

Abstract
We previously reported that TLR4-/- mice are refractory to mouse-adapted A/PR/8/34 (PR8) influenza-induced lethality and that therapeutic administration of the TLR4 antagonist, Eritoran, blocked PR8-induced lethality and acute lung injury (ALI) when given starting 2 days post-infection. Herein, we extend these findings: anti-TLR4- or TLR2-specific IgG therapy also conferred significant protection of wild-type (WT) mice from lethal PR8 infection. If treatment is initiated 3 h prior to PR8 infection and continued daily for 4 days, Eritoran failed to protect WT and TLR4-/- mice, implying that Eritoran must block a virus-induced, non-TLR4 signal that is required for protection. Mechanistically, we determined that (i) Eritoran blocks HMGB1-mediated, TLR4-dependent signaling in vitro and circulating HMGB1 in vivo, and an HMGB1 inhibitor protects against PR8; (ii) Eritoran inhibits pulmonary lung edema associated with ALI, (iii) IL-1β contributes significantly to PR8-induced lethality, as evidenced by partial protection by IL-1 receptor antagonist (IL-1Ra) therapy. Synergistic protection against PR8-induced lethality was achieved when Eritoran and the anti-viral drug, oseltamivir, were administered starting 4 days post-infection. Eritoran treatment does not prevent development of an adaptive immune response to subsequent PR8 challenge. Overall, our data support the potential of a host-targeted therapeutic approach to influenza infection.
Introduction
Influenza continues to evolve with new antigenic variants emerging annually, as exemplified by the last several influenza seasons in which the recommended vaccine was considerably less efficacious than predicted1-4. Therefore, there remains a pressing need to develop alternatives to the annual influenza vaccines and anti-viral agents currently used to mitigate the effects of influenza infection. Multiple Pattern Recognition Receptors (PRRs), including TLR3, TLR4, TLR7, TLR8, TLR10, and the intra-cytosolic sensor, RIG-I, have been implicated in influenza-induced disease, although TLR10 is not functional in mice5-12. CD14 is required for influenza-induced cytokine production by mouse macrophages, independent of TLR2 and TLR413. In addition, influenza-infected MyD88-/- and MyD88/TRIF doubly deficient mice show a dramatic reduction of pulmonary cytokine production when compared to WT mice5,11,12, indicating the important role of these TLR signaling pathways in disease.
Imai et al.14 proposed that chemical or microbial insults trigger NADPH-dependent reactive oxygen species that generate a host-derived oxidized phospholipid, oxidized 1-palmitoyl-2-arachidonyl-phosphatidylcholine (OxPAPC), in lungs. They concluded that regardless of the initial sensing involved in pathogen recognition, OxPAPC initiates a common TLR4-, TRIF-, and IL-6-dependent pathway in macrophages that leads to ALI. We showed that treatment of influenza-infected mice with Eritoran, the most potent, synthetic lipid A analog known15, blocked influenza-induced lethality and ALI. When administered daily to WT mice for 5 days, starting on days 2, 4, or 6 post-infection, Eritoran treatment significantly improved survival and clinical symptoms, while decreasing ALI, OxPAPC accumulation, the cytokine storm, and systemic inflammation16. Herein, we extend our previous findings by characterizing the role of TLR4 and TLR2 in influenza-induced lethality and ALI and provide new insights into the molecular mechanisms underlying protection by modulators of these important TLR-mediated inflammatory pathways.
Results
Eritoran in Influenza models
We previously reported that TLR4-/- mice were refractory to mouse-adapted influenza, PR816,17, and have now confirmed that TLR4-/- mice are also refractory to a more pathogenic, mouse-adapted pandemic H1N1 strain, ma.Ca/0418 (Supplementary Figure 1A). We also showed previously that therapeutic treatment of PR8-infected, WT mice therapeutically with Eritoran, significantly protected against lethality, and attenuated ALI, findings now reproduced in C57BL/6 mice infected with the ma.Ca/04 strain (Supplementary Figure 1B and 1C). Eritoran also protected PR8-infected BALB/c mice (data not shown). We found that an additional daily dose of Eritoran (i.e., administered once vs. twice daily), starting four days post-infection, failed to improve the protection achieved with a single dose daily (data not shown). Together, these data expand our previous observations that therapeutic treatment of mice with Eritoran protects against lethality with additional influenza strains and newly explored conditions of infection. The following experiments were designed to provide mechanistic insights into Eritoran-mediated protection and to identify the pathways that contribute to influenza-induced disease that are affected by Eritoran treatment.
Elucidation of signaling requirements underlying influenza-induced lethality and protection by Eritoran
Eritoran blocks TLR4 signaling by binding in the deep hydrophobic pocket of its co-receptor, MD2, thereby blocking ligand-induced dimerization19 .To validate the role of TLR4 in PR8-induced lethality, mice were treated with a highly specific anti-TLR4 antibody20 (Figure 1A). Anti-TLR4 IgG, but not an isotype-matched control IgG, administered i.v. on day 2 only or on days 2 and 4 post-PR8 infection, protected mice against lethal infection (Figure 1B) and elicited significantly improved clinical scores (p < 0.004; Figure 1C). This result confirms that TLR4 signaling is, indeed, central to influenza-induced lethality and clinical symptoms.
Figure 1.

Anti-TLR4 IgG treatment protects mice from lethal influenza challenge. (A) C57BL/6J mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.). Mice received either control IgG or a highly specific anti-TLR4 IgG (2 mg/mouse; i.v.) once (day 2 only) or twice (days 2 and 4). Survival (B) and clinical scores (C) were monitored daily. Each graph represents the combined results of 2 separate experiments (5 mice/treatment group/experiment).
TLR4 activates both the MyD88- and TRIF-dependent signaling pathways8. One of the central conclusions of Imai et al.14 was that TLR4-mediated ALI induced by inactivated H5N1 influenza or the host-derived oxidized phospholipid, OxPAPC, is entirely TRIF-dependent. However, MyD88 has been implicated in the host response to influenza9,12. IRAK4, the first enzyme recruited to MyD88, initiates signaling leading to IKKα/β/γ complex activation, lκBα phosphorylation, and ultimately, NF-κB activation. The TRIF pathway drives IRF3 activation and results in delayed NF-κB activation, independent of IRAK421. To delineate the downstream pathway(s) underlying the host response to influenza and the protective mechanisms of Eritoran, we compared PR8-induced lethality and the efficacy of Eritoran in IRAK4 kinase dead knock-in (IRAK4KDKI) mice that have a catalytically inactive form of IRAK4 that blocks MyD88-dependent signaling, vs. TRIF-/- mice. IRAK4KDKI mice exhibited a slightly delayed mean time to death compared to WT mice, and Eritoran therapy resulted in ∼60% survival compared to ∼90% in WT mice (Figure 2A). Interestingly, TRIF-/- mice were more resistant to PR8 infection than WT or IRAK4KDKI mice (∼50% survival), but not as refractory as TLR4-/- mice16,17. However, treatment with Eritoran significantly improved the survival of TRIF-/- mice to WT levels (p < 0.001; Figure 2B). VIPER is peptide TLR4-inhibitory peptide derived from the A46 protein of vaccinia virus that has been shown to inhibit both MyD88-and TRIF-dependent TLR4 signaling by binding to and targeting the sorting adaptors TIRAP and TRAM22. When WT mice were infected with PR8 and treated therapeutically with either a cell-permeating VIPER peptide, 9R-VIPER, or Eritoran, 9R-VIPER treatment resulted in partial protection (∼50%), consistent with a role for TIRAP and/or TRAM in protection (Supplemental Figure 2). Thus altogether, both MyD88- and TRIF-dependent pathways contribute to influenza-mediated disease and Eritoran-induced protection.
Figure 2.
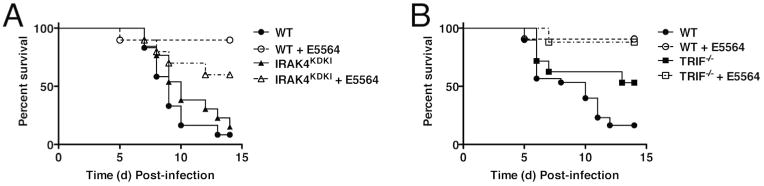
Effect of Eritoran on IRAK4KDKI and TRIF-/- mice. WT C57BL/6J (A and B), IRAK4KDKI (A) and TRIF-/- (B) mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.). Mice received vehicle (saline; i.v.) or Eritoran (E5564; 200 μg/mouse; i.v) daily from day 2 to day 6 post-infection. Survival was monitored for 14 days. Data shown is combined results of 2-3 separate experiments (5-10 mice/treatment group/experiment).
We reported previously that TLR2-/- mice were similarly sensitive to WT mice for PR8-induced lethality. However, unlike WT mice, Eritoran therapy failed to protect TLR2-/- mice; thus, TLR2 was presumed to be a direct or indirect target for Eritoran16. To confirm the role of TLR2 in influenza-induced disease, we used a monoclonal antibody (mAb) directed against TLR2 (clone T2.5) that blocks TLR2-mediated signaling in vivo23. Groups of mice were either treated with anti-TLR2 or with an isotype control antibody 3 h prior and 1 day post-PR8 infection, while two other groups of mice received anti-TLR2 or control antibody on days 2 and 4 post-PR8 infection (Figure 3A). Similar to the protection achieved with anti-TLR4 IgG (Figure 1), treatment of PR8-infected WT mice with anti-TLR2 antibody significantly protected against lethality when administered on days 2 and 4 post-infection (p < 0.001; Figure 3B); however, anti-TLR2 treatment was not effective when administered earlier. These results suggest the presence of a TLR2 agonist released late after PR8 infection contributes to lethality.
Figure 3.
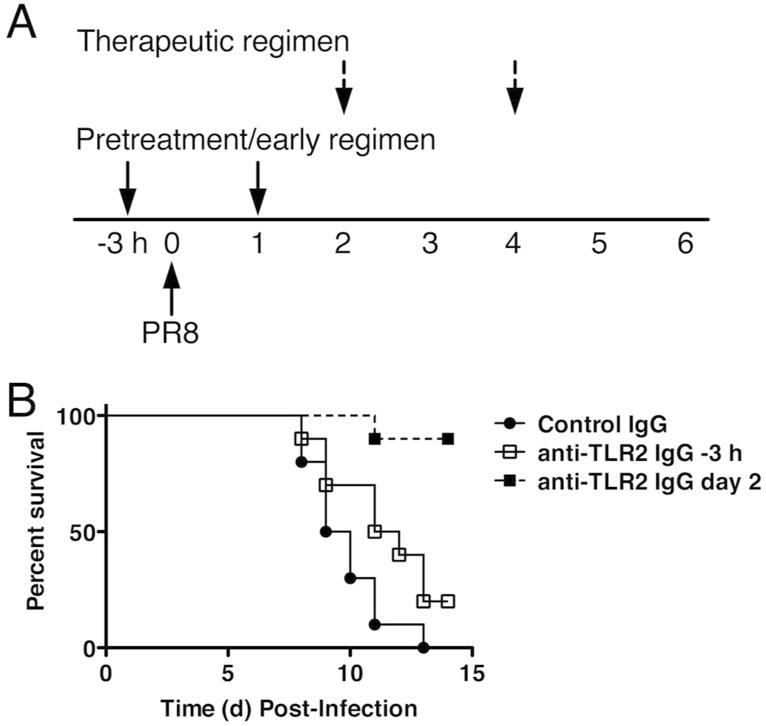
Anti-TLR2 IgG treatment protects mice from lethal influenza challenge. (A) Experimental protocol. C57BL/6J mice were either treated with isotype control IgG or anti-TLR2 (T2.5; 100 μg/ms; i.v.) 3 h prior to and 1 day post-infection or on days 2 and 4 post-infection. Survival (B) was monitored daily. Data shown is combined results of 2 separate experiments (5 mice/treatment group/experiment).
To extend these findings, WT, TLR2-/-, TLR4-/-, and TLR2/4 double knockout mice were infected with a sub-lethal dose (LD10) of PR8 and monitored for 14 days. The TLR2/4 double knockout mice were much more susceptible than the WT or individual knockout mice (Supplementary Figure 3A). ALI was significantly worse in TLR2/4 double-knockout mice than in WT, with inflammatory infiltrates throughout the parenchyma and alveolar spaces (composed of neutrophils and lymphocytes) (Supplementary Figure 3B). These findings suggest that a TLR2 agonist induced early during virus infection is necessary for the resistance of TLR4-/- mice to lethal PR8 infection.
Timing of Eritoran treatment is critical for protection
Neither differential influenza replication (Figure 5A, left panel) nor the levels of inducible IFN-β mRNA (Figure 4A, right panel) accounted for the resistance of the TLR4-/- mice to PR8 infection. Eritoran therapy protected PR8-infected WT mice (Figure 4B and 4C, open circle, left panel), but did not affect the resistance of TLR4-/- mice (Figure 4B and 4D; open circle, right panel), as we reported previously16. However, when Eritoran treatment was initiated prophylactically (3 h prior to PR8 infection) and continued daily for an additional 4 days (Figure 4B, pretreatment/early regimen; closed squares), WT mice were not protected from lethality (Figure 4C, closed square, left panel). This finding implies that an early influenza-inducible, but late-acting mediator of lethality and ALI must be the target of Eritoran in WT mice. Surprisingly, this identical regimen rendered TLR4-/- mice susceptible to PR8 infection (Figure 4B and 4D, closed square, right panel), indicating that a non-TLR4 target of Eritoran is necessary for the resistance of TLR4-/- mice to PR8 infection, consistent with our data obtained with the TLR2/4 double knockout mice. Pre-/early treatment with Eritoran had no effect on the susceptibility of CD14-/- or TLR2-/- mice (data not shown).
Figure 5.

Eritoran blunts HMGB1-induced TLR4 signaling in vitro and influenza-induced HMGB1 release and lethality in vivo. (A) Thioglycollate-elicited C57BL/6J macrophages were treated with medium alone (M) or Eritoran (E; 10 ng/ml) for 1 h, and then with LPS (L; 10 ng/ml) or HMGB1 (H; 1 μg/ml) for 2 h. Total RNA was processed and subjected to qRT-PCR for detection of TNFα or IFN-β mRNA expression. (B) Cotton rats (6-8/time point/treatment) were infected with pdH1N1 A/California/04/09 (9 × 105 TCID50 by i.n.), then treated with Eritoran or saline starting at 2 days post-infection. Serum HMGB1 levels were measured by ELISA (* p < 0.05). (C) C57BL/6J mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.). Mice received control peptide (500 μg/mouse; i.p.), Eritoran (E5564; 200 μg/mouse; i.v.), or the HMGB1 peptide, P5779 (500 μg/mouse; i.p.) from day 2 to day 6 post-infection. Survival (C) and clinical scores (D) were monitored daily for 14 days. Data shown is combined from 2 experiments (5-10 mice/treatment group/experiment).
Figure 4.
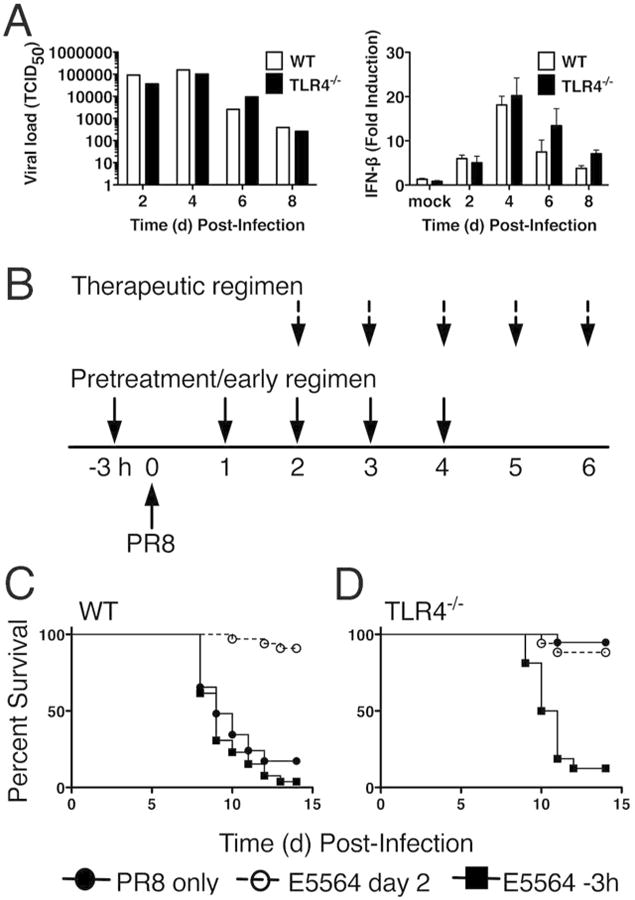
Pretreatment of WT or TLR4-/- mice with Eritoran does not protect from lethal influenza challenge. (A) WT and TLR4-/- mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.) and sacrificed on days 2, 4, 6 and 8 post-infection. Lungs were harvested and assayed for viral titers and IFN-β mRNA. Data shown is combined results of 2 separate assays (4-5 mice/group/experiment). (B) Basic experimental protocol used to compare pretreatment/early vs. therapeutic treatment with Eritoran in PR8-infected mice. C57BL/6J WT and TLR4-/- mice were either untreated (solid circles), or treated with Eritoran starting 3 h prior to infection and on day 1 for 4 successive days (3 h; solid squares; pretreatment/early regimen), or starting 2 days after infection for 5 successive days (open circles; therapeutic regimen). (C and D) Mice were monitored daily for survival. The data represent the combined results of two separate experiments (5-6 mice/treatment/experiment).
P5779, an HMGB1 antagonist, blocks influenza-mediated lethality
We previously reported that lungs of Eritoran-treated, PR8-infected mice (as shown in Figure 1A) showed blunted cytokine induction as well as accumulation of OxPAPC16, a danger associated molecular pattern (DAMP) shown by Imai et al. to mediate ALI by its action on macrophages through TLR414. High Mobility Group B1 (HMGB1), a DAMP first implicated in endotoxicity and Gram negative sepsis24, has been reported to be released during severe influenza infection25 and to activate TLR4 by binding to the TLR4 co-receptor, MD226. HMGB1-stimulated WT murine macrophages induced MyD88- (TNFα) and TRIF- (IFN-β) dependent gene expression that was inhibited by Eritoran in vitro (Figure 5A). Mice infected with PR8 (data not shown) and cotton rats infected with a non-adapted human influenza pdH1N1 strain, exhibited increased circulating HMGB1 that was inhibited by Eritoran treatment in vivo (Figure 5B). Thus, HMGB1, like OxPAPC, may represent a DAMP that is released relatively late after infection that contributes to influenza-induced ALI through TLR4 activation. P5779 is a small molecule inhibitor of HMGB1 that was shown recently to prevent MD-2/HMGB1 interaction and block HMGB1-induced TLR4 signaling, while not interfering with LPS-induced cytokine/chemokine induction27. P5779 protected mice against hepatic ischemia/reperfusion injury, APAP chemical toxicity, and sepsis27. To assess the efficacy of P5779 in influenza infection, WT C57BL/6J mice were infected with PR8 and, two days later mice were treated with either Eritoran (E5564), an inactive control peptide, or with P5779 for 5 consecutive days. Both Eritoran- and P5579-treated mice showed significant survival and lowered clinical scores, while mice treated with the control inhibitor showed higher clinical scores and succumbed to infection (Figure 5C and 5D).
A PAR2 antagonist blocks influenza-induced lethality and lung leak
A host-derived protein, originally termed zonulin (now known to be pre-haptoglobin 2)28, was found to increase intestinal permeability by phosphorylation of tight junction proteins29. Signaling was dependent upon protease-activated receptor 2 (PAR2)28, a signaling protein that we have shown previously to interact physically and functionally with TLR430. A zonulin analog peptide antagonist, AT-1001 (larazotide acetate), is well-tolerated in humans and attenuates gut inflammation associated with celiac disease31. Recently, AT-1001 was also reported to attenuate ALI in mice induced by intrapulmonary deposition of IgG immune complexes or LPS by inhibiting phosphorylation of the tight junction protein, ZO-1, reducing the number of leukocytes and myeloperoxidase activity in bronchoalveolar lavage fluid (BALF)32. Since both PAR2-/- and TLR4-/- mice are comparably refractory to lethal PR8 infection17, we compared the efficacy of AT-1001 vs. Eritoran therapy during a lethal influenza challenge. WT mice were infected with PR8 and treated with vehicle (saline), Eritoran, or AT-1001 for 5 consecutive days starting on day 2 post-infection. Treatment of mice with AT-1001 resulted in significant protection and lowered clinical scores, comparably to Eritoran treatment (Figures 6A, B). Since AT-1001 reduced the pulmonary edema associated with LPS- or immune complex-induced ALI32, we tested whether Eritoran and AT-1001 mediated a decrease in lung edema caused by PR8 infection as measured by wet-to-dry (W/D) weight ratio. Mice infected with PR8 and treated with vehicle showed significantly higher W/D ratios than mice infected with PR8 and treated with either Eritoran or AT-1001 (Figure 6C), suggesting that an additional protective effect of Eritoran during influenza infection is to attenuate ALI by blunting pulmonary edema.
Figure 6.
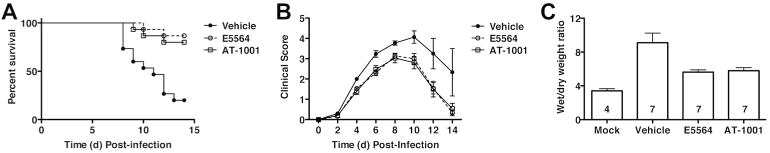
Effect of AT-1001 against lethal influenza challenge. (A) C57BL/6J mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.; ∼LD90). Mice received vehicle (saline; i.v.), Eritoran (E5564; 200 μg/mouse; i.v.), or AT-1001 (150 μg/mouse; i.v.) from day 2 to day 6 post-infection. Survival (A) and clinical scores (B) were monitored daily for 14 days. Data shown is combined from 3 separate experiments (5 mice/treatment group/experiment). (C) Lung wet-to-dry (W/D) weight ratio as an index for pulmonary edema after infection. C57BL/6J mice were infected and treated as described above. On day 7 post-infection, lungs were harvested and and lung weights were measured immediately after excision and recorded as wet weight. Lung tissue was air dried for 5-6 days and re-weighed until a stable dry weight obtained. The W/D weight ratio was calculated by dividing the wet by dry weight. The N for each group is indicated in each bar. Each vertical bar represents the mean (+ s.e.m.).
Contribution of IL-1 to influenza-induced lethality
We previously showed that IL-1β mRNA is strongly inhibited in the lungs of PR8-infected, Eritoran-treated mice16. Subsequently, Teijero et al. reported that influenza-infected IL-1R-/- mice exhibited diminished cytokine levels in the BALF 2 days p.i.12, but neither ALI nor lethality were evaluated. rIL-1 receptor antagonist (IL-1Ra; generic anakinra) is used clinically to treat highly inflammatory diseases (e.g., rheumatoid arthritis, cryopyrin-associated periodic syndromes and Macrophage Activation Syndrome)33. PR8-infected C57BL/6J WT mice treated therapeutically with rIL-1Ra showed significant, but intermediate, survival (Figure 7A), and clinical scores (Figure 7B), in contrast to the protection afforded by Eritoran. This suggests that while IL-1β participates in mediating influenza-induced disease, other mediators, whose action is inhibited by Eritoran, are likely involved.
Figure 7.
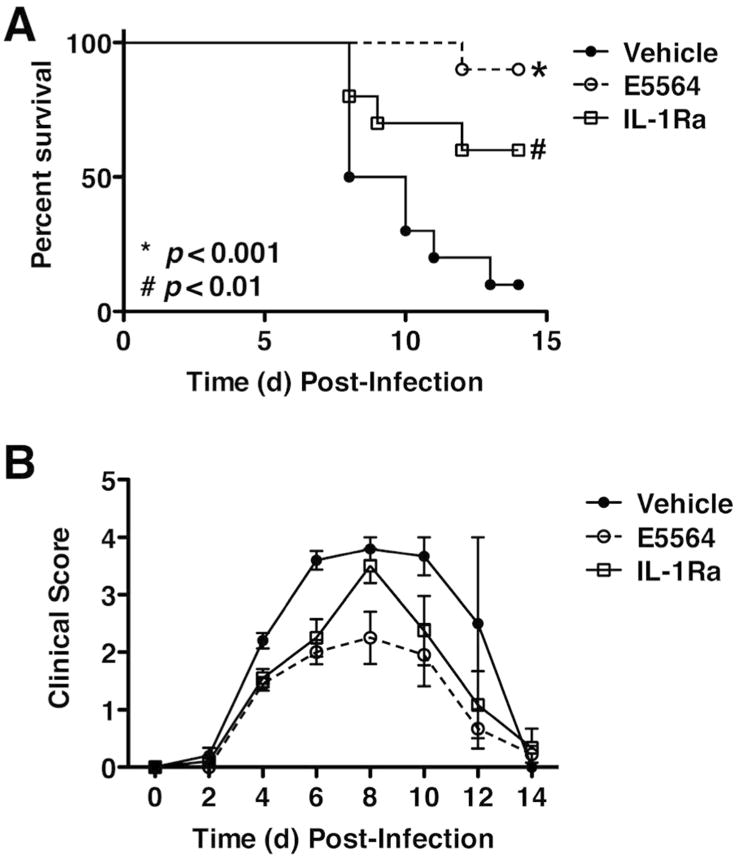
Effect of IL-1Ra against lethal influenza challenge. C57BL/6J mice were infected with mouse-adapted influenza strain PR8 (∼7500 TCID50, i.n.). Mice received vehicle (saline; i.v.), Eritoran (E5564; 200 μg/mouse; i.v.), or the IL-1Ra (150 μg/mouse; i.v.) from day 2 to day 6 post-infection. Survival (top panel) and clinical scores (bottom panel) were monitored daily for 14 days. Data shown is combined from 2 separate experiments (5 mice/treatment group/experiment).
Eritoran treatment improves the efficacy of oseltamivir therapy against influenza
The efficacy of Eritoran administered alone or combined with the approved neuraminidase inhibitor antiviral therapy, Tamiflu™ (oseltamivir), was assessed. To be effective, oseltamivir treatment is recommended within 2 days of the onset of symptoms34,35. Since, in our model, mice show clinical symptoms starting at day 3 post-infection16, we compared mice treated with vehicle, Eritoran alone, oseltamivir alone, or both Eritoran and oseltamivir starting on days 2, 4, or 6 post-infection for 5 consecutive days. When treatment was initiated on day 2 post-infection, both agents were highly protective, with no benefit when administered together (Supplementary Figure 4). However, when administration of treatment was delayed until 4 or 6 days post-infection, treatment with oseltamivir alone showed little protective effect (particularly when administered starting at Day 6), while Eritoran still elicited a significant degree of protection from lethality, as we reported previously16. Importantly, when initiated on day 4, the combined Eritoran/oseltamivir treatment resulted in a significant improvement in survival (p < 0.05; Supplemental Figure 4A), as well as significant reduction of viral titers (p < 0.05; Supplemental Figure 4B). This is consistent with Zheng et al. who showed co-administration of celecoxib, a COX-2 inhibitor, and zanamivir improved survival of influenza-infected mice better than treatment with zanamivir alone36. Our previous study showed that Eritoran blunts COX-2 induction during influenza infection16.
Survivors of PR8 infection after Eritoran treatment are protected from lethal re-infection
Importantly, mice that were protected from PR8 by Eritoran survived secondary PR8 challenge 4 weeks after initial infection, without additional Eritoran therapy (Supplementary Figure 5A). The half-life of Eritoran is approximately 5 h in rodents37, so it would not be expected to be involved in the protective effect seen when survivors were re-infected 28 days after the first PR8 infection. To test this hypothesis, mice were treated with Eritoran for five successive days. Five days after the last Eritoran treatment, mice were infected with PR8. A control group was treated with Eritoran starting 2 days post-infection for 5 consecutive days (days 2-6). As shown in Supplementary Figure 5B, the lethality seen in the mice given Eritoran early is equivalent to mice that were given vehicle (saline), supporting the idea that the half-life of Eritoran is too short to mediate protection observed upon re-challenge of survivors (Supplementary Figure 5A). To confirm that the Eritoran-treated, PR8-infected surviving mice develop an adaptive memory response, we repeated the assay from Supplementary Figure 5A and carried out hemagglutination inhibition assays using the sera from mice infected and treated with Eritoran. In mice that were PR8-infected, followed by Eritoran treatment (days 2-6), strong hemagglutination inhibition titers were observed four weeks after infection (ranging from 640 to 2560; n =5). Thus, the anti-inflammatory effect of Eritoran during primary infection does not prevent development of an adaptive immune response against influenza.
Discussion
Influenza is a major health concern globally. The virus mutates rapidly, leading to anti-viral resistance or altered expression of immunogenic epitopes such that extant vaccines are rendered ineffective. Based on our previous studies16, we demonstrated that Eritoran (E5564), a well-tolerated, synthetic TLR4 antagonist38,39, represents a novel therapeutic approach to ameliorate influenza-induced ALI by blocking TLR-mediated signaling in response to host-derived DAMPs. Herein, we delineate further the cellular and molecular underpinnings for both induction of ALI and its abatement by Eritoran therapy.
Our data show that Eritoran, administered after infection, does not alter the refractoriness of TLR4-/- mice to influenza infection16; both Eritoran and anti-TLR4 antibody therapy protected WT mice, and, Eritoran binds to both CD14 and MD2 in vitro16, both of which are required for TLR4 signaling19. In addition to 9R-VIPER (Suppl. Fig. 2), the cell-permeating TLR2 TIR decoy peptide, 2R9, that blocks TLR4, 7, and 9 signaling by blocking recruitment of TIRAP to the TLR TIR domain40, protected WT mice from PR8-induced lethality comparably to Eritoran. Together, these findings strongly implicate TLR4 in both influenza-induced disease and as a target for Eritoran-mediated protection. Since Eritoran pre/early treatment renders TLR4-/- mice susceptible to PR8 infection, Eritoran must also interact with a non-TLR4 PRR that is required early for induction of resistance. We postulate that Eritoran, administered by the pre/early treatment, binds CD14 and inhibits transfer of specific PAMPs or DAMPs to TLR241, TLR342, TLR7, and/or TLR943 since CD14 has been shown to act as a co-receptor for each of these PRRs, and since the latter three have been implicated in the host response to influenza 5-12 and are capable of inducing IFN-β21 . This hypothesis is supported by our observation that WT and TLR4-/- mice, when treated with Eritoran by the early regimen, are as susceptible as IFN-β-/- mice to PR8 infection16, and that CD14-/- mice could not be protected by Eritoran pre/early or therapeutic regimens16, presumably because they cannot produce cytokines as suggested by Pauligk et al13. Our previous work showed that 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a potent inducer of IFN-β, protected WT, but not IFN-β-/- mice, from PR8-induced lethality when administered 3 h prior to and on day 1 post-PR8 infection44, again supporting the need for IFN-β early in infection for survival.
The role of TLR2 in Eritoran-mediated protection of PR8-infected mice is enigmatic. Despite the fact that we observed that anti-TLR2 mAb, but not control IgG, protected WT mice from PR8 when administered therapeutically, the same anti-TLR2 mAb, when administered -3 h prior to and 1 day after PR8 infection, provided only minimal protection. This observation strongly suggests that TLR2, like TLR4, also plays a damaging role later in infection. OxPAPC45 and HMGB126 have been reported to be both TLR2 and TLR4 agonists that are induced later in infection. These DAMPs may synergize to increase TLR2 expression and/or TLR2-dependent signaling. Our observation that TLR2/4 double knockout mice are highly susceptible to sublethal PR8 infection may suggest that the absence of both TLR2 and TLR4 would leave MyD88 more available for IL-1 and IL-18 signaling. Alternatively, it is also possible that TLR2 is required to produce something that mediates resistance to PR8 in TLR4-/- mice.
Eritoran blunts both the influenza-induced “cytokine storm” and the accumulation of OxPAPC16, an oxidized phospholipid TLR4 DAMP14. Mechanistically, these findings have now been extended by showing that (i) Eritoran blocked HMGB1-mediated TLR4-dependent signaling in vitro, HMGB1 release into serum in vivo, and protected comparably to P5779, a highly selective HMGB1 inhibitor; and (ii) inhibited pulmonary lung edema equivalently to AT-1001, an inhibitor of zonulin-induced pulmonary edema32. Others have shown that influenza induces necroptosis in lung epithelial cells46,47, and that HMGB1 is released from necroptotic cells48. Therefore, it is possible that treatment of mice with Eritoran blocks release of HMGB1 by blocking necroptosis as well as the subsequent signaling through the TLR4/MD2 complex. We previously reported that lung sections from placebo-treated mice showed significant pathology, including epithelial necrosis, and that Eritoran-treated mice had intact lung epithelia compared to the placebo group16.
Eritoran acts on cells that express TLR4/MD2. At this time, we do not know if stromal cells and/or cells of myeloid lineage are involved in the protective effect. Zhang et al. recently reported that, in addition to its profound effects on the lung, influenza infection affects mucosal epithelium in the intestinal tract49, consistent with our report of systemic inflammation induced by PR8 infection16 and our results showing that administration of AT-1001 (larazotide acetate), an inhibitor of intestinal and lung leakage28,31,32, reduced PR8-induced mortality. Thus, it is possible that influenza-mediated intestinal leakage underlies changes in gut microbiota that are required for development of adaptive immunity50. In a model of trauma/hemorrhagic shock, Sodhi et al. reported that ALI was induced in WT mice, but not in mice selectively engineered to lack TLR4 on gut epithelial cells51. In this model, ALI in WT mice could be blocked by neutralizing HMGB1 or by treatment with a small molecule TLR4 inhibitor51. Additional experiments will be required to test the hypothesis that DAMP release by influenza-induced necrosis of lung epithelial cells acts locally to induce ALI, as well systemically to cause gut leak and an altered microbiota50.
Additionally, the partial protection achieved by treatment of PR8-infected mice supports a role for IL-1 signaling in PR8-induced lethality, as suggested by Tejeiro et al.12; however, the data also suggest a role for additional inflammatory mediators in PR8-induced disease since the protection is partial. Nonetheless, the IL-1α precursor (pro-IL-1α) is expressed in the nuclei of most tissues bound to chromatin52. During an acute ischemic event, the loss of oxygenation and increased acidosis triggers release of the (pro-IL-1α into the cytosol and then into the milieu upon cellular necrosis. Pro-IL-1α binds to the IL-1R1 and is biologically active53, causing the induction of chemokines that mediate neutrophilic infiltration. Thus, the partial protection elicited by IL-1Ra (Figure 7) may be attributable to blockade of the interaction of pro-IL-1α with the IL-1R1, in addition to the down-regulation of IL-1β mRNA induced in PR8-infected, Eritoran-treated mice.
Our observation that Eritoran-treated mice that survived initial infection and exhibited blunting of the cytokine storm were protected from secondary influenza infection suggests that Eritoran does not prevent development of an adaptive immune response. During primary influenza infection, antibody levels are detectable by day 10 post-infection and persist above baseline for weeks54. We observed that significant levels of anti-HA antibodies were induced in mice infected with PR8 followed by Eritoran therapy on days 2-6, even 4 weeks after infection. Thus, Eritoran does not prevent induction of an adaptive immune response to infection. While such mice remain refractory to secondary PR8 infection 28 days after the primary infection, future studies will be required to determine if Eritoran treatment alters the enhanced sensitivity to secondary bacterial infections seen in influenza-infected mice.
Overall, our findings underscore the complex nature of microbe-host inflammatory cell interactions that control the host's ability to respond to invading virus. We provide evidence that multiple receptors on innate immune cells are likely involved; however, it is also likely that unknown interactions among these receptors in response to microbial and host ligands significantly affect, both qualitatively and quantitatively, the host response.
Methods
Reagents
Eritoran (E5564) was provided by Eisai Inc. (Andover, MA) and prepared as described previously16. Escherichia coli K235 LPS was prepared as previously described55. The anti-TLR2 IgG and isotype control IgG were purchased from Affymetrix (Santa Clara, CA). Recombinant HMGB1 was provided by Kevin Tracey (Feinstein Institute for Medical Research, Manhasset, NY). 9R-VIPER was synthesized by GenScript (Piscataway, NJ). P5779 and control peptide were provided by Yousef Al-Abed (Feinstein Institute for Medical Research, Manhasset, NY). The anti-TLR4 IgG and isotype control IgG were provided by Thierry Roger and Thierry Calandra (Infectious Diseases Service, Centre Hospitalier Universitaire Vaudois and University of Lausanne, Lausanne, Switzerland). AT-1001 was kindly provided by Alessio Fassano (Division of Pediatric Gastroenterology and Nutrition, MGH, Boston, MA). IL-1Ra (anakinra) was obtained from Sweden Orphan BioVitrum (SOBI), Stockholm, Sweden.
Mice and cotton rats
Six to 8-week old, WT C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice with targeted mutations were bred onto or derived directly from a C57BL/6J background. TLR4-/- mice (provided by Shizuo Akira, Osaka, Japan; bred at UMB (Baltimore, MD) and Univ. Massachusetts Medical School, (Worcester, MA), TRIF-/- (bred at U. Mass. Med. School). IRAK4KDKI (provided by Lilly Research Laboratories, Indianapolis, IN, bred at UMB), TLR2-/- mice (provided by Shizuo Akira; bred at U. Mass. Med. School), and TLR2/TLR4 double knockout mice (bred at U. Mass. Medical School (Worcester, MA). BALB/cByJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mouse strains were housed and bred in specific pathogen-free conditions. Experiments were conducted in accordance with the guidelines set forth by the University of Maryland, Baltimore and the University of Massachusetts Medical School Department of Animal Medicine and approved by each institute's Institutional Animal Care and Use Committees (IACUC).
Inbred young adult (4-8 weeks old) cotton rats (Sigmodon hispidus) were bred at Sigmovir Biosystems, Inc. (Rockville, MD). All cotton rat experiments were conducted with Institutional Animal Care and Use Committee (IACUC) approval from Sigmovir Biosystems, Inc.
Virus
Mouse-adapted H1N1 influenza A/PR/8/34 virus (“PR8”) (ATCC, Manassas, VA) was grown previously described56 and was kindly provided by Dr. Donna Farber (Columbia University, NY). Mouse-adapted H1N1 influenza maCa.04 was provided by Daniel Perez (University of Georgia, Athens, GA). Non-adapted human influenza virus strain pH1N1was obtained and grown as previously described57.
Virus challenge and treatments
Mice were infected with mouse-adapted influenza virus, strains A/PR/8/34 intranasally (i.n.) (PR8; ∼7500 TCID50, 25 μl/nares) or maCa.04 (∼2200 TCID50, i.n.). Two days after infection, mice received either vehicle or E5564 intravenously (Eritoran; 200 μg/mouse in 100 μl; i.v.), anti-TLR4 IgG (2 mg/mouse, i.v.) or its isotype control IgG, anti-TLR2 (T2.4 clone; 100 μg/mouse, i.v.) or its isotype control IgG, 9R-VIPER (200 nmol; i.p.), P5779 or its control peptide (500 μg/mouse; i.p.), AT-1001 (150 μg/mouse, i.v.), IL-1Ra (150 μg/mouse, i.v.), or Oseltamivir orally (p.o.) (1 mg/mouse) daily (Day 2 to Day 6). In some experiments, groups of mice were treated with Eritoran or Oseltamivir starting at day 4 or day 6 post-infection and treated for 5 successive days. In some experiments, some groups of mice were treated with E5564 (200 μg/mouse, i.v.) 3 h prior to infection with PR8 and then treated for 4 consecutive days starting day 1 post-infection (days 1-5). In some experiments, groups of mice were treated with anti-TLR2 (T2.5 clone, 100 μg/mouse, i.v.) or its control isotype IgG 3 h prior to infection with PR8 and then treated for a second time at day 1 post-infection for a total of two treatments. Mice were monitored daily for survival, weight loss, and clinical signs of illness (e.g., lethargy, piloerection, ruffled fur, hunched posture, rapid shallow breathing, audible crackling) for 14 days. A clinical score ranging from 0 (no symptoms) to 5 (moribund) was ascribed to each mouse daily16,58.
Histopathology
Lungs were inflated and perfused and fixed with 4% PFA. Fixed sections (8 μm) of paraffin-embedded lungs were stained with hematoxylin and eosin (H&E). Slides were randomized, read blindly, and examined for tissue damage, necrosis, apoptosis, and inflammatory cellular infiltration.
TLR2/4 double knockout mouse studies
C57BL/6J WT, TLR4-/-, TLR2-/-, and TLR2/4 double knockout mice were 8-12 weeks of age at the time of infection. Influenza A/PR/8/34 (Charles River Laboratories, Wilmington, MA) virus stocks were diluted in sterile phosphate buffered saline (PBS) and kept on ice prior to use. Mice were anesthetized with isoflurane and infected 40,000 pfu (30 μl; i.n.) per mouse. In two assays, mice were monitored for survival for 14 days post-infection. In another assay, mice were sacrificed on day 5 post-infection and the lungs harvested for pathology. Lungs were inflated in situ with 1 ml of formalin (10% formaldehyde) obtained from the U. Mass. Med. School Morphology Core. The inflated lungs were fixed for 24-48 h, after which they were transferred to PBS. Lungs were bisected vertically and processed for paraffin embedding by the University of Massachusetts Medical School Morphology Core. Slides were prepared and H&E stained for histological analysis.
Lung wet-to-dry weight ratio
The lung wet-to-dry (W/D) weight ratio was used as an index of pulmonary edema after infection with influenza in mice that were untreated or treated with either E5564 or AT-1001. On day 7 post-infection, mice were euthanized and dissected for the total lung, and the lung weight was measured immediately after excision (wet weight). Lung tissue was then air dried for 5-6 days and re-weighed everyday until a dry weight was stable and this acted as the final dry weight. The W/D weight ratio was calculated by dividing the wet by the final dry weight59.
Quantitative real-time PCR (qRT-PCR)
Total RNA isolation and qRT-PCR were performed as previously described16. Levels of mRNA for specific genes are reported as relative gene expression normalized to mock-infected lungs.
Macrophage cell cultures and treatment
Thioglycollate-elicited peritoneal macrophages from C57BL/6J WT mice were enriched as described16 after plating in 12-well (2×106 cells/well) tissue culture plates. Macrophages were pre-treated with Eritoran (10 ng/ml) for 1 h and then stimulated with LPS (10 ng/ml) or HMGB1 (1 μg/ml) for 2 h.
HMGB1 serum levels
Cotton rats were infected with 9 × 104 TCID50 of pH1N1 intranasally on day 0. On day 2 post-infection, animals were treated with saline (mock) or with 37.33 mg/kg of Eritoran i.v., daily. Blood samples were collected on days 0, 4, and 6 post-infection and serum was used for measuring HMGB1 levels using an ELISA kit according to manufacturer's protocol (IBL International, Toronto, Ontario, Canada).
Viral titration
Virus titers were obtained from supernatants of lung homogenates of PR8-infected mice that were either left untreated (NT) or treated with Eritoran only, Oseltamivir only, or both Eritoran and Oseltamivir and harvested on day 7 post-infection and expressed at TCID50/ml as described previously44.
Hemagglutination Inhibition (HAI) assay
Prior to serological analysis, mice sera were treated with receptor-destroying enzyme (RDE; Denka Seiken, Cat. # 370013). Serum was mixed with RDE and incubated at 37°C for 20 h, followed by heat inactivate at 56°C for 30 min. Finally, 0.6 ml of 1× PBS was added to give a 1:10 dilution of the initial sample. To quantify HAI activity, two fold serial dilutions of treated sera in 96-well plates were incubated with 4 hemagglutinating units of PR8 virus at room temperature for 15 min. Turkey red blood cells at 0.5% in PBS were added to each dilution and gently mixed. Plates were incubated for 30 min-1 h at room temperature. The HAI titer was reported as the reciprocal of the highest dilution of serum that inhibited hemagglutination.
Statistics
Statistical differences between two groups were determined using an unpaired, two-tailed Student's t test with significance set at p < 0.05. For comparisons between >3 groups, analysis was done by one-way ANOVA followed by a Tukey's multiple comparison test with significance determined at p < 0.05. For survival studies, a Log-Rank (Mantel-Cox) test was used.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI104541 (SNV and JCGB), DK048373 (AF), AI15614 (CD), Swiss National Science Foundation grants 320030_149511 (TR), 310030_138488 and 149511 (TC), by Science Foundation Ireland grant 11/PI/1056 (AGB).
Footnotes
Conflict of Interest: With the exception of FG, none of the authors have a competing interest. FG is an employee of Eisai Inc.
Author Contributions: KAS and SNV carried out the study design, with advice from JCGB, AF, CD, TR, KJT, AY, and EKJ. KAS, WL, MCP, LMP, CP, and EKJ performed experiments. FG, TR, TC, AB, AG, CD, KJT, AY, and EKJ provided crucial reagents and advice for this study. KAS and SNV prepared the manuscript with input and approval of all other co-authors.
References
- 1.Belongia EA, Sundaram ME, McClure DL, Meece JK, Ferdinands J, VanWormer J. Waning vaccine protection against influenza A (H3N2) illness in children and older adults during a single season. Vaccine. 2015;33:246–51. doi: 10.1016/j.vaccine.2014.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flannery B, et al. Early estimates of season influenza vaccine effectiveness-United States, January 2015. MMWR Morb Mortal Wkly Rep. 2015;64:10–15. [PMC free article] [PubMed] [Google Scholar]
- 3.Pebody RG, et al. Low effectiveness of season influenza vaccine in preventing laboratory-confirmed influenza in primary care in the United Kingdom : 2014/15 mid-season results. Euro Surveil. 2015;20 [PubMed] [Google Scholar]
- 4.McLean HQ, et al. Influenza vaccine effectiveness in the United States during 2012-2013: variable protection by and virus type. J Infect Dis. 2015;211:1529–40. doi: 10.1093/infdis/jiu647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leung YH, et al. Highly pathogenic avian influenza A H5N1 and pandemic H1N1 virus infections have different phenotypes in Toll-like receptor 3 knockout mice. J Gen Virol. 2014;95:1870–1879. doi: 10.1099/vir.0.066258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 7.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 9.Seo SU, et al. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J Virol. 2010;84:12713–12722. doi: 10.1128/JVI.01675-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai SY, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 2014;10:e1003848. doi: 10.1371/journal.ppat.1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci U S A. 2014;111:3799–3804. doi: 10.1073/pnas.1400593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauligk C, Nain M, Reiling N, Gemsa D, Kaufmann A. CD14 is required for influenza A virus-induced cytokine and chemokine production. Immunobiology. 2004;209:3–10. doi: 10.1016/j.imbio.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Imai Y, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lien E, et al. A novel synthetic acyclic lipid A-like agonist activates cells via the lipopolysaccharide/toll-like receptor 4 signaling pathway. J Biol Chem. 2001;276:1873–1880. doi: 10.1074/jbc.M009040200. [DOI] [PubMed] [Google Scholar]
- 16.Shirey KA, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nhu QM, et al. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;3:29–39. doi: 10.1038/mi.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye J, et al. Variations in the hemagglutinin of the 2009 H1N1 pandemic virus: potential for strains with altered virulence phenotype? PLoS Pathog. 2010;6:e1001145. doi: 10.1371/journal.ppat.1001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HM, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Roger T, et al. Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A. 2009;106:2348–2352. doi: 10.1073/pnas.0808146106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 22.Lysakova-Devine T, et al. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J Immunol. 2010;185:4261–71. doi: 10.4049/jimmunol.1002013. [DOI] [PubMed] [Google Scholar]
- 23.Meng G, et al. Antagonistic antibody prevents toll-like receptor 2-driven lethal shock-like syndromes. J Clin Invest. 2004;113:1473–1481. doi: 10.1172/JCI20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 25.Alleva LM, Budd AC, Clark IA. Systemic release of high mobility group box 1 protein during severe murine influenza. J Immunol. 2008;181:1454–1459. doi: 10.4049/jimmunol.181.2.1454. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14. doi: 10.1084/jem.20141318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tripathi A, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci U S A. 2009;106:16799–804. doi: 10.1073/pnas.0906773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldblum SE, et al. The active Zot domain (aa 288-293) increases ZO-1 and myosin 1C serine/threonine phosphorylation, alters interaction between ZO-1 and its binding partners, and induces tight junction disassembly through proteinase activated receptor. FASEB J. 2011;25:144–158. doi: 10.1096/fj.10-158972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rallabhandi P, et al. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem. 2008;283:24314–24325. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddling JB. The safety, tolerance, pharmacokinetic and pharmacodynamics effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–66. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 32.Rittirsch D, et al. Zonulin as prehaptoglobin2 regulates lung permeability and activates the complement system. Am J Physiol Lung Cell Mol Physiol. 2013;304:L863–72. doi: 10.1152/ajplung.00196.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khazeni N, Bravata DM, Holty JE, Uyeki TM, Stave CD, Gould MK. Systematic review: safety and efficacy of extended-duration antiviral chemoprophylaxis against pandemic and seasonal influenza. Ann Intern Med. 2009;151:464–473. doi: 10.7326/0003-4819-151-7-200910060-00143. [DOI] [PubMed] [Google Scholar]
- 35.Freund B, Gravenstein S, Elliott M, Miller I. Zanamivir: a review of clinical safety. Drug Saf. 1999;21:267–281. doi: 10.2165/00002018-199921040-00003. [DOI] [PubMed] [Google Scholar]
- 36.Zheng BJ, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci U S A. 2008;105:8091–8096. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaneko K, Ueda R, Nemoto H, Iijima H, Yoshimura T. Disposition of a synthetic analog of lipid A (E5564) in rats. Xenobiotica. 2003;33:323–339. doi: 10.1080/0049825021000058097. [DOI] [PubMed] [Google Scholar]
- 38.Kalil AC, et al. Influence of severity of illness on the effects of eritoran tetrasodium (E5564) and on other therapies for severe sepsis. Shock. 2011;36:327–331. doi: 10.1097/SHK.0b013e318227980e. [DOI] [PubMed] [Google Scholar]
- 39.Mullarkey M, et al. Inhibition of endotoxin response by E5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304:1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- 40.Piao W, et al. A decoy peptide that disrupts TIRAP recruitment to TLRs is protective in a murine model of influenza. Cell Rep. 2015;11:1941–1952. doi: 10.1016/j.celrep.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for Toll-like receptors. J Immunol. 1999;163:6748–6755. [PubMed] [Google Scholar]
- 42.Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA mediated TLR3 activation is enhanced by CD14. Immunity. 2006;24:153–163. doi: 10.1016/j.immuni.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Baumann CL, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207:2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirey KA, et al. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol. 2011;89:351–357. doi: 10.1189/jlb.0410216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadl A, et al. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. 2011;51:1903–1909. doi: 10.1016/j.freeradbiomed.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodrigue-Gervais IG, et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe. 2014;15:23–35. doi: 10.1016/j.chom.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol. 2014;22:199–207. doi: 10.1016/j.tim.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 48.Qing DY, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med. 2014;190:1243–1254. doi: 10.1164/rccm.201406-1095OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S, et al. Small intestinal injury in mice infected with respiratory influenza A virus: evidence for virus induced gastroenteritis. Biotechnol Lett. 2015;37:1585–1592. doi: 10.1007/s10529-015-1847-8. [DOI] [PubMed] [Google Scholar]
- 50.Ichinohe T, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sodhi CP, et al. Intestinal epithelial TLR-4 activation is required for development of acute lung injury after trauma/hemorrhagic shock via the release of HMGB1 from the gut. J Immunol. 2015;194:4931–4939. doi: 10.4049/jimmunol.1402490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen I, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim B, et al. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol. 2013;4:391. doi: 10.3389/fimmu.2013.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swain SL, Dutton RW, Woodlan DL. T cell responses to influenza virus infection: effector and memory cells. Viral Immunol. 2004;17:197–209. doi: 10.1089/0882824041310577. [DOI] [PubMed] [Google Scholar]
- 55.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee Y. Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry. 1967;6:2363–2376. doi: 10.1021/bi00860a011. [DOI] [PubMed] [Google Scholar]
- 56.Teijaro JR, et al. Costimulation modulation uncouples protection from immunopathology in memory T cell response to influenza virus. J Immunol. 2009;82:6834–43. doi: 10.4049/jimmunol.0803860. [DOI] [PubMed] [Google Scholar]
- 57.Blanco JC, et al. Receptor characterization and susceptibility of cotton rats to avian and 2009 pandemic influenza virus strains. J Virol. 2013;87:2036–45. doi: 10.1128/JVI.00638-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tate MD, Brooks AG, Reading PC. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection in mice. Respiratory Res. 2008;9:57–70. doi: 10.1186/1465-9921-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng C, et al. Neuraminidase reprograms lung tissue and potentiates lipolysaccharide-induced acute lung injury in mice. J Immunol. 2013;191:4828–37. doi: 10.4049/jimmunol.1202673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.