

Abstract
Background:
Decoy receptor 3 (DcR3) binds to Fas ligand (FasL) and inhibits FasL-induced apoptosis. The receptor is overexpressed in hepatocellular carcinoma (HCC), and it is associated with the growth and metastatic spread of tumors. DcR3 holds promises as a new target for the treatment of HCC, but little is known regarding the molecular mechanisms underlying the oncogenic properties of DcR3. The present work, therefore, examined the role of DcR3 in regulating the growth and invasive property of liver cancer cell HepG2.
Methods:
HepG2 cells were stably transfected with lentivirus-based short hairpin RNA vector targeting DcR3. After the knockdown of DcR3 was confirmed, cell proliferation, clone formation, ability of migrating across transwell membrane, and wound healing were assessed in vitro. Matrix metalloproteinase-9 (MMP 9) and vascular epithelial growth factor (VEGF)-C and D expressions of the DcR3 knockdown were also studied. Comparisons between multiple groups were done using one-way analysis of variance (ANOVA), while pairwise comparisons were performed using Student's t test. P < 0.05 was regarded statistically significant.
Results:
DcR3 was overexpressed in HepG2 compared to other HCC cell lines and normal hepatocyte Lo-2. Stable knockdown of DcR3 slowed down the growth of HepG2 (P < 0.05) and reduced the number of clones formed by 50% compared to those without DcR3 knockdown (P < 0.05). The knockdown also reduced the migration of HepG2 across transwell matrix membrane by five folds compared to the control (P < 0.05) and suppressed the closure of scratch wound (P < 0.05). In addition, the messenger RNA levels of MMP 9, VEGF-C, and VEGF-D were significantly suppressed by DcR3 knockdown by 90% when compared with the mock control (P < 0.05).
Conclusions:
Loss of DcR3 impaired the growth and invasive property of HCC cell line of HepG2. Targeting DcR3 may be a potential therapeutic approach for the treatment of HCC.
Keywords: Decoy Receptor 3, Hepatocellular Carcinoma, Matrix Metalloproteinase 9, Neoplasm Metastasis, Vascular Endothelial Growth Factor C, Vascular Endothelial Growth Factor D
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies, ranking the 3rd leading cause of cancer-related deaths worldwide.[1] Its incidence has risen significantly in the past decades, partly due to the increasing rate of hepatitis virus infection.[2,3,4,5] Early HCC can be treated by surgical resection, radiofrequency ablation, transarterial chemoembolization, liver transplantation, and chemotherapy. However, because of the asymptomatic nature of HCC, at the first presentation in clinic, many patients are at advanced stages and are not eligible for most of the treatment options. Prognosis of advanced HCC is very poor, and the 5-year survival rate can be <5%.[6] It is imperative to nominate new therapeutic targets of HCC for new drug development. Decoy receptor 3 (DcR3 or TNFRSF6B) was first discovered by Pitti et al. in 1998.[7] It binds to Fas ligand (FasL) and inhibits FasL-induced apoptosis. Recent studies reported that expression of the DcR3 was minimal in normal tissues, but it was highly expressed in various types of tumors such as lung cancer,[7,8] esophageal cancer,[7] gastric cancer,[7] colon cancer,[7] colorectal cancer,[8] pancreatic cancer,[8] HCC,[8,9,10] kidney cancer,[11] breast cancer,[8,12] ovarian cancer,[13] and skin cancer.[14] The gene was found amplified in about half of the primary lung and colon tumors studied.[7] In HCC, about 60% of the tumor tissues had a positive expression of DcR3.[8,9,10] Expressions of the gene were not detected in adjacent normal tissues. A report also indicated that expression of DcR3 was correlated with tumor size, clinical stages, tumor invasiveness, and metastasis of HCC.[8] It has been suggested that tumor cells might escape from tumor necrosis factor receptor mediated-apoptosis by expressing DcR3, which blocks FasL function.[15,16] Knocking down DcR3 in colon cancer cell line SW780 suppressed the metastatic ability of the cells with concomitant reductions in matrix metalloproteinase-9 (MMP 9), and vascular epithelial growth factor (VEGF)-C and D.[17] Despite these interesting findings, the molecular mechanism underlying the oncogenic property of DcR3 in liver cancer remains to be fully deciphered. In this context, we used lentivirus-based short hairpin RNA (shRNA) vector to silence DcR3 expression in HepG2 cells and then investigate its effect on the growth and invasiveness of the cells.
Methods
Cell lines and reagents
Human metastatic HCC cell lines such as MHCC97H and MHCC97L were bought from the cell bank of Zhongshan Hospital, Fudan University (Shanghai, China), while human primary HCC cell lines such as Huh751, HepG2, PLC, and human embryonic liver cell Lo-2 were purchased from Cancer Hospital of Chinese Academy of Medical Sciences (Beijing, China). Rabbit DcR3 polyclonal antibody, rabbit monoclonal antibodies against VEGF-C, VEGF-D, MMP9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and horseradish peroxidase-conjugated secondary antibody (rabbit) were all obtained from Abcam (Cambridge, UK).
Lentivirus-mediated stable knockdown of decoy receptor 3 in HepG2
There were two groups of HepG2 receiving lentivirus transfection. To perform lentivirus transfection, HepG2 cells were seeded onto 96-well plates in a cell density of 3 × 103 cells/well. One group was transfected with lentivirus harboring Lv-DcR3-EGFP-shRNA, which was a shRNA plasmid targeting DcR3. Another group was transfected with lentivirus harboring a control plasmid Lv-DcR3-EGFP-shRNA. All lentivirus transfections were done in the presence of 5 mg/L polybrene. After culture with complete medium for 96 h, the transfected HepG2 cells were examined for green fluorescent protein (GFP) expression under an inverted fluorescence microscope. The number of GFP-positive cells was then used to calculate the transfection efficiency. Successfully transfected HepG2 cells were then selected by puromycin (1 mg/L) for 2 weeks. Stable knockdown of DcR3 in the selected HepG2 cells was confirmed using real-time polymerase chain reaction (RT-PCR) and Western blotting.
In all in vitro assays, HepG2 cell group stably transfected with control plasmid and DcR3-targeting shRNA was designated as negative control (NC) and DcR3-siRNA, respectively. HepG2 receiving no lentivirus transfection was also included as blank control group (CON).
Real-time polymerase chain reaction
Total RNAs were harvested from HCC cell lines using TRIzol (Invitrogen, Carlsbad, CA, USA). First-strand complementary DNA was synthesized from total RNAs using Prime Script RT Reagent Kit (Takara, Dalian, China) as per the manufacturer's instruction. DcR3, MMP9, VEGF-C, and VEGF-D were amplified using SYBR Prime Script RT-PCR Kit (Takara, Dalian, China) with GAPDH as the internal control. Expression levels of target-of-interest were calculated as relative expression using 2−ΔΔCT approach.
Western blotting
Phosphate-buffered saline (PBS)-washed cells were sonicated in ice-cold lysis buffer. Whole-cell lysate obtained was then centrifuged at 12,000 rounds/min for 10 min. The supernatant obtained was resolved in 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane, and separately probed with DcR3 (diluted at 1:1000), MMP9 (diluted at 1:1000), VEGF-C (diluted at 1:1000), VEGF-D (diluted at 1:1000), and GAPDH (diluted at 1:10000) antibodies. Western blotting image acquisition was performed using imaging software from Bio-Rad (Berkeley, California, USA). Gray values of DcR3 bands were normalized with those of GAPDH.
Clone formation assay
Untransfected and lentivirus-transfected HepG2 cells were seeded into 6-well plates in a cell density of 800 cells/well, and they were allowed to grow for 5–7 days until clones were visible. PBS-washed cells were fixed with 4% paraformaldehyde and then stained with 1% crystal violet. The stained clones were counted, and the rate of clone formation was calculated as follows: (number of stained clones/number of seeded cells) × 100%.
Transwell assay
The invasiveness of untransfected and lentivirus-transfected HepG2 cells was studied using transwell assay (Axygen Scientific, Union City, CA, USA). Briefly, before the addition of HepG2 cells into transwell chambers, the membrane of each chamber was coated with BD Biosciences membrane matrix (50 mg/L) (1:8 dilution, Franklin Lakes, NJ, USA). After that, cell suspensions (1 × 105 cells) prepared in serum-free medium were added into the upper chambers of transwells, with the lower chambers filled with complete medium supplemented with 10% fetal bovine serum (FBS). After incubation at 37°C for 24 h, residual HepG2 cells in the upper chambers were wiped off with a cotton swab, while those migrated onto the lower surface of the membrane were fixed with formaldehyde and stained with crystal violet for 20 min. Stained HepG2 cells were then counted from 5 random microscopic views. For each group, experiment was done in triplicate. The relative invasion rate was determined as follows: (number of migrated transfected HepG2/number of migrated untransfected HepG2) × 100%.
Wound healing assay
Untransfected and lentivirus-transfected HepG2 cells were seeded into 6-well plates in a cell density of 1 × 105/ml and cultured. Once confluence was achieved, five scratches were made in each well. After washing away the detached cells by sterile PBS, cells were cultured in medium supplemented with 2% FBS under standard condition. At 0, 24, and 48 h after scratch, images of the wounds were captured for the measurement of wound width.
Statistical analysis
All data were statistically analyzed using IBM SPSS Statistics for Windows, version 20.0 (IBM Corp., Armok, NY, USA). Values were expressed as mean ± standard deviation (SD). Comparisons between multiple groups were done using one-way analysis of variance, while pairwise comparisons were performed using Student's t-test. P < 0.05 was regarded statistically significant.
Results
Decoy receptor3 expression in different liver cancer cell lines
Before we studied the functional role of DcR3 in liver cancer, we first examined its gene and protein expressions in primary liver cancer (Huh751, HepG2, and PLC) and metastatic (MHCC97H and MHCC97L) cells. RT-PCR revealed that compared to normal hepatocyte Lo-2, DcR3 gene expression was upregulated in liver cancer cells, in which the most elevated level was seen in PLC cells [Figure 1a]. DcR3 protein expression was also upregulated in liver cancer cell lines compared to normal hepatocyte as shown by Western blotting [Figure 1b]. Besides, HepG2 but not PLC was illustrated to express the highest level of DcR3 protein. Based on these data, HepG2 was chosen as the model cell line for the subsequent functional characterization of DcR3.
Figure 1.
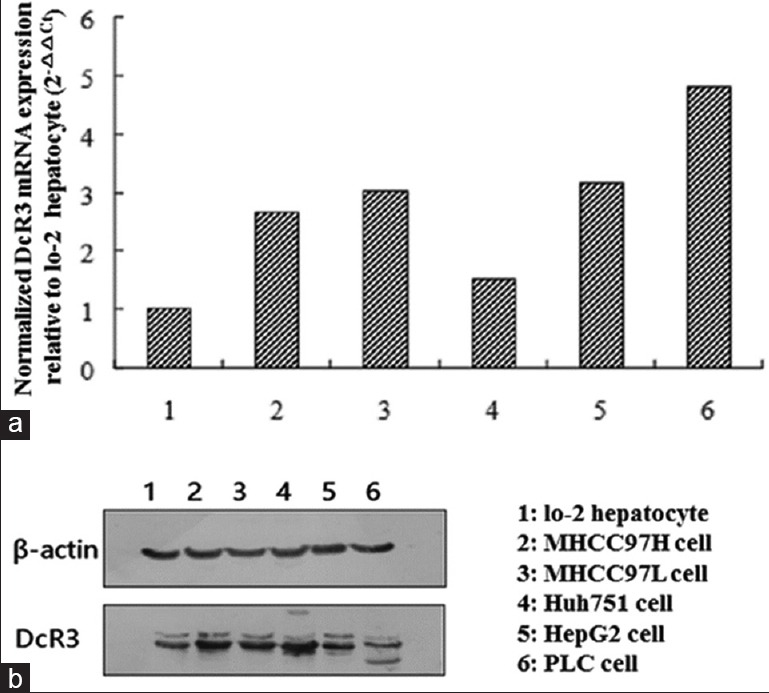
DcR3 expressions in different hepatocellular carcinoma cell lines, and normal hepatocytes were profiled. (a) Real-time PCR determined the upregulated mRNA levels of DcR3 in hepatocellular carcinoma cell lines compared to normal hepatocyte Lo-2. (b) Western blotting was employed to show the differential DcR3 protein levels in hepatocellular carcinoma cell lines and normal hepatocyte. DcR3: Decoy receptor 3; mRNA: Messenger RNA; PCR: Polymerase chain reaction.
Generation of stable decoy receptor 3 knockdown cell line
To study the functional role of DcR3 in liver cancer, we stably knocked down DcR3 expression in HepG2 cells by transfecting the cells with DcR3 shRNA using lentivirus. The plasmid harbored a reporter-enhanced GFP (EGFP) so that the transfection efficiency could be determined using fluorescent microscopy [Figure 2]. Results illustrated that positive EGFP signal could be detected in NC and DcR3-siRNA groups but not in CON group, indicating the successful transfection of plasmids into HepG2 cells by lentivirus. We then selected the transfected HepG2 cells by addition of puromycin, and after that, examined the protein and messenger RNA (mRNA) levels of DcR3 after lentivirus transfection. Western blotting clearly showed that DcR3 protein was suppressed in HepG2 transfected with DcR3 shRNA [Figure 3a]. In accordance with this result, RT-PCR demonstrated that DcR3 gene expression was significantly downregulated in DcR3-siRNA group compared to NC and CON groups, of which the differences were statistically significant (all P < 0.05) [Figure 3b].
Figure 2.

Transfection of HepG2 with lentivirus-based shRNA vector was examined using fluorescence microscopy. (a-c) Cell morphology images captured by inverted microscope. (d-f) Images captured by fluorescence microscope showing the fluorescent signal of the enhanced green fluorescent protein. All images were captured with a magnification of ×200. shRNA: Short hairpin RNA. CON: The blank control group; NC: Negative control group.
Figure 3.
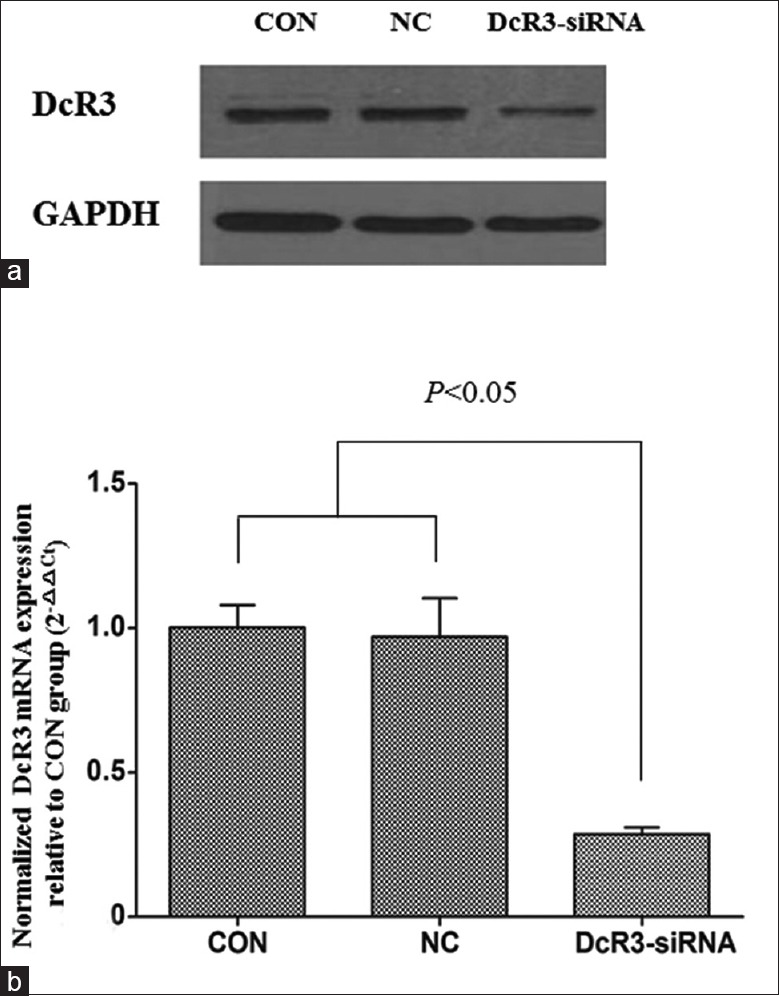
DcR3 expression in HepG2 transfected with and without DcR3 siRNA was assessed. (a) Western blotting examined DcR3 protein level in mock (CON), and HepG2 transfected with empty vector (NC) and DcR3 shRNA (DcR3-siRNA). House-keeping target GAPDH was included as the loading control. (b) RT-PCR determined significant downregulation of DcR3 mRNA in DcR3-siRNA when compared to CON and NC groups. The pairwise comparisons between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups were statistically significant (P < 0.05). DcR3: Decoy receptor 3; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; shRNA: Short hairpin RNA; mRNA: Messenger RNA; RT-PCR: Real-time polymerase chain reaction. CON: The blank control group; NC: Negative control group.
In vitro oncogenic properties of decoy receptor 3 in HepG2
We next examined whether stable knockdown of DcR3 would affect the oncogenic properties of HepG2 cells in vitro. Clone formation assay revealed that the number of clones formed in DcR3-siRNA group was smaller than those of in CON and NC groups by 50% [Figure 4a]. Cell proliferation assay showed that DcR3-siRNA group exhibited slower growth rate compared with CON and NC groups. The growth of DcR3-siRNA group plateaued 3 days after the start of the experiment, while both CON and NC groups continued to proliferate [Figure 4b]. We studied also the invasiveness of the cells using transwell assay [Figure 4c]. Analysis showed that the number of cells transmigrated across the membranes was substantially reduced in DcR3-siRNA group when compared to CON and NC groups (P < 0.05) [Figure 4d]. In addition, we performed wound healing assay to demonstrate that at 24 and 48 h after scratch, the wound of DcR3 knockdown was significantly wider than those of CON and NC groups (P < 0.05) [Figure 5a and 5b], suggesting the loss of DcR3 would reduce the migration of HepG2.
Figure 4.

Effect of DcR3 knockdown on the oncogenic properties of HepG2 was examined. (a) Clone formation assay. (b) Cell proliferation assay. Asterisk (*) represents significant differences (P < 0.05) between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups at respective time points. (c) Transwell assay. Arrow shows the migration of HepG2 cells across transwell membranes (original magnification ×200). (d) Graph showing the number of cell colonies counted on the bottom of transwell membranes. The pairwise comparisons between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups were statistically significant (P < 0.05). DcR3: Decoy receptor 3. CON: The blank control group; NC: Negative control group.
Figure 5.

Effect of DcR3 knockdown on the wound healing ability of HepG2 was studied. (a) Representative set of images showing the migration of HepG2 cells toward scratch wound (×200). (b) Graph presenting the normalized scratch width relative to CON group at 0, 24, and 48 h after the scratch. At time points of 24 and 48 h, the pairwise comparisons between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups were statistically significant (P < 0.05). DcR3: Decoy receptor 3; CON: The blank control group; NC: Negative control group.
Effect of decoy receptor 3 knockdown on matrix metalloproteinase-9 and vascular epithelial growth factor expression
With the effect of DcR3 on the oncogenic properties of HepG2 characterized, we investigated whether the expressions of MMP9 and VEGF (i.e., VEGF-C and VEGF-D) would be altered in the absence of DcR3. Western blotting showed the knockdown of DcR3-suppressed protein levels of MMP9, VEGF-C, and VEGF-D in HepG2 cells [Figure 6a]. In accordance with these results, RT-PCR determined the downregulation of gene expressions of MMP9 [Figure 6b], VEGF-C [Figure 6c], and VEGF-D [Figure 6d] in HepG2 cells with DcR3 be stably knocked down. In DcR3-siRNA group, the mRNA levels of MMP9, VEGF-C, and VEGF-D were <10% of those of CON group, and for all of them, the pairwise comparison between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups were statistically significant (P < 0.05).
Figure 6.

Effect of DcR3 knockdown on the expressions of MMP9, VEGF-C, and VEGF-D of HepG2 was studied using Western blotting and RT-PCR. (a) Western blotting examined MMP9, VEGF-C, and VEGF-D protein levels in CON, NC, and DcR3-siRNA groups. House-keeping target GAPDH was included as the loading control. (b-d) RT-PCR determined significant downregulation of MMP9, VEGF-C, and VEGF-D mRNAs in HepG2 transfected with DcR3 shRNA group when compared to CON and NC groups. The pairwise comparisons between DcR3-siRNA and CON groups and between DcR3-siRNA and NC groups were statistically significant (P < 0.05). DcR3: Decoy receptor 3; RT-PCR: Real-time polymerase chain reaction; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; shRNA: Short hairpin RNA; mRNA: Messenger RNA; MMP6: Matrix metalloproteinase-9; VEGF: Vascular epithelial growth factor; CON: The blank control group; NC: Negative control group.
Discussion
DcR3 has been reported to be highly expressed in a wide variety of solid malignancies including HCC, of which the overexpressions in HCC tumors were associated with tumor growth, cancer metastasis, and more importantly, dismal survivals of patients.[8,9,10] DcR3 is, therefore, believed to be a key oncogenic driver of HCC. The mechanism underlying the oncogenic properties of DcR3, however, has remained to be fully deciphered. To fill the knowledge gap, the present work characterized the functional role of DcR3 in liver cancer cell line in vitro. By loss-of-function approach, we revealed that DcR3 was important to the growth, clonogenicity, and invasiveness of HepG2 cells. Knockdown of DcR3 in HepG2 cells also downregulated the expressions of MMP9, VEGF-C, and VEGF-D.
Surgical resection of tumor is a curative approach for the treatment of HCC. With the recent advances in cancer biology and therapy, radiofrequency ablation, transarterial chemoembolization, and molecular-targeted therapies have emerged as promising therapeutic approaches for patients with small HCC (i.e., ≤2 cm). Unfortunately, most HCC tumors, when first diagnosed, are so advanced for surgery and other therapeutic approaches.[6] Our experiments showed that knockdown of DcR3 could significantly reduce the proliferation and anchorage-independent growth of HepG2, suggesting DcR3 as a key oncogenic driver of HCC progression. Based on these findings, targeting DcR3 is a potential approach to reduce tumor burden, so allowing patients to be managed by surgery and other therapies.
Metastatic spread of cancer cells from the primary site to distinct organs significantly worsens the prognosis of patients with HCC. Based on the long-standing “Seed and Soil” hypothesis of cancer metastasis, the metastatic potential of most solid malignancies including HCC is largely determined by the invasiveness and migration ability of cancer cells.[18,19] Here, we clearly illustrated that stable suppression of DcR3 expression could impair the invasion and migration of HepG2. In addition, loss of DcR3 resulted in remarkable decreases in the expressions of MMP 9, VEGF-C, and VEGF-D. MMP 9, VEGF-C, and VEGF-D, which are the essential mediators of metastasis. MMP 9 is a metalloproteinase that facilitates the detachment of cancer cells from the primary tumor and the later extravasation at the secondary site.[20] Dynamic expressions of MMP 9 and other proteinases were reported to regulate the early invasion of HCC.[21] Studies also suggested that MMP 9 would serve as a prognostic marker for the risk of metastasis in cancer patients.[22,23] VEGF-C and VEGF-D activate the signaling pathways of tumor angiogenesis, a process important in the establishment of metastatic foci and the subsequent tumor growth at the secondary site.[24] Taken together, suppressing DcR3 would be an attractive way for the prevention of metastasis.
To conclude, the present work provided evidences to support that DcR3 is essential to the growth and invasive behavior of liver cancer cells. Targeting DcR3 may be a potential therapeutic approach for the treatment of HCC. More in-depth functional characterization of DcR3 in animal models is warranted.
Financial support and sponsorship
This study was supported by grants from the Natural Science Foundation of China (No. 81550033), and the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (No: ZYLX201612).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Ning-Ning Wang
References
- 1.Zhao JJ, Yan T, Zhao H, Zhou JG, Huang Z, Zhang YF, et al. Evaluation of eight different clinical staging systems associated with overall survival of chinese patients with hepatocellular carcinoma. Chin Med J. 2015;128:316–21. doi: 10.4103/0366-6999.150095. doi: 10.4103/0366-6999.150095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block T, Mehta AS, London WT. Hepatocellular carcinoma of the liver. Cancer Biomark. 2010;9:375–83. doi: 10.3233/CBM-2011-0165. doi: 10.3233/CBM-2011-0165. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Li C, Wen TF, Peng W, Chen LP. Postoperative low absolute lymphocyte counts may predict poor outcomes of hepatocellular carcinoma after liver resection. Chin Med J. 2016;129:536–41. doi: 10.4103/0366-6999.176982. doi: 10.4103/0366-6999.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang XP, Liu L, Wang P, Ma SL. Human sulfatase-1 improves the effectiveness of cytosine deaminase suicide gene therapy with 5-fluorocytosine treatment on hepatocellular carcinoma cell line hepg2 in vitro and in vivo. Chin Med J. 2015;128:1384–90. doi: 10.4103/0366-6999.156800. doi: 10.4103/0366-6999.156800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HY, Kim CW, Choi JY, Park CH, Lee CD, Yim HW. A decade-old change in the screening rate for hepatocellular carcinoma among a hepatitis B virus-infected population in Korea. Chin Med J. 2016;129:15–21. doi: 10.4103/0366-6999.172551. doi: 10.4103/0366-6999.172551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu XS, Chen W, Miao RC, Zhou YY, Wang ZX, Zhang LQ, et al. Survival analysis of hepatocellular carcinoma: A comparison between young patients and aged patients. Chin Med J. 2015;128:1793–800. doi: 10.4103/0366-6999.159356. doi: 10.4103/0366-6999.159356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitti RM, Marsters SA, Lawrence DA, Roy M, Kischkel FC, Dowd P, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396:699–703. doi: 10.1038/25387. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Han B, Sheng H, Lin M, Moore PA, Zhang J, et al. Clinical significance of detecting elevated serum DcR3/TR6/M68 in malignant tumor patients. Int J Cancer. 2003;105:724–32. doi: 10.1002/ijc.11138. doi: 10.1002/ijc.11138. [DOI] [PubMed] [Google Scholar]
- 9.Chen G, Luo D. Expression of decoy receptor 3 in liver tissue microarrays. Natl Med J India. 2008;21:275–8. [PubMed] [Google Scholar]
- 10.Shen HW, Gao SL, Wu YL, Peng SY. Overexpression of decoy receptor 3 in hepatocellular carcinoma and its association with resistance to Fas ligand-mediated apoptosis. World J Gastroenterol. 2005;11:5926–30. doi: 10.3748/wjg.v11.i38.5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macher-Goeppinger S, Aulmann S, Wagener N, Funke B, Tagscherer KE, Haferkamp A, et al. Decoy receptor 3 is a prognostic factor in renal cell cancer. Neoplasia. 2008;10:1049–56. doi: 10.1593/neo.08626. doi: 10.1593/neo.08626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge Z, Sanders AJ, Ye L, Wang Y, Jiang WG. Expression of death decoy receptor-3 (DcR3) in human breast cancer and its functional effects on breast cancer cells in vitr o. J Exp Ther Oncol. 2011;9:109–18. doi: 10.3892/or.2013.2259. [PubMed] [Google Scholar]
- 13.Connor JP, Felder M. Ascites from epithelial ovarian cancer contain high levels of functional decoy receptor 3 (DcR3) and is associated with platinum resistance. Gynecol Oncol. 2008;111:330–5. doi: 10.1016/j.ygyno.2008.07.012. doi: 10.1016/j.ygyno.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 14.Maeda T, Hao C, Tron VA. Ultraviolet light (UV) regulation of the TNF family decoy receptors DcR2 and DcR3 in human keratinocytes. J Cutan Med Surg. 2001;5:294–8. doi: 10.1007/s102270000030. doi: 10.1007/s102270000030. [DOI] [PubMed] [Google Scholar]
- 15.Curtin JF, Cotter TG. Live and let die: Regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003;15:983–92. doi: 10.1016/s0898-6568(03)00093-7. doi: 10.1016/S0898-6568(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 16.Ge Z, Sanders AJ, Ye L, Jiang WG. Aberrant expression and function of death receptor-3 and death decoy receptor-3 in human cancer. Exp Ther Med. 2011;2:167–72. doi: 10.3892/etm.2011.206. doi: 10.3892/etm.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu W, Xu YC, Tao Y, He P, Li Y, Wu T, et al. DcR3 regulates the growth and metastatic potential of SW480 colon cancer cells. Oncol Rep. 2013;30:2741–8. doi: 10.3892/or.2013.2769. doi: 10.3892/or.2013.2769. [DOI] [PubMed] [Google Scholar]
- 18.Mathias RA, Gopal SK, Simpson RJ. Contribution of cells undergoing epithelial-mesenchymal transition to the tumour microenvironment. J Proteomics. 2013;78:545–57. doi: 10.1016/j.jprot.2012.10.016. doi: 10.1016/j.jprot.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18:43–73. doi: 10.1615/critrevoncog.v18.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Dou C, Jia Y, Li Q, Zheng X, Yao Y, et al. RIG-I suppresses the migration and invasion of hepatocellular carcinoma cells by regulating MMP9. Int J Oncol. 2015;46:1710–20. doi: 10.3892/ijo.2015.2853. doi: 10.3892/ijo.2015.2853. [DOI] [PubMed] [Google Scholar]
- 21.Chen RX, Song HY, Dong YY, Hu C, Zheng QD, Xue TC, et al. Dynamic expression patterns of differential proteins during early invasion of hepatocellular carcinoma. PLoS One. 2014;9:e88543. doi: 10.1371/journal.pone.0088543. doi: 10.1371/journal.pone.0088543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goos JA, Coupé VM, van de Wiel MA, Diosdado B, Delis-Van Diemen PM, Hiemstra AC, et al. A prognostic classifier for patients with colorectal cancer liver metastasis, based on AURKA, PTGS2 and MMP9. Oncotarget. 2016;7:2123–34. doi: 10.18632/oncotarget.6188. doi: 10.18632/oncotarget.6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren Z, Liang S, Yang J, Han X, Shan L, Wang B, et al. Coexpression of CXCR4 and MMP9 predicts lung metastasis and poor prognosis in resected osteosarcoma. Tumour Biol. 2016;37:5089–96. doi: 10.1007/s13277-015-4352-8. doi: 10.1007/s13277-015-4352-8. [DOI] [PubMed] [Google Scholar]
- 24.Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med. 2011;17:347–62. doi: 10.1016/j.molmed.2011.01.015. doi: 10.1016/j.molmed.2011.01.015. [DOI] [PubMed] [Google Scholar]