

Summary
Argonaute proteins are important effectors in RNA silencing pathways, but they must interact with other machinery to trigger silencing. Ago hooks have emerged as a conserved motif responsible for interaction with Argonaute proteins, but little is know about the sequence surrounding Ago hooks that must restrict or enable interaction with specific Argonautes.
Here we investigated the evolutionary dynamics of an Argonaute-binding platform in NRPE1, the largest subunit of RNA Polymerase V. We compared NRPE1 sequences from more than 50 species, including dense sampling of two plant lineages.
This study demonstrates that the Argonaute-binding platform of NRPE1 retains Ago-hooks, intrinsic disorder, and repetitive character while being highly labile at the sequence level. We reveal that loss of sequence conservation is due to relaxed selection and frequent expansions and contractions of tandem repeat arrays. These factors allow a complete restructuring of the Ago-binding platform over 50-60 million years. This evolutionary pattern is also detected in a second Ago-binding platform, suggesting it is a general mechanism.
The presence of labile repeat arrays in all analyzed NRPE1 Ago-binding platforms indicates that selection maintains repetitive character, potentially to retain the ability to rapidly restructure the Ago-binding platform.
Keywords: Argonaute, Ago hook, Intrinsic disorder, Polymerase V, Relaxed selection, RNA-directed DNA methylation, Repeat expansion, Tandem Repeat
Introduction
Eukaryotes share three canonical RNA polymerases (Pol I - III) that mediate transcription of ribosomal, messenger, and transfer RNAs in the cell, respectively. All plant genomes encode two additional RNA polymerases (Pol IV and V), which facilitate small RNA-directed DNA methylation (Huang et al., 2015; Coruh et al., 2015). Pol IV initiates production of 24 nucleotide small interfering RNAs, which are bound by ARGONAUTE4 (AGO4) and direct AGO4 to non-coding transcripts produced by Pol V. AGO4 associates with both the scaffold transcript and Pol V itself and recruits additional factors to initiate and maintain epigenetic silencing of Pol V transcribed loci (Matzke & Mosher, 2014).
The unique activities of Pol IV and V are enabled primarily through the largest subunits, NRPD1 for Pol IV and NRPE1 for Pol V. While these proteins share extensive similarity with other Pol subunits throughout the catalytic regions, NRPD1 and NRPE1 possess distinctive carboxy-terminal domains. Both carboxy-terminal domains have a DeCL domain with unknown function (Pontier et al., 2005), but only NRPE1 contains a region that is rich in GW, WG, and GWG peptides, also known as Ago hooks (Till et al., 2007) (Fig. 1a). Together these peptides create an Argonaute (Ago)-binding platform that is required for AGO4 association with NRPE1 and small RNA-directed DNA methylation (El-Shami et al., 2007).
Figure 1. NRPE1 contains a non-conserved Ago-binding platform.

(a) Schematic of NRPD1 and NRPE1 genes illustrates the difference in carboxy terminal domains. (b) Identity between NRPE1 orthologs is lowest in the Ago-binding platform. After MUSCLE alignment columns with >20% gaps were stripped; a 10-amino acid sliding window of identity is displayed. (c) The Ago-binding platform is intrinsically disordered. IUPred disorder prediction for four NRPE1 orthologs (Citrus clementina, Brachypodium distachyon, Arabidopsis thaliana, and Linus usitatissimum); a 10-amino acid rolling average is plotted.
Ago hooks mediate interactions between Argonaute proteins and their diverse protein partners, including SPT5L in Arabidopsis thaliana (also known as KTF1), GW182 proteins in animals, Tas3 in fission yeast, and Wag1p and CnjBp in the ciliate Tetrahymena thermophila (Pfaff & Meister, 2013). The Ago hooks in NRPE1 can recruit human Ago2 (El-Shami et al., 2007), demonstrating their function as a conserved short linear motif. Ago-binding platforms vary greatly in number of Ago hooks, from 3 in Tas3 to 45 in SPT5L (Lahmy et al., 2009; Huang et al., 2009; Bies-Etheve et al., 2009). Despite the presence of other conserved domains in Argonaute-interacting proteins, sequence conservation is weak among Ago-binding platforms (Partridge et al., 2007; Till et al., 2007; Lian et al., 2009). There is also no conservation of secondary structure; indeed some Ago-binding platforms are intrinsically disordered (Bednenko et al., 2009; Pfaff & Meister, 2013). Intrinsically disordered regions do not form stable folds, but might adopt a secondary structure upon binding a cofactor (Tompa, 2002).
In Arabidopsis thaliana NRPE1, the Ago-binding platform contains a 17 amino acid tandem repeat that includes an Ago hook (Pontier et al., 2005; El-Shami et al., 2007). The NRPE1 Ago-binding platform is highly diverse across the land plant lineage, showing a variety of type and number of Ago hooks (Huang et al., 2015). Interestingly, nearly all NRPE1 orthologs contain tandem repeats in the Ago-binding platform, although these vary in sequence and repeat number (El-Shami et al., 2007; Huang et al., 2015).
To better understand how evolution shapes Ago-binding platforms, we studied the evolution of the NRPE1 Ago-binding platform in flowering plants. We discovered a very different mode of evolution in the NRPE1 Ago-binding platform relative to conserved domains surrounding this region. This domain exhibits highly relaxed selection coupled with maintenance of three correlated characteristics: intrinsic disorder, presence of Ago hooks, and tandem repeats. Detailed evolutionary analyses in two plant lineages permit estimation of the tempo of divergence and elucidate mechanisms underlying sequence variation in the Ago-binding platform. Parallel analysis of a second Ago-binding protein demonstrates that these mechanisms are general features of Ago-binding platforms. These analyses illuminate an unusual pattern of protein evolution that allows rapid sequence divergence while maintaining key functional features.
Materials and Methods
Identification of orthologs
Previously published NRPE1 sequences (Huang et al., 2015; Wang & Ma, 2015) were retrieved from public databases and assessed for orthology with reciprocal BLAST searches (Table S1). To ensure Ago-binding platforms were full-length, only orthologs with a DeCL domain were included. Orthologs in Oryza, Brassicaceae, and their closest relatives were retrieved through BLAST or TBLASTX searches against whole genomes in CoGe (Lyons et al., 2008) or Ensembl Plants with the Or. sativa or Ar. thaliana nucleotide sequence used as query, respectively (Table S1). Upstream and downstream genes were identified to confirm synteny among orthologs. We inferred phylogeny of the catalytic domain to confirm orthology. SPT5L orthologs were retrieved through BLAST searches of Phytozome and were confirmed by their domain structure.
In unannotated genomes NRPE1 coding sequence was predicted using FGENESH+ with Ar. thaliana or Or. sativa protein sequences at http://www.softberry.com (Softberry, Inc., New York, USA) and then manually corrected. Oryza orthologs with missing sequence data were amplified from genomic DNA using primers GGGAAGAGGATCAAATGGAGGTTCC and CTCAGCACAGTGGGTTCAATTTCTCC. The resulting amplicons were cloned and at least two independent clones were sequenced.
The Ar. arenosa NRPE1 sequence was retrieved from short read data (Hollister et al., 2012) by aligning to the Ar. lyrata genome using Bowtie-2.2.1 (Langmead & Salzberg, 2012) on the iPlant Collaborative Discovery Environment (Oliver et al., 2013). The Ar. arenosa individual with the deepest coverage was designated the reference sequence.
De novo nucleotide sequences were deposited in Genbank (accession numbers KU738145-KU738148) and all NRPE1 and SPT5L coding sequences used for analysis are included in the Supplemental Information (Notes S1-S4).
Characterization of Ago-binding platforms
All protein sequences were visualized and annotated in Geneious 6.1.8 (Biomatters Ltd., Auckland, New Zealand). NRPE1 catalytic domains were annotated as in (Pontier et al., 2005); SPT5L domains were retrieved from the Uniprot (www.uniprot.org) and Pfam (www.pfam.xfam.org) databases. To identify conservation, protein sequences were aligned with MUSCLE in Geneious and positions were removed from downstream analysis when >20% of the sequences lacked data.
Disorder predictions for individual Ago-binding platforms were generated with IUPred at http://iupred.enzim.hu/ (Dosztányi et al., 2005). Disorder tendency for individual residues were averaged to calculate disorder for each Ago-binding platform.
Tandem repeats were identified using three different techniques with different sensitivity to detect repeats. The most conservative approach was T-REKS, which utilizes a k-means algorithm (Jorda & Kajava, 2009). Ago-binding platforms with a T-REKS score ≥ 0.85 were placed in the highest repeat category (+++).Ago-binding platforms with a lower T-REKS score were placed in the second repeat category (++). When T-REKS failed to detect a repeat (score <0.7), Ago-binding platforms were further categorized by self-similarity with RADAR (Heger & Holm, 2000) and dot plots (Geneious 6.1.8). Ago-binding platforms possessing RADAR scores ≥200 were placed in the third category (+) and all others in the last category (-). Importantly, repeats were detectable in all NRPE1 Ago-binding platforms using both self-similarity approaches. For Ago-binding platforms with multiple repeat arrays composed of different repeat units, orthologs were categorized based on the highest scoring tandem repeat.
Phylogenetic assessment of tandem repeats
All nucleotide alignments were performed with MUSCLE (Edgar, 2004) aided by translated amino acid sequence in Geneious 6.1.8. Where necessary, alignments were manually corrected to minimize partial repeat units. Maximum likelihood trees were inferred with RAxML version 7.7.1 (Stamatakis et al., 2008) or PHYML in Geneious 6.1.8 (Guindon & Gascuel, 2003) under a general time reversible (GTR) model with gamma distributed rate heterogeneity. Bootstrap support values were calculated from 100 replicate datasets.
Tests for selection
Population level single nucleotide polymorphism data for Ar. thaliana and Or. sativa were obtained from the 1001 Genomes Project (Cao et al., 2011) and 3000 Rice Genomes Project (Alexandrov et al., 2015), respectively. Accessions containing ambiguous bases or internal stop codons were excluded from analyses. Tajima's D, Ka/Ks, and McDonald-Kreitman tests were performed with DnaSP v 5.10.01 (Librado & Rozas, 2009). For the McDonald-Kretiman test, repeat units without clear co-linearity based on phylogenetic assessment of individual repeats were removed from the alignment.
Results
Ago-binding platforms conserve Ago hooks, disorder, and repetitive character
To assess variation in the NRPE1 Ago-binding platform, we retrieved 30 NRPE1 orthologs from the Phytozome database (Table S1, Fig. S1) and aligned these orthologs with NRPE1 from the model plant Ar. thaliana and the early diverging angiosperm Amborella trichopoda. There was readily detectable conservation of amino acid sequence in the catalytic portion of NRPE1 and the DeCL domain (average identity = 57% and 55%, respectively), but conservation in the Ago-binding platform was poor (average identity = 15%, Fig. 1b, S2). Another indication of the poor alignment within the Ago-binding platform is the high incidence of gaps; the alignment is significantly longer than the longest sequence (1047 vs 690). Pairwise comparisons between closely related species confirm that sequence divergence in the Ago-binding platform is due both to substitutions and insertion/deletion events (Fig. S2).
Despite this lack of sequence identity, all NRPE1 orthologs contained >200 amino acids of sequence between the catalytic and DeCL domains, including ≥1 Ago hook (Table 1), indicating the presence of a bona fide Ago-binding platform. The number of Ago hooks was positively correlated with length of the Ago-binding platform (R2 = 0.78, p < 3×10-10, Fig. S3a). All NPRE1 orthologs also displayed repetitive character, ranging from weak signals in a self-alignment matrix (Heger & Holm, 2000), to highly similar tandem repeats (Table 1) (Jorda & Kajava, 2009). Ago-binding platforms with higher repeat scores tended to be longer and contained more Ago hooks (Fig. S3b). All NRPE1 orthologs also contained an extended region of intrinsic disorder that overlapped the Ago-binding platform (Table 1, Fig. 1c). Intrinsic disorder was specifically located between the catalytic and DeCL domains, with some NRPE1 orthologs also containing a small region of disorder at the extreme carboxy terminus (Fig. 1c). Disorder was correlated with length of the Ago-binding platform (R2 = 0.39, p < 0.001, Fig. S3b), with longer platforms possessing higher average disorder.
Table 1. Characteristics of NRPE1 AGO platforms in angiosperm taxa.
| length1 | AGO hooks | average disorder2 | repeat score3 | |||
|---|---|---|---|---|---|---|
| GW-WG-GWG | ||||||
| Am.tricopoda | 538 | 9 | 6 | 10 | 0.71 | + |
| Aq.coerulea, a | 628 | 2 | 7 | 4 | 0.59 | +++ |
| Aq.coerulea, b | 557 | 9 | 11 | 1 | 0.62 | ++ |
| Ar.thaliana | 502 | 1 | 17 | 1 | 0.75 | ++ |
| Br.distachyon, a | 508 | 3 | 11 | 2 | 0.73 | ++ |
| Br.distachyon, b | 255 | 2 | 1 | 0 | 0.63 | – |
| Ci.clementina | 495 | 5 | 13 | 4 | 0.64 | +++ |
| Ci.sinensis | 494 | 5 | 13 | 4 | 0.63 | +++ |
| Cu.sativus | 576 | 7 | 7 | 5 | 0.69 | + |
| Eu.grandis | 442 | 5 | 7 | 3 | 0.72 | + |
| Fr.vesca | 610 | 2 | 23 | 2 | 0.72 | ++ |
| Gl.max, a | 644 | 3 | 14 | 1 | 0.69 | +++ |
| Gl.max, b | 687 | 4 | 12 | 7 | 0.69 | ++ |
| Go.raimondii | 551 | 10 | 6 | 3 | 0.64 | +++ |
| Li.usitatissimum | 508 | 2 | 4 | 7 | 0.73 | +++ |
| Me.truncatula | 573 | 4 | 17 | 1 | 0.74 | +++ |
| Mi.guttatus | 690 | 2 | 19 | 8 | 0.74 | +++ |
| Or.sativa, a | 533 | 5 | 7 | 2 | 0.73 | ++ |
| Or.sativa, b | 241 | 0 | 1 | 0 | 0.61 | – |
| Pa.virgatum, a | 518 | 4 | 10 | 1 | 0.70 | + |
| Pa.virgatum, b | 234 | 1 | 1 | 0 | 0.50 | – |
| Ph.vulgaris | 667 | 4 | 19 | 4 | 0.72 | ++ |
| Po.trichocarpa, a | 549 | 7 | 7 | 10 | 0.71 | + |
| Po.trichocarpa, b | 266 | 2 | 5 | 2 | 0.59 | – |
| Pr.persica | 466 | 1 | 11 | 0 | 0.62 | +++ |
| Sa.purpurea | 553 | 8 | 6 | 10 | 0.71 | + |
| Se.italica, a | 533 | 3 | 11 | 2 | 0.75 | ++ |
| Se.italica, b | 235 | 1 | 1 | 0 | 0.54 | – |
| So.bicolor | 268 | 2 | 1 | 0 | 0.58 | – |
| So.lycopersicum | 259 | 3 | 4 | 0 | 0.67 | – |
| Th.cacao | 440 | 2 | 10 | 2 | 0.55 | ++ |
| Vi.vinifera | 446 | 5 | 5 | 3 | 0.69 | + |
Length of AGO platform as measured from the end of the H domain to the beginning of the DeCL domain.
Average disorder based on IUPred predictions for each residue in the AGO platform
–, RADAR score <200; +, RADAR score >200 and T-REKS <0.7; ++, T-REKS 0.7-0.85; +++, T-REKS >0.85 (see Materials and Methods)
Ar. thaliana SPT5L also contains an Ago-binding platform with repetitive sequence (Bies-Etheve et al., 2009). We retrieved SPT5L orthologs from 32 angiosperm taxa (Table S1) and consistently identified repetitive structure and Ago-hooks in each sequence. As we previously observed at NRPE1 orthologs (Huang et al., 2015), SPT5L orthologs possess a variety of unique tandem repeats, including multiple repeat units in the same Ago-binding platform (Fig. S4a). Alignment of these sequences demonstrates reduced sequence conservation and increased structural disorder specifically in the Ago-binding platform (Fig. S4bc), similar to the patterns detected in NPRE1.
Known Ago-binding platforms across eukaryotes also evolve repetitive character, as demonstrated in a self-alignment matrix (El-Shami et al., 2007), and specific amino acids are enriched or excluded from predicted Ago-binding domains (Karlowski et al., 2010). We cross-referenced the abundance of amino acids in predicted Ago-binding platforms from diverse proteins with the tendency of specific sidechains to promote order or disorder generally (Radivojac et al., 2007). Setting aside tryptophan due to its necessary presence in Ago hooks, six other order-promoting amino acids are among the eight most underrepresented amino acids in Ago-binding platforms (Fig. S5). This observation suggests that intrinsic disorder is a defining characteristic of Ago-binding platforms, and that selection has acted against the incorporation of order-promoting amino acids within these domains.
Taken together, these observations indicate that the Ago-binding platform in NRPE1 is a suitable representative of Ago-binding platforms from diverse proteins – it is mutable at the sequence level, but diverse orthologs possess Ago hooks, intrinsic disorder, and repetitive nature.
Tandem repeat units in Ago-binding platforms are hypervariable
To assess the evolution of the NRPE1 Ago-binding platform on a finer evolutionary scale, we identified the NRPE1 sequences from 10 species in the genus Oryza (Fig. S6, Table S1), representing 9-10 million years of evolution (Guo & Ge, 2005). Grasses such as Or. sativa contain two NRPE1 paralogs with different Ago-binding platforms; in this analysis we focused on NRPE1a, the paralog with the longer Ago-binding platform. For several species the available genome sequences contained gaps in the NRPE1a locus. We generated full-length genomic sequences for these species before predicting coding sequence. All Oryza NRPE1a proteins possessed the expected RNA polymerase catalytic domains followed by a carboxy terminus containing an Ago-binding platform and a DeCL domain, supporting their identity as NRPE1a homologs. Neighboring genes were also identified to confirm synteny and support orthology (Fig. S6).
All NRPE1a orthologs contained repeated sequence in the Ago-binding platform, as identified by self-alignment analysis, and the repeat unit was nearly identical in each taxon (Fig. 2a). Leersia perrieri, which diverged from species in Oryza ∼14 million years ago (Guo & Ge, 2005), also contained the same repeat unit (Fig. 2b), but more distantly related members of the family Poaceae, such as Brachypodium distachon and Triticum aestivum (40-50 million years diverged from species in Oryza (International Brachypodium Initiative, 2010), do not possess the Oryza repeat in their Ago-binding platforms (Fig. 2c). Brac. distachyon and Tr. aestivum, which diverged from each other 32-39 MYA (International Brachypodium Initiative, 2010), are also dissimilar from each other in the NRPE1a Ago-binding platform (data not shown).
Figure 2. NRPE1 contains a non-conserved Ago-binding platform.

(a) Schematic of the Ago-binding platform in Oryza (Or.) species demonstrates conservation of the tandem repeat array. Cladogram (left) is based on a ML tree of the catalytic regions. (b) Individual repeat units are conserved for Or. sativa to Leersia (Le). perrieri. (c) Brachypodium (Br.) distachyon does not contain sequence with similarity to Oryza repeats. Br. distachyon and Or. sativa carboxy terminal regions were aligned with MUSCLE and the Br. distachyon regions corresponding to Or. sativa repeats were aligned and shaded by similarity to consensus.
To assess conservation of the NRPE1 Ago-binding platform within a second family, we also identified NRPE1 sequences from 16 species in the family Brassicaceae, and from Tarenya hassleriana, a member of the sister family Cleomaceae (diverged from Brassicaceae ∼65 MYA (Beilstein et al., 2010), Fig. S7). Within each Brassicaceae Ago-binding platform we identified a repeat array with similarity to the Ar. thaliana Ago-binding platform (Fig. 3a,b), including in the earliest-diverging taxon, Aethionema arabicum (∼54 million years diverged from Ar. thaliana, (Beilstein et al., 2010). This indicates a common origin of the Ago-binding platform in all Brassicaceae. Although a clearly-defined repeat was not evident in the Ta. hasseleriana Ago-binding platform, there was repetitive structure to this region suggesting the presence of highly decayed repeats (Fig. S8). However, identity between the Brassicaceae and Ta. hassleriana Ago-binding platforms was lowest in the repeat array and it is not clear whether the decayed repeats detected in the Ta. hassleriana Ago-binding platform share an evolutionary origin with the repeats in Brassicaceae Ago-binding platforms. Combined with analysis of Oryza and Poaceae above, this indicates that the Ago-binding platform in NRPE1 can be completely reshaped over 30-65 million years.
Figure 3. The NRPE1 tandem repeat is conserved in Brassicaceae.
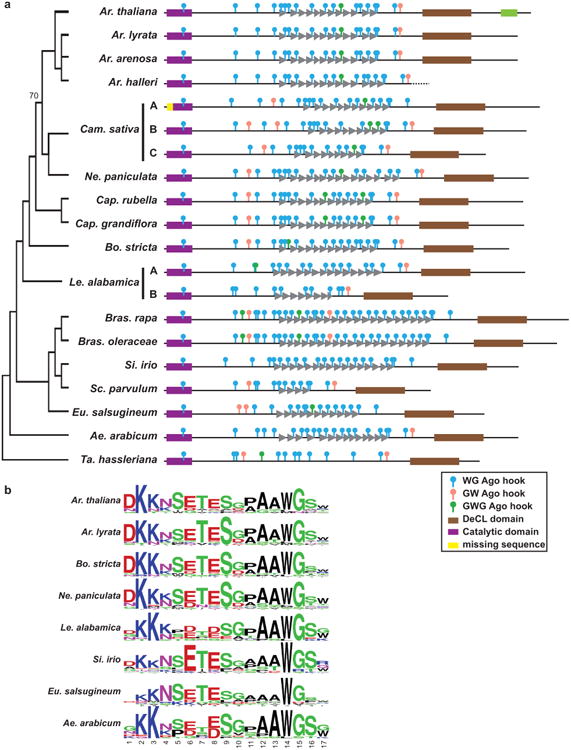
(a) Schematic of the Ago-binding platform in Brassicaceae species demonstrates conservation of the tandem repeat array within Brassicaceae, but not in the sister family Cleomaceae (Ta. hassleriana). Cladogram (left) is a ML tree of the NRPE1 catalytic regions, which matches the expected species tree. Branches with <100 support are marked. Full species names are listed in Supplemental Table 1. (b) Repeat arrays have similar repeat units, but vary in overall consensus sequences.
We next characterized variation in the tandem repeats found in the SPT5L Ago-binding platform. The Ar. thaliana SPT5L is reported to contain two related motifs surrounding its many Ago-hooks (Bies-Etheve et al., 2009). We identified two tandem repeat arrays that share a similar but distinct repeat unit (Fig. 4a). Interestingly, these arrays are encoded by separate exons, suggesting they might represent two discrete Ago-binding platforms. The pattern of two separate tandem repeat encoded on separate exons is found across the Brassicaceae family, including in Ae. arabicum. The sequence of the first repeat array is shared between Ae. arabicum and Ar. thaliana, but the repeat sequence found in the second array is divergent between these species (Fig. 4b), indicating restructuring of this region since their divergence ∼54 million years ago (Beilstein et al., 2010).
Figure 4. SPT5L contains two tandem repeats in the Ago-binding platform.
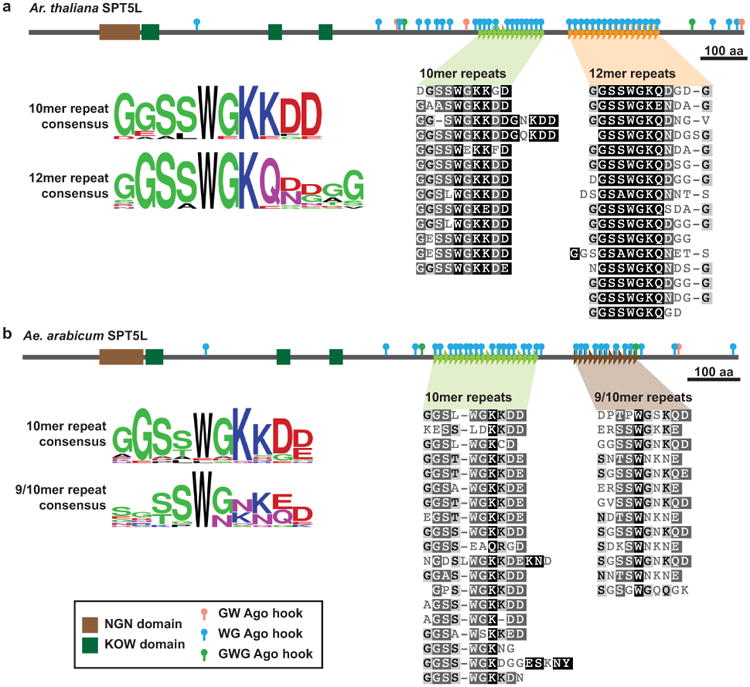
(a) Arabidopsis (Ar.) thaliana SPT5L contains two distinct but related tandem repeat arrays. (b) SPT5L from Aethionema (Ae.) arabicum, the earliest-diverging member of Brassicaceae, also possesses two distinct tandem repeat arrays. The second array has diverged from Ar. thaliana, while the first array remains similar. Repeat units were aligned with MUSCLE and shaded by similarity to consensus.
Repeat arrays in Ago-binding platforms frequently expand and contract
For each NRPE1 repeat array individual repeat units were identified and annotated. As described above, the same basic repeat unit is present in each Oryza or Brassicaceae NRPE1 Ago-binding platform, although there are small differences in the consensus sequence of the repeat between species (Fig. 3b). Most sequence divergence in the Ago-binding platform was due to variation in the number of repeat units contained in each array (Fig. 2a and 3a). The variation was particularly striking in Brassicaceae, with NRPE1 Ago-binding platforms varying from four to twenty copies of the repeat (Fig. 3a). Similar variation in the number of repeat units was found among Brassicaceae SPT5L orthologs. The first array contains 14-21 copies of the repeat, while the second varies from 2-22 copies (Fig. S9). There is no apparent correlation between the sizes of the two arrays in SPT5L, nor between the Ago-binding platforms of NRPE1 and SPT5L.
The variation in size of the repeat array in these Ago-binding platforms suggests that these repeats undergo expansion and contraction due to incorrect pairing during meiosis or repair of double-strand breaks (Paques et al., 1998; Richard & Paques, 2000). To assess this, we aligned individual repeat units from a single NRPE1 repeat array and inferred phylogeny of the repeats (Kane et al., 2010; Schaper et al., 2014). By comparing phylogeny with linear arrangement in the sequence, we infer that single- and multi-repeat duplications have occurred (Fig. 5). For example, in Ar. thaliana sequence similarity between repeat units 1 – 2 – 3 and 10 – 11 –12 supports a duplication involving the three-unit block. This duplication is further supported by the presence of a short interrupting sequence following repeats 1 and 10. Similar analysis in Br. rapa uncovers multiple duplications of a 2-unit segment and other single-unit duplications. The duplication events that characterize Br. rapa and Ar. thaliana are independent and lineage-specific within Brassicaceae.
Figure 5. The NRPE1 tandem repeat arrays are built through duplication.

Phylogenetic analysis of individual repeat units from Arabidopsis (Ar.) thaliana and Brassica (Br.) rapa indicates repeat arrays contain duplications of single or multiple repeat units. (top) repeat array diagrams with related repeats colored the same. Red bars indicated degraded repeat units that were not included in phylogenetic assessment, but further support a duplication. (bottom) Cladograms are ML trees of nucleotide sequence from individual repeats. Given the length of the sequence (51 nucleotide) and possible duplications of partial repeats, we expected the topology to be poorly supported, however some nodes are supported with strong bootstrap values (values ≥50 from 100 replicates are reported).
To acquire further evidence for expansion/contraction at the NRPE1 Ago-binding platform, we tested relationships between repeats from multiple closely-related species. For the most part, the twelve repeat units of Ar. thaliana, the twelve repeat units in Ar. lyrata, and the twelve repeat units of Ar. arenosa were co-linear. Repeat units in the same linear location form a clade within our phylogenetic analyses, suggesting there has been no restructuring of the repeat array since the divergence of these species (Fig. S10). Ar. lyrata and Ar. arenosa also contains the two interrupting sequences at the same position as Ar. thaliana, further supporting the hypothesis of no rearrangement of repeats since the divergence of these species.
Capsella rubella also contains twelve copies of the Brassicaceae repeat unit, however phylogenetic analysis of individual repeat units indicated that a three-repeat segment underwent a duplication independent of that in the Arabidopsis species, and this duplication was coincident with the deletion of three repeats from later in the array (Fig. 6a). Reconstruction of the duplication history of Cap. rubella repeats supported this model of a three-repeat duplication (Fig. S11). Boechera stricta is sister to Cap. rubella in organismal trees (Beilstein et al., 2008), and phylogenetic assessment of Bo. stricta demonstrated that its repeat structure is similar to that of the Arabidopsis species, indicating that this structure was present in the common ancestor of the Arabidopsis-Boechera-Capsella clade (Fig. S12). Analysis of another species closely related to Cap. rubella, Neslia paniculata, identified an additional, more complex expansion event (Fig. 6b), confirming that expansions and contractions of the repeat array are frequent and often lineage-specific. The presence and placement of the interrupting sequence within the Cap. rubella, Bo. stricta, and Ne. paniculata tandem repeat arrays fully supports our conclusions regarding expansion and contraction events in these arrays (Fig. 6, S12).
Figure 6. The NRPE1 tandem repeat array undergoes lineage-specific repeat expansions.

(a) Phylogenetic comparison of individual repeat units from Ar. thaliana (Ar. thal.), Ar. lyrata (Ar. lyr.) and Cap. rubella (Cap. rub.) reveals a tandem duplication of repeats 4-5-6 in the Cap. rubella lineage. (b) Similar analysis with Ne. paniculata (Ne. pani.) demonstrates an independent and complex duplication of a single repeat unit. Cladograms are ML trees with bootstrap support values from 100 replicates listed when ≥50. Branches supporting the duplications are marked with red dots. In the diagrams, repeat units (triangles) are numbered based on phylogenetic relationship to Arabidopsis repeats and colored to match clades above. Duplications are underlined and degenerated repeats that were not included in phylogenetic analysis are shown as red boxes.
The high frequency and complex nature of the expansions and contractions in the tandem repeat array makes it difficult to reconstruct a full duplication history for each sequence. Assessment of phylogenetic relationships between SPT5L tandem repeats is particularly difficult due to the short repeat unit. However other examples of clear expansion or contraction events include the reduction of repeats in Sc. parvulum NRPE1 (Fig. 3) and Cap. rubella SPT5L (Fig. S9), and expansion of repeats in Brassica species NRPE1 (Fig. 3). Further evidence that expansion/contraction events occur frequently can be seen in recent polyploidy species with multiple homeologous sequences. The recent triplication giving rise to Camelina sativa left three copies of NRPE1, which have 9, 10, and 11 copies of the repeat in their Ago-binding platforms, despite diverging only 5.4 MYA (Kagale et al., 2014) (Fig. 3). Similarly, both SPT5L Ago-binding platforms show expansion/contraction events among the three Cam. sativa SPT5L copies (Fig. S9) highlighting the swift evolution of Ago-binding platforms.
Purifying selection is relaxed in the NRPE1 Ago-binding platform
Expansion and contraction of repeats can explain differences in length between the Ago-binding platforms from different taxa, but without divergence between repeat units, this process cannot alter the sequence of a repeat array. The expansion event detected in Ne. paniculata NRPE1, which resulted in 4 copies of what was a single repeat in the ancestral state, demonstrates how small polymorphisms between repeats can be amplified to influence the consensus repeat in a given species and might help explain small differences in the NRPE1 repeat consensus between Brassicaceae species (Fig. 3b).
To investigate the rate of substitution in NRPE1, we first compared the protein sequences of Ar. thaliana, Ar. lyrata, Ar. arenosa, and Bo. stricta. Our previous analysis demonstrates that recombination has not shuffled the repeats between these species (Fig. 5, S11), and therefore differences in nucleotide sequence must be due to substitution events. In the catalytic region amino acid identity ranged from 95-99% for pairwise comparisons, while identity decreased to 87-90% in the Ago-binding platform. This observation supports our hypothesis that the loss of sequence conservation in the Ago-binding platform is not due solely to expansion and contraction of the repeat array.
Amino acid substitutions could arise through neutral processes or might occur through positive selection in the Ago-binding platform. To examine signatures of selection at the population level in the Ar. thaliana or Or. sativa NRPE1 Ago-binding platform, we calculated Tajima's D. Haplotypes were compared from 800 Ar. thaliana individuals and 1134 Or. sativa individuals (Cao et al., 2011; Alexandrov et al., 2015). Due to the low divergence between individuals in the Or. sativa population, the Tajima's D value was not significantly different from neutrality in this species (data not shown). In the Ar. thaliana population, D was -2.19 (p < 0.05), suggesting directional selection. However 15% of the Ar. thaliana genome has a Tajima's D below -2 (Nordborg et al., 2005), and the calculated value was not substantially different from neighboring genes, suggesting that this weakly-significant value could be due to the “hitchhiker effect” or that it might be a remnant of Ar. thaliana population history.
To investigate signatures of selection that occurred during the evolution of the Ago-binding platform in Brassicaceae, we first measured the rate of synonymous (Ks) and non-synonymous (Ka) substitutions between between Ar. thaliana and Bo. stricta in a sliding window across the coding sequence (Fig. 7). There is a clear elevation of the Ka/Ks ratio in the Ago-binding platform driven by an increase in non-synonymous substitutions (average Ka/Ks = 0.45 in Ago-binding platform versus 0.19 in catalytic domain). A similar pattern was detected between Ar. thaliana and Ar. lyrata, albeit with lower substitution rates due to more recent divergence (catalytic = 0.22; Ago-binding = 0.59). Although Ka/Ks remains below 1 throughout NRPE1, indicating that positive selection is not occurring, the elevated Ka/Ks ratio in the Ago-binding platform suggests that relaxed negative selection contributes to diversification of this region.
Figure 7. The Ago-binding platform is under relaxed selection.

Synonymous (green) and non-synonymous (orange) substitutions were calculated between Ar. thaliana and Bo. stricta in 300 nt sliding windows. The Ka/Ks ratio (grey bars) is elevated in the Ago-binding platform relative to the catalytic and DeCL domains.
We also performed a McDonald-Kreitman test of adaptive evolution between the Ar. thaliana population and NRPE1 sequences for which we could confirm homology of repeats phylogenetically. For all comparisons we were unable to reject neutrality in the Ago-binding platform, however the low number of substitutions between these species weakens our ability to detect selective forces. Indeed, we were also unable to reject neutrality within the catalytic region where we anticipate negative selection (Table S2). Earlier-diverging members of the Brassicaceae family display significant negative selection in the catalytic domain, but expansions and contractions of the repeat array make it impossible to measure substitution rates in the Ago-binding platform of these taxa. This pattern further highlights the rapid and specific restructuring of the Ago-binding platform within NRPE1.
Discussion
Our phylogenetic analysis of NRPE1 and SPT5L orthologs across closely-related taxa indicates that the rapid diversification of the Ago-binding platform occurs through frequent expansion and contraction of a tandem repeat array coupled with relaxed negative selection. As substitutions arise in individual repeat units, they are spread through the repeat array by repeat expansions (Kane et al., 2010). Combined with contraction of the repeats, these expansions and substitutions shift the consensus repeat sequence and create sequence diversity while retaining repetitive character (Fig. 8). Our analysis indicates that this iterative process can completely reshape the Ago-binding platform of a lineage in as little as 35 million years.
Figure 8. A model for extreme divergence in the NRPE1 Ago-binding domain.

A tandem repeat array (brown arrows) experiences amino acid substitution (colored bars) and small insertion/deletions. This process degrades the repeats until only very weak similarity remains (middle lineage). Expansion of the repeats through duplication of single or multiple repeat units generates Ago-binding domains with no detectable homology but conservation of repetitive character (compare left and right lineages).
Although evolution is highly relaxed in the NRPE1 Ago-binding platform, selection acts to maintain several characteristics in this domain. Interestingly, these characteristics are also present in SPT5L and other Ago-binding platforms, suggesting that they are fundamental to Ago-binding domains.
The most obviously maintained feature of Ago-binding platforms is the presence of Ago hooks. While we know that Argonaute interaction is mediated through Ago hooks, many aspects of this interaction are unclear (Pfaff & Meister, 2013). Are all Ago hooks in a single platform functional? Do multiple Ago hooks function redundantly, additively, or synergistically? In vitro, a single Ago hook is sufficient to bind AGO4, but eight Ago hooks cause greater AGO4 recovery (El-Shami et al., 2007). Yet if more Ago hooks increase AGO4 association, how do taxa tolerate contractions of their repeat array that reduce the number of Ago hooks? Conversely, if a minimum number of Ago hooks are sufficient, why do some taxa maintain large repeat arrays with many Ago hooks? Our description of the diversity of NRPE1 and SPT5L Ago-binding platforms between closely-related taxa is the first step to answering these questions.
It is also unclear how a specific Ago-binding platform discriminates between diverse Argonaute proteins. The Ago-binding platform from human GW182 can functionally replace the Ago-binding platform in Ar. thaliana NRPE1, suggesting residues surrounding the Ago hooks are dispensable for function (El-Shami et al., 2007). However, this NRPE1-GW182 fusion protein does not fully complement an nrpe1 mutation (El-Shami et al., 2007), indicating that sequence context influences Argonaute binding. It is possible that the Ago-binding platform of NRPE1 is co-evolving with a rapidly changing Argonaute, which is itself evolving to avoid association with viral silencing suppressors (Azevedo et al., 2010).
The second feature that is maintained in Ago-binding platforms is intrinsic disorder. As many as half of all eukaryotic proteins contain an intrinsically-disordered region that does not form an autonomous structure (Tompa, 2002) and these regions have several characteristics that are favorable for an Ago-binding platform. First, the lack of a rigid secondary structure in an intrinsically disordered region reduces evolutionary constraint (Brown et al., 2010; Nilsson et al., 2011; Szalkowski & Anisimova, 2011; Khan et al., 2015), suggesting that IDRs are an important means of evolutionary flexibility. The extreme sequence variation we observed in NRPE1 is a prime example of this flexibility. Intrinsically disordered regions are also frequent sites of Short Linear motifs (SLiMs) such as Ago hooks (Neduva & Russell, 2005; Fuxreiter et al., 2007) and many intrinsically disordered domains function in molecular recognition (van der Lee et al., 2014). Finally, there are a number of advantages to disorder with respect to protein binding. Due to their unfolded nature, disordered regions can contact a larger area of a binding partner to enhance specificity, or to assemble several proteins into a complex (Tompa, 2002). Some intrinsically disordered regions also undergo synergistic folding and adopt a folded structure upon binding their partner. The loss of entropy associated with synergistic folding balances binding affinity and allows reversible but specific interactions (Tompa, 2002).
Finally, it is clear that Ago-binding platforms also maintain repetitive character. This is not altogether surprising, as tandem repeats (including well-structured motifs such as Armadillo, Ankyrin, WD40, and leucine-rich repeats) are a major component of eukaryotic genomes (Kajava, 2012). Expansion and contraction of repeats offers an opportunity for rapid evolution of regulatory or functional roles (Gemayel et al., 2010). Given the high rate of modification of the tandem repeat array NRPE1 and SPT5L Ago-binding platforms, it is particularly surprising that repetitiveness is maintained in these regions. One might expect that at some frequency a repeat array would contract to a single unit and/or become too heterogeneous due to substitution to allow new expansions. In these situations repetitive character would be lost while intrinsic disorder and Ago hooks would be maintained. While we find examples of NRPE1 orthologs with very weak repetitive signals (Table 1), these are rare and appear to be taxon-specific. For example, Ta. hassleriana (65 MY diverged from Brassicaceae, (Beilstein et al., 2010) has heavily degraded repeats while Carica papaya (72 MYA diverged, (Ming et al., 2008) has a strong repeat score but does not contain the Brassicaceae repeat unit. While it is possible that these disordered tandem repeats have an as yet undiscovered purpose, our evidence indicates that the tandem repeat array exists as a mechanism to enable rapid sequence alterations. This raises the provocative hypothesis that Ago-binding platforms conserve of the ability to diversify and that any platform that becomes frozen due to lack of repetitiveness is eventually selected against. Alternatively, selection might promote the generation of tandem repeats from non-repetitive Ago-binding platforms, although there is no clear the mechanism for this selection. Taken together, our analyses build on findings from other eukaryotic systems and highlight an unusual pattern of protein evolution that promotes rapid sequence divergence while retaining key functional features.
Supplementary Material
Acknowledgments
We wish to thank Drs. Dario Copetti and Rod Wing for supplying genomic DNA from Oryza species and members of the PaBeBaMo group for helpful discussions. This work is supported by National Science Foundation Grant MCB-1243608 to RAM.
Footnotes
Author Contributions: RAM and MAB designed and oversaw the experiments; JTT and RAM performed experiments; RAM, JTT, and MAB wrote the manuscript.
References
- Alexandrov N, Tai S, Wang W, Mansueto L, Palis K, Fuentes RR, Ulat VJ, Chebotarov D, Zhang G, Li Z, et al. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic acids research. 2015;43:D1023–7. doi: 10.1093/nar/gku1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo J, Garcia D, Pontier D, Ohnesorge S, Yu A, Garcia S, Braun L, Bergdoll M, Hakimi MA, Lagrange T, et al. Argonaute quenching and global changes in Dicer homeostasis caused by a pathogen-encoded GW repeat protein. Genes & Development. 2010;24:904–915. doi: 10.1101/gad.1908710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednenko J, Noto T, DeSouza LV, Siu KWM, Pearlman RE, Mochizuki K, Gorovsky MA. Two GW Repeat Proteins Interact with Tetrahymena thermophila Argonaute and Promote Genome Rearrangement. Molecular and Cellular Biology. 2009;29:5020–5030. doi: 10.1128/MCB.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilstein MA, Al-Shehbaz IA, Mathews S, Kellogg EA. Brassicaceae phylogeny inferred from phytochrome A and ndhF sequence data: tribes and trichomes revisited. American Journal of Botany. 2008;95:1307–1327. doi: 10.3732/ajb.0800065. [DOI] [PubMed] [Google Scholar]
- Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bies-Etheve N, Pontier D, Lahmy S, Picart C, Vega D, Cooke R, Lagrange T. RNA-directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO reports. 2009;10:649–654. doi: 10.1038/embor.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Johnson AK, Daughdrill GW. Comparing models of evolution for ordered and disordered proteins. Molecular biology and evolution. 2010;27:609–621. doi: 10.1093/molbev/msp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature genetics. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Coruh C, Cho SH, Shahid S, Liu Q, Wierzbicki A, Axtell MJ. Comprehensive Annotation of Physcomitrella patens Small RNA Loci Reveals That the Heterochromatic Short Interfering RNA Pathway Is Largely Conserved in Land Plants. The Plant Cell. 2015;27:2148–2162. doi: 10.1105/tpc.15.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztányi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics (Oxford, England) 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shami M, Pontier D, Lahmy S, Braun L, Picart C, Vega D, Hakimi MA, Jacobsen SE, Cooke R, Lagrange T. Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes & Development. 2007;21:2539–2544. doi: 10.1101/gad.451207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics (Oxford, England) 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annual review of genetics. 2010;44:445–477. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Guo YL, Ge S. Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. American Journal of Botany. 2005;92:1548–1558. doi: 10.3732/ajb.92.9.1548. [DOI] [PubMed] [Google Scholar]
- Heger A, Holm L. Rapid automatic detection and alignment of repeats in protein sequences. Proteins. 2000;41:224–237. doi: 10.1002/1097-0134(20001101)41:2<224::aid-prot70>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Hollister JD, Arnold BJ, Svedin E, Xue KS, Dilkes BP, Bomblies K. Genetic adaptation associated with genome-doubling in autotetraploid Arabidopsis arenosa. In: Mauricio R, editor. PLoS Genetics. Vol. 8. 2012. p. e1003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Jones AME, Searle IR, Patel K, Vogler H, Hubner NC, Baulcombe DC. An atypical RNA polymerase involved in RNA silencing shares small subunits with RNA polymerase II. Nature Structural & Molecular Biology. 2009;16:91–93. doi: 10.1038/nsmb.1539. [DOI] [PubMed] [Google Scholar]
- Huang Y, Kendall T, Forsythe ES, Dorantes-Acosta A, Li S, Caballero-Perez J, Chen X, Arteaga-Vázquez M, Beilstein MA, Mosher RA. Ancient Origin and Recent Innovations of RNA Polymerase IV and V. Molecular biology and evolution. 2015;32:1788–1799. doi: 10.1093/molbev/msv060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- Jorda J, Kajava AV. T-REKS: identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics (Oxford, England) 2009;25:2632–2638. doi: 10.1093/bioinformatics/btp482. [DOI] [PubMed] [Google Scholar]
- Kagale S, Koh C, Nixon J, Bollina V, Clarke WE, Tuteja R, Spillane C, Robinson SJ, Links MG, Clarke C, et al. The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nature Communications. 2014;5:3706. doi: 10.1038/ncomms4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava AV. Tandem repeats in proteins: from sequence to structure. Journal of structural biology. 2012;179:279–288. doi: 10.1016/j.jsb.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Kane J, Freeling M, Lyons E. The Evolution of a High Copy Gene Array in Arabidopsis. Journal of molecular evolution. 2010;70:531–544. doi: 10.1007/s00239-010-9350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlowski WM, Zielezinski A, Carrère J, Pontier D, Lagrange T, Cooke R. Genome-wide computational identification of WG/GW Argonaute-binding proteins in Arabidopsis. Nucleic acids research. 2010;38:4231–4245. doi: 10.1093/nar/gkq162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T, Douglas GM, Patel P, Nguyen Ba AN, Moses AM. Polymorphism Analysis Reveals Reduced Negative Selection and Elevated Rate of Insertions and Deletions in Intrinsically Disordered Protein Regions. Genome biology and evolution. 2015;7:1815–1826. doi: 10.1093/gbe/evv105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmy S, Pontier D, Cavel E, Vega D, El-Shami M, Kanno T, Lagrange T. PolV(PolIVb) function in RNA-directed DNA methylation requires the conserved active site and an additional plant-specific subunit. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:941–946. doi: 10.1073/pnas.0810310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian SL, Li S, Abadal GX, Pauley BA, Fritzler MJ, Chan EKL. The C-terminal half of human Ago2 binds to multiple GW-rich regions of GW182 and requires GW182 to mediate silencing. RNA (New York, NY) 2009;15:804–813. doi: 10.1261/rna.1229409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics (Oxford, England) 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch D, et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiology. 2008;148:1772–1781. doi: 10.1104/pp.108.124867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nature Reviews Genetics. 2014;15:394–408. doi: 10.1038/nrg3683. [DOI] [PubMed] [Google Scholar]
- Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KLT, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452:991–996. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V, Russell RB. Linear motifs: evolutionary interaction switches. FEBS letters. 2005;579:3342–3345. doi: 10.1016/j.febslet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Grahn M, Wright APH. Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins. Genome Biol. 2011;12:R65. doi: 10.1186/gb-2011-12-7-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, Bakker E, Calabrese P, Gladstone J, Goyal R, et al. The pattern of polymorphism in Arabidopsis thaliana. In: Mitchell-Olds T, editor. PLoS Biology. Vol. 3. 2005. pp. e196–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver SL, Lenards AJ, Barthelson RA, Merchant N, McKay SJ. Using the iPlant collaborative discovery environment. Current protocols in bioinformatics. 2013;Chapter 1 doi: 10.1002/0471250953.bi0122s42. editoral board, Andreas D. Baxevanis … [et al.] Unit1.22–1.22.26. [DOI] [PubMed] [Google Scholar]
- Paques F, Leung WY, Haber JE. Expansions and contractions in a tandem repeat induced by double-strand break repair. Molecular and Cellular Biology. 1998;18:2045–2054. doi: 10.1128/mcb.18.4.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JF, DeBeauchamp JL, Kosinski AM, Ulrich DL, Hadler MJ, Noffsinger VJP. Functional separation of the requirements for establishment and maintenance of centromeric heterochromatin. Molecular Cell. 2007;26:593–602. doi: 10.1016/j.molcel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Pfaff J, Meister G. Argonaute and GW182 proteins: an effective alliance in gene silencing. Biochemical Society transactions. 2013;41:855–860. doi: 10.1042/BST20130047. [DOI] [PubMed] [Google Scholar]
- Pontier D, Yahubyan G, Vega D, Bulski A, Saez-Vasquez J, Hakimi MA, Lerbs-Mache S, Colot V, Lagrange T. Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes & Development. 2005;19:2030–2040. doi: 10.1101/gad.348405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophysical Journal. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard GF, Paques F. Mini- and microsatellite expansions: the recombination connection. EMBO reports. 2000;1:122–126. doi: 10.1093/embo-reports/kvd031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper E, Gascuel O, Anisimova M. Deep conservation of human protein tandem repeats within the eukaryotes. Molecular biology and evolution. 2014;31:1132–1148. doi: 10.1093/molbev/msu062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Systematic Biology. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Szalkowski AM, Anisimova M. Markov models of amino acid substitution to study proteins with intrinsically disordered regions. In: Uversky VN, editor. PLoS One. Vol. 6. 2011. p. e20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till S, Lejeune E, Thermann R, Bortfeld M, Hothorn M, Enderle D, Heinrich C, Hentze MW, Ladurner AG. A conserved motif in Argonaute-interacting proteins mediates functional interactions through the Argonaute PIWI domain. Nature Structural & Molecular Biology. 2007;14:897–903. doi: 10.1038/nsmb1302. [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends in Biochemical Sciences. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, et al. Classification of intrinsically disordered regions and proteins. Chemical reviews. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ma H. Step-wise and lineage-specific diversification of plant RNA polymerase genes and origin of the largest plant-specific subunits. The New phytologist. 2015;207:1198–1212. doi: 10.1111/nph.13432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.