

Abstract
Physiologically based pharmacokinetic (PBPK) modeling and simulation can be used to predict the pharmacokinetic behavior of drugs in humans using preclinical data. It can also explore the effects of various physiologic parameters such as age, ethnicity, or disease status on human pharmacokinetics, as well as guide dose and dose regiment selection and aid drug–drug interaction risk assessment. PBPK modeling has developed rapidly in the last decade within both the field of academia and the pharmaceutical industry, and has become an integral tool in drug discovery and development. In this mini-review, the concept and methodology of PBPK modeling are briefly introduced. Several case studies were discussed on how PBPK modeling and simulation can be utilized through various stages of drug discovery and development. These case studies are from our own work and the literature for better understanding of the absorption, distribution, metabolism and excretion (ADME) of a drug candidate, and the applications to increase efficiency, reduce the need for animal studies, and perhaps to replace clinical trials. The regulatory acceptance and industrial practices around PBPK modeling and simulation is also discussed.
KEY WORDS: PBPK, PK prediction, Absorption, Metabolism, Drug–drug interaction, Special population
Graphical Abstract
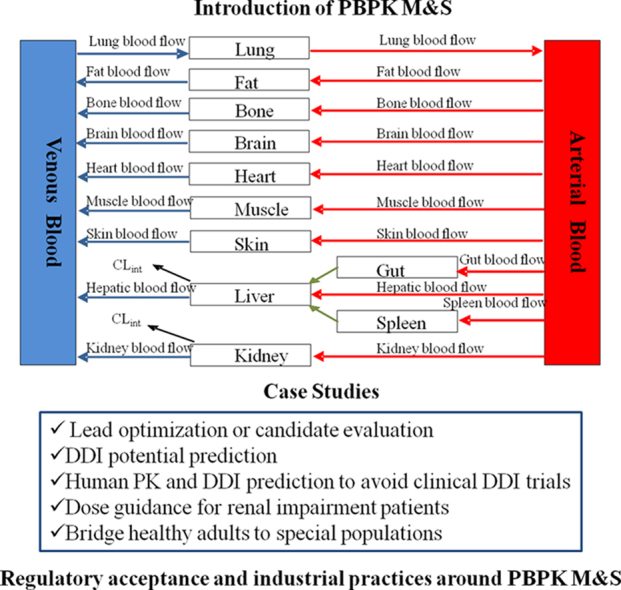
Physiologically based pharmacokinetic (PBPK) modeling has developed rapidly in the last decade within both the field of academia and the pharmaceutical industry, and has become an integral tool in drug discovery and development. In this review, the concept and methodology of PBPK modeling are briefly introduced. Several case studies were discussed based on our work about how PBPK modeling and simulation (M&S) can be utilized through various stages of drug discovery and development. The regulatory acceptance and industrial practices around PBPK modeling and simulation is also discussed.
1. Introduction
The concept of physiologically based pharmacokinetic (PBPK) models was first introduced by Teorell in 19371. For several decades, growing efforts have been made to refine PBPK models so that they can be applied in drug development2. Thanks to the advancement in computing power and increasing access to preclinical data, especially in vitro data, on absorption, distribution, metabolism and excretion (ADME). PBPK modeling and simulation currently receives extensive attention during drug discovery and development3, 4, and in submissions for regulatory filing and reviews5, 6. As a unique tool, PBPK models can be used to estimate the pharmacokinetic (PK) profile of a compound based on its preclinical ADME data and can be used to assess the exposure in a target organ after the administration of a drug by taking into account the rate of absorption and disposition in that organ, as well as metabolism within that organ if it is applicable. Based on the PK data generated from one dose schedule, the PBPK model can be used to evaluate the PK profile of different dose schedules and/or dose routes. Based on the PK data from one ethnic population, the PBPK model can be used to predict the PK profile in different ethnic populations as well as populations of various age and disease stages. This mini-review describes the PBPK methodology used in drug discovery and development and specific examples of its application together with the regulatory acceptance and industrial common applications.
2. PBPK methodology
PBPK models are made up of compartments corresponding to the different physiological organs of the body, linked by the circulating blood system. Each compartment is exactly described by a tissue volume and blood flow rate that is specific to the species of interest. Each tissue is defined with assumptions of either perfusion-rate-limited or permeability-rate-limited. Perfusion-rate-limited kinetics tends to exist for small lipophilic molecules where the blood flow to the tissue proved to be the limiting process of the absorption. Permeability-rate-limited kinetics occurs for more hydrophilic and larger molecules where the permeability across the cell membrane becomes the limiting process of absorption7. Drug is disposed via the exile blood flow after being metabolized in the organ, if applicable.
A schematic of a PBPK model is shown in Fig. 1. The mass balance differential equations used in these models have been described previously8 and follow the principles shown below.
Figure 1.

Schematic of a PBPK model.
Non-Eliminating tissues:
| (1) |
where Q is blood flow (L/h), C is concentration (mg/L), V is volume (L); and T represents tissues, A represents arterial, V represents venous.
| (2) |
where Kp is tissue to plasma partition coefficient of the compound and B:P is the ratio of blood to plasma.
Eliminating tissues:
| (3) |
where CLint is the intrinsic clearance of the compound (L/h), and u is unbound.
Different from the conventional PK models, PBPK model is composed of two main parts—an anatomical “backbone” which contains species specific physiological parameters that are independent of the drug and hence can be applied to any compounds, and a drug-specific part which consists of the individual drug׳s ADME properties applied to the relevant processes within each tissue compartment. Parameters for incorporating into PBPK models are either drug-dependent (e.g., binding to blood, fub; tissue-to-plasma distribution coefficient, KPT; tissue permeability–surface area product, PST; enzymatic activity, Vmax/Km) or drug-independent (e.g. blood flows, QT; tissue volumes, VT; tissue composition). The accuracy of the PBPK prediction of ADME parameters by the model not only depends on the present knowledge of animal or human physiology, but also on the physiochemical and biochemical properties of the test compounds.
3. The key points in PBPK model construction
3.1. Acquisition of drug dependent parameters
PBPK modeling is a bottom-up approach that integrates a large number of drug specific data, parameters on species physiology (system data), and a good understanding of all active processes affecting the pharmacokinetic properties of a drug. System-dependent parameters (e.g., tissue volume, blood flow, glomerular filtration rate, amount of microsomal protein/hepatocytes per gram of liver, plasma protein, enzyme, and transporter abundance) for human and preclinical species are available in the literature and have been compiled in the commercial PBPK platforms, including GastroPlus (www.simulations-plus.com), PKSIM (www.systemsbiology.com/products/pk-sim.html), Simcyp (www.simcyp.com), ADMEWORKS DDI Simulator (http://www.fqs.pl/chemistry_materials_life_science/products/ddi_simulator), CLOEPK (http://www.cyprotex.com/insilico/), and many other modeling software. For example, PBPK models in humans specify ethnic population (specific system parameters) account for variability (standard deviation or range) and the covariation between these parameters in that ethnic population. Drug dependent parameters include physicochemical properties (molecular weight, pKa, basic or acidic nature of the drug), solubility (logD) and permeability, blood cell and plasma protein binding (e.g. fraction unbound in plasma (fu,p), blood plasma partitioning [B:P]), transporter contribution to drug disposition, and in vitro data on the metabolism by hepatic or ex-hepatic enzymes (e.g., intrinsic clearance (CLint)). A lack of sufficient in vitro and in vivo data may hamper the use of this approach. These compound-specific parameters are often determined using in vitro assays or sometimes in silico models. Table 1 lists the input data required for building a basic PBPK model, in this case, for Simcyp simulation.
Table 1.
Data requirement for building a PBPK model in Simcyp®.
| Parameter | Unit convert to | In vitro test system |
|---|---|---|
| Molecular weight | g/mol | Physicochemistry property measurement, less prefer an in silico prediction |
| logP | Octanol:water partition coefficient | |
| pKa (s) | Physicochemistry property measurement, less prefer to use an in silico prediction | |
| Compound type | Base, acid, neutral | Based on the chemical structure or pH-dependent solubility test |
| pH-dependent solubility | µg/mL | Measured in buffer with different pH |
| Plasma protein binding | fu | In vitro in human plasma (pay attention to whether compound binds to AGP) |
| Blood–plasma partitioning | B:P | In vitro in human blood |
| Apparent permeability | 10−6 cm/s | Caco-2 , MDCK |
| Intrinsic clearance in microsomes, or S9, or hepatocytes, or rhCYP | µL/min/mg for microsomes and S9, uL/min/million cells for hepatocytes, uL/min/pmol for rhCYP | In vitro assay, or use in vivo clearance if available |
| Protein concentration in in vitro test | mg/mL | In vitro assay for intrinsic clearance |
| In vitro test matrix binding | fu | Measure the free fraction using the same protein concentration in the in vitro test system |
| Vmax and Km (if study for saturable PK; study metabolic-mediated DDI as a victim) | pmol/min/mg, µmol/L | The same in vitro system where intrinsic clearance was determined |
| Percent of enzyme (e.g. CYP) contribution to the metabolism (study DDI as a victim; study metabolic-mediated DDI as a perpetrator) | fm | In vitro reaction phenotyping |
| Reversible inhibition, IC50 | µmol/L | Human liver microsomes or suitable in vitro system |
| Mechanism-based CYP inhibition, kinact, KI | h–1, µmol/L | |
| CYP Induction, Jmax, EC50 | fold induction, µmol/L | Human hepatocytes with positive controls in 3 donors |
Some transporter data can be incorporated; when clinical data become available, CL, Vss, fa, Ka, etc., can be incorporated to refine the initial model.
3.2. Combination of “bottom-up” and “middle-out” methods to create and refine a PBPK model
PBPK modeling typically uses a “bottom-up” approach and is initially constructed based on preclinical data during the early drug discovery stage. Compound-specific parameters generated using in vitro models are used to predict in vivo PK profile in preclinical species and humans. For example, a “bottom-up” methodology for the clinical PK profile prediction proposed by Jones et al.9 is described as following:
-
(1)
Verification of intravenous disposition prediction in preclinical species, for example, assessment of most appropriate Kp prediction methodology taking into account method assumptions, assessment of the prediction accuracy and the physicochemical properties of the particular compound;
-
(2)
Verification of oral absorption prediction in preclinical species over a range of doses to further assess prediction accuracy;
-
(3)
Simulation of disposition and absorption in humans—using appropriate CL and Kp prediction methods selected based on the preclinical verification step. Once preclinical or clinical in vivo data are available, the mechanistic PBPK models can be further refined and updated (“middle-out” approach) and applied prospectively to simulate unstudied scenarios and, when appropriate, these predictions can be incorporated into regulatory submissions, product labels, additional post-approval studies, and next generation follow-on drugs10.
During this stage, mismatches between simulation and observation may frequently occur and parameter sensitivity analysis is critical to identify the inputs that have the most influence on a simulated profile. The selection of which parameters to focus upon for the parameter sensitivity analysis requires a good understanding of the nature of each input data, as well as how they may impact the simulated profile. It is also important to have an understanding of how the input data are generated and the associated errors, and also an awareness of the reasonable range of input values.
4. Applications of PBPK modeling during drug research and development
PBPK models are routinely applied from the early discovery stage, where there is limited data captured for any compound of interest, to late drug development, where large amounts of data are available9. PBPK modeling can be categorized into three major roles, that can be used to inform regulatory communications, that have impacted clinical development decisions and that promote the mechanistic understanding of clinical observations11.
5. Lead optimization or candidate evaluation, a case study
Unlike late development stages where PK data from animal can be used to refine the PBPK model built on in vitro data, in drug discovery, these processes mainly rely on the use of physicochemical properties, in vitro data, and increasingly in silico data. This example illustrates the use of a PBPK absorption model (GastroPlus v. 8.5) in the prediction of human oral bioavailability from preclinical studies for a candidate compound. YQA-14 is a novel and selective dopamine D3 receptor antagonist, with the potential to treat drug addiction. Earlier compounds in its structural class tend to have poor oral bioavailability in humans due to the pronounced metabolism from aldehyde oxidase (AO). The aim of this study was to simulate the clinical pharmacokinetic behavior of YQA-14 using a PBPK model to assess the likelihood of developing YQA-14 as a clinical candidate12. YQA-14 is a lipophilic and basic compound with three pKa values (6.91, 9.30, and 10.91) and a logD7.4 value of 2.15. At pH 6.5, the solubility of YQA-14 was 0.004 mg/mL. It was stable in human liver cytosolic fractions (less AO metabolism liability compared to the previous candidates), and the liver microsomal clearances and in vivo clearances were moderate in rats, dogs (in vitro and in vivo) and humans (in vitro only). It also had moderate bioavailability in preclinical species. For human PK prediction, a “bottom-up” full PBPK model was first built by inputting the main parameters obtained from in vitro studies (Table 2). This model was then validated and modified by in vivo PK profiles of rats and dogs (Fig. 2). After oral administration, YQA-14 was rapidly absorbed in preclinical species with a Tmax around 0.5–1 h; this is consistent with the high permeability obtained from the Caco-2 assessment. Oral bioavailability in rats and dogs were 15.6% and 45.9%, respectively. Because rats have a higher hepatic clearance, both in vitro and in vivo, then might have a higher pre-system metabolism. Poor solubility could be another reason for the lower bioavailability in rats because a higher dose was given to rats compared to dogs. After the preclinical model was validated, physicochemical properties, models/modules used to predict tissue distribution, compound dissolution and precipitation information, combined with respective in vitro human data (clearance, plasma protein and microsomal binding, and RBC partitioning) were utilized to simulate human plasma concentration vs. time profiles of YQA-14 at 287 mg QD, a therapeutic dose extrapolated from the rat pharmacology study. A bioavailability of 16.9% was predicted in humans. However, after decreasing the oral dose from 287 mg to 57.4 mg (the low end of the projected human efficacious dose), the predicted bioavailability increased from 16.9% to 35.1%, whereas no change in elimination parameters such as t1/2 was observed, suggesting that solubility did play a role in the absorption of YQA-14 in humans. These acceptable PK properties make YQA-14 an improved candidate for further development as a potential dopamine D3R antagonism for the treatment of drug addiction in clinic.
Table 2.
Input data used in the GastroPlus™ PBPK model.
| Parameter | Value |
|---|---|
| Molecular weight (g/mol) | 442.95 |
| pKa | 6.91, 9.30, 10.91 |
| logD at pH 7.4 | 2.15 |
| Caco-2 permeability (10–6 cm/s) | 19.90, 22.13 |
| (propranolol, control) | |
| Aqueous solubility at pH 6.5 (mg/mL) | 0.004 |
| Rbp in rat, dog, human | 0.70, 0.67, 0.65 |
| %Fu in rat, dog, human plasma and human liver microsomes | 1.29, 1.05, 0.96, 64 |
| CLint in rat, dog and human liver microsomes (mL/min/kg) | 57.60, 6.42, 13.46 |
| In vitro predicted hepatic clearance in rat, dog and human (mL/min/kg) | 31.60, 5.49, 8.05 |
| In vivo clearance, rat, dog (mL/min/kg) | 29.7, 8.3 |
| Dose (rat, dog, and human, mg/kg, QD) | 25, 5, 4.1 |
This table is adapted from Ref. 12 with permission.
Figure 2.

Observed (□) and PBPK model–simulated (-) plasma concentration–time profiles of YQA-14 in rats (A and B) and dogs (C and D) after a single i.v. (A and C) or (p.o.) (B and D) administration. Observed plasma concentration–time profiles (OBS) were obtained for rats and dogs after single i.v. and p.o. administration of YQA-14 at 25 and 5 mg/kg, respectively (n=3 rats/group; n=4 dogs/group). This figure is adapted from Ref. 12 with permission.
6. Drug–drug interaction (DDI) potential prediction, a case study
This example demonstrates the potential of using PBPK modeling in the prediction of DDI risk13. Naturally occurring furanocoumarin compounds psoralen (PRN) and isopsoralen (IPRN) are bioactive constituents in herbaceous plants. They are widely used as active ingredients in many Chinese herbal medicines. Both PRN and IPRN showed potent reversible inhibition of CYP1A2 in human liver microsomes (HLMs). In addition, time-dependent inhibition of CYP1A2 was observed with IPRN but not PRN. In an attempt to assess the potential DDI risk, Simcyp simulations were conducted to predict phenacetin (a CYP1A2 substrate) AUC changes under the co-administration of PRN or IPRN by allowing perpetrator and victim dosed at the same time once a day for 10 days. A reduced PBPK model was built using the basic physicochemical data listed in Table 314. Simulations were performed in healthy subjects (n=100, 50% men, aged 40–65 years) by using a Simcyp population-based simulator (version 11, Simcyp Ltd., Sheffield, UK). A population of smokers was constructed by modifying the CYP1A2 abundance in healthy subjects from 52 to 94 pmol/mg microsomal protein to mimic individuals who smoke 20 cigarettes per day14. The Simcyp default phenacetin profile was used without further modification. The maximum allowed daily doses of PRN and IPRN (60 mg, Chinese Pharmacopoeia Commission, 2010) were used to predict the worst-case scenario of DDI. Fig. 3 presents the 10-day simulations of plasma concentration–time profiles of a 1500 mg daily dose of phenacetin with a 60 mg daily dose of PRN or IPRN. The results showed that PRN increased the AUC of phenacetin by 1.71-fold and 2.12-fold in healthy volunteers and smokers, respectively, whereas IPRN increased the AUC of phenacetin by 3.24-fold and 5.01-fold in healthy volunteers and smokers, respectively. It is worth noting that in this simulation, the smoker population has lower basal AUC because of their high CYP1A2 activity. Co-administration of the moderate reversible inhibitor PRN was not able to bring the AUC back to the level of a healthy volunteer, suggesting an incomplete balance of the higher CYP1A2 activity induced by the smoke. However, when a more potent inhibition IPRN was applied (both reversible and time-dependent CYP1A2 inhibitor), the AUC in the smoke population was comparable to the level of healthy volunteer suggesting that the inhibition and inactivation effects by IPRN balanced off the CYP1A2 activity induced by smoke. On the other hand, the change of clearance and AUC were more profound when the co-administration IPRN or PRN in smoke population.
Table 3.
Input data of PRN and IPRN for Simcyp® simulation.
| Parameter | PRN | IPRN |
|---|---|---|
| Molecular weight | 186.17 | 186.17 |
| logD7.4 | 1.63 | 1.32 |
| Blood–plasma partition co-efficient (B/P) | 0.82 | 0.65 |
| Plasma protein binding (fu) | 0.283 | 0.126 |
| Microsomal protein binding at 0.5 mg/mL (fu) | 0.745 | 0.906 |
| Apparent permeability value: Papp (10–6 cm/s) Caco-2 | 51.6 | 44.6 |
| (calibration compound atenolol Papp = 1.40×10−6 cm/s) | ||
| Microsomal clearance (μL/min/mg) | 14.5 | 8.0 |
| CYP1A2 IC50 (μmol/L) | 0.26 | 0.22 |
| CYP1A2 KI (μmol/L) | 0.40 | |
| CYP1A2 kinact (min-1) | 0.05 |
For reversible inhibition, Ki were estimated using IC50/2;
Both compounds are in neutral condition under physiological pH, thus pKa was not available;
1400 mg phenacetin QD×10 and 60 mg PRN or IPRN QD×10 were applied;
This table is adapted from Ref. 14 with permission.
Figure 3.

Simcyp simulation results of phenacetin AUC0–24 at 1400 mg daily×10 days in the presence of IPRN (60 mg daily×10 days) and absence of IPRN in healthy subjects (A) and smokers (B), or the presence of PRN (60 mg daily×10 days) and absence of PRN in healthy subjects (C) and smokers (D). The outer curves represent phenaceitn concentration in the presence of PRN or IPRN. This figure is adapted from Ref. 14 with permission. IPRN, isopsoralen; PRN, psoralen.
7. Human PK and DDI prediction to avoid clinical DDI trials, a case study
Orteronel (TAK-700) is an oral, nonsteroidal, reversible, selective 17,20-lyase inhibitor that was in development for the treatment of patients with metastatic castration-resistant prostate cancer. In vitro CYP inhibition study in human liver microsomes showed that orteronel is a moderate inhibitor in CYP1A2, 2C8, 2C9, and 2C19, with IC50 values of 17.8, 27.7, 30.8 and 38.8 µmol/L, respectively. However, it showed no inhibition in CYP2B6, 2D6 or 3A4/5 (IC50>100 µmol/L, Table 4)15. The Cmax of orteronel in patients who had consumed a high-fat meal was at average of 9.18 µmol/L and thus, the [I]/IC50 ratio calculated using a basic static model showed that orteronel could cause as high as 1.84-fold of DDI. Following the FDA DDI guidance16, if basic static models show that a perpetrator has the potential of causing DDI (i.e. [I]/IC50>0.1), following up DDI assessment using a PBPK model under the dynamic conditions with both substrate and inhibitor is recommended. A PBPK model was then built with physicochemical and preclinical data and oral clearance from a human phase I trial because in vitro metabolic clearance does not reflect the total body clearance (Table 5). The resulting model well described the observed clinical PK (Fig. 4). This model was then used to simulate DDI potential with a set of sensitive CYP probe substrates, theophylline, repaglinide, (S)-warfarin, and omeprazole for CYP1A2, 2C8, 2C9, and 2C19, respectively (built in compound profiles within the Simcyp software, no further modification was made). The DDI potential of orteronel toward these 4 CYPs at the dynamic concentration scenario was simulated. As shown in Table 6, orteronel would not cause DDI with any of the 4 CYPs with AUC changes all less than 1.25-fold, with the criteria considered as no DDI by the FDA16.
Table 4.
Orteronel [I]/Ki values and predicted AUC ratio using static model.
| Parameter | CYP1A2 | CYP2C8 | CYP2C9 | CYP2C19 |
|---|---|---|---|---|
| Orteronel IC50 (μmol/L) | 17.8 | 27.7 | 30.8 | 38.8 |
| [I]/Ki | 1.03 | 0.66 | 0.60 | 0.47 |
| Substrate (fm) | Theophylline (0.90) | Repaglinide (0.64) | (S)-warfarin (1.00) | Omeprazole (0.87) |
| AUC ratio | 1.84 | 1.34 | 1.60 | 1.39 |
Abbreviations: CYP, cytochrome P450; [I], inhibitor concentration that is the total plasma maximum concentration (Cmax); IC50, 50% inhibitory concentration; Ki, inhibition dissociation constant.
Note: The mean Cmax in the subjects with the high-fat meal was 9.18 μmol/L. Ki=IC50/2, assuming competitive inhibition. The fm was adapted from Simcyp® v 11. AUC ratio was calculated using the basic static equation: AUCR=1/(fm/((1+[I]/Ki)+(1–fm))).
This table is adapted from Ref. 15 with permission.
Table 5.
Orteronel input data for PBPK M&S.
| Parameter | Value |
|---|---|
| Compound type | Monoprotic base |
| Molecular weight | 307.35 |
| logD7.4 | 1.322 |
| pKa | 6.600 |
| Blood–plasma partition coefficient (B/P) | 1.39 |
| Plasma protein binding (fu) | 0.403 |
| Main binding protein | HSA |
| Microsomal protein binding at 0.5 mg/mL (fu,mic) | 0.961 |
| fu (gut) | 1 |
| fa | 0.86 |
| Ka (L/h) | 0.79 |
| Qgut (L/h) | 8.394 |
| Apparent intrinsic permeability value: Papp (10–6 cm/s) Caco-2 | 9.05 |
| Calibration compound (propranolol) value: Papp (10–6 cm/s) Caco-2 | 25.1 |
| Clinical oral clearance (CL/F), (L/h) | 16.9 |
| Human ADME clearance routes (renal, hepatic, other) | 53%, 28%, 19% |
| Clinical oral clearance, %CV | 15.7 |
| Clinical volume of distribution (Vd/F), (L/kg) | 1.4 |
| Clinical volume of distribution, %CV | 30.2 |
| CYP1A2 Ki (µmol/L)a | 8.9 |
| CYP2C8 Ki (µmol/L)a | 13.8 |
| CYP2C9 Ki (µmol/L)a | 15.4 |
| CYP2C19 Ki (µmol/L)a | 19.4 |
Abbreviations: %CV, percent coefficient of variation; ADME, absorption, distribution, metabolism, excretion; fa, fraction absorbed; fu, fraction unbound; fu (gut), apparent unbound fraction in enterocytes; HSA, human serum albumin; IC50, 50% inhibitory concentration; Ka, first-order absorption rate constant; Ki, reversible inhibition constant; logD7.4, logarithm of the octanol–water partition coefficient at pH 7.4; Papp, apparent passive permeability; pKa, logarithmic acid dissociation constant; Qgut, hypothetical blood flow term that is used to indicate complex interplay among passive intestinal permeability, active transport, enterocyte drug binding, blood flows to enterocytes, and gut metabolism.
This table is adapted from Ref. 15 with permission.
All inhibition was assumed conservatively to be reversible; Ki values were calculated: IC50/2.
Figure 4.

Simulated and actual mean orteronel concentration-versus-time curves. The line represents the simulated mean area under the concentration-versus-time curve after a single dose of orteronel at 400 mg; the circles represent the actual data points from the high-fat diet group (n=42) treated with a single dose of orteronel 400 mg. This figure was adapted from Ref. 15 with permission.
Table 6.
DDI analysis: simulated area under the concentration–time curve ratios for orteronel.
| CYP/substrate | Dose | Orteronel IC50 (µmol/L) |
|---|---|---|
| CYP1A2/theophylline (SV) | 125 mg TID | 17.8 |
| CYP2C8/repaglinide (SV) | 0.25 mg BID | 27.7 |
| CYP2C9/(S)-warfarin (Sim) | 10 mg QD | 30.8 |
| CYP2C19/omeprazole, enteric-coated (SV) | 20 mg BID | 38.8 |
Abbreviations: BID, twice daily; CYP, cytochrome P450; DDI, drug–drug interaction; IC50, 50% inhibitory concentration; QD, once daily; Sim, profile based on in vitro data; SV, profile based on in vivo data; TID, 3 times daily.
This table was adapted from Ref. 15 with permission.
8. Dose guidance for renal impairment patients, a case study
This case illustrates how PBPK modeling can inform appropriate dosing of renal impairment (RI) patients in phase I/III studies and thereby enable characterization of safety and efficacy in the RI patients during the late stage of drug development17. Data from human ADME study revealed that orteronel (see last example) is a drug that is primarily cleared by kidney excretion. The extent of orteronel biotransformation is minimal, with cytochrome P450 isozymes having only a minor role. Thus, patients with renal impairment may have increased exposure to orteronel because of their impaired urinary excretion capability. A PBPK model was built as described in the last case study. The predicted PK profile was then validated using clinical PK data before applying the model to simulate PK profile of orteronel in moderate (glomerular filtration rate (GFR), 30–60 mL/min) or severe (GFR, <30 mL/min) RI patients. By comparing the PBPK model outputs with the population PK analysis results from phase 2 trials, it was demonstrated that PBPK modeling can accurately predict the effect of moderate and severe RI on the PK profile of orteronel (i.e. fold increase in AUC). In this model, exposure to orteronel increased as a reversed function of the estimated proportion of orteronel cleared by the kidney, aligning with the degree of renal impairment. The AUC for orteronel, when given at the clinical dose of 400 mg BID, was predicted to increase by 52% in patients with moderate RI and 83% in patients with severe RI compared with the healthy population group. Furthermore, the PBPK simulation also predicted that a reduced dose of orteronel of 220 mg BID (or a rounded dose of 200 mg BID) would achieve exposures in severely impaired subjects comparable to those seen in subjects with normal renal function treated at 400 mg BID (Fig. 5). Then a PopPK model was built to determine if dose adjustments might be required for renal RI patients in the clinical setting. Results of the PopPK suggested that patients with mild RI may not require dose adjustments as they were predicted to have only a 20% higher exposure compared to the healthy subjects (a scenario not included in the PBPK modeling). Patients with severe RI given orteronel 200 mg BID were predicted to have similar orteronel plasma concentrations as control subjects given 400 mg BID. In summary, this case demonstrates that for a drug being eliminated primarily via renal route, the PBPK modeling approach can play a key role for guiding dose selection. This analysis helped the inclusion of patients with RI in phase III trials with appropriate dose adjustment. That could serve as an alternative to a dedicated RI study, or suggests that a reduced-size study in severe RI patients may be sufficient to assess the exposure risk in other RI patients.
Figure 5.

Physiologically based pharmacokinetic (PBPK) simulation of orteronel in (A) healthy subjects (observed and simulated values), subjects with moderate renal impairment (simulated values), and subjects with severe renal impairment (simulated values), and (B) regression of orteronel clearance vs. glomerular filtration rate (GFR) based on PBPK simulations in healthy subjects, subjects with moderate renal impairment, and subjects with severe renal impairment. Observed data for healthy subjects (high-fat diet group, n=42) were obtained from clinical study C21007. The clinical scenario assumed 100% bioavailability with all uncharacterized metabolism treated as hepatic clearance (orteronel dose: 400 mg BID for 10 days). CL, total clearance; RI, renal impairment. This figure was adapted from Ref. 17 with permission.
9. Bridge healthy adults to special populations
PBPK models can be utilized to extrapolate the drug pharmacokinetic behavior in healthy volunteer to patient populations that are a challenge to obtain PK profiles for, such as predicting doses and drug exposures in children and infants18 and patients suffered from impaired renal or liver function11, 15. The work of Parrott and colleagues19 exemplified the usage of a mechanistic PBPK model in predicting the pharmacokinetics of a neuraminidase inhibitor oseltamivir and its active metabolite oseltamivir carboxylate (OC) which are for the treatment and prophylaxis of influenza A and B infections in infants and neonates. In their strategy (Fig. 6), the simulation of pharmacokinetics in adult animal species was first conducted. After a reasonable simulation of pharmacokinetics in the adult animal is achieved with a refinement, prediction of human pharmacokinetics was performed using information captured during the refinement of the animal model. For prediction of juvenile PK profile in humans, the same methodology is followed. First, a juvenile animal model is generated, which accounts for age-dependent differences that are known to impact the PK behavior of the drug, then the model was verified by comparison with the data obtained from a juvenile animal study. Finally, the prediction of juvenile humans was done using a PBPK model that accounted for age dependency in humans and information gathered from adult human and juvenile animal studies for refinement. This provided the first insight of drug exposure in juveniles since a PK study in this population is hard to come by.
Figure 6.

PBPK modeling strategy employed to predict exposure in neonates and infants. A stepwise approach is followed with verification against in vivo data at each step. Simulations in juveniles are based on a model incorporating age dependencies in physiology and incorporating data from relevant in vitro systems. Verification in juvenile animals allows for model refinement before prediction in children. This figure was adapted from Ref. 19 with permission.
10. Regulatory submission
PBPK modeling has been gaining acceptance at various regulatory bodies as part of submission package. Discussion of modeling and simulation approaches can be found in the updated DDI guidance from both the European Medicines Agency (EMA)20 and the U.S. Food and Drug Administration (FDA)16. Recently, The FDA hosted a workshop at their White Oak Campus in Silver Spring, MD, USA. At the workshop, the director of the Center of Drug Evaluation and Research (CDER), Dr. Janet Woodcock, concluded that “the modeling work performed thus far at CDER has contributed tremendously to overall drug development, in terms of safety and efficacy, which ultimately result in patient benefits”6. Both FDA scientists and industrial and academic representatives agreed that the current advance in PBPK modeling enable us to predict investigational drugs as a substrate of drug metabolizing enzyme with high confidence, especially when the drug is primarily metabolized by CYP3A and 2D621, 22. Among compounds in the BCS classification, the PK profile of type I compounds with high solubility and high permeability usually can be predicted quite well from their preclinical data. On the other hand, prediction of exposure changes due to CYP induction has not been well validated, neither had the compounds of mix of CYP inducer and time-dependent inhibitor. PK prediction involving transporters is still not reliable due to poor understanding of the scaling factors used to extrapolate in vitro data to in vivo disposition. The same is true for metabolism and disposition in the gut. Due to the complexity of the metabolism, absorption, and transporter activity involved at the different segments of the gastrointestinal tract, and the unique nature of the of individual compound, PBPK modeling for gut absorption (Fg) has yet to be optimized22, 23. Drug metabolizing enzyme-transporter interplay, PK prediction in organ impairment population, and allometry scaling down to children younger than 2 years of age (ontogeny and maturation), are among the areas that still need more research. There are much experiences lacked in the area of pregnancy, obesity, and the geriatric population, as well as food effect, formulation, and pH effects. The prediction of intracellular concentration is also often a challenge24, 25.
As PBPK modeling advances, the FDA has seen an increase in modeling work in submission packages. Much of the modeling work is cited in drug product labels to illustrate the degree or lack of DDI risk with co-administration of market drugs5, 6, 22, 26, 27, 28, 29, 30. From July 1, 2008 to December 31, 2013, there were 112 PBPK packages submitted to the FDA including 5 run by the agency. Among these packages, most of the studies (76/112) were DDI-related. Most of the DDI simulation (45/76) had no clinical data available for comparison, and of these eight studies were for perpetrators. Most of the DDI submissions were for CYP inhibition risk evaluation, only 8 cases were for CYP induction and 1 was for transporter inhibition. A few cases (3) had clinical data available for building and optimizing the final models22. In most situations, the PBPK models were included in the submission of IND or NDA.
11. Common industrial application for PBPK modeling
In the pharmaceutical industry, PBPK modeling is used for purposes, such as mechanistic studies, aiding internal drug discovery or clinical development decisions, and informing regulatory communication including filing at various stages (e.g., IND and NDA). It is mostly applied at the development stage. Below are a few outlines of its routine applications:
-
1)
At the lead optimization stage of drug discovery, PBPK modeling can provide human PK prediction at clinical dose and dose schedule. A high projected dose (e.g., >1 g/day) may discourage further investment in that drug candidate if it is not a first-in-class drug candidate. For a similar reason, if a drug needs to be administrated multiple times a day, it may face challenge in marketing if it is neither a first-in-class nor a best-in-class drug candidate.
-
2)
At the candidate selection stage of drug discovery, should a drug candidate be partially metabolized by polymorphic enzymes, such as CYP2D6 or 2C19, a PBPK model can be applied to simulate the exposure in population including poor metabolizer to determine whether poor metabolizers need to be excluded in the first-in-human (FIH) trials. The model can be further refined with the data from FIH healthy volunteer trials to help to design a DDI study (e.g., dose adjustment) in the poor metabolizer population.
-
3)
At the drug development stage, DDI risk simulation is the most popular application for PBPK modeling. Whether a drug candidate is a substrate of drug metabolizing enzymes or a perpetrator, a DDI risk simulation with standard care medications can assess the risk of co-medication with these standard care market drugs. Oftentimes, the most potent perpetrator or the most sensitive substrate is used in the initial simulations to assess the worst case scenario. If enough safety margins are presented at the worst case scenario, then clinical trials with moderately sensitive substrates or perpetrator may get waivered.
-
4)
Many drugs are cleared via hepatic metabolism. Some may be excreted via renal excretion. Thus, exposure simulation in organ impairment patients is important to know. There are successful examples of such a simulation described above and in the literature11, 15.
-
5)
In DDI trial, dependent on the half-life of the substrate, the dose frequency and dose duration of the inhibitor need to be optimized to cover the duration of the exposure of the substrate as much as possible. The more exposure overlap between the substrate and the inhibitor, the better we can capture the DDI potential between these two drugs31. In a crossover study design, the washout period is changed upon the application of an inhibitor and therefore needs to be simulated prior the study. A lengthy washout period is costly and also not convenient to the patients, whereas a washout period too short would affect the data quality.
-
6)
For many high clearance compounds and substrate of CYP3A4/5, the CYP3A4/5 inhibitor ritonavir is often used to enhance the exposure. PBPK modeling can be used to simulate whether, in the presence of ritonavir, the exposure of the drug candidate can reach the level of efficacious exposure. When DDI liability cannot be avoided, for example part of the chemical structure is responsible for the pharmacological activity but also carries DDI liability, modeling and simulation can be applied to evaluate whether alternative routes of administration (e.g., intravenous) would reduce the DDI liability. Drugs target for inhalation or other non-conventional routes are better evaluated using PBPK modeling to get the feasibility test.
12. Conclusions
PBPK modeling is a useful tool for the prediction of human PK profile from preclinical data. Once FIH PK data or human ADME data becomes available, the model can be further fine-tuned as illustrated in Fig. 715. It is a good tool for evaluating and optimizing clinical trial design, for example, to select the dose and dose schedule. It helps to understand the individual variability and parameters that have the most impact on human PK profile through sensitivity analysis. Hence, PBPK modeling provides a practical solution for extrapolating PK profile from healthy population to some ethnical, special age, or disease populations where clinical PK study is the hardest to conduct. In the DDI prediction area, PBPK modeling can help to determine the washout period in a crossover study design to set the minimal but sufficient clinical trial duration. It can also be applied as an alternative to DDI trials in some special populations, such as pediatrics and organ-impairment patients where the actual DDI trial is hard to conduct due to logistical or ethical issues. Thus, it can sometimes provide waiver for conducting unnecessary clinical DDI trials which then speeds up the drug development process and put fewer burdens on patients. Conducting DDI trials with multiple perpetrators in patients is also not ethical and practical, the PBPK modeling, in this case, can provide information about “what if” all of those drugs are co-administered together. On the other hand, as discussed earlier, PBPK modeling is a bottom-up approach, its results dependent on the quality of the input data. Although software are available for the prediction of physicochemical properties of compounds, such as logP and pKa, in authors experience, it is critical to use measured values to get a reliable PBPK prediction, especially when predicting human PK profile, rather than the AUC ratio for DDI purpose. For example, for a set of clinical candidates (about 40 compounds), the number of compounds for which the predicted PK profile within two fold of observed clinical values dropped from about 70% to half of that when in silico predicted logP and pKa were used (unpublished data). Transporter is another emerging area of PBPK modeling, however, most of the data generated are qualitative to answer the question of yes or no of whether a compound is a substrate of a transporter. PBPK modeling relies on kinetic data, such as the clearance of the compound via that transporter. Thus, additional data of transporter clearance are needed for PBPK modeling.
Figure 7.

Application of physiologically based pharmacokinetic modeling and simulation in various stages of drug discovery and development. Models were initially built with preclinical data, and later refined with available clinical information. This figure was adapted from Ref. 15 with permission.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Teorell T. Kinetics of distribution of substances administered to the body. I. The extravascular modes of administration. Arch Int Pharmacodyn Ther. 1937;57:205–225. [Google Scholar]
- 2.Reddy M., Yang R.S.H., Clewell H.J., III, Andersen M.E. Physiologically based pharmacokinetic modeling: science and applications. Wiley; Hoboken, NJ: 2005. [Google Scholar]
- 3.Rowland M., Peck C., Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 4.Jones H.M., Chen Y., Gibson C., Heimbach T., Parrott N., Peters S.A. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247–262. doi: 10.1002/cpt.37. [DOI] [PubMed] [Google Scholar]
- 5.Sinha V., Zhao P., Huang S.M., Zineh I. Physiologically based pharmacokinetic modeling: from regulatory science to regulatory policy. Clin Pharmacol Ther. 2014;95:478–480. doi: 10.1038/clpt.2014.46. [DOI] [PubMed] [Google Scholar]
- 6.Wagner C., Zhao P., Pan Y., Hsu V., Grillo J., Huang S.M. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacomet Syst Pharmacol. 2015;4:226–230. doi: 10.1002/psp4.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown R.P., Delp M.D., Lindstedt S.L., Rhomberg L.R., Beliles R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- 8.Jones H.M., Parrott N., Jorga K., Lavé T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45:511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 9.Jones H.M., Dickins M., Youdim M., Gosset J.R., Attkins N.J., Hay T.L. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica. 2012;42:94–106. doi: 10.3109/00498254.2011.627477. [DOI] [PubMed] [Google Scholar]
- 10.Peters S.A., Ungell A.L., Dolgos H. Physiologically based pharmacokinetic (PBPK) modeling and simulation: applications in lead optimization. Curr Opin Drug Discov Dev. 2009;12:509–518. [PubMed] [Google Scholar]
- 11.Shardlow C.E., Generaux G.T., Patel A.H., Tai G.Y., Tran T., Bloomer J.C. Impact of physiologically based pharmacokinetic modeling and simulation in drug development. Drug Metab Dispos. 2013;41:1994–2003. doi: 10.1124/dmd.113.052803. [DOI] [PubMed] [Google Scholar]
- 12.Liu F., Zhuang X.M., Yang C.P., Li Z., Xiong S., Zhang Z.W. Characterization of preclinical in vitro and in vivo ADME properties and prediction of human PK using a physiologically based pharmacokinetic model for YQA-14, a new dopamine D3 receptor antagonist candidate for treatment of drug addiction. Biopharm Drug Dispos. 2014;35:296–307. doi: 10.1002/bdd.1897. [DOI] [PubMed] [Google Scholar]
- 13.Plowchalk D.R., Rowland Yeo K. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Clin Pharmacol. 2012;68:951–960. doi: 10.1007/s00228-011-1189-y. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang X.M., Zhong Y.H., Xiao W.B., Li H., Lu C. Identification and characterization of psoralen and isopsoralen as potent CYP1A2 reversible and time-dependent inhibitors in human and rat preclinical studies. Drug Metab Dispos. 2013;41:1914–1922. doi: 10.1124/dmd.113.053199. [DOI] [PubMed] [Google Scholar]
- 15.Lu C., Suri A., Shyu W.C., Prakash S. Assessment of cytochrome P450-mediated drug–drug interaction potential of orteronel and exposure changes in patients with renal impairment using physiologically based pharmacokinetic modeling and simulation. Biopharm Drug Dispos. 2014;35:543–552. doi: 10.1002/bdd.1919. [DOI] [PubMed] [Google Scholar]
- 16.FDA, Center for Drug Evaluation and Research (CDER). Guidance for industry: drug interaction studies-study design, data analysis, implications for dosing, and labeling recommendations [Draft Guidance]. U.S. Department of Health and Human Services, Food and Drug Administration: Rockville, MD; 2012. Available from: 〈http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf〉.
- 17.Suri A., Chapel S., Lu C., Venkatakrishnan K. Physiologically based and population PK modeling in optimizing drug development: a predict-learn-confirm analysis. Clin Pharmacol Ther. 2015;98:336–344. doi: 10.1002/cpt.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manolis E., Pons G. Proposals for model-based paediatric medicinal development within the current European Union regulatory framework. Br J Clin Pharmacol. 2009;68:493–501. doi: 10.1111/j.1365-2125.2009.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parrott N., Davies B., Hoffmann G., Koerner A., Lave T., Prinssen E. Development of a physiologically based model for oseltamivir and simulation of pharmacokinetics in neonates and infants. Clin Pharmacokinet. 2011;50:613–623. doi: 10.2165/11592640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.EMA, Committee for Human Medicinal Products (CHMP). Guideline on the investigation of drug interactions [Final]. European Medicines Agency: London; 2012. Available from: 〈http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf〉.
- 21.Vieira M.D., Kim M.J., Apparaju S., Sinha V., Zineh I., Huang S.M. PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther. 2014;95:550–557. doi: 10.1038/clpt.2014.43. [DOI] [PubMed] [Google Scholar]
- 22.Wagner C., Pan Y., Hsu V., Grillo J.A., Zhang L., Reynolds K.S. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54:117–127. doi: 10.1007/s40262-014-0188-4. [DOI] [PubMed] [Google Scholar]
- 23.Peters S.A., Jones C.R., Ungell A.L., Hatley O.J.D. Predicting drug extraction in the human gut wall: assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin Pharmacokinet. 2016 doi: 10.1007/s40262-015-0351-6. Available from: 〈 http://dx.doi.org/10.1007/s40262-015-0351-6〉. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu X., Korzekwa K., Elsby R., Fenner K., Galetin A., Lai Y., Matsson P. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther. 2013;94:126–141. doi: 10.1038/clpt.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu C., Li P., Gallegos R., Uttamsingh V., Xia C.Q., Miwa G.T. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34:1600–1605. doi: 10.1124/dmd.106.010793. [DOI] [PubMed] [Google Scholar]
- 26.Huang S.M. PBPK as a tool in regulatory review. Biopharm Drug Dispos. 2012;33:51–52. doi: 10.1002/bdd.1777. [DOI] [PubMed] [Google Scholar]
- 27.Leong R., Vieira M.L.T., Zhao P., Mulugeta Y., Lee C.S., Huang S.M. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91:926–931. doi: 10.1038/clpt.2012.19. [DOI] [PubMed] [Google Scholar]
- 28.Wagner C., Pan Y., Hsu V., Sinha V., Zhao P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin Pharmacokinet. 2016;55:475–483. doi: 10.1007/s40262-015-0330-y. [DOI] [PubMed] [Google Scholar]
- 29.Zhao P., Rowland M., Huang S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin Pharmacol Ther. 2012;92:17–20. doi: 10.1038/clpt.2012.68. [DOI] [PubMed] [Google Scholar]
- 30.Zhao P., Vieira M.L.T., Grillo J.A., Song P.F., Wu T.C., Zheng J.H. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J Clin Pharmacol. 2012;52(Suppl 1):91S–108S. doi: 10.1177/0091270011415528. [DOI] [PubMed] [Google Scholar]
- 31.Zhao P., Ragueneau-Majlessi I., Zhang L., Strong J.M., Reynolds K.S., Levy R.H. Quantitative evaluation of pharmacokinetic inhibition of CYP3A substrates by ketoconazole: a simulation study. J Clin Pharmacol. 2009;49:351–359. doi: 10.1177/0091270008331196. [DOI] [PubMed] [Google Scholar]