

Introduction
It has become clear that reactive oxygen species (ROS) contribute to the development of hypertension via myriad effects. ROS are essential for normal cell function, however they mediate pathological changes in the brain, the kidney and blood vessels that contribute to the genesis of chronic hypertension. There is also emerging evidence that ROS contribute to immune activation in hypertension. In this review, we will discuss these events and how they coordinate to contribute to hypertension and its consequent end-organ damage.
Reactive oxygen species
Reactive oxygen species (ROS) are formed by oxidation-reduction reactions in which one molecule is reduced by removal of an electron, which is then transferred to a recipient molecule. ROS can be divided into two major groups: free radicals and non-radical derivatives. Free radicals possess an unpaired electron in their outer orbital, which make them highly reactive. These include superoxide (O2·−), the hydroxyl radical (OH.), lipid peroxy-radicals (LOO.) and alkoxy- radicals (LO.). Nitric oxide (NO) is also a free radical, and often referred to as a reactive nitrogen species. Non-radical ROS include hydrogen peroxide (H2O2), peroxynitrite (ONOO−), hypochlorous acid (HOCl−) and reactive carbonyls. These do not possess unpaired electrons, and are more stable with a longer half-life but have strong oxidant properties.
Physiological roles of ROS
Although originally considered toxic by-products of cellular metabolism, ROS are now recognized to have signaling roles that are critical for normal cell function, including proliferation, differentiation, aging, host defense and repair processes. Recent studies show that ROS, including H2O2, may drive pro-survival signaling and protect from the aging process.1 As a part of innate immunity, ROS not only contribute to host defense via respiratory bursts in phagocytes, but also by signaling chemotaxis of inflammatory cells to sites of infection or injury. Related to this, ROS also participate in tissue repair and remodeling by inducing expression of matrix metalloproteinases (MMPs).2 These responses, which are vital for normal cell function, become exaggerated in disease states and promote pathological processes.
Oxidative stress
The term oxidative or oxidant stress traditionally refers to an imbalance between the production of reactive oxygen species and antioxidant defenses. This can lead to an increase in ambient levels of ROS that can damage various cellular components including DNA, proteins and lipids. This traditional definition of oxidant stress has been modified, because it is now clear that such an imbalance might be localized to subcellular compartments such the mitochondria, the nucleus or localized at the cellular membrane. Localized alterations of ROS production in the mitochondria can affect energy homeostasis, while localized ROS production in the nucleus can affect transcriptional events and epigenetic control. Extracellular ROS can participate in outside in signaling and affect cellular function.
Major ROS molecules
Superoxide radical
Superoxide, produced by 1-electron reduction of molecular oxygen, can act both as an oxidant and as a reductant in biological systems, depending on the redox potential of the molecule with which it is reacting. Superoxide is important as it serves as the progenitor for many other biologically relevant ROS, including hydrogen peroxide (H2O2), the hydroxyl radical (HO·) and peroxynitrite (OONO−), which forms upon reaction of O2·− with NO.
Hydrogen Peroxide
Hydrogen peroxide is formed by dismutation of O2·−, which can occur either spontaneously or can be catalyzed by the superoxide dismutases (SODs). In contrast to O2·−, H2O2 is relatively stable under physiological conditions. Because it is uncharged and lipophilic, H2O2 can readily diffuse across membranes and thus can react with targets in organelles and cells apart from where it is formed. In this regard, H2O2 has been implicated as a signaling molecule that can, among other actions, promote vasodilatation, activate gene transcription, modify phosphatase activity and activate other sources of ROS. As part of the antioxidant defense mechanisms, catalase and glutathione peroxidase (Gpx) can further reduce H2O2 to H2O. Myeloperoxidase catalyzes the reaction of H2O2 with the chloride ion to generate hypochlorous acid (HOCl−), which is a strong oxidant with high reactivity. Other peroxidases use alternate anions to generate other oxidants. Some of these reactions are illustrated in Figure 1.
Figure 1.
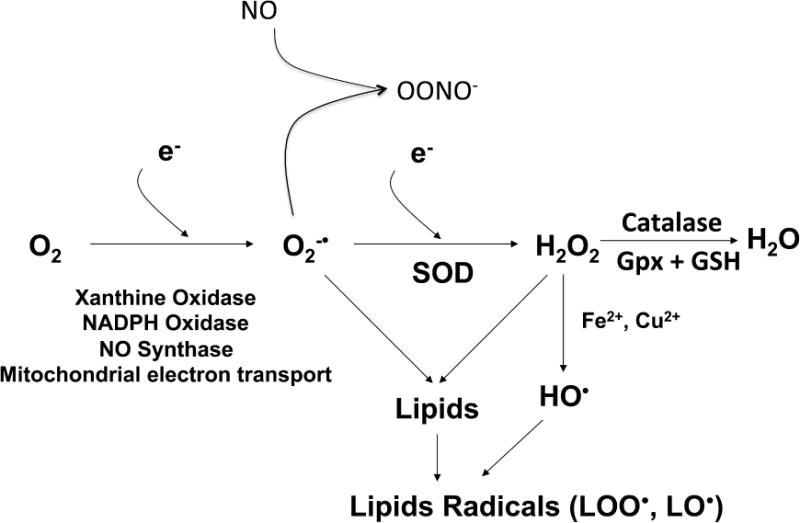
Sources and formation of reactive oxygen species in mammalian cells that are relevant to hypertension.
Hydroxyl radical
Hydroxyl is formed when O2·− reacts with H2O2 in the Haber-Weiss reaction where O2·− donates one electron to H2O2. Hydrogen peroxide can also accept one electron from the Ferrous cation (Fe2+) in the Fenton reaction to generate OH. and a Ferric cation (Fe3+). The hydroxyl radical is a highly reactive oxidant that can attack a variety of biomolecules including lipids, proteins and DNA.
Peroxynitrite
As mentioned above, OONO− is the product of the spontaneous reaction between O2·− and NO. This reaction is essentially diffusion limited, with a rate that has been estimated to be 9 × 109 mols × sec−1. At physiological pH, OONO− exists in the protonated form, HOONO or peroxynitrous acid, which is uncharged and can diffuse across cell membranes. Moreover, HONOO undergoes homolysis to yield hydroxyl, and in fact might serve as a more important source of this radical that the Fenton reaction mentioned above. Like hydroxyl, OONO− is a very strong oxidant and can react with lipids, DNA and proteins. Peroxynitrite can react with and modify proteins and other cellular structures causing oxidative damage to these macromolecules. In particular, OONO− modifies protein tyrosine residues to form 3-nitrotyrosine, a biomarker for OONO− in tissues and blood.
Reactive carbonyls
Carbonyls are highly reactive molecules that contain a carbon atom double bonded to an oxygen atom (C=O). These are formed both enzymatically and non-enzymatically from lipids, sugars and proteins, and include reactive aldehydes and advanced glycation end products. These have been extensively reviewed elsewhere,3 but are highly reactive and can modify macromolecules such as proteins and DNA, forming cross-links and altering both structure and function. Reactive carbonyls accumulate with aging, increased oxidative stress and in the setting of various diseases, and likely underlie the pathogenesis of these diseases. An example is isolevuloglandin-modification of lysines, which as noted below, produces protein modifications that lead to immune activation in hypertension.4
Sources of ROS
NADPH oxidase
The NADPH oxidases are major sources of ROS in mammalian cells. The NADPH oxidase was first identified in professional phagocytes as a multi-subunit enzyme complex responsible for the oxidative burst used to kill invading micro-organisms. The catalytic subunit of the neutrophil oxidase is also referred to as gp91phox, due to its apparent molecular weight and the fact that it is extensively glycosylated. Subsequently, it was discovered that there are 6 related proteins, which have been named the Nox enzymes, termed Nox 1 to 5 and the two Duox enzymes. These vary in tissue distribution, function and regulatory mechanisms. Nox1 exists in colon, muscle, prostate, uterus and blood vessels and plays important roles in host defense and blood pressure regulation. As noted above, Nox2 is found in phagocytes, where it is responsible for the oxidative burst. It is also present in endothelial cells, tubular cells of the kidney and in other immune cells. Nox3 is present in fetal tissue and in the inner ear where it is essential for vestibular function. Nox4, expressed in kidneys, vessels and bone, is involved in vasoregulation and erythropointin synthesis. Nox5, not present in rodents, is a Ca2+-dependent homolog that is activated in response to intracellular Ca2+ mobilization in lymph nodes, testes and blood vessels. Nox5 is also expressed in atherosclerotic lesions and seems to accumulate in more complex lesions.5 Duox1/2 are distant Nox homologues involved in hormone biosynthesis in the thyroid.
Nox 1 through 4 require the small docking subunit p22phox for function and stability. In addition the various Nox enzymes are activated upon translocation of cytoplasmic subunits. For Nox2, these include p47phox, p67phox, p40phox and the small g protein rac. For Nox1, alternate subunits, termed NoxA1 and NoxO1 can substitute for p47phox and p67phox. Poldip2 associates with Nox4 and positively regulates its activity and cellular functions.6 Factors that cause assembly of these cytoplasmic units with the membrane components include inflammatory cytokines, growth factors, mechanical forces and various G protein coupled receptor agonists. Of particular importance to cardiovascular disease, ang II activates the NADPH oxidases via the AT1 receptor and stimulation of a signaling pathway involving c-Src, protein kinase C (PKC), phospholipase D (PLD) and phospholipase A2 (PLA2).7
The NADPH oxidases utilize a Fe2+ containing heme group in their catalytic center as a means of electron transfer. This center should be able to only perform one-electron reductions of oxygen, and thus the major initial product of these enzymes should be O2·−. Surprisingly, the most measurable product of Nox4 and the Duox enzymes seems to be H2O2. In the case of Nox4, this has been attributed to structural characteristics that retard the release of O2·− until it spontaneously dismutes to H2O2.8 We have also found that Nox5 seems to predominately release H2O2.5
Studies of rodents lacking or overexpressing components of the NADPH oxidase have been most revealing in demonstrating a role of these enzymes in hypertension. Mice lacking p47phox, a cytosolic component essential for activation of Nox2 are protected against ang II and DOCA-salt hypertension.9, 10 Overexpression p22phox, the small docking subunit of the Nox enzymes in vascular smooth muscle enhanced the hypertensive response to ang II aging. Mice overexpressing Nox1 develop augmented hypertension when given a chronic infusion of ang II, and mice lacking this enzyme are protected against ang II-induced hypertension.11, 12 Recently, Nox4 has been shown to play a role in salt-sensitive hypertension in rats.13 The role of Nox4 of modulating renal sodium transport is discussed more completely below. Overexpression of human Nox5 in the podocytes of increases albuminuria and causes a proportional increase in baseline blood pressure.14
Nitric oxide synthase uncoupling and tetrahydrobiopterin
The nitric oxide synthase (NOS) enzymes are the endogenous sources of NO in mammalian cells. NO has myriad effects on cardiovascular function, including modulation of vascular tone, blood pressure, sympathetic outflow, renal renin release and renal sodium excretion. In the absence of their critical cofactor tetrahydrobiopterin (BH4), or their substrate L-arginine, the NOS enzymes become uncoupled, such that they produce O2·− rather than NO. NOS uncoupling has been documented as a source of ROS in diseases such as hypertension, atherosclerosis, diabetes and following ischemia and reperfusion injury. In several of these diseases, oral supplementation of BH4 reverses NOS uncoupling, and improves endothelial function. Importantly, oral BH4 blunts the elevation of blood pressure in angiotensin II- and salt-induced hypertension in animals and has been shown to lower blood pressure in humans with hypertension.15–17 There seems to be interplay between overproduction of NO by inflammatory monocytes and uncoupling of the endothelial cell isoform of NOS.18 Treatment with a tetrahydrobiopterin precursor reduces the pressor response and increase in sympathetic outflow that occurs in response to hand grip in subjects with chronic kidney disease.19 Moreover, administration of BH4 also prevents the development of endothelium dysfunction, vascular inflammation and atherosclerosis induced by disturbed flow.20, 21 Acute and short-term administration of BH4 improves endothelial function in humans with rheumatoid arthritis.22 There is also convincing evidence that eNOS uncoupling occurs in humans with obstructive sleep apnea, leading to increased vascular superoxide production and endothelial dysfunction.23 Thus, reversing NOS uncoupling is an attractive approach to improve endothelium function and prevent the pathogenesis of vascular diseases.
A major cause of NOS uncoupling is oxidation of tetrahydrobiopterin by oxidants such as peroxynitrite. Interestingly, ROS produced by the NADPH oxidase play a role in this process, and mice lacking the NADPH oxidase are protected against tetrahydrobiopterin oxidation in the setting of hypertension.10
Xanthine Oxidase
Xanthine oxidoreductase (XOR) is another important source of ROS in mammalian cells. XOR exists in two forms, including xanthine dehydrogenase (XDH), and xanthine oxidase (XO). XDH transfers electrons from hypoxanthine and xanthine to NAD+ yielding NADH and uric acid, whereas XO transfers electrons to oxygen from these same substrates to generate O2·− and H2O2. The cellular ratio of XO to XDH is therefore critical in modulating ROS production by these enzymes. XDH is converted to XO when a critical cysteine residue is oxidized by peroxynitrite.24 This conversion is also favored in several pathophysiological settings including inflammation,25 hypoxia,26 and radiation exposure,27 and likely contributes to increased ROS production and in these situations. XO has been shown to contribute to experimental hypertension in animal models,28–30 and there is accumulating evidence supporting a role of xanthine oxidase in human hypertension.31–33 While a recent study found no significant effect of the xanthine oxidase inhibitor allopurinol in blood pressure control in hypertensive humans,34 a prior retrospective analysis of a large database showed that treatment with allopurinol was associated with a small (2.1 mmHg) reduction of systolic blood pressure.35 A carefully controlled randomized trial showed that 4 weeks of treatment of adolescent with allopurinol reduced systolic pressure by 6 mmHg while the placebo group had no change in pressure.35 These effects of allopurinol could be due to lowering of uric acid, which is known to activate the inflammasome, or due to inhibition of xanthine oxidase. In one study, allopurinol markedly lowered blood pressure in obese adolescents with pre-hypertension, while the uricosuric agent probenecid had no effect.36 These findings implicate xanthine oxidase, and potential ROS derived from this enzyme, as having an important role in adolescent pre-hypertension.
Mitochondria
The mitochondria are responsible for the majority of ATP production in the cell. These organelles contain 5 enzyme complexes that comprise the electron transport chain. Electrons are transported sequentially from NADH through complexes I to V, the site of ATP generation. During normal mitochondrial function, this electron transfer from one complex to the next is efficient and there is minimal loss or leak from electron transport, however in various disease states, electron leak is increased and can lead to reduction of oxygen and formation of O2·− and H2O2. Electron leak can occur at complexes I to IV, but occurs predominantly at complexes I and III due to defects in these complexes. These defects can include changes in their levels, post-translational modifications like glutathionylation or nitration. A recurring paradigm is that oxidative damage to mitochondrial DNA promotes deficiency in components of the electron transport chain, promoting electron leak. An important phenomenon is reverse electron transport, which occurs particularly in complex I in various pathophysiological states. Mitochondrial dysfunction is a major source of cellular ROS production in various pathophysiological states. Importantly, ROS from the NADPH oxidase have been shown to enter the mitochondria and promote electron leak and ROS production from the electron transport chain in hypertension.37 Of note, antioxidants target the mitochondria and have proven effective in both preventing and reversing experimental hypertension.38 Mitochondrial cyclophilin D (CypD) plays a critical role in opening of membrane transition pore, which depolarizes the mitochondria and enhances ROS production by this organelle. Recently it was found that pharmacological inhibition or deletion of CypD in mice prevents ang II induced hypertension.39 Mitochondrial DNA fragments, indicative of mitochondrial damage, are increased in the urine of hypertensive humans and directly correlate with markers of renal injury in these individuals.40
Antioxidant defense mechanisms
Antioxidants are molecules that prevent the oxidation of other molecules often by being oxidized themselves. Due to our oxygen rich atmosphere, all living organisms, including bacteria, plants and animals have adapted and developed enzymatic and non-enzymatic defenses against chronic oxidative stress. In mammalian cells, the major intracellular antioxidant enzymes include superoxide dismutase, catalase and glutathione peroxidase. As mentioned above, SOD catalyzes the dismutation of O2·− to H2O2 and molecular O2. Catalase and glutathione peroxidase further decompose H2O2 into H2O and O2. Glutathione peroxidase requires glutathione as a co-substrate, yielding oxidized glutathione (GSSG) upon reaction. In addition to H2O2, glutathione peroxidase also reduces lipid hydroperoxides to their respective alcohols and thus protects the cell membrane from lipid peroxidation. Glutathione peroxidase is also protective against OONO−, which it efficiently reduces to nitrite.41 Unlike O2·− or H2O2, the there are no enzymatic antioxidants that scavenge hydroxyl, but a variety of non-enzymatic antioxidants including vitamin C (ascorbic acid), vitamin E (α-tocopherol) and glutathione react with and eliminate this radical.
Mammalian cells have the capacity to modulate their antioxidant levels by transcriptional modulators such as Nrf2 and DJ-1. The former is a transcription factor that binds to the antioxidant response element of many genes, including the glutathione-S transferases, heme oxygenase 1 and glutamate-cysteine ligase which controls glutathione synthesis.42 Nrf2 is activated in the setting of oxidative stress, and moves to the nucleus to induce these and other important antioxidant genes. DJ-1 is a recently discovered protein that has antioxidant properties, and translocates to either the nucleus or the mitochondria upon oxidative stress.43 As discussed below, DJ-1 plays a major role in the response of proximal tubular cells to dopamine, and ultimately regulates blood pressure.44
Limitations of Antioxidant Therapy
Although basic studies have strongly supported a role of ROS in cell dysfunction and animal models of disease, clinical trials with high dose antioxidants have been disappointing. Numerous large clinical trials have failed to show beneficial effects of either vitamin C or vitamin E supplementation in cancer, cardiovascular disease and neurodegenerative diseases.45 A very recent meta-analysis of 50 randomized trials including almost 300,000 patients confirmed the futility of treatment with a variety of antioxidants in cardiovascular disease.46 In hypertensive subjects, a few small trials initially showed benefit,47,48 but, later large clinical trials, including the SU.VI.MAX study, showed no improvement in blood pressure with antioxidant therapy.49, 50 Surprisingly, large doses of beta-carotene, vitamin A and vitamin E have paradoxically worsened cardiovascular outcomes in some studies.51, 52 The failure of antioxidants in humans might reflect the low rate constant of vitamins such as E and C with superoxide and related ROS, the inability to target subcellular sites where ROS are formed, and the fact that some ROS have beneficial effects. Prevention of ROS generation by inhibiting specific enzymatic sources might be more efficient than nonspecific antioxidants such as vitamin C and E. In this regard, a recent study showed that a Nox 1 inhibitor is effective in reducing atherosclerosis in mice with experimental diabetes.53 It is also possible that these trials were negative because they did not target patients that actually have oxidative stress. As an example, the effect of vitamin E vs. placebo was studied on cardiovascular outcomes in almost 1500 middle-aged diabetic patients with the haptaglobin 2/2 genotype.54 The 2/2 genotype is associated with a reduction of haptaglobin’s antioxidant properties and therefore these patients were deemed to be at risk for oxidative stress. Vitamin E reduced all cardiovascular events, myocardial infarction and stroke by 50% in this population. Because of this striking benefit of vitamin E, the study was stopped prematurely. It is also possible that more potent, catalytic agents such as Tempol or targeted agents such as mitoTempol, which scavenges radicals specifically in the mitochondria, would have greater efficacy. This topic has been reviewed recently.55
Another drawback to the use of individual antioxidants is that individual agents are unlikely able to replenish deficiencies in multiple endogenous antioxidants that might be encountered in a disease like hypertension or caused by long-standing dietary indiscretion. For example, treatment with vitamin E would not be expected to restore selenium or ascorbate levels. In contrast, consumption of a diet rich in antioxidants might have a beneficial effect. As an example, the dietary approach to stop hypertension (DASH) study showed that a diet high in fruits and vegetables had a striking benefit in control of blood pressure.56 Likewise a recent study showed that grape seed extract, which contains numerous potential nutrients, was also effective in lowering blood pressure in subjects with pre-hypertension.57
Potential roles of reactive oxygen species in hypertension
A large body of literature has shown that excessive production of ROS contributes to hypertension and that scavenging of ROS decreases blood pressure. Despite the evidence that oxidative stress contributes to hypertension, the mechanisms involved are not well understood. Hypertension is associated with increased ROS production by multiple organs, including the brain, the vasculature, and the kidney, all of which are likely important. A major problem is that we currently lack a complete understanding of which of these organs or cell types predominate in the genesis of hypertension or if there is important interplay between them that causes this disease. In the following section, we discuss evidence that ROS in the central nervous system (CNS), the kidney, and the vasculature contribute to hypertension and review recent data showing that the adaptive immunity is activated by oxidative events and can contribute to hypertension by interacting with these organs.
Renal oxidative stress and hypertension
There is ample evidence supporting that ROS generated in the kidney and its blood vessels contribute to the development and maintenance of hypertension. Virtually all cells in the kidney, including vessels, glomeruli, podocytes, interstitial fibroblasts, the medullary thick ascending limb (mTAL), the macula densa, the distal tubule, and the collecting duct express components of the NADPH oxidase, and various stimuli have been shown to activate these. Several of the effects of ROS in the kidney are summarized in Fig. 2. For purposes of discussion, we first focus on oxidative events in the renal cortex and then in the medulla.
Figure 2.
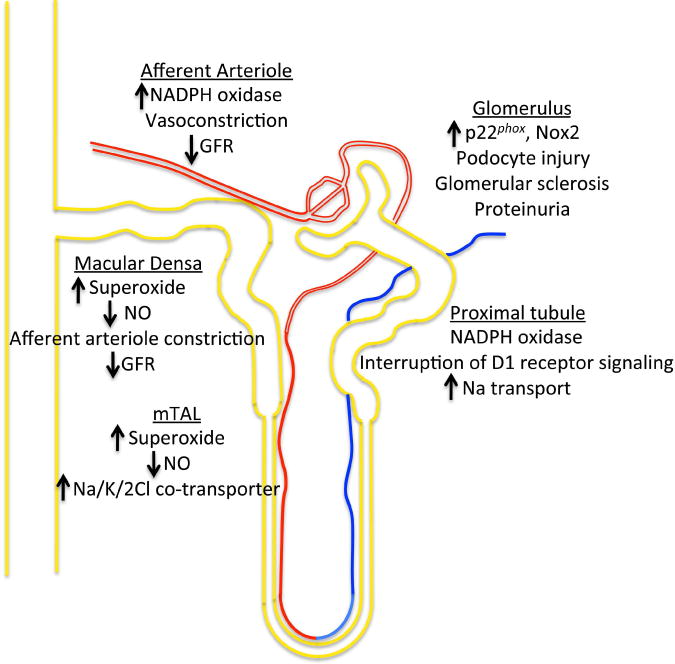
Renal effects of ROS and NO.
Several studies have examined the effect of various hypertensive stimuli on the renal cortex and how these are modulated by ROS. The structures that are targets of oxidant stress include the afferent arteriole, the glomerulus, the proximal tubule, and the cortical collecting duct (CCD). As with other vessels, an increase in O2·− in the afferent arteriole can oxidatively degrade NO, which would enhance afferent arteriolar vasoconstriction and reduce glomerular filtration rate. Indeed, studies in rabbits have shown that ang II-induced hypertension increases expression of the NADPH oxidase subunit p22phox activates the NADPH oxidase, and causes endothelial dysfunction in afferent arterioles.58 Studies of isolated afferent arterioles have also shown that O2·− generated by the NADPH oxidase potentiates intracellular calcium.59 ROS are also generated in the afferent arterioles of spontaneous hypertensive rats and in the kidney of animals in response to other vasoconstrictors such as endothelin-1 (ET-1) and thromboxane prostanoids.60, 61 A recent study has shown that O2·− enhances afferent arteriolar myogenic tone, while H2O2 inhibits this via actions on voltage-gated potassium channels.62 These findings illustrate the complexity of attempting to scavenge all ROS to improve renal perfusion.
Podocyte injury causes proteinuria and is a precursor to glomerulosclerosis. Moreover, proteinuria seems to promote further podocyte injury in a feed-forward fashion. Dahl-salt sensitive rats demonstrate up-regulation of glomerular p22phox and Nox2 and the antioxidant tempol reduces glomerular sclerosis and proteinuria in these animals.63,64 Recent evidence has implicated a role of plasminogen in stimulating expression of Nox2 and Nox4 in podocytes and causing podocyte injury.65 Ang II also stimulates mitochondria ROS generation and induces podocyte autophagy.66 Oxidative injury impairs the crosstalk between nephrin and caveolin-1 in podocytes, leading to disruption of glomerular filtration barrier.67 ROS also mediate mesangial cell proliferation, migration and extracellular matrix deposition, features characteristic of glomerulosclerosis induced by angiotensin II or aldosterone.68 Mice lacking p47phox are protected against the development of glomerulosclerosis and albuminuria following either Adriamycin injection or 5/6 nephrectomy, supporting a role of the NADPH oxidase in this common form of glomerular injury.69
Components of the NADPH oxidase exist in lipid rafts within epithelial cells of the proximal tubule, where they are maintained in an inactive state. Dopamine-1 (D1) receptor agonists inhibit, whereas disruption of lipid rafts and angiotensin II stimulate the proximal tubule NADPH oxidase function.70, 71 ROS from the NADPH oxidase modulate sodium transport via altering Na/K ATPase and Na/H exchange-3 (NHE-3) function on the basal and apical membranes of the proximal tubular cells, respectively.72, 73 Deletion of the dopamine D2 receptor (D2R) inhibits ROS production in renal proximal tubular cells and leads to hypertension, supporting a role of ROS in the proximal tubule in blood pressure control.44
An important mechanism by which ROS in the cortex modulates sodium reuptake and blood pressure is via tubuloglomerular feedback. This is a mediated by an interaction of the macula densa of the thick ascending limb as it makes contact with its own glomerulus in the cortex. Sodium concentration is sensed by the macula densa via its apical Na/K/2Cl co-transporter, which in turn stimulates NO produced by the neuronal nitric oxide synthase. This promotes dilatation of afferent arterioles and increases glomerular filtration.74, 75 Increases in O2·− in macula densa can inactivate NO, leading to afferent arteriolar vasoconstriction and a reduction of glomerular filtration rate.76 An elegant study of isolated, single nephrons by Nouri and colleagues showed that in vivo RNA silencing of the NADPH oxidase subunit p22phox enhances single tubular glomerular filtration in angiotensin II-treated rats but not in control rats. By either including or excluding the distal tubule, these authors showed that this effect was likely mediated by ROS produced in the macula densa.77
The epithelial Na+ channel (ENaC) mediates final tubular adjustment of Na+ reabsorption in the cortical collecting duct (CCD).78 Angiotensin II activates the NADPH oxidase in the CCD, stimulating ENaC activity.79 This is likely mediated by aldosterone, the principle regulator of ENaC activity. Aldosterone induces superoxide generation in A6 epithelial cells, which in turn reduces inhibition of ENaC by NO.80 A similar pathway modulates ENAC activity in response to insulin, the insulin-like growth factor and the epidermal growth factor.81
Oxidative stress also modulates sodium reabsorption and blood pressure in the renal medulla. As in the blood vessel, there is a balance between O2·− and NO produced by epithelial cells of the medullary thick ascending limb (mTAL) and the pericytes of the vasa recta. NO synthase activity is higher in the renal medulla compared than in the cortex, likely contributing to independent regulation of medullary and cortical perfusion.82 Cowley’s group has shown that NO released by cells of the mTAL promotes dilation of the adjacent vasa recta, increasing medullary flow. This augments interstitial Starling forces and promotes sodium movement to the tubule and thus natriuresis and diuresis.83, 84 Activation of the NADPH oxidase in the medulla has the opposite effect; promoting vasa recta vasoconstriction, sodium movement into the vasa recta, reducing natriuresis and increasing blood pressure. A growing body of evidence indicates that medullary O2·− affects sodium transport.85, 86 Superoxide enhances Na/K/2Cl cotransporter activity via a protein kinase C activation in mTAL preparations.87 Infusion of angiotensin II in vivo mimics this effect and administration of the .O2− scavenger tempol prevents this.88 Nitric oxide has the opposite effect on NaCl transport in the mTAL. NO generated by eNOS inhibits the Na/K/2Cl cotransporter and NHE-3 in isolated thick ascending limb.89 This inhibitory effect on the Na/K/2Cl cotransporter is mediated by phosphodiesterase-mediated degradation of cyclic AMP while NO directly inhibits NHE-3.90 Of interest, increased sodium delivery to cells of the mTAL stimulates H2O2 production by the mitochondria, and this is linked to vasodilatation of the nearby vasa recta.91
The role of ROS in control of renal perfusion and sodium handling likely illustrates a normal physiological role of these molecules and show that they are not uniformly deleterious. The kidney must retain sodium and water during times of salt deprivation and in land-dwelling mammals survival would be impossible without this. It is therefore likely that generation of O2·− and other ROS within the kidney play a crucial role in this important physiological process.
Reactive oxygen species, the central nervous system and hypertension
It is now obvious that the CNS is essential for production and maintenance of most forms of experimental hypertension, principally by enhancing sympathetic efferent outflow. Even the hypertension caused by hormones like angiotensin II and aldosterone, which have myriad systemic effects, is importantly mediated by actions in the central nervous system.92, 93 As an illustration of this, the development of many forms of experimental hypertension is prevented by destruction of a region of the forebrain surrounding the anteroventral third cerebral ventricle (AV3V) in rodents.94, 95 This region includes the median preoptic eminence, the organum vasculosum of the lateral terminalis, and the preoptic periventricular nucleus (Fig. 3).96 Following AV3V lesioning, virtually all of the central actions of angiotensin II, including drinking behavior, vasopressin secretion and increased sympathetic outflow, are diminished or eliminated. These portions of the forebrain are connected to other regions involved in central cardiovascular regulation. Important among these are the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT), which are circumventricular organs (CVO) lacking a blood-brain barrier. Other CVOs include the median eminence and the area prostrema. Hormones in the periphery can act on these regions, modulating neuronal input into cardiovascular control centers in the mid- and hindbrain, including the parabrachial nucleus, the nucleus tractus solitarius (NTS) and the rostral ventral lateral medulla (VLM) (see Fig. 3).
Figure 3.
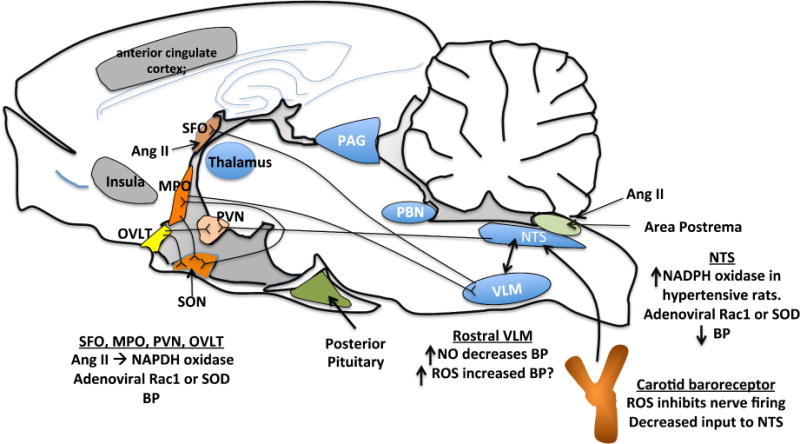
Sites in the brain implicated in hypertension. Shown also are documented actions of ROS and NO to modulate central control of blood pressure.
There is strong evidence that signaling in these brain centers and their influence on blood pressure is modulated by local production of ROS.97 Intracerebroventricular (ICV) injection of an adenovirus encoding SOD attenuates the hypertension caused by either local injection or systemic infusion of angiotensin II.98 Zimmerman and colleagues showed that angiotensin II stimulates O2·− production in cultured neurons and that this increases intracellular calcium.99 Peterson et al, using selective siRNA expressing adenoviruses, have shown that Nox2 and Nox4 have different roles in the SFO, such that both are linked to blood pressure regulation, but only Nox2 modulates drinking behavior.100 Lob et al used Cre-Lox technology to delete p22phox in the SFO and showed that this completely abrogated the hypertensive response to long-term infusion of angiotensin II.101
There is also evidence that ROS derived from the NADPH oxidase enhances nerve firing within the hypothalamus. ICV injection of angiotensin II increases NADPH oxidase-mediated O2·− production not only in the SFO but also in anterior hypothalamic nuclei such as the median preoptic eminence and in the paraventricular nucleus of the hypothalamus.102 These effects were blocked by the NADPH oxidase inhibitor apocynin as were the hemodynamic effects of centrally administered angiotensin II. Thus, angiotensin II and its effects on the NADPH oxidase seem to coordinate activation of several forebrain centers to promote a hypertensive response.
Signaling between the forebrain and the hindbrain pontomedullary cardiovascular control centers is also important for blood pressure control. The nucleus tractus solitarius (NTS) is such a hindbrain nucleus that receives input from the CVO and relays inhibitory stimuli from baroreceptors. Angiotensin II augments ROS production by the NADPH oxidase in neurons from the NTS, and this in turn activates L-type calcium channel activity and neuronal firing.103 NADPH oxidase is increased in the NTS of stroke-prone, spontaneously hypertensive rats.104 H2O2 causes delayed hyperexcitability of NTS neurons via actions on barium sensitive potassium channels. This might be expected to enhance sympathetic outflow in vivo.105
As discussed above, NO can be consumed by O2·− and thus can act as a radical scavenger. NO produced in response to ang II stimulation of the AT2 receptor limits the effects of O2·− in the medial NTS and thus inhibits L-type channel activation.106 NO has a similar role in the ventral lateral medulla, which lies below the NTS and receives and sends signals to the NTS and thus regulates sympathetic tone.107 Experimental interventions that increase NO in the rostral VLM lower blood pressure, whereas increase in oxidative stress in this region raises blood pressure.108, 109
ROS modulate baroreflex function, which is commonly abnormal in the setting of chronic hypertension. Studies by Li and colleagues have shown that ROS generated in the carotid bulb of atherosclerotic rabbits reduce carotid sinus nerve responses to elevations of pressure and that this could be mimicked by exogenous administration of ROS and prevented by ROS scavenging.110
Efferent sympathetic renal nerves govern both the vasculature and tubular segments of the kidney. Stimulation of the renal sympathetic nerves promotes afferent arteriolar vasoconstriction, renin release and increases Na+ reabsorption.111 Alpha1 adrenergic receptor activation enhances ROS generation and constriction of the afferent arterioles, reducing renal blood flow (RBF) in angiotensin II-infused rabbits.112 In contrast, β1 adrenergic receptor activation inhibits ROS generation and promotes vasodilation.113 In keeping with these important roles of renal sympathetic nerves in controlling renal ROS production, renal denervation blunts blood pressure elevation in multiple experimental hypertensive models, including angiotensin II-induced, DOCA-salt and two kidney one clip hypertension as well as SHR and SHRSP rats.114 The role of renal denervation to control human hypertension remains a topic of debate.
A consequence of renal denervation is ablation of the renal afferent nerves. These are mainly located in the renal pelvis and are activated by various physical and chemical stimuli.115 During kidney injury, increased input from these afferent nerves activates central sympathetic nuclei in a ROS-dependent manner.116 For instance, renal denervation prevents release of norepinephrine from the posterior hypothalamic nuclei and blood pressure elevation following kidney injury induced intrarenal phenol injection.117 Intrarenal phenol injection increases NADPH oxidase expression in the central nuclei and this is prevented by intracerebroventricular (ICV) injection of the SOD mimetic tempol or PEG-SOD. Interestingly, a vitamin E-fortified diet blunts the increase of sympathetic nerve activity and hypertension in response to intrarenal phenol injection.118 These data indicate that ROS modulate both efferent and afferent renal nerve activity and therefore promote development of hypertension.
Vascular oxidative stress and hypertension
Elevated vascular ROS promote the development of hypertension in various animal models, including angiotensin II-induced and DOCA-salt hypertension,9, 119, 120 Dahl salt-sensitive hypertension and the spontaneous hypertensive rat (SHR).121–123 Humans with hypertension have alterations of vascular reactivity. The several mechanisms by which ROS modulate the vasculature are illustrated in figure 4.
Figure 4.
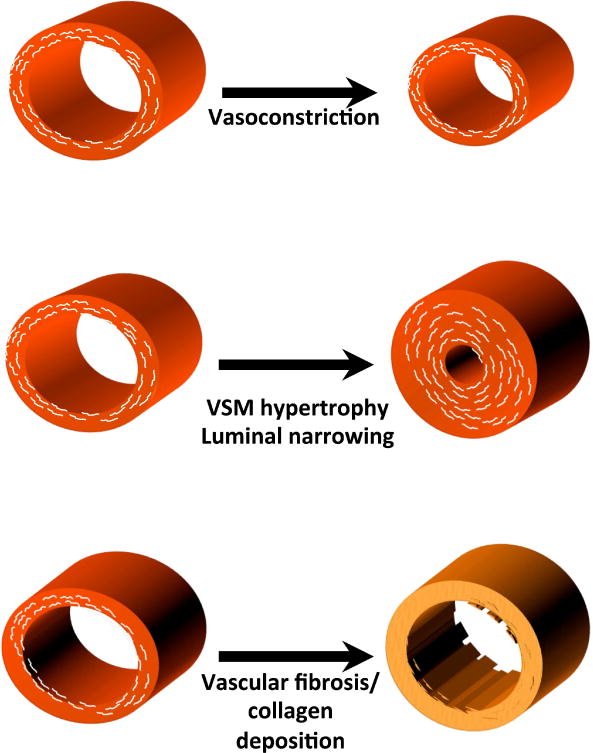
Mechanisms by which ROS and NO modulate vascular function and structure in hypertension.
The endothelium regulates vascular tone via releasing of endothelium-derived relaxing factors (EDRFs) and endothelium-derived contractile factors (EDCFs). Oxidative stress causes imbalanced production and bioavailability of these molecules leading to endothelial dysfunction. NO is one of the most important mediators of endothelium-dependent relaxation in blood pressure regulation. The major mechanism for alteration of NO bioavailability when O2·− is increased is that this radical rapidly reacts with NO, as discussed above. Uncoupling of endothelial NOS, due to deprivation of its cofactor tetrahydrobiopterin, results in reduced NO release, increased O2·− production, impaired endothelium-dependent relaxation and elevated arterial pressure. Similarly, a lack of the eNOS substrate L-arginine also reduces NO bioavailability and impairs NO-induced dilation. L-arginine is the substrate for both eNOS and arginase, and increased arginase activity reduces the local bioavailability of L-arginine.124 Interestingly, peroxinitrite and hydrogen peroxide increase the expression and activity of arginase in endothelial cells, potentially contributing to endothelial dysfunction.125
Oxidation of membrane fatty acids and in particular arachidonic acid can lead to formation of F2-isoprostanes, which are present in the blood of patients with oxidative stress (e.g. those with hypercholesterolemia, diabetics, and cigarette smokers). Importantly, plasma F2-isoprostanes are increased in animals with experimental hypertension and in humans with renovascular hypertension.126, 127 These oxidatively modified fatty acids act on prostaglandin H/thromboxane receptors to enhance vasoconstriction.
Superoxide and other ROS can also affect the structure and function of the vascular media. An important consequence of ROS formation is vascular smooth muscle hypertrophy. Eutrophic vascular remodeling leads to an increase in medial thickness at the expense of a reduction in luminal diameter. This reduces the cross-sectional area of the effective vasculature, increasing systemic vascular resistance.128 H2O2 seems to be a major mediator of the hypertrophic effect of angiotensin II in cultured cells,129 and vascular smooth muscle hypertrophy is strikingly increased in mice overexpressing the NADPH oxidase in the vascular smooth muscle.130
Vascular ROS also stimulate the production of collagen and fibronectin both in vitro and in vivo,131, 132 and removal of vascular ROS prevents these fibrotic changes.133, 134 Structural changes such as these in resistance vessels could add to the increased systemic vascular resistance that occurs in established hypertension and, therefore, worsen the disease.135, 136 In larger central arteries, this leads to loss of the Windkessel effect, and increases pulse wave velocity. In population studies, increased pulse wave velocity precedes the development of hypertension by several years. There is substantial evidence that ROS contribute to vascular stiffening. Plasma levels of myeloperoxidase and F2-isoprostanes, markers of oxidation, correlate with arterial stiffening in humans.137 Studies in experimental animals have shown that ROS contribute to fibrotic changes in several tissues including blood vessels.138, 139
Endothelium-dependent hyperpolarization represents an NO-independent mechanism of vasodilatation that is particularly important in resistance vessels.140 The nature of the endothelium-derived hyperpolarizing factor (EDHF) remains a topic of study, but H2O2 produced by the mitochondria is clearly one EDHF that acts by activating the Ca++-dependent potassium (BK) channel.141 This seems to be an important mechanism of flow mediated vasodilatation in human coronary arterioles. The production of H2O2 is mediated by calcium entry into endothelial cells through the TRP vanilloid type 4 (TRPV4) channel in several vascular beds.142 Another EDHF is likely a cytochrome p450 epoxide metabolite of arachidonic acid, which also opens the TRPV4 channel.143, 144
H2O2 also affects the NO-cGMP pathway. H2O2 acutely stimulates NO production and over the long term induces expression of the endothelial nitric oxide synthase (NOS3).145–147 H2O2 also activates protein kinase G (PKG) by inducing cross-linking two alpha subunits of this enzyme via disulfide bond formation, and therefore promotes vasodilation independent of NO.
Reactive oxygen species, inflammation and hypertension
Diverse stimuli common to the hypertensive milieu, including angiotensin II, aldosterone, catecholamines, increased vascular stretch, and endothelin promote ROS production, which then increases expression of proinflammatory molecules that cause rolling, adhesion, and transcytosis of inflammatory cells.148, 149 As a result, there is a striking accumulation of inflammatory cells in the vessel and kidney.150–152 In keeping with this, there is an increase in plasma markers of inflammation in hypertensive humans.153, 154 Although macrophages are commonly considered important in the genesis of cardiovascular disease, increasing evidence has accumulated suggesting that the adaptive immune response and, in particular, T lymphocytes are important in hypertension. Mice and rats lacking the Rag1 gene are partly protected against the blood pressure elevation and renal damage caused by angiotensin II or salt.155–157 Effector T cells enter the kidney and the perivascular fat, where they release cytokines that promote salt retention and vasoconstriction, ultimately promoting hypertension. These cells mediate their effect by producing cytokines that potently affect vascular and renal function, including TNFα, IFN-γ and IL-17A (Figure 5). Mice lacking IL-17A do not sustain angiotensin II-induced hypertension, and treatment with soluble IL17C lowers blood pressure and prevents oxidative stress in experimental pre-eclampsia.158, 159 IL17A directly evokes endothelial dysfunction and ROS production in isolated vessels and causes hypertension when infused in mice.160 Recent data indicate that IL17A modulates expression and function of the sodium hydrogen exchanger 3 and sodium chloride cotransporter in renal tubular epithelial cells, thus promoting sodium reuptake by these cells.161 IL-17A also promotes aortic stiffening and directly stimulates production of collagen by aortic fibroblasts.162
Figure 5.
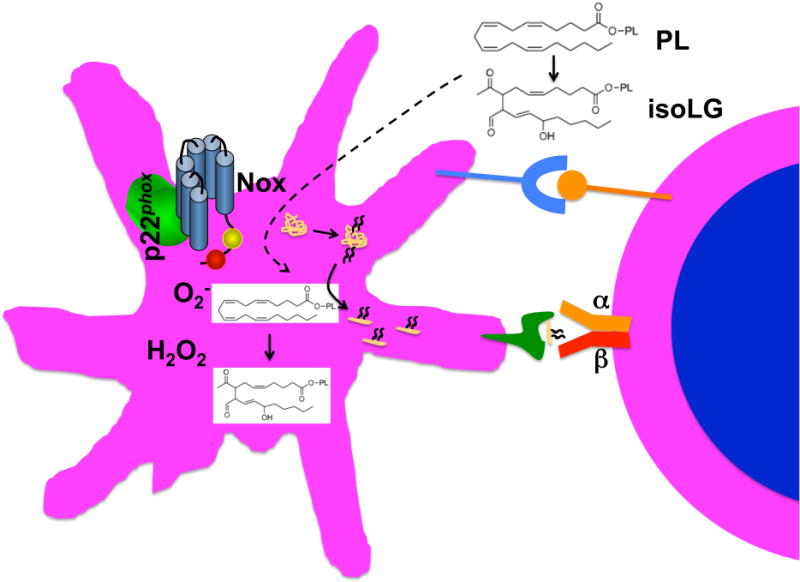
Formation of isolevuloglandins in antigen presenting cells and modification of self-proteins leading to T cell activation.
Recently, we identified a novel mechanism linking oxidative events and immune activation in hypertension. This involves formation of isolevuloglandins (isoLGs), which are a form of reactive carbonyl produced upon oxidation of fatty acids and phospholipids. These react rapidly with lysines on proteins and we found that hypertension increases formation of isoLG-modified proteins in monocyte-derived dendritic cells of mice and in monocytes of humans. These modified proteins seem immunogenic (Figure 6).4 We have found that iso-LG-modified peptides are presented in major histocompatibility complexes and can promote T cell activation. Chronic vascular oxidant stress also seems to enhance formation of these and their accumulation in dendritic cells.163
Figure 6.
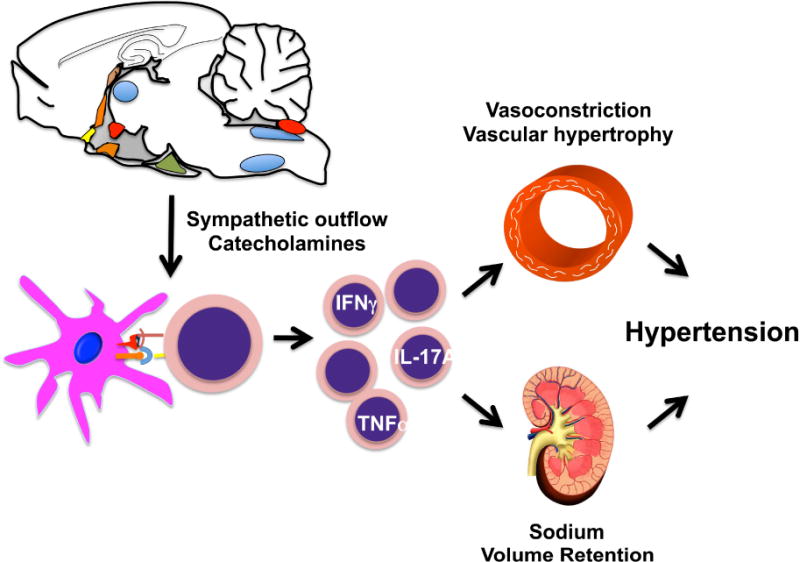
Interplay between immune activation, the central nervous system, the vasculature and the kidney in hypertension.
The observation that circulating cells, including T cells and monocytes, are important in the genesis of hypertension might help provide a unifying link between oxidative events in the CNS, the vasculature, and the kidney. These interactions, illustrated in Fig. 6, are dependent on ROS formation in all of these sites. Centrally, in the SFO, and other CVOs, NADPH oxidase activation increases neuronal firing and sympathetic nerve outflow to peripheral lymphoid tissues, leading to T cell activation.164 In DCs, ROS lead to isoLG formation and modification of self-proteins. ROS in the kidney and vessels initiates signal that cause T cell homing and infiltration.150 Finally, cytokines released by T cells diffuse to renal and vascular cells, promoting further NADPH oxidase activation, sodium retention, and vasoconstriction, leading to overt hypertension.
Summary
This review has summarized some of the data supporting a role of ROS and oxidant stress in hypertension. There is evidence that hypertensive stimuli, such as high salt and angiotensin II, promote production of ROS in the brain, the kidney, and the vasculature and that each of these sites contributes either to hypertension or to the untoward sequelae of this disease. Although the NADPH oxidase in these various organs is a predominant source, other enzymes likely contribute to ROS production and signaling in these tissues. A major clinical challenge is that the routinely used antioxidants are ineffective in preventing or treating cardiovascular disease and hypertension. This is likely because these drugs are either ineffective or act in a non-targeted fashion, such that they remove not only injurious but also beneficial ROS involved in normal cell signaling. An important and relatively new concept is that inflammatory cells contribute to hypertension in a ROS-dependent fashion. Future studies are needed to understand the interaction of inflammatory cells with the CNS, the kidney, and the vasculature and how this might be interrupted to provide therapeutic benefit.
Key Points.
Oxidative stress is considered a major mechanism in hypertension.
Formation of reactive oxygen species contribute to dysfunction in the vasculature, the kidney and the central nervous system.
Recent evidence supports a role of reactive oxygen species in inflammation in hypertension.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Shin MH, Moon YJ, Seo JE, Lee Y, Kim KH, Chung JH. Reactive oxygen species produced by NADPH oxidase, xanthine oxidase, and mitochondrial electron transport system mediate heat shock-induced MMP-1 and MMP-9 expression. Free Radic Biol Med. 2008;44:635–645. doi: 10.1016/j.freeradbiomed.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 3.Semchyshyn HM. Reactive carbonyl species in vivo: generation and dual biological effects. ScientificWorldJournal. 2014;2014:417842. doi: 10.1155/2014/417842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J, 2nd, Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–4656. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52:1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 8.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 12.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 13.Cowley AW, Jr, Yang C, Zheleznova NN, Staruschenko A, Kurth T, Rein L, Kumar V, Sadovnikov K, Dayton A, Hoffman M, Ryan RP, Skelton MM, Salehpour F, Ranji M, Geurts A. Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension. 2016;67:440–450. doi: 10.1161/HYPERTENSIONAHA.115.06280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holterman CE, Thibodeau JF, Towaij C, Gutsol A, Montezano AC, Parks RJ, Cooper ME, Touyz RM, Kennedy CR. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol. 2014;25:784–797. doi: 10.1681/ASN.2013040371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison DG, Chen W, Dikalov S, Li L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv Pharmacol. 2010;60:107–132. doi: 10.1016/B978-0-12-385061-4.00005-2. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Li L, Brod T, Saeed O, Thabet S, Jansen T, Dikalov S, Weyand C, Goronzy J, Harrison DG. Role of increased guanosine triphosphate cyclohydrolase-1 expression and tetrahydrobiopterin levels upon T cell activation. J Biol Chem. 2011;286:13846–13851. doi: 10.1074/jbc.M110.191023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porkert M, Sher S, Reddy U, Cheema F, Niessner C, Kolm P, Jones DP, Hooper C, Taylor WR, Harrison D, Quyyumi AA. Tetrahydrobiopterin: a novel antihypertensive therapy. J Hum Hypertens. 2008;22:401–407. doi: 10.1038/sj.jhh.1002329. [DOI] [PubMed] [Google Scholar]
- 18.Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, Brahler M, Knorr M, Brandt M, Karbach SH, Becker C, Oelze M, Bauersachs J, Widder J, Munzel T, Daiber A, Wenzel P. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin AM, Liao P, Millson EC, Quyyumi AA, Park J. Tetrahydrobiopterin ameliorates the exaggerated exercise pressor response in patients with chronic kidney disease: a randomized controlled trial. Am J Physiol Renal Physiol. 2016;310:F1016–1025. doi: 10.1152/ajprenal.00527.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, Chen W, Rezvan A, Jo H, Harrison DG. Tetrahydrobiopterin deficiency and nitric oxide synthase uncoupling contribute to atherosclerosis induced by disturbed flow. Arterioscler Thromb Vasc Biol. 2011;31:1547–1554. doi: 10.1161/ATVBAHA.111.226456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofmeister LH, Lee SH, Norlander AE, Montaniel KR, Chen W, Harrison DG, Sung HJ. Phage-display-guided nanocarrier targeting to atheroprone vasculature. ACS Nano. 2015;9:4435–4446. doi: 10.1021/acsnano.5b01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maki-Petaja KM, Day L, Cheriyan J, Hall FC, Ostor AJ, Shenker N, Wilkinson IB. Tetrahydrobiopterin Supplementation Improves Endothelial Function But Does Not Alter Aortic Stiffness in Patients With Rheumatoid Arthritis. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varadharaj S, Porter K, Pleister A, Wannemacher J, Sow A, Jarjoura D, Zweier JL, Khayat RN. Endothelial nitric oxide synthase uncoupling: a novel pathway in OSA induced vascular endothelial dysfunction. Respir Physiol Neurobiol. 2015;207:40–47. doi: 10.1016/j.resp.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakuma S, Fujimoto Y, Sakamoto Y, Uchiyama T, Yoshioka K, Nishida H, Fujita T. Peroxynitrite induces the conversion of xanthine dehydrogenase to oxidase in rabbit liver. Biochem Biophys Res Commun. 1997;230:476–479. doi: 10.1006/bbrc.1996.5983. [DOI] [PubMed] [Google Scholar]
- 25.Friedl HP, Till GO, Ryan US, Ward PA. Mediator-induced activation of xanthine oxidase in endothelial cells. Faseb J. 1989;3:2512–2518. doi: 10.1096/fasebj.3.13.2806779. [DOI] [PubMed] [Google Scholar]
- 26.Sohn HY, Krotz F, Gloe T, Keller M, Theisen K, Klauss V, Pohl U. Differential regulation of xanthine and NAD(P)H oxidase by hypoxia in human umbilical vein endothelial cells. Role of nitric oxide and adenosine. Cardiovasc Res. 2003;58:638–646. doi: 10.1016/s0008-6363(03)00262-1. [DOI] [PubMed] [Google Scholar]
- 27.Kale RK. Post-irradiation free radical generation: evidence from the conversion of xanthine dehydrogenase into xanthine oxidase. Indian J Exp Biol. 2003;41:105–111. [PubMed] [Google Scholar]
- 28.Suzuki H, DeLano FA, Parks DA, Jamshidi N, Granger DN, Ishii H, Suematsu M, Zweifach BW, Schmid-Schonbein GW. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc Natl Acad Sci U S A. 1998;95:4754–4759. doi: 10.1073/pnas.95.8.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swei A, Lacy F, Delano FA, Parks DA, Schmid-Schonbein GW. A mechanism of oxygen free radical production in the Dahl hypertensive rat. Microcirculation. 1999;6:179–187. [PubMed] [Google Scholar]
- 30.Shirakura T, Nomura J, Matsui C, Kobayashi T, Tamura M, Masuzaki H. Febuxostat, a novel xanthine oxidoreductase inhibitor, improves hypertension and endothelial dysfunction in spontaneously hypertensive rats. Naunyn Schmiedebergs Arch Pharmacol. 2016 doi: 10.1007/s00210-016-1239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardillo C, Kilcoyne CM, Cannon RO, 3rd, Quyyumi AA, Panza JA. Xanthine oxidase inhibition with oxypurinol improves endothelial vasodilator function in hypercholesterolemic but not in hypertensive patients. Hypertension. 1997;30:57–63. doi: 10.1161/01.hyp.30.1.57. [DOI] [PubMed] [Google Scholar]
- 32.Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- 33.Kohagura K, Tana T, Higa A, Yamazato M, Ishida A, Nagahama K, Sakima A, Iseki K, Ohya Y. Effects of xanthine oxidase inhibitors on renal function and blood pressure in hypertensive patients with hyperuricemia. Hypertens Res. 2016 doi: 10.1038/hr.2016.37. [DOI] [PubMed] [Google Scholar]
- 34.Segal MS, Srinivas TR, Mohandas R, Shuster JJ, Wen X, Whidden E, Tantravahi J, Johnson RJ. The effect of the addition of allopurinol on blood pressure control in African Americans treated with a thiazide-like diuretic. J Am Soc Hypertens. 2015;9:610–619 e611. doi: 10.1016/j.jash.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300:924–932. doi: 10.1001/jama.300.8.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60:1148–1156. doi: 10.1161/HYPERTENSIONAHA.112.196980. [DOI] [PubMed] [Google Scholar]
- 37.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 38.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, Dikalov SI. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension. 2016;67:1218–1227. doi: 10.1161/HYPERTENSIONAHA.115.07085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, Textor SC, Lerman LO. Urinary Mitochondrial DNA Copy Number Identifies Chronic Renal Injury in Hypertensive Patients. Hypertension. 2016 doi: 10.1161/HYPERTENSIONAHA.116.07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arteel GE, Briviba K, Sies H. Protection against peroxynitrite. FEBS Lett. 1999;445:226–230. doi: 10.1016/s0014-5793(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 42.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan X, Kelsen SG, Merali S. Proteomic analysis of oxidative stress-responsive proteins in human pneumocytes: insight into the regulation of DJ-1 expression. J Proteome Res. 2008;7:4955–4961. doi: 10.1021/pr800295j. [DOI] [PubMed] [Google Scholar]
- 44.Cuevas S, Zhang Y, Yang Y, Escano C, Asico L, Jones JE, Armando I, Jose PA. Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension. 2012;59:446–452. doi: 10.1161/HYPERTENSIONAHA.111.185744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. Jama. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 46.Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346:f10. doi: 10.1136/bmj.f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF, Jr, Vita JA. Treatment of hypertension with ascorbic acid. Lancet. 1999;354:2048–2049. doi: 10.1016/s0140-6736(99)04410-4. [DOI] [PubMed] [Google Scholar]
- 48.Mullan BA, Young IS, Fee H, McCance DR. Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension. 2002;40:804–809. doi: 10.1161/01.hyp.0000039961.13718.00. [DOI] [PubMed] [Google Scholar]
- 49.Czernichow S, Bertrais S, Blacher J, Galan P, Briancon S, Favier A, Safar M, Hercberg S. Effect of supplementation with antioxidants upon long-term risk of hypertension in the SU.VI.MAX study: association with plasma antioxidant levels. J Hypertens. 2005;23:2013–2018. doi: 10.1097/01.hjh.0000187259.94448.8a. [DOI] [PubMed] [Google Scholar]
- 50.Kim MK, Sasaki S, Sasazuki S, Okubo S, Hayashi M, Tsugane S. Lack of long-term effect of vitamin C supplementation on blood pressure. Hypertension. 2002;40:797–803. doi: 10.1161/01.hyp.0000038339.67450.60. [DOI] [PubMed] [Google Scholar]
- 51.Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 52.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Jama. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 53.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 54.Milman U, Blum S, Shapira C, Aronson D, Miller-Lotan R, Anbinder Y, Alshiek J, Bennett L, Kostenko M, Landau M, Keidar S, Levy Y, Khemlin A, Radan A, Levy AP. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2–2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28:341–347. doi: 10.1161/ATVBAHA.107.153965. [DOI] [PubMed] [Google Scholar]
- 55.Dikalov SI, Dikalova AE. Contribution of mitochondrial oxidative stress to hypertension. Curr Opin Nephrol Hypertens. 2016;25:73–80. doi: 10.1097/MNH.0000000000000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, 3rd, Simons-Morton DG, Karanja N, Lin PH, Group DA-SCR Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 57.Park E, Edirisinghe I, Choy YY, Waterhouse A, Burton-Freeman B. Effects of grape seed extract beverage on blood pressure and metabolic indices in individuals with pre-hypertension: a randomised, double-blinded, two-arm, parallel, placebo-controlled trial. Br J Nutr. 2016;115:226–238. doi: 10.1017/S0007114515004328. [DOI] [PubMed] [Google Scholar]
- 58.Wang D, Chen Y, Chabrashvili T, Aslam S, Borrego Conde LJ, Umans JG, Wilcox CS. Role of oxidative stress in endothelial dysfunction and enhanced responses to angiotensin II of afferent arterioles from rabbits infused with angiotensin II. J Am Soc Nephrol. 2003;14:2783–2789. doi: 10.1097/01.asn.0000090747.59919.d2. [DOI] [PubMed] [Google Scholar]
- 59.Fellner SK, Arendshorst WJ. Angiotensin II, reactive oxygen species, and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol. 2005;289:F1012–1019. doi: 10.1152/ajprenal.00144.2005. [DOI] [PubMed] [Google Scholar]
- 60.Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R117–124. doi: 10.1152/ajpregu.00476.2002. [DOI] [PubMed] [Google Scholar]
- 61.Araujo M, Wilcox CS. Oxidative Stress in Hypertension: Role of the Kidney. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li L, Lai EY, Wellstein A, Welch WJ, Wilcox CS. Differential effects of superoxide and hydrogen peroxide on myogenic signaling, membrane potential, and contractions of mouse renal afferent arterioles. Am J Physiol Renal Physiol. 2016;310:F1197–1205. doi: 10.1152/ajprenal.00575.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagase M, Shibata S, Yoshida S, Nagase T, Gotoda T, Fujita T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 64.Meng S, Cason GW, Gannon AW, Racusen LC, Manning RD., Jr Oxidative stress in Dahl salt-sensitive hypertension. Hypertension. 2003;41:1346–1352. doi: 10.1161/01.HYP.0000070028.99408.E8. [DOI] [PubMed] [Google Scholar]
- 65.Raij L, Tian R, Wong JS, He JC, Campbell KN. Podocyte Injury: The Role of Proteinuria, Urinary Plasminogen and Oxidative Stress. Am J Physiol Renal Physiol. 2016 doi: 10.1152/ajprenal.00162.2016. ajprenal 00162 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia J, Ding G, Zhu J, Chen C, Liang W, Franki N, Singhal PC. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol. 2008;28:500–507. doi: 10.1159/000113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren Z, Liang W, Chen C, Yang H, Singhal PC, Ding G. Angiotensin II induces nephrin dephosphorylation and podocyte injury: role of caveolin-1. Cell Signal. 2012;24:443–450. doi: 10.1016/j.cellsig.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hua P, Feng W, Rezonzew G, Chumley P, Jaimes EA. The transcription factor ETS-1 regulates angiotensin II-stimulated fibronectin production in mesangial cells. Am J Physiol Renal Physiol. 2012;302:F1418–1429. doi: 10.1152/ajprenal.00477.2011. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Chen X, Su Y, Paueksakon P, Hu W, Zhang MZ, Harris RC, Blackwell TS, Zent R, Pozzi A. p47(phox) contributes to albuminuria and kidney fibrosis in mice. Kidney Int. 2015;87:948–962. doi: 10.1038/ki.2014.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Banday AA, Lokhandwala MF. Loss of biphasic effect on Na/K-ATPase activity by angiotensin II involves defective angiotensin type 1 receptor-nitric oxide signaling. Hypertension. 2008;52:1099–1105. doi: 10.1161/HYPERTENSIONAHA.108.117911. [DOI] [PubMed] [Google Scholar]
- 71.Han W, Li H, Villar VA, Pascua AM, Dajani MI, Wang X, Natarajan A, Quinn MT, Felder RA, Jose PA, Yu P. Lipid rafts keep NADPH oxidase in the inactive state in human renal proximal tubule cells. Hypertension. 2008;51:481–487. doi: 10.1161/HYPERTENSIONAHA.107.103275. [DOI] [PubMed] [Google Scholar]
- 72.Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-kappaB and protein kinase C. J Am Soc Nephrol. 2007;18:1446–1457. doi: 10.1681/ASN.2006121373. [DOI] [PubMed] [Google Scholar]
- 73.Banday AA, Lau YS, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and salt-sensitive hypertension in Sprague-Dawley rats. Hypertension. 2008;51:367–375. doi: 10.1161/HYPERTENSIONAHA.107.102111. [DOI] [PubMed] [Google Scholar]
- 74.Deng A, Baylis C. Locally produced EDRF controls preglomerular resistance and ultrafiltration coefficient. Am J Physiol. 1993;264:F212–215. doi: 10.1152/ajprenal.1993.264.2.F212. [DOI] [PubMed] [Google Scholar]
- 75.Vallon V, Traynor T, Barajas L, Huang YG, Briggs JP, Schnermann J. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J Am Soc Nephrol. 2001;12:1599–1606. doi: 10.1681/ASN.V1281599. [DOI] [PubMed] [Google Scholar]
- 76.Liu R, Ren Y, Garvin JL, Carretero OA. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int. 2004;66:268–274. doi: 10.1111/j.1523-1755.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 77.Nouri P, Gill P, Li M, Wilcox CS, Welch WJ. p22phox in the macula densa regulates single nephron GFR during angiotensin II infusion in rats. Am J Physiol Heart Circ Physiol. 2007;292:H1685–1689. doi: 10.1152/ajpheart.00976.2006. [DOI] [PubMed] [Google Scholar]
- 78.Sun Y, Zhang JN, Zhao D, Wang QS, Gu YC, Ma HP, Zhang ZR. Role of the epithelial sodium channel in salt-sensitive hypertension. Acta Pharmacol Sin. 2011;32:789–797. doi: 10.1038/aps.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun P, Yue P, Wang WH. Angiotensin II stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol. 2012;302:F679–687. doi: 10.1152/ajprenal.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu L, Bao HF, Self JL, Eaton DC, Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am J Physiol Renal Physiol. 2007;293:F1666–1677. doi: 10.1152/ajprenal.00444.2006. [DOI] [PubMed] [Google Scholar]
- 81.Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am J Physiol Cell Physiol. 2013;304:C102–111. doi: 10.1152/ajpcell.00231.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu F, Park F, Cowley AW, Jr, Mattson DL. Quantification of nitric oxide synthase activity in microdissected segments of the rat kidney. Am J Physiol. 1999;276:F874–881. doi: 10.1152/ajprenal.1999.276.6.F874. [DOI] [PubMed] [Google Scholar]
- 83.Dickhout JG, Mori T, Cowley AW., Jr Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res. 2002;91:487–493. doi: 10.1161/01.res.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 84.Mattson DL, Roman RJ, Cowley AW., Jr Role of nitric oxide in renal papillary blood flow and sodium excretion. Hypertension. 1992;19:766–769. doi: 10.1161/01.hyp.19.6.766. [DOI] [PubMed] [Google Scholar]
- 85.Mori T, Cowley AW., Jr Angiotensin II-NAD(P)H oxidase-stimulated superoxide modifies tubulovascular nitric oxide cross-talk in renal outer medulla. Hypertension. 2003;42:588–593. doi: 10.1161/01.HYP.0000091821.39824.09. [DOI] [PubMed] [Google Scholar]
- 86.Beltowski J, Marciniak A, Jamroz-Wisniewska A, Borkowska E. Nitric oxide – superoxide cooperation in the regulation of renal Na(+), K(+)-ATPase. Acta Biochim Pol. 2004;51:933–942. [PubMed] [Google Scholar]
- 87.Silva GB, Ortiz PA, Hong NJ, Garvin JL. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension. 2006;48:467–472. doi: 10.1161/01.HYP.0000236646.83354.51. [DOI] [PubMed] [Google Scholar]
- 88.Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension. 2008;52:1091–1098. doi: 10.1161/HYPERTENSIONAHA.108.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu Rev Physiol. 2011;73:359–376. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 90.Garvin JL, Hong NJ. Nitric oxide inhibits sodium/hydrogen exchange activity in the thick ascending limb. Am J Physiol. 1999;277:F377–382. doi: 10.1152/ajprenal.1999.277.3.F377. [DOI] [PubMed] [Google Scholar]
- 91.Ohsaki Y, O’Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW., Jr Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol. 2012;302:F95–F102. doi: 10.1152/ajprenal.00469.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 93.Peterson JR, Sharma RV, Davisson RL. Reactive oxygen species in the neuropathogenesis of hypertension. Curr Hypertens Rep. 2006;8:232–241. doi: 10.1007/s11906-006-0056-1. [DOI] [PubMed] [Google Scholar]
- 94.Gordon FJ, Haywood JR, Brody MJ, Johnson AK. Effect of lesions of the anteroventral third ventricle (AV3V) on the development of hypertension in spontaneously hypertensive rats. Hypertension. 1982;4:387–393. doi: 10.1161/01.hyp.4.3.387. [DOI] [PubMed] [Google Scholar]
- 95.Brody MJ. Central nervous system and mechanisms of hypertension. Clin Physiol Biochem. 1988;6:230–239. [PubMed] [Google Scholar]
- 96.Whyte DG, Johnson AK. Thermoregulatory role of periventricular tissue surrounding the anteroventral third ventricle (AV3V) during acute heat stress in the rat. Clin Exp Pharmacol Physiol. 2005;32:457–461. doi: 10.1111/j.1440-1681.2005.04211.x. [DOI] [PubMed] [Google Scholar]
- 97.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 98.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 99.Zimmerman MC, Sharma RV, Davisson RL. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension. 2005;45:717–723. doi: 10.1161/01.HYP.0000153463.22621.5e. [DOI] [PubMed] [Google Scholar]
- 100.Peterson JR, Burmeister MA, Tian X, Zhou Y, Guruju MR, Stupinski JA, Sharma RV, Davisson RL. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54:1106–1114. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension. 2013;61:382–387. doi: 10.1161/HYPERTENSIONAHA.111.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Erdos B, Broxson CS, King MA, Scarpace PJ, Tumer N. Acute pressor effect of central angiotensin II is mediated by NAD(P)H-oxidase-dependent production of superoxide in the hypothalamic cardiovascular regulatory nuclei. J Hypertens. 2006;24:109–116. doi: 10.1097/01.hjh.0000198026.99600.59. [DOI] [PubMed] [Google Scholar]
- 103.Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J Neurosci. 2004;24:5516–5524. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nozoe M, Hirooka Y, Koga Y, Sagara Y, Kishi T, Engelhardt JF, Sunagawa K. Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension. 2007;50:62–68. doi: 10.1161/HYPERTENSIONAHA.107.087981. [DOI] [PubMed] [Google Scholar]
- 105.Ostrowski TD, Hasser EM, Heesch CM, Kline DD. H(2)O(2) induces delayed hyperexcitability in nucleus tractus solitarii neurons. Neuroscience. 2014;262:53–69. doi: 10.1016/j.neuroscience.2013.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang G, Coleman CG, Glass MJ, Zhou P, Yu Q, Park L, Anrather J, Pickel VM, Iadecola C. Angiotensin II type 2 receptor-coupled nitric oxide production modulates free radical availability and voltage-gated Ca2+ currents in NTS neurons. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1076–1083. doi: 10.1152/ajpregu.00571.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guyenet PG, Darnall RA, Riley TA. Rostral ventrolateral medulla and sympathorespiratory integration in rats. Am J Physiol. 1990;259:R1063–1074. doi: 10.1152/ajpregu.1990.259.5.R1063. [DOI] [PubMed] [Google Scholar]
- 108.Kishi T, Hirooka Y, Kimura Y, Sakai K, Ito K, Shimokawa H, Takeshita A. Overexpression of eNOS in RVLM improves impaired baroreflex control of heart rate in SHRSP. Rostral ventrolateral medulla. Stroke-prone spontaneously hypertensive rats. Hypertension. 2003;41:255–260. doi: 10.1161/01.hyp.0000050649.30821.cb. [DOI] [PubMed] [Google Scholar]
- 109.Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- 110.Suzuki M, Mori M, Miura S, Suematsu M, Fukumura D, Kimura H, Ishii H. Omeprazole attenuates oxygen-derived free radical production from human neutrophils. Free Radic Biol Med. 1996;21:727–731. doi: 10.1016/0891-5849(96)00180-3. [DOI] [PubMed] [Google Scholar]
- 111.Grassi G, Seravalle G, Brambilla G, Mancia G. The sympathetic nervous system and new nonpharmacologic approaches to treating hypertension: a focus on renal denervation. Can J Cardiol. 2012;28:311–317. doi: 10.1016/j.cjca.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 112.Wang D, Jose P, Wilcox CS. beta(1) Receptors protect the renal afferent arteriole of angiotensin-infused rabbits from norepinephrine-induced oxidative stress. J Am Soc Nephrol. 2006;17:3347–3354. doi: 10.1681/ASN.2006030212. [DOI] [PubMed] [Google Scholar]
- 113.Boivin V, Jahns R, Gambaryan S, Ness W, Boege F, Lohse MJ. Immunofluorescent imaging of beta 1- and beta 2-adrenergic receptors in rat kidney. Kidney Int. 2001;59:515–531. doi: 10.1046/j.1523-1755.2001.059002515.x. [DOI] [PubMed] [Google Scholar]
- 114.DiBona GF, Esler M. Translational medicine: the antihypertensive effect of renal denervation. Am J Physiol Regul Integr Comp Physiol. 2010;298:R245–253. doi: 10.1152/ajpregu.00647.2009. [DOI] [PubMed] [Google Scholar]
- 115.Johns EJ, Kopp UC, DiBona GF. Neural control of renal function. Compr Physiol. 2011;1:731–767. doi: 10.1002/cphy.c100043. [DOI] [PubMed] [Google Scholar]
- 116.Chan SH, Tai MH, Li CY, Chan JY. Reduction in molecular synthesis or enzyme activity of superoxide dismutases and catalase contributes to oxidative stress and neurogenic hypertension in spontaneously hypertensive rats. Free Radic Biol Med. 2006;40:2028–2039. doi: 10.1016/j.freeradbiomed.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 117.Ye S, Zhong H, Yanamadala S, Campese VM. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension. 2006;48:309–315. doi: 10.1161/01.HYP.0000231307.69761.2e. [DOI] [PubMed] [Google Scholar]
- 118.Campese VM, Ye S. A vitamin-E-fortified diet reduces oxidative stress, sympathetic nerve activity, and hypertension in the phenol-renal injury model in rats. J Am Soc Hypertens. 2007;1:242–250. doi: 10.1016/j.jash.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 119.Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, Dudley SC, Jr, Harrison DG. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002;277:48311–48317. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 120.Beswick RA, Dorrance AM, Leite R, Webb RC. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension. 2001;38:1107–1111. doi: 10.1161/hy1101.093423. [DOI] [PubMed] [Google Scholar]
- 121.Suzuki H, Swei A, Zweifach BW, Schmid-Schonbein GW. In vivo evidence for microvascular oxidative stress in spontaneously hypertensive rats. Hydroethidine microfluorography. Hypertension. 1995;25:1083–1089. doi: 10.1161/01.hyp.25.5.1083. [DOI] [PubMed] [Google Scholar]
- 122.Zhou X, Bohlen HG, Miller SJ, Unthank JL. NAD(P)H oxidase-derived peroxide mediates elevated basal and impaired flow-induced NO production in SHR mesenteric arteries in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H1008–H1016. doi: 10.1152/ajpheart.00114.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Swei A, Lacy F, DeLano FA, Schmid-Schonbein GW. Oxidative stress in the Dahl hypertensive rat. Hypertension. 1997;30:1628–1633. doi: 10.1161/01.hyp.30.6.1628. [DOI] [PubMed] [Google Scholar]
- 124.Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, Humphrey JD, Kuo L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension. 2004;44:935–943. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]
- 125.Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol. 2012;165:506–519. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ortiz MC, Sanabria E, Manriquez MC, Romero JC, Juncos LA. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II. Hypertension. 2001;37:505–510. doi: 10.1161/01.hyp.37.2.505. [DOI] [PubMed] [Google Scholar]
- 127.Minuz P, Patrignani P, Gaino S, Degan M, Menapace L, Tommasoli R, Seta F, Capone ML, Tacconelli S, Palatresi S, Bencini C, Del Vecchio C, Mansueto G, Arosio E, Santonastaso CL, Lechi A, Morganti A, Patrono C. Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation. 2002;106:2800–2805. doi: 10.1161/01.cir.0000039528.49161.e9. [DOI] [PubMed] [Google Scholar]
- 128.Folkow B, Grimby G, Thulesius O. Adaptive structural changes of the vascular walls in hypertension and their relation to the control of the peripheral resistance. Acta Physiol Scand. 1958;44:255–272. doi: 10.1111/j.1748-1716.1958.tb01626.x. [DOI] [PubMed] [Google Scholar]
- 129.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 130.Laude K, Cai H, Fink B, Hoch N, Weber DS, McCann L, Kojda G, Fukai T, Schmidt HH, Dikalov S, Ramasamy S, Gamez G, Griendling KK, Harrison DG. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am J Physiol Heart Circ Physiol. 2005;288:H7–12. doi: 10.1152/ajpheart.00637.2004. [DOI] [PubMed] [Google Scholar]
- 131.Patel R, Cardneau JD, Colles SM, Graham LM. Synthetic smooth muscle cell phenotype is associated with increased nicotinamide adenine dinucleotide phosphate oxidase activity: effect on collagen secretion. J Vasc Surg. 2006;43:364–371. doi: 10.1016/j.jvs.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 132.Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22:535–542. doi: 10.1097/00004872-200403000-00016. [DOI] [PubMed] [Google Scholar]
- 133.Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens. 2006;24:757–766. doi: 10.1097/01.hjh.0000217860.04994.54. [DOI] [PubMed] [Google Scholar]
- 134.Zaw KK, Yokoyama Y, Abe M, Ishikawa O. Catalase restores the altered mRNA expression of collagen and matrix metalloproteinases by dermal fibroblasts exposed to reactive oxygen species. Eur J Dermatol. 2006;16:375–379. [PubMed] [Google Scholar]
- 135.Park JB, Intengan HD, Schiffrin EL. Reduction of resistance artery stiffness by treatment with the AT(1)-receptor antagonist losartan in essential hypertension. J Renin Angiotensin Aldosterone Syst. 2000;1:40–45. doi: 10.3317/jraas.2000.009. [DOI] [PubMed] [Google Scholar]
- 136.Schiffrin EL. Vascular stiffening and arterial compliance. Implications for systolic blood pressure. Am J Hypertens. 2004;17:39S–48S. doi: 10.1016/j.amjhyper.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 137.Kals J, Kampus P, Kals M, Pulges A, Teesalu R, Zilmer K, Kullisaar T, Salum T, Eha J, Zilmer M. Inflammation and oxidative stress are associated differently with endothelial function and arterial stiffness in healthy subjects and in patients with atherosclerosis. Scand J Clin Lab Invest. 2008;68:594–601. doi: 10.1080/00365510801930626. [DOI] [PubMed] [Google Scholar]
- 138.Stephen EA, Venkatasubramaniam A, Good TA, Topoleski LD. The effect of oxidation on the mechanical response and microstructure of porcine aortas. J Biomed Mater Res A. 2013 doi: 10.1002/jbm.a.34998. [DOI] [PubMed] [Google Scholar]
- 139.Soskel NT, Watanabe S, Sandberg LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest. 1984;85:70S–73S. doi: 10.1378/chest.85.6_supplement.70s. [DOI] [PubMed] [Google Scholar]
- 140.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18:286–290. doi: 10.1038/nm.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- 143.Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 144.Zheng X, Zinkevich NS, Gebremedhin D, Gauthier KM, Nishijima Y, Fang J, Wilcox DA, Campbell WB, Gutterman DD, Zhang DX. Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J Am Heart Assoc. 2013;2:e000080. doi: 10.1161/JAHA.113.000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cai H, Li Z, Davis ME, Kanner W, Harrison DG, Dudley SC., Jr Akt-Dependent Phosphorylation of Serine 1179 and Mitogen-Activated Protein Kinase Kinase/Extracellular Signal-Regulated Kinase 1/2 Cooperatively Mediate Activation of the Endothelial Nitric-Oxide Synthase by Hydrogen Peroxide. Mol Pharmacol. 2003;63:325–331. doi: 10.1124/mol.63.2.325. [DOI] [PubMed] [Google Scholar]
- 146.Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, Dudley SC, Jr, Harrison DG. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002;277:48311–48317. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 147.Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86:347–354. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- 148.Landmesser U, Harrison DG. Oxidative stress and vascular damage in hypertension. Coron Artery Dis. 2001;12:455–461. doi: 10.1097/00019501-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 149.Theuer J, Dechend R, Muller DN, Park JK, Fiebeler A, Barta P, Ganten D, Haller H, Dietz R, Luft FC. Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord. 2002;2:3. doi: 10.1186/1471-2261-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23:776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 151.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582–593. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- 152.Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008;52:256–263. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. Jama. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 154.Preston RA, Ledford M, Materson BJ, Baltodano NM, Memon A, Alonso A. Effects of severe, uncontrolled hypertension on endothelial activation: soluble vascular cell adhesion molecule-1, soluble intercellular adhesion molecule-1 and von Willebrand factor. J Hypertens. 2002;20:871–877. doi: 10.1097/00004872-200205000-00021. [DOI] [PubMed] [Google Scholar]
- 155.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295:F515–524. doi: 10.1152/ajprenal.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–414. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, Wallukat G, Dechend R, Lamarca B. Administration of Interleukin-17 Soluble Receptor C Suppresses TH17 Cells, Oxidative Stress, and Hypertension in Response to Placental Ischemia During Pregnancy. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.113.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II-Induced Hypertension. Hypertension. 2016;68:167–174. doi: 10.1161/HYPERTENSIONAHA.116.07493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and Mechanical Stretch Promote Aortic Stiffening in Hypertension Through Activation of p38 Mitogen-Activated Protein Kinase. Circ Res. 2014;114:616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, Chen W, Mernaugh RL, Cai H, Bernstein KE, Goronzy JJ, Weyand CM, Curci JA, Barbaro NR, Moreno H, Davies SS, Roberts LJ, 2nd, Madhur MS, Harrison DG. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:50–67. doi: 10.1172/JCI80761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]