

Abstract
Gargantulide A (1), an extremely complex 52-membered macrolactone, was isolated from Streptomyces sp. A42983 and displayed moderate activity against MRSA. The planar structure of 1 was determined using 2D NMR, and its stereochemistry was partially established on the basis of NOESY correlations, J-based configuration analysis, and Kishi’s universal NMR database.
Graphical abstract
The bacterial membrane protein ATPase SecA facilitates the transfer of preproteins across the cell membrane and is essential for bacterial survival.1,2 There have been various SecA inhibitors identified, but none have advanced to clinical trials.2,3 A 384-well high throughput bioassay that used BIOMOL Green to measure free phosphate (Pi) release during the transport of SecA preprotein, proOmpA, through the SecYEG complex located on inverted membrane vesicles was developed to identify SecA inhibitors. An active MeOH extract derived from Streptomyces sp. A42983 was identified that displayed moderate activity in the SecA assay and whole cell activity against methicillin-resistant Staphylococcus aureus (MRSA) ATCC25923. Bioassay-guided fractionation led to the isolation of a novel and exceptionally complex macrolactone, gargantulide A (1), possessing a 52-membered ring.
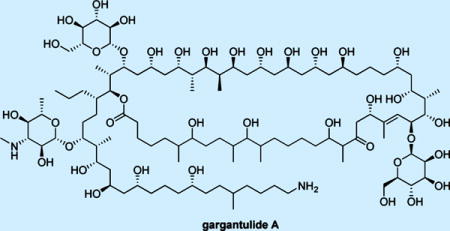
Gargantulide A (1) was isolated as a white powder (Figure 1) and was assigned a molecular formula of C105H200N2O38 based on the high resolution ESI-FT-MS ([M + 2H]2+ = 1049.6974, Δ = 1.2), which was consistent with seven degrees of unsaturation. The 1H NMR spectrum showed severely overlapped resonances from 0.5–5.5 ppm due to the presence of numerous oxymethines, methylenes associated with saturated hydrocarbon chains, and methyl groups. The 13C NMR spectrum similarly suffered from extensive signal degeneracy, but clearly contained one ketone group (δC 214.4), one ester or amide-type carbonyl (δC 175.1), two olefinic carbons (δC 123.3 and 146.5), and three anomeric carbons (δC 96.8, 102.3 and 104.3) as identified by their typical 13C NMR chemical shifts. Fortunately, a multiplicity-edited HSQC experiment allowed us to assign nearly all carbon signals to their corresponding proton(s) and resulted in the identification of 50 methine (including 39 oxymethine), 38 methylene, and 14 methyl carbons. Further HSQC analysis allowed identification of five partially overlapped carbon resonances near δC 34.0 [δC 34.22 (C-5, CH2) – δH 1.19/1.48; δC 34.01 (C-13, CH2) – δH 1.08/1.76; δC 34.04 (C-67, CH2) – δH 1.13/1.51; δC 34.07 (C-68, CH) – δH 1.43; δC 34.18 (C-1′, CH2) – δH 1.07/1.35] as well as three oxymethines in the cluster of δC 66.2 and δH 4.10 which were deduced from the molecular formula and proton integration data.
Figure 1.
Structure of Gargantulide A (1).
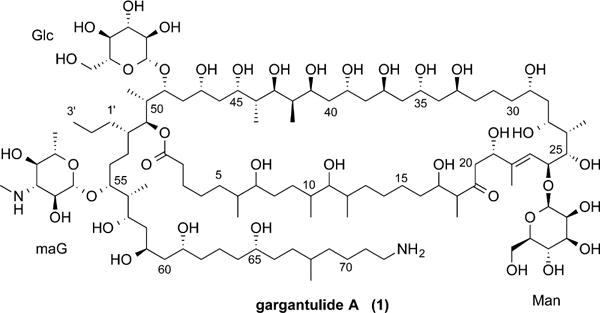
Elucidation of the structure of gargantulide A (1) was initiated by considering the strong HMBC correlations between the protons of 14 methyl groups and their nearby carbons, thus allowing the identification of 12 subunits (A–L) as shown in Figure 2. Nine subunits were nicely revealed by well-separated signals in the expanded HMBC spectrum, and these assignments were confirmed by COSY and TOCSY data. Subunits B, I, and L which contained the nearly indistinguishable carbon resonances around δC 34.0 could not be unambiguously assigned by HMBC, but the separated resonances of their attached protons allowed these latter NMR resonances to be confidently assigned by the combination of COSY, TOCSY, and HSQC-TOCSY data (Figures S6–S14).
Figure 2.
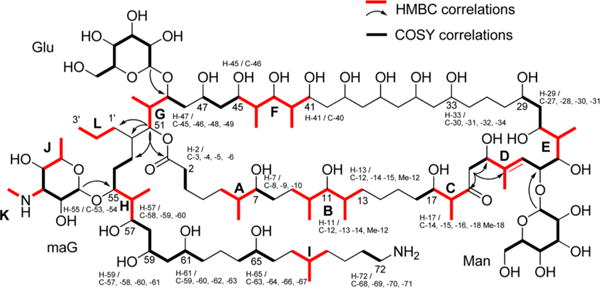
Twelve subunits and their linkages by HMBC or HSQC-TOCSY correlations.
Next, the structures of the two conventional pyranose residues were established as β-glucose (Glc) and β-mannose (Man) in a straightforward manner using 3JHH coupling constants from DQF-COSY, along with TOCSY and NOE correlations. Furthermore, these sugar components, cleaved by acid hydrolysis, were recognized as D-Glc and D-Man by comparison of retention times with the authentic sugars using GC/MS of their trimethylsilyl derivatives (Figure S1). One remaining sugar moiety was elucidated as 3,6-deoxy-3-methylamino pyranose by sequential COSY correlations from an anomeric proton to a methyl doublet, and HMBC correlations between subunit K of a methylamino group (δC 31.4; δH 2.78) and maG-3 carbon, and the anomeric proton and maG-5 carbon. The large coupling constants between protons in this moiety and NOEs of maG-1/maG-3, maG-1/maG-5, and mag-2/maG-4 led to 3,6-deoxy-3-methylamino glucose (maG).
Following the assignment of the three sugar components, the linkage of the subunits defined above was accomplished by intensive inspection of 2D NMR spectra (COSY, TOCSY, HSQC, HMBC, and HSQC-TOCSY). As detailed below, HSQC-TOCSY correlations from oxymethine protons or other well-separated protons to neighboring carbons verified the connection of the subunits as shown in Figure 2. COSY and HSQC-TOCSY correlations from H2-2 allowed connection to H-5 in subunit A and allowed assignments of 13C resonances for C-3 to C-6. The subunits A and B were connected by two nearly overlapping resonances for C-7 (δC 32.7) and C-9 (δC 32.4), indicated by COSY correlation of H-7/H-8 and HSQC-TOCSY correlations from H-7 to C-10. Further connection of this spin system from H-13 in subunit B was prevented by TOCSY correlations overlapped with signals associated with other subunits. On the other hand, the linkages of subunits C, D, and E were readily determined by COSY and HMBC correlations due to distinct resonances as shown in Figure 2. Fortunately, the same resonances were located in the two TOCSY correlation sets propagating from each terminal proton of subunits B and C. HSQC-TOCSY correlations allowed assignment of three aliphatic methylene carbons between subunits B and C (C-14 to C-16). In a similar way, the methine H-47, which was recognized by the COSY and TOCSY correlations with H-46 in subunit F, played a crucial role in the connectivity of subunits F and G by the HSQC-TOCSY correlations. Successive connection from subunit G could be conducted by the extensive HMBC correlations of H-51/C-1, H-51/C-52, H-51/C-53, and H-51/C-1′. The assignment of the overlapped C-1′ resonance in subunit L was confirmed by NOE correlation between the corresponding proton and H-51 multiplet. Linkage of subunit G to H was evident from a combination of COSY correlations between two relatively distinct protons (Ha-53 and Hb-54), and weak HSQC-TOCSY correlations from the H-55 multiplet to the C-53 and C-54 resonances. Following subunit H, the consecutive methylene-oxymethine groups from C-58 to C-62 were apparent from a mutual sharing of TOCSY and HSQC-TOCSY correlations with three oxymethine 1H signals (H-57, H-59, and H-61). HSQC-TOCSY correlations associated with yet another oxymethine proton, H-65, enabled connection of subunits H and I. The terminal amino propyl moiety was attached to the distal side of subunit I by HSQC-TOCSY correlations starting with H-72.
The assignment of the C-29 to C-33 segment adjacent to subunit E was enabled by three common HSQC-TOCSY correlations initiated from two different protons (H-29 and H-33). Their carbon chemical shifts were similar to those of C-62 to C-64 which were also involved in a 1,5-diol unit. However, extending COSY or TOCSY correlations from the H-33 signal was not possible because of both 1H and 13C spectral overlap. Interestingly, these overlapped signals corresponded to three unassigned oxymethines (around δC 66.2; δH 4.10) and three methylenes (around δC 46.5; δH 1.56), reminiscent of chemical shift and functionality to the alternating C-57 to C-62 methylene-oxymethine section. The insertion of these alternating groups between C-33 and C-40 was subsequently confirmed by COSY and TOCSY spectra and allowed connection between subunits E and F. Finally, HMBC correlation between the signal for H-2 and an ester carbonyl C-1 (δC 175.1), and placement of the three sugar components based on HMBC correlations as given in Figure 2, completed the planar structure of 1 as a triglycosylated 52-membered macrolactone, thereby accounting for the 7 degrees of unsaturation.
Gargantulide A (1) contains 34 stereocenters on the macrolactone ring and the side chain, together with three different and fully substituted pyranose sugar residues, for a total of 49 chiral centers. A combination of NOE (NOESY) and coupling constant analysis (DQF-COSY and HECADE) was used to establish the configurations of the relatively rigid segments close to the three sugar residues (C-21–27 and C-47–57). Extending from the two stereochemically defined sugars (Man and Glc), the configurational assignment of the 1,3-diol (C-27–C-29), 1,3,5-triol (C-45–C-49 and C-57–C-61), 1,3,5,7-pentol (C-33–C-41) and a contiguous hydroxy/methyl/hydroxy/methyl set of substituents in the flexible chain (C-41–45) could be successfully accomplished by application of Kishi’s universal NMR database.4–7
Based on the stereochemically defined β-D-mannose unit, NOE correlations between Man-1/C-24 and Man-5/Me-22 allowed assignment of the absolute configuration of C-24 as S. Strong NOE correlations of H-23/H-25, H-23/H26, Me-22/H-24, Me-22/H-27, and H-24/H-27 allowed assignment of configurations for the C-23 to C26 segment (Figure 3A), and this was additionally supported by homo- and heteronuclear coupling constants between these proximal atoms.5 A small coupling constant between H-27 and Ha-28 (JHH ∼ 2.4 Hz) and an NOE correlation between H-26 and Ha-28 defined the configuration of C-27 as R. The configuration of C-29 could then be assigned as S by its carbon chemical shift value which matched typical values in Kishi’s NMR database for an oxymethine carbon in a 1,3-anti diol relationship [δC = 69.2, in CD3OD].6 Accordingly, the stereochemistry of the C-23 to C-29 fragment was completely assigned as 24S, 25S, 26S, 27R, and 29S. Additionally, a weak NOE signal between Man-1 and H-20 (appearing consistently in three repeated NOESY experiments) along with a strong NOE between H-21 and H-23 allowed deduction of C-21 as S.
Figure 3.
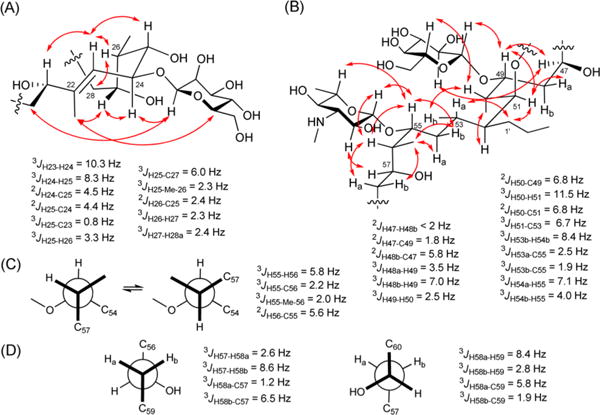
Crucial NOE correlations observed near the three sugar residues and corresponding J-based configuration analyses.
In a similar manner, the absolute configuration of C-49 was defined as R based on strong NOE correlations between Glc-1/H-49, Glc-1/H-50, and weak NOE correlations of Glc-2/H-47 and Glc-5/H-50. Despite the fact that the chemical shifts of Glc-2 (δH 3.18) and Glc-5 (δH 3.20) were nearly isochronous, their NOE correlations could be distinguished from one another by NOESY. Additional NOE correlations of H-49/H-50, H-49/H-51, H-50/Hb-53, H-51/Ha-48 and H-51/H-1′, H-52/Me-50 led to assignments of 47R, 49R, 50R, 51S, and 52S. Homo- and heteronuclear couplings also supported this determination as shown in Figure 3B. In particular, the S configuration of H-47 was supported by an NOE correlation with H-49 and a small coupling constant with Hb-48; this latter signal was present in a gauche orientation with the hydroxy group on C-47 as revealed by a large heteronuclear coupling with C-47.
Furthermore, a large coupling constant between the well-resolved protons Hb-53 and Hb-54, and NOE interaction between Ha-53/H-1′, allowed deduction of the configuration at C-52. Small heteronuclear coupling constants between Ha-53/C-55 and Hb-53/C-55 indicated a gauche relationship with C-55 and these two protons, and also strong NOE correlations of H-55/Hb-54, Me-56/Ha-54, Me-56/Hb-54, maG-1/H-55, maG-1/H-56, and maG-1/H-57 defined C-55 as R (Figure 3B). Based on this configuration, the stereochemistry of the next carbon C-56 was apparent from J-based configuration analysis for the pair of interconverting rotamers (Figure 3C), revealing a 56R configuration.8 In turn, the configuration of C-57 could be established, as for C-27, by NOE correlation of H-56/Ha-58, the latter of which was gauche to H-57 (3JH58a-H57 = 2.2 Hz). Similarly, the configuration of the oxymethine carbon C-59 was assigned anti to C-57 by J-based analysis (Figure 3D). The configuration of C-61 as syn with C-59 was deduced by Kishi’s NMR database for a 1,3,5-triol moiety: the assignment of the carbon chemical shift of C-59 at δC 68.8 (CD3OD) indicated that it must possess either an anti/syn or syn/anti configuration.6 Additionally, the configuration of C-65 was suggested by the empirical rule for 1,5-diol systems in which the two central methylene protons are equivalent in 1,5-anti diols and nonequivalent in 1,5-syn diols.9 Because the H-63 signals were nonequivalent (δH 1.38 and 1.61), this implied that C-61 and C-65 are in a syn orientation, thus assigning C-65 as S. On the other hand, the maG pyranose moiety attached to C-55 could be established as L by weak NOE interactions of maG-1/H-57, maG-5/H-55, and maG-2/H-57 as well as a stronger NOE between maG-1 and H-55.
Next, relative configurations for the C-33 to C-45 segment were assigned by Kishi’s NMR database values. On the basis of the 45, 47-syn diol moiety identified from the carbon chemical shift of C-47 (δc 71.42), the configuration of the C-45 to C-41 hydroxy/methyl/hydroxy/methyl/hydroxy sequence was determined following a previously reported procedure.10 In graphs comparing carbon chemical shifts of C-44 and Me-44 with the corresponding Kishi’s NMR database values (1a–h, s), the possible configuration of the first hydroxy/methyl/hydroxy/methyl sequence [C-45 to C-42 with Me-44] was indicated as 1a (ααββ), 1e (βαββ), 1f (βααα), 1g (βαβα), or 1h (βααβ). By applying the same method to the second hydroxy/methyl/hydroxy/methyl sequence [C-44 to C-41 with Me-42], stereoisomer 2d (αβββ) was selected as the best match. By combining these two results, the relative configuration of the C-45 to C-41 section was identified as either ααβββ or βαβββ. Of these two, the ααβββ possibility for C-45 to C-41 was preferred by measurement of small coupling constants for 3JH44–H45 and, 2JH44–H45, and a strong HMBC correlation of H-45/Me-44, thus indicating a threo relationship between the C-45 hydroxy group and Me-44. Furthermore, the relative tetraol configuration in the C-33 to C-41 section was assignable by the distinctive chemical shifts of the central carbons in the two overlapping 1,3,5-triol units. These depend on the relative orientation of the three hydroxy groups in each triol [δC 70.4 for 1,3-syn/3,5-syn; δC 66.3 for 1,3-anti/3,5-anti; δC 68.3 for 1,3-anti/3,5-syn or 1,3-syn/3,5-anti in CD3 OD].4 Comparison of these NMR database values with those observed (C-35 δC 66.35; C-39 δC 66.4) suggested a continuous anti/anti configuration in the C-33 to C-41 segment. Using the empirical rule described above for 1,5-diols, relay of this stereochemical information to C-29 was possible through observation of the equivalence of the methylene protons at H-31, thus suggesting a 29,33-anti-diol substructure. Finally, even though C-47 is the central carbon of a modified 1,3,5-triol moiety, its 13C NMR chemical shift is very close to that for the syn/syn configuration noted above, indicating a syn relationship to C-45, and thus completing the configurational assignments for the C-29 to C-51 segment.
Compared with monazomycin,10 desertomycin,11 and mathemycin,12 glycosylated macrolactones with an amino group at their termini, gargantulide A is larger in ring size and features an unusual amino sugar residue.
Gargantulide A (1) had an IC50 of 8 μg/mL in the HTS assay that measured the release of phosphate using BIOMOL Green from the reaction of inverted membrane vesicles, proOmpA, a precursor protein, and the SecA enzyme. In a secondary screening, 1 was not active at concentrations up to 160 μg/mL against the ATPase domain of SecA and the chaperone GroEL, which indicated that 1 might not be specifically inhibiting SecA. Gargantulide A (1) had an MIC of 2 μg/mL against S. aureus ATCC25923 that was not affected by the presence of 40% horse serum (MIC 1 μg/mL). Although 1 was not active against the E. coli DC2 (MIC >32 μg/mL), which is a mutant deficient in osmoregulation of periplasmic oligosaccharide synthesis, activity was rescued against an E. coli Imp (MIC 1 μg/mL), which is a mutant with increased membrane permeability. Gargantulide A (1) also had an MIC of 0.5 μg/mL against Clostridium difficile 6196 HMC and 4 μg/mL against C. difficile 6671 HMC. Gargantulide A (1) was profiled for antifungal activity and was found to be weakly active against Candida albicans ATCC90028 (IC50 64 μg/mL) and inactive against Saccharomyces cerevisiae SKY54 (MIC >128 μg/mL). No hemolysis was observed at concentrations up to 200 μg/mL. Overall this was a promising profile for a Gram-positive antibiotic, strengthened by the observation of excellent activity against multiple strains of methicillin-resistant S. aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), vancomycin-resistant enterococci (VRE), and penicillin-resistant Streptococcus pneumonia (PRSP) (Table S2).
Given this biological profile, the high molecular weight, and other physicochemical properties of gargantulide A (1), it was envisaged that 1 might become an intravenously dosed Gram-positive antibiotic or an orally administered treatment for the anaerobe C. difficile. Gargantulide A (1) was administered to female CD-1 mice using a lateral tail injection; however, the mice rapidly died in a state of rigid paralysis at a dose of 5 mg/kg. Mice treated with 5 mg/kg of 1 via subcutaneous injection showed no sign of immediate distress but died within 12 h. Unfortunately, this severe toxicity precluded any further development of 1 as an antibiotic.
In summary, gargantulide A (1), discovered using a combination of a SecA HTS assay and whole cell screening, was deduced as a polyketide with a 52-membered macrolactone ring. The structure of this antibiotic was exceptionally complex in that it contained 105 carbon atoms, of which nearly half were chiral centers. Although the configuration of several chiral centers remains unassigned, most were determined using diverse methodologies. This compound displayed promising activity against pathogenic Gram-positive bacteria (MRSA, MRSE, VRE, PRSP, and C. difficile) but was found to be highly toxic to mice, precluding further development.
Supplementary Material
Acknowledgments
We acknowledge NIH NS053398 and CA100851 (W.H.G.) and S. R. Whitton and other members of the MerLion microbiology team for fermentation and extract generation.
Footnotes
Supporting Information
Experimental procedures and spectral data of 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Rao CVS, De Waelheyns E, Economou A, Anné J. Biochim Biophys Acta. 2014;1843:1762–1783. doi: 10.1016/j.bbamcr.2014.02.004. [DOI] [PubMed] [Google Scholar]; (b) Gouridis G, Karamanou S, Sardis MF, Schärer MA, Capitani G, Economou A. Mol Cell. 2013;52:655–666. doi: 10.1016/j.molcel.2013.10.036. [DOI] [PubMed] [Google Scholar]; (c) Lycklama A, Nijeholt JA, Driessen AJ. Philos Trans R Soc London, Ser B: Biol Sci. 2012;367:1016–1028. doi: 10.1098/rstb.2011.0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Chatzi KE, Sardis MF, Economou A, Karamanou S. Biochim Biophys Acta. 2014;1843:1466–1474. doi: 10.1016/j.bbamcr.2014.02.014. [DOI] [PubMed] [Google Scholar]; (b) Segers K, Anné J. Chem Biol. 2011;18:685–698. doi: 10.1016/j.chembiol.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 3.(a) Chen W, Chaudhary A, Cui J, Jin J, Hsieh Y, Yang H, Huang Y, Tai PC, Wang B. J Chin Pharm Sci. 2012;21:526–530. [Google Scholar]; (b) Cui J, Jin J, Hsieh YH, Yang H, Ke B, Damera K, Tai PC, Wang B. ChemMedChem. 2013;8:1384–1393. doi: 10.1002/cmdc.201300216. [DOI] [PubMed] [Google Scholar]; (c) Akula N, Trivedi P, Han FQ, Wang N. Eur J Med Chem. 2012;54:919–924. doi: 10.1016/j.ejmech.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi Y, Tan CH, Kishi Y. Helv Chim Acta. 2000;83:2562–2571. [Google Scholar]
- 5.Hayashi N, Kobayashi Y, Kishi Y. Org Lett. 2001;3:2249–2252. doi: 10.1021/ol010109r. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi Y, Hayashi N, Kishi Y. Org Lett. 2001;3:2253–2255. doi: 10.1021/ol010110q. [DOI] [PubMed] [Google Scholar]
- 7.Bifulco G, Dambruoso P, Gomez-Paloma L, Riccio R. Chem Rev. 2007;107:3744–3779. doi: 10.1021/cr030733c. [DOI] [PubMed] [Google Scholar]
- 8.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 9.Miyata Y, Matsunaga S. Tetrahedron Lett. 2008;49:6334–6336. [Google Scholar]
- 10.Nakayama H, Furihata K, Seto H, Otake N. Tetrahedron Lett. 1981;22:5217–5220. [Google Scholar]
- 11.Bax A, Aszalos A, Dinya Z, Sudo K. J Am Chem Soc. 1986;108:8056–8063. [Google Scholar]
- 12.Mukhopadhyay T, Vijayakumar EKS, Nadkarni SR, Fehlhaber HW, Kogler H, Petry S. J Antibiot. 1998;51:582–585. doi: 10.7164/antibiotics.51.582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.