentially in neurons (Sugino et al., 2014, Gabel et al., 2015). GO analysis of genes upregulated in the KO (n = 197) indicates that they are significantly enriched for GO terms related to cytoskeleton organization and cellular/neuronal projection morphogenesis (Fig. 3d), which is consistent with previous GO analysis with commonly upregulated genes from multiple published microarray and RNA-sequencing datasets from various brain regions (Gabel et al., 2015). In contrast, GO analysis of genes downregulated in the KO (n = 66) did not reveal any significant biological processes or pathways. To visualize the known interactions among proteins encoded by upregulated genes, the data were analyzed using the GeNets algorithm (Fig. 3e). This analysis revealed clustering of affected genes into six distinct PPI communities, suggesting that some aberrantly upregulated genes in Mecp2 KO mice may participate in the assembly of macromolecular complexes that contribute synergistically to RTT pathology. Further comparison of the PPI network of MeCP2-repressed genes with the SFARI human autism gene database indicated that 32 of 197 genes upregulated in the KO have been identified as genes mutated in ASD patients (Fisher's exact test, p = 2.38 × 10−9, see Fig. 3e). Therefore, many MeCP2-repressed genes identified in area CA1 by TRAP-seq are independently implicated as risk alleles in ASDs.
Recent studies suggest that the RNA-binding protein FMRP preferentially stalls ribosomes on translating mRNAs of long length (Darnell et al., 2011, Gabel et al., 2015). We compared the genes upregulated in the Mecp2 KO (197 genes) with FMRP-bound mRNA targets (735 expressed in hippocampal CA1 neurons of the original 842 genes) and observed a significant overlap of 49 genes (24.9% of MeCP2 repressed genes, p = 1.1 × 10−22, Fisher's exact test; Fig. 4a; representative validations by qPCR in Fig. 4b). In contrast, only 7 genes downregulated in the KO are also FMRP targets (10.6%, p = 0.033; Fig. 4a). The significant overlap between genes upregulated in the Mecp2 KO and FMRP direct targets raises the possibility that MeCP2 and FMRP might regulate a common group of mRNAs at the transcriptional and translational levels, respectively. Among the 49 genes that are FMRP direct targets and upregulated in Mecp2 KO, 14 genes have been identified as ASD risk genes in the SFARI human autism gene database (Fisher's exact test, p = 3.14 × 10−9). Interestingly, whereas the FMRP-bound genes reported by Darnell et al. (2011) were significantly longer than non-FMRP target genes, the 49 MeCP2-repressed/FMRP-bound genes were even longer still (Fig. 4c).
Figure 4.
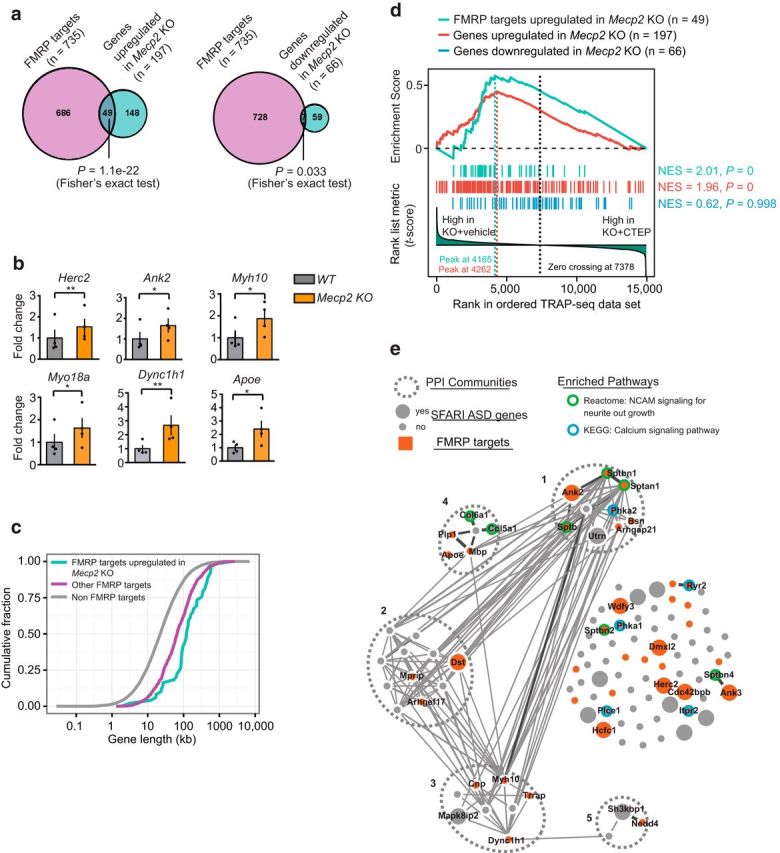
Genes upregulated in Mecp2 KO have a significant overlap with FMRP direct mRNA targets. The list of FMRP directly associated mRNAs from the mouse brain tissue is from published datasets (Darnell et al., 2011). a, Venn diagrams showing a significant overlap between FMRP direct mRNA targets and mRNAs upregulated in Mecp2 KO (Fisher's exact test, p = 1.1 × 10−22) and a fewer number of genes overlapping between FMRP direct targets and mRNAs downregulated in Mecp2 KO (Fisher's exact test p = 0.033). b, Validation of TRAP-seq identified target genes that were shared between genes upregulated in Mecp2 KO and FMRP direct mRNA targets by qPCR. Two-tailed paired t tests: Herc2 p = 0.0099 **, Ank2 p = 0.0296 *, Myh10 p = 0.0335 *, Apoe p = 0.0117 *, Myo18a p = 0.02 *, Dync1h1 p = 0.0039 **, n = 4 animals. c, Cumulative distribution of gene lengths for FMRP targets upregulated in Mecp2 KO (cyan), other FMRP direct targets (magenta), and the rest of mRNAs that are non-FMRP targets (gray). Two-sample KS test: FMRP targets upregulated in Mecp2 KO versus other FMRP targets p = 6.3 × 10−5, other FMRP targets versus non-FMRP targets p = 1.2 × 10−67. d, GSEA of genes upregulated in Mecp2 KO (red vertical bars, n = 197), upregulated in Mecp2 KO/FMRP-bound mRNAs (cyan vertical bars, n = 49), or genes downregulated in Mecp2 KO (blue vertical bars, n = 66 genes) on a ranked list of genes differentially expressed between vehicle-treated and CTEP-treated Mecp2 KO hippocampal CA1 neurons. Genes that are relatively downregulated in CTEP-treated Mecp2 KO are displayed on the left side of the plot. The red and cyan line in the upper part of the plot indicates the GSEA enrichment running scores for the list of genes upregulated or downregulated in Mecp2 KO, respectively. The vertical dash lines indicate the peaks corresponding to enrichment score (ES). e, PPI network analysis of a subset of genes upregulated in Mecp2 KO that are downregulated by the CTEP treatment (n = 108 leading edge genes). This analysis identified five distinct PPI communities. In addition, genes that are identified as SFARI ASD genes are highlighted as larger nodes, whereas those overlapping with FMRP-bound targets are highlighted in red. Genes significantly enriched in two functionally relevant pathways are labeled with green outer-circle (NCAM signaling for neurite out growth, adjusted-p = 5.8 × 10−7) or blue outer-circle (Calcium signaling pathway, adjusted-p = 1.7 × 10−2), respectively. Gene names are indicated for those that are enriched in the two pathways.
Studies of the Fmr1−/y mouse have shown that the increased protein synthesis caused by loss of FMRP in the hippocampus is downstream of a signaling pathway recruited by activation of mGluR5 (Osterweil et al., 2010, Bhakar et al., 2012). Chronic inhibition of mGluR5 with a negative allosteric modulator can correct a wide array of disease phenotypes in Fmr1 KO mice (Michalon et al., 2012). Because a significant portion of genes upregulated in Mecp2 KO are FMRP targets, we wondered whether they also might be subjected to regulation by the mGluR5 signaling pathway. We therefore treated Mecp2 KO mice with either vehicle or an mGluR5-negative allosteric modulator (NAM), 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP) from P30 to P60 and investigated the molecular changes in Mecp2 KO (n = 3 animals for KO treated with CTEP, n = 2 animals for KO treated with vehicle) hippocampal CA1 neurons by TRAP-seq. Using GSEA (Mootha et al., 2003, Subramanian et al., 2005), we ranked genes according to their differential expression between vehicle- versus CTEP-treated conditions. Interestingly, both the 49 shared MeCP2-repressed/FMRP targets and the total set of 197 genes upregulated in the Mecp2 KO showed a significant shift toward the left side (vehicle-treated) of the ranked gene list, indicating that CTEP treatment tends to reduce their expression levels in Mecp2 KO. In contrast, the 66 genes downregulated in Mecp2 KO as a gene set showed a random distribution (Fig. 4d). These analyses indicate that MeCP2-repressed genes, particularly those genes regulated by both MeCP2 and FMRP, might be specifically subject to regulation by CTEP treatment.
PPI analysis of the genes that responded most to CTEP treatment (108 leading edge genes, which are those on the left side of the peak of enrichment score, indicated by red dashed vertical lines in Fig. 4d) identified five distinct PPI communities, of which community 1 is enriched for actin binding, calmodulin binding, and cytoskeleton binding genes and community 2 is enriched for actin cytoskeleton organization (DAVID GO analysis of biological processes). Additional pathway enrichment analysis (using the Reactome and KEGG pathway database) indicated that these genes were enriched in two pathways: calcium signaling and NCAM signaling for neurite out growth (Fig. 4e). For example, Ryr2 and Itpr2 are both involved in internal calcium release regulation upon mGluR activation. Downregulation of both with CTEP treatment may therefore tune down aberrant calcium signaling. Together, these results suggest that CTEP may have potential to ameliorate Mecp2 KO symptoms phenotypically by modulating the expression of a subset of MeCP2-repressed genes.
We next investigated the effect of chronic CTEP treatment on a battery of RTT phenotypes. One of the neurostructural hallmarks of RTT mice highly relevant to cellular health is reduced neuronal soma size, which had been reproducibly observed across multiple loss-of-function Mecp2 mutant mouse lines and cellular RTT models (Taneja et al., 2009, Goffin et al., 2012, Yazdani et al., 2012, Li et al., 2013, Wang et al., 2013). Consistent with the observation that many cytoskeleton genes were aberrantly upregulated in Mecp2 KO and tuned down by CTEP treatment, we found that chronic treatment with CTEP significantly ameliorated the CA1 soma size deficit observed in Mecp2 KO mice at P45–P60 (Fig. 5a,b). This effect appears to constitute a “rescue” of the mutant phenotype because treatment was without any significant effect in the WT.
Figure 5.
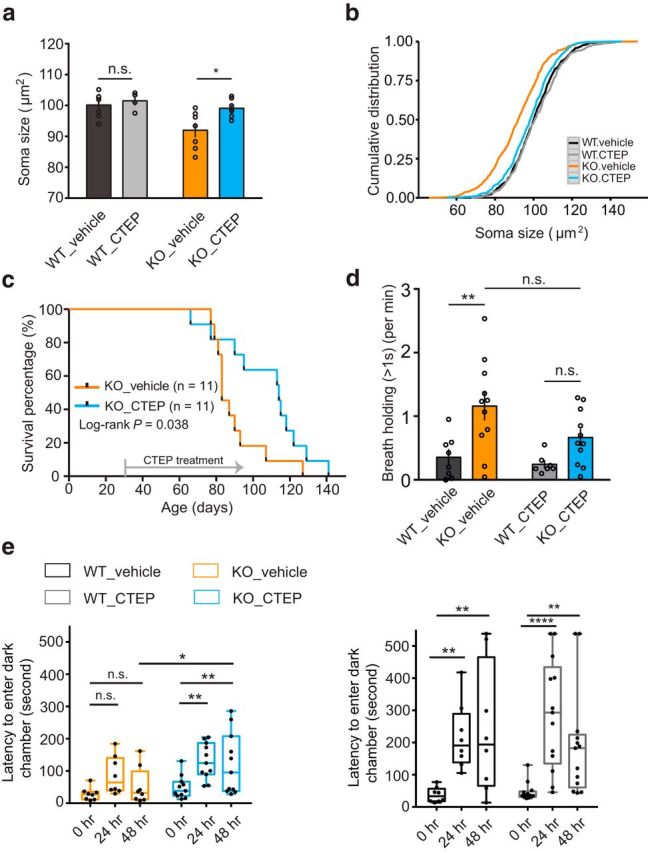
Chronic CTEP treatments of Mecp2 KO mice ameliorates hippocampal reduced soma size, prolongs lifespan, partially reduces long breath holdings, and ameliorates IA deficits. a, CTEP treatment ameliorates reduced neuronal soma size shown by averaged soma size by animals (WT_vehicle 100.17 ± 1.52 n = 7, WT_CTEP 101.51 ± 1.48 n = 4, KO_vehicle 91.99 ± 2.3 n = 7, KO_CTEP 99.11 ± 1.09 n = 7, 2-way ANOVA, interaction p = 0.123, drug treatment p = 0.028, genotype p = 0.008, Sidak's multiple-comparisons test WT_vehicle vs WT_CTEP n.s., KO_vehicle vs KO_CTEP *). b, CTEP treatment ameliorates reduced neuronal soma size in hippocampal CA1 neurons shown in cumulative distribution by cell numbers [KS test: WT_CTEP (n = 322 cells) vs WT_vehicle (n = 500 cells) p = 0.346, KO_vehicle vs WT_vehicle p = 0, KO_vehicle (n = 419 cells) vs KO_CTEP (n = 483 cells) p = 9.08 × 10−12, KO_CTEP vs WT_vehicle p = 0.042]. c, Kaplan–Meier survival curve showed that Mecp2 KO mice with treated CTEP survived longer than littermates treated with vehicle (KO_vehicle: n = 11 median survival is 83 d, KO_CTEP: n = 11, median survival is 113 d. Log-rank test, p = 0.038). d, Whole-body plethysmography analyses showed that CTEP treatment may partially reduce long breath holdings (>1 s) in Mecp2 KO mice (WT_vehicle 0.355 ± 0.118, n = 8; WT_CTEP 0.244 ± 0.056, n = 7; KO_vehicle 1.159 ± 0.218, n = 11; KO_CTEP 0.663 ± 0.129, n = 11; 2-way ANOVA, interaction p = 0.251, treatment p = 0.0748, genotype p = 0.0008, Tukey's multiple-comparisons test WT_vehicle vs KO_vehicle **, KO_vehicle vs WT_CTEP **, WT_CTEP vs KO_CTEP n.s., WT_vehicle vs KO_CTEP ns, WT_vehicle vs WT_CTEP n.s., KO_vehicle vs KO_CTEP n.s.). e, CTEP treatment ameliorated IA deficits in Mecp2 KO mice. CTEP treatment did not alter acquisition of IA in WT mice (WT_vehicle: 0 h 37.26 ± 8.26 n = 8, 24 h 217.36 ± 36.32 n = 8; 48 h 237.95 ± 71.17; WT_CTEP: 0 h 46.45 ± 7.92, 24 h 288.48 ± 47.71, 48 h 193.61 ± 46.36, n = 13, 2-way RM-ANOVA, interaction p = 0.286, treatment p = 0.794, time p < 0.0001; Sidak's multiple-comparisons test: 0 h vs 24 h **, 0 h vs 48 h **, 24 h vs 48 h ns, WT_vehicle n = 8; 0 h vs 24 h ****, 0 h vs 48 h **, 24 h vs 48 h n.s., WT_CTEP n = 13). CTEP treatment ameliorated acquisition deficits in Mecp2 KO mice (KO_vehicle: 0 h 28.59 ± 7.04, 24 h 86.6 ± 20.62, 48 h 52.21 ± 20.00 n = 8; KO_CTEP: 0 h 49.16 ± 10.73, 24 h 130.22 ± 16.80, 48 h 121.29 ± 28.81, n = 11; 2-way RM-ANOVA, interaction p = 0.383, treatment p = 0.036, time p = 0.001; Sidak's multiple-comparisons test: KO_vehicle n = 8, 0 h vs 24 h n.s., 0 h vs 48 h n.s., 24 h vs 48 h n.s.; KO_CTEP n = 11, 0 h vs 24 h **, 0 h vs 48 h **, 24 h vs 48 h n.s.; KO_vehicle vs KO_CTEP 48 h *).
Chronic treatment with low-dose CTEP starting at P30 also prolonged the lifespan of RTT mice significantly (Fig. 5c). Mecp2 KO mice have severe cardiorespiratory deficits, with irregular breath patterns and breath holding, recapitulating human patients symptoms and likely contributing to premature mortality (Katz et al., 2009, Tarquinio et al., 2015). Therefore, we evaluated the breathing patterns of awake, freely moving mice at a symptomatic age (∼P45-P60) with whole-body plethysmography. Analysis of the interbreath intervals revealed that CTEP treatment partially reduced long breath holdings in the Mecp2 KO (>1 s) compared with vehicle-treated KO mice (Fig. 5d), potentially contributing to the improvement in lifespan.
Next, we tested the mice in a hippocampus-dependent behavioral assay, inhibitory avoidance (IA), which had been shown to reveal cognitive deficits in mouse models of FX and 16p11.2 microdeletion (Dölen et al., 2007, Tian et al., 2015) and requires proper protein synthesis (Quevedo et al., 1999). The IA training apparatus comprises a box with two chambers separated by a door. One chamber is brightly lit and the other dark. Mice are placed in the lit chamber before the door is opened to allow entry into the dark side. Upon entry to the dark side, the animals receive a foot shock. They later demonstrate memory for this experience by avoiding the dark side when given the opportunity to enter. Memory acquisition and strength are estimated by the latency to enter the dark chamber at 24 and 48 h after training. Mecp2 KO mice and WT littermates show comparable sensitivity to the foot shock as measured by the amount of wild running 2 s after the shock. However, the mutants display a severe cognitive deficit, with no significant increase in latency when tested 24 or 48 h later (Fig. 5e), even when tested at P30, before symptom onset. CTEP treatment from P16–P30 may have modestly improved cognitive performance because significant increases in latency were observed at 24 and 48 h, although this improvement fell far short of performance in WT mice (Fig. 5e).
Assays of several other RTT phenotypes failed to reveal an effect of CTEP treatment. Weekly manual scoring of body weight, tremor, hindlimb clasping, and mobility on an open bench showed no significant improvement (Fig. 6a,b). Open-field tests showed that, regardless of drug treatment, Mecp2 KO mice traveled less distance, particularly in the first 5 min of the test session (Fig. 6c,d). They also spent less time in the center versus the margin of the arena, indicating possible decreased locomotor activity or increased anxiety. However, Mecp2 KO mice explored less on the second day compared with the first day in the same open arena, indicating normal habituation to the arena. We further tested anxiety using the elevated plus maze. Interestingly, Mecp2 KO mice spent more time on the open arm than the closed arm regardless of drug treatment, which is usually interpreted as a reduced level of anxiety (Fig. 6e). In the marble-burying assay, CTEP treatment had a small and statistically nonsignificant amelioration effect on the numbers of marbles that had been displaced from their original positions, although the numbers of marbles that were completely buried did not increase significantly (Fig. 6f). Furthermore, CTEP treatment had little effect in contextual fear conditioning tests in which Mecp2 KO mice also had been shown to have severe deficits (Fig. 6g). Together, these behavioral analyses indicate that the beneficial effects of CTEP treatment were restricted to a narrow range of phenotypes, possibly reflecting regional variations in the brain pathophysiology responsible for the diverse manifestations of the disease. Unlike fragile X, excessive consequences of mGluR5 activation are not a thread that connects a wide array of phenotypes in RTT.
Figure 6.
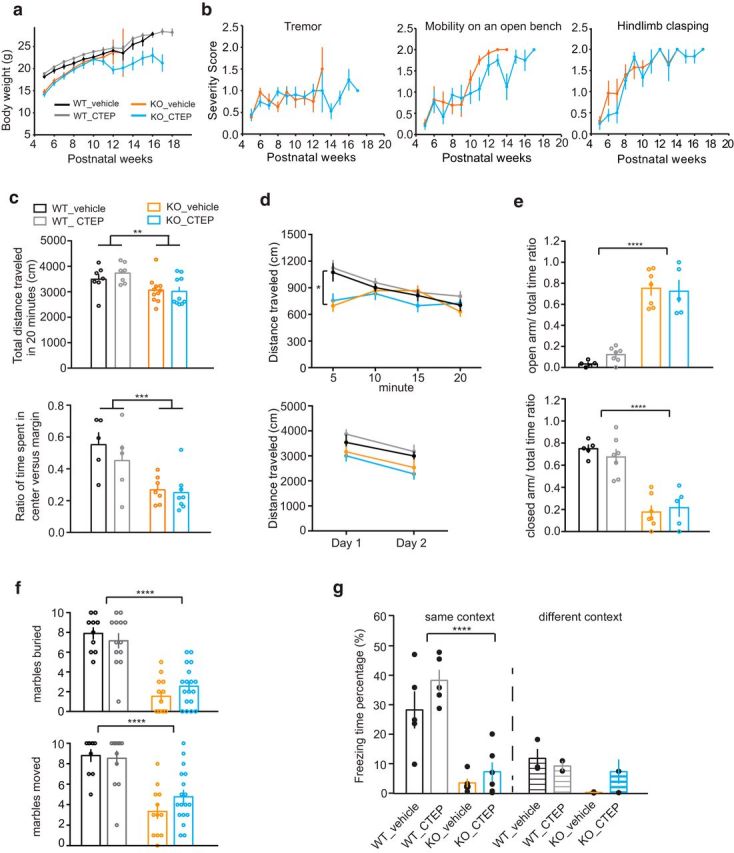
CTEP treatment of Mecp2 KO mice does not improve body weight, tremor, hindlimb clasping, mobility, or anxiety deficits. a, b, CTEP treatment does not improve body weight, tremor, hindlimb clasping, or mobility on an open bench (severity score: 0 equals WT, 1 is partially impaired, 2 is severely impaired). c, Open-field test showing that Mecp2 KO mice traveled less than WT littermates in a 20 min window regardless of vehicle or CTEP treatment (2-way ANOVA, genotype p = 0.0025, treatment and interaction ns, WT_V n = 7, WT_CTEP n = 7, KO_V n = 11, KO_CTEP n = 10). The same open-field test also showed that Mecp2 KO mice spent less time in the center versus margin of the test arena compared with WT littermates regardless of vehicle or CTEP treatment (2-way ANOVA, genotype p = 0.0006, treatment and interaction not significant, WT_V n = 5, WT_CTEP n = 5, KO_V n = 7, KO_CTEP n = 8). d, Open-field test on the first day divided into bins of 5 min showed that, whereas WT littermate ran the most distance during the first 5 min and then gradually reduced exploration, Mecp2 KO mice did not reach peak exploration until 10 min later (2-way RM-ANOVA, WT_V vs KO_V: interaction p = 0.0004, time p < 0.0001, genotype p = 0.103; KO_V vs KO_CTEP: interaction p = 0.0591, time p = 0.0093, treatment p = 0.8284). Open-field test on 2 consecutive days showed that both WT and KO mice regardless of drug treatment habituated to the environment (2-way RM-ANOVA, WT_V vs KO_V: interaction p = 0.776, time p = 0.003, genotype p = 0.22; KO_V vs KO_D: interaction p = 0.737, time p = 0.0003, treatment p = 0.477). e, Regardless of CTEP treatment, compared with WT, Mecp2 KO mice spent more time in the open arm than in the closed arm in the elevated plus maze (WT_vehicle_open arm: 0.033 ± 0.013 n = 5; WT_CTEP_open arm: 0.121 ± 0.027 n = 7; KO_vehicle_open arm: 0.752 ± 0.065 n = 7; KO_CTEP_open arm: 0.725 ± 0.102 n = 5; WT_vehicle_closed arm: 0.749 ± 0.034 n = 5; WT_CTEP_closed arm: 0.675 ± 0.067 n = 7; KO_vehicle_closed arm: 0.177 ± 0.056 n = 7; KO_CTEP_closed arm: 0.216 ± 0.078 n = 5, 2-way ANOVA, genotype p < 0.0001). f, Mecp2 KO mice were impaired in a marble-burying test and CTEP treatment did not significantly improve the displacement of marbles (2-way ANOVA, only genotype is significant for both marbles that are moved and marbles that are displaced, p < 0.0001). g, WT mice regardless of CTEP treatment showed robust freezing behavior in the same context but little freezing in a different context (WT_vehicle_same context: 28.26 ± 6.3% n = 5; WT_CTEP_same conext: 38.26 ± 3.64% n = 5; WT_vehicle_different context: 11.84 ± 3.18% n = 3; WT_CTEP_different context: 9.23 ± 1.72% n = 2). Mecp2 KO mice regardless of CTEP treatment showed little freezing behavior in either the same or different context (KO_vehicle_same context: 3.48 ± 1.44% n = 5; KO_CTEP_same context: 7.29 ± 3.18% n = 6; KO_vehicle_different context: 0.25 ± 0.15% n = 2; KO_CTEP_different context: 7.34 ± 4.12% n = 3). Comparison of WT and KO in the same context condition: 2-way ANOVA, interaction p = 0.45, treatment p = 0.099, genotype p < 0.0001.
Discussion
Dysregulated hippocampal mGluR-LTD and protein synthesis have been demonstrated in several mouse lines carrying gene mutations that cause human ASD and ID, including the models for FXS, TSC, and 16p11.2 microdeletion (Huber et al., 2002, Auerbach et al., 2011, Tian et al., 2015). In the current study, we demonstrate that protein-synthesis-dependent mGluR-LTD is also affected in a mouse model of RTT. Although hippocampal mGluR-LTD per se may not be a core mechanism for the expression of ASD or ID, it has proven to be a very sensitive assay to study postsynaptic protein translation regulation. Together, these LTD studies support the hypothesis that altered translation regulation may be a shared consequence of diverse gene mutations that manifest in humans as ASD and intellectual disability. Because the dependence of mGluR-LTD on protein synthesis varies with age in mouse and rat, whether various mGluR-LTD phenotypes in ASD mouse models reflect neuronal maturation deficits needs further investigation.
In addition to bulk protein synthesis analyses, here we analyzed mRNAs associated with ribosomes specifically in hippocampal CA1 neurons in an unbiased manner for the first time. Compared with WT littermates, the majority of altered mRNAs were upregulated in Mecp2 KO mice, consistent with an increase in active translation. It is likely that the increased bulk protein synthesis is a downstream effect of loss of transcription repression in the absence of MeCP2, preferentially on long genes. It is both interesting and reassuring to observe that our TRAP-seq results from hippocampal CA1 neurons, revealing robust upregulation of long genes, are remarkably consistent with the findings from multiple gene expression datasets using various crude brain tissue lysates (Gabel et al., 2015). Our data support the conclusion that the derepression of long genes occurs specifically in excitatory hippocampal neurons in RTT mice. This underscores the power of studying cell-type-specific translating mRNAs, which bypasses the difficulty of deducing neuronal specific signals from mixed cell populations from brain tissue.
The significant overlap between MeCP2-repressed genes and FMRP direct targets suggests that MeCP2 and FMRP might regulate a common group of mRNAs at the transcriptional and translational levels, respectively. Functional annotations of MeCP2-repressed/FMRP direct target genes revealed multiple genes involved in cytoskeleton organization and cell morphogenesis. For example, Myh10, Myo5a, Myo18a, Dync1h1, Kif1a, Ank2, and Ank3 play key roles in activities such as transport of synaptic vesicles, spine morphology, and dynamics. Importantly, a significant part of MeCP2-repressed/FMRP direct target genes belong to SFARI genes that have direct implications in ASDs (e.g., Ank2, Ank3, Dscam, Dst, Herc2, Reln, and Wdyf3, etc.). Signaling via mGluR5 has been implicated in the pathophysiology of fragile X and a number of disorders that manifest as ASD (Wenger et al., 2016). We find that the majority of MeCP2-repressed genes (both FMRP targets and non-FMRP targets) as a gene set may be tuned down by chronic CTEP treatment, establishing a potential mechanistic link between MeCP2-dependent transcription repression and mGluR-dependent translation regulation. Further investigation is needed to address the biological functions of those individual target genes.
Our TRAP-seq result provides a snapshot of mRNAs actively engaged with ribosomes and presumably undergoing active translation, likely reflecting the transcriptional level regulation by MeCP2. The data support the conclusion that MeCP2 represses transcription preferentially on a group of long genes in hippocampal CA1 neurons and offers an explanation for the slight increase in basal translation observed in hippocampal slices from the KO. The TRAP-seq result by itself, however, cannot be taken as indicative of translational efficiency because it does not provide quantitative ribosome occupancy information as ribosome profiling does in cultured cells, which requires much larger input materials (Li et al., 2014). Furthermore, protein synthesis regulation is tightly regulated by neural activity (Ebert and Greenberg, 2013) and, in addition to the increased protein synthesis observed here, the hippocampus from Mecp2 KO mice shows increased network excitability (Calfa et al., 2011, McLeod et al., 2013). In contrast, hESC-derived MeCP2 mutant neurons in culture show a global downregulation of transcription and translation and hypoactivity (Li et al., 2013). Because Mecp2 KO mice display increased or decreased activity level among different brain regions as measured by the immediate early gene c-fos (Kron et al., 2012), it is likely that protein synthesis level may also exhibit brain region variation.
Recent work reported that the mTOR/AKT signaling pathway was impaired in Mecp2 KO mice as well as in cultured human embryonic stem cell-derived RTT neurons (Ricciardi et al., 2011, Li et al., 2013). Insulin-like growth factor 1 (IGF-1) was reported to alleviate a variety of symptoms in Mecp2 KO mice by enhancing the mTOR/AKT pathway (Tropea et al., 2009, Castro et al., 2014). Although we did not observe downregulation of the AKT signaling pathway in our Mecp2 mouse colony, it is interesting to note that increasing mTOR signaling can ameliorate excessive mGluR5-dependent protein synthesis in fragile X (Auerbach et al., 2011, Bhakar et al., 2012) and could function similarly in RTT.
Excessive protein synthesis in fragile X is also corrected by chronic inhibition of mGluR5 using CTEP (Michalon et al., 2012). In the Mecp2 KO mice, we found that chronic low-dose CTEP treatment prolongs lifespan, mildly ameliorates IA deficits, and rescues the reduced soma size of hippocampal CA1 neurons. Further work needs to address the possible effects of CTEP treatment beyond the hippocampus; for example, the cardiorespiratory system, which has more direct relevance to general health. However, the limited effect of CTEP treatment on most of the other behavioral and cognitive aspects of the RTT mouse model highlights the challenge of identification and correction of the widespread core deficits of RTT.
Footnotes
This work was supported by the Howard Hughes Medical Institute, the Simons Foundation (Grant SFARI 275050 to M.F.B.). J.T. received a postdoctoral fellowship from Rett Syndrome Research Trust. H.W. received postdoctoral fellowships from the Jane Coffin Childs Memorial Fund for Medical Research, and the National Human Genome Research Institute (Grant K99HG007982). Y.Z. is an investigator of the Howard Hughes Medical Institute. We thank Nathaniel Heintz and Myriam Heiman for assistance with the TRAP experiments and Arnold Heynen, Robert Komorowski, Rujie Yao, Alana H. Peters, Tyrone Lee, Rodrigo Garcia, Erik Sklar, and Suzanne Meagher for technical and administrative assistance.
M.F.B. is coinventor on a patent for a method to treat autistic disorder with inhibitors of signaling by mGluR5. The remaining authors declare no competing financial interests.
References
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SA, Wijetunge LS, Jackson AD, Katsanevaki D, Osterweil EK, Komiyama NH, Grant SG, Bear MF, Nägerl UV, Kind PC, Wyllie DJ. Convergence of hippocampal pathophysiology in Syngap+/− and Fmr1-/y mice. J Neurosci. 2015;35:15073–15081. doi: 10.1523/JNEUROSCI.1087-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet. 2009;18:2431–2442. doi: 10.1093/hmg/ddp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dölen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Dorr N, Pilorge M, Takahashi N, Buxbaum JD. Haploinsufficiency of Cyfip1 produces fragile X-like phenotypes in mice. PLoS One. 2012;7:e42422. doi: 10.1371/journal.pone.0042422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfa G, Hablitz JJ, Pozzo-Miller L. Network hyperexcitability in hippocampal slices from Mecp2 mutant mice revealed by voltage-sensitive dye imaging. J Neurophysiol. 2011;105:1768–1784. doi: 10.1152/jn.00800.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J, Garcia RI, Kwok S, Banerjee A, Petravicz J, Woodson J, Mellios N, Tropea D, Sur M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2014;111:9941–9946. doi: 10.1073/pnas.1311685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, Zoghbi HY. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci U S A. 2015;112:5509–5514. doi: 10.1073/pnas.1505909112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Lacey CJ, Lee T, Bowden HA, Graybiel AM. Severe drug-induced repetitive behaviors and striatal overexpression of VAChT in ChAT-ChR2-EYFP BAC transgenic mice. Front Neural Circuits. 2014;8:57. doi: 10.3389/fncir.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM. Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat Protoc. 2006;1:122–124. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Allen M, Zhang L, Amorim M, Wang IT, Reyes AR, Mercado-Berton A, Ong C, Cohen S, Hu L, Blendy JA, Carlson GC, Siegel SJ, Greenberg ME, Zhou Z. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat Neurosci. 2012;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, Zhu H, Chang Q, Gao Y, Ming GL, Song H. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP) Nat Protoc. 2014;9:1282–1291. doi: 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan C, Li HH, Kwan HC, Francke U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med Genet. 2007;8:36. doi: 10.1186/1471-2350-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DM, Dutschmann M, Ramirez JM, Hilaire G. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168:101–108. doi: 10.1016/j.resp.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, Katz DM. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 2012;32:13860–13872. doi: 10.1523/JNEUROSCI.2159-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-O. [DOI] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, Kwok SM, Feldman DA, Bateup HS, Gao Q, Hockemeyer D, Mitalipova M, Lewis CA, Vander Heiden MG, Sur M, Young RA, Jaenisch R. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13:446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod F, Ganley R, Williams L, Selfridge J, Bird A, Cobb SR. Reduced seizure threshold and altered network oscillatory properties in a mouse model of Rett syndrome. Neuroscience. 2013;231:195–205. doi: 10.1016/j.neuroscience.2012.11.058. [DOI] [PubMed] [Google Scholar]
- Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo J, Vianna MR, Roesler R, de-Paris F, Izquierdo I, Rose SP. Two time windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem. 1999;6:600–607. doi: 10.1101/lm.6.6.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, Pizzorusso T, Giustetto M, Broccoli V. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- Samaco RC, McGraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2(+/-) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet. 2013;22:96–109. doi: 10.1093/hmg/dds406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, Nelson SB. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci. 2014;34:12877–12883. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, Santistevan NJ, Li W, Zhao X, Jin P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarquinio DC, Hou W, Neul JL, Kaufmann WE, Glaze DG, Motil KJ, Skinner SA, Lee HS, Percy AK. The Changing Face of Survival in Rett Syndrome and MECP2-Related Disorders. Pediatr Neurol. 2015;53:402–411. doi: 10.1016/j.pediatrneurol.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci. 2015;18:182–184. doi: 10.1038/nn.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IT, Reyes AR, Zhou Z. Neuronal morphology in MeCP2 mouse models is intrinsically variable and depends on age, cell type, and Mecp2 mutation. Neurobiol Dis. 2013;58:3–12. doi: 10.1016/j.nbd.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger TL, Kao C, McDonald-McGinn DM, Zackai EH, Bailey A, Schultz RT, Morrow BE, Emanuel BS, Hakonarson H. The role of mGluR copy number variation in genetic and environmental forms of syndromic autism spectrum disorder. Sci Rep. 2016;6:19372. doi: 10.1038/srep19372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Tao J, Chen PJ, Shahab A, Ge W, Hart RP, Ruan X, Ruan Y, Sun YE. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18161–18166. doi: 10.1073/pnas.1005595107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani M, Deogracias R, Guy J, Poot RA, Bird A, Barde YA. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells. 2012;30:2128–2139. doi: 10.1002/stem.1180. [DOI] [PubMed] [Google Scholar]