

Abstract
Introduction
Genetic factors and environmental exposures, including pesticides, contribute to the risk of Parkinson’s disease (PD). There have been few studies of gene and pesticide exposure interactions in PD, and all of the prior studies used a candidate gene approach.
Methods
We performed the first genome-wide gene-environment interaction analysis of pesticide exposure and risk of Parkinson’s disease. Analyses were performed using data on >700,000 single nucleotide polymorphisms (SNPs) in 364 discordant sibling pairs. In addition to testing for SNP-pesticide interaction effects, we also performed exploratory analyses of gene-pesticide interactions at the gene level.
Results
None of the gene-environment interaction results were significant after genome-wide correction for multiple testing (α=9.3E-07 for SNP-level tests; α=2.1E-06 for gene-level tests). Top results in the SNP-level tests provided suggestive evidence (P<5.0E-06) that the effect of pesticide exposure on PD risk may be modified by SNPs in the ERCC6L2 gene (P=2.4E-06), which was also supported by suggestive evidence in the gene-level analysis (P=4.7E-05). None of the candidate genes assessed in prior studies of gene-pesticide interactions reached statistical support in this genome-wide screen.
Conclusion
Although no significant interactions were identified, several of the genes with suggestive evidence of gene-environment interaction effects have biological plausibility for PD risk. Further investigation of the role of those genes in PD risk, particularly in the context of pesticide exposure, in large and carefully recruited samples is warranted.
Keywords: Genome Wide Association Study, Gene-Environment Interaction, Parkinson’s disease, Pesticide exposure
1. INTRODUCTION
Studies indicate that both environmental exposures and genetic risk factors contribute to the development of Parkinson’s disease (PD) [1]. Although a number of genetic risk factors associated with PD have been confirmed [2,3], the underlying mechanisms and causes of the disease are largely unknown. In particular, little is known about specific gene-environment (GE) interactions that influence PD susceptibility. One of the well-established environmental contributors to PD risk is pesticide exposure [4]. Because of genetically-determined inter-individual differences in metabolizing specific substances, the effect that pesticide exposure has on PD risk is presumably at least partially modified by genetic variation.
PD gene-pesticide interactions have been reported for a number of candidate genes including ABCB1, ALDH2, CYP2D6, SKP1, NQO1, PON1, and SLC6A3 [5]. However, overall, prior research has provided limited evidence of gene-pesticide interactions in PD: the findings from candidate gene studies were often non-significant, or significant with small effects and limited or no replication. Importantly, there have been no comprehensive studies that investigated interactions between genes and pesticide exposure in PD at the genome-wide level. The genome-wide GE interaction studies that have begun to emerge for complex traits are generally thought to be underpowered for detecting most GE interaction effects [6]. Therefore, novel statistical approaches, such as gene-level GE interaction analyses have been proposed to improve power of genome-wide GE interaction studies [7].
Here we analyze data from a prior genome-wide association study (GWAS) of PD [8], with the aim of detecting effects of interactions between pesticide exposure and genetic variation on the risk of PD. In addition to performing SNP-pesticide interaction analyses across the genome, we also evaluated the evidence for gene-pesticide interactions at the gene level.
2. Methods
2.1. Subjects
The Institutional Review Board of the Mayo Clinic approved the study, and all subjects provided written informed consent. As part of the discovery phase (“tier 1”) of a prior GWAS, 443 cases with PD and 443 sibling controls were genotyped using a genome-wide SNP platform [8]. Cases were enrolled from the clinical practice of the Department of Neurology of the Mayo Clinic in Rochester, MN, between June 1996 and May 2004, and underwent a standardized clinical assessment performed by a neurologist specialized in movement disorders. Cases had at least two of four cardinal signs of parkinsonism (rest tremor, rigidity, bradykinesia, and/or postural instability) and no features atypical for PD (such as unexplained upper motor neuron signs or cerebellar signs) [9]. Controls were siblings of cases who screened negative for PD via telephone interview (i.e. had no prior diagnosis of PD, no prior treatment with levodopa, and did not have three or more of nine PD symptoms), or who were confirmed free of PD by neurological examination. When possible, controls were matched to cases first by sex, and then by closest age at enrollment. The details of case and control recruitment and data and specimen collection were previously described [8].
2.2. Environmental Exposure Assessment
The collection of data on pesticide exposures was previously described [10,11]. Exposures data were obtained by telephone via direct or proxy (for incapacitated subjects) interviews using a structured risk factors questionnaire administered by specifically trained study assistants. To reduce interview and recall biases, interviewers were kept unaware of the case and control status of subjects, and the subjects (or their proxy) were kept unaware of the study hypothesis. A reliability study of the risk factors questionnaire was conducted, as previously described [11]. Supplemental Text File S1 provides the items from the risk factors questionnaire that assessed occupational and hobby-related pesticides exposures (including herbicides, insecticides and fungicides). For the analyses of this study, pesticide exposure (occupational or hobby use) was coded as yes (ever) or no (never). The questions and responses used to code pesticides exposures as yes (ever) are highlighted in the Supplemental File.
2.3. Genotyping, quality control, and imputation
The genotyping and quality control have been previously described [8]. In total, 443 discordant sib-pairs were genotyped using the Perlegen platform. Of the 248,535 genotyped SNPs with unique positions on National Center for Biotechnology Information (NCBI) build 34, the genotyping call rate was >80% for 220,143 SNPs, including 205,031 SNPs that were polymorphic within the study sample. After removing SNPs showing departures from Hardy-Weinberg Equilibrium (P<0.001), 198,345 SNPs remained. Prior to imputation, additional data cleaning was performed using PLINK v1.07 (http://pngu.mgh.harvard.edu/~purcell/plink/). In particular, all SNPs with a MAF <0.01 or missing rates >2% and individual samples with genotyping efficiency <95% were excluded. This resulted in 149,817 analyzable SNPs in 433 PD cases and 428 sibling controls. Autosomal SNPs were imputed using IMPUTE v2.0 [12], with the precompiled “HapMap 3 + 1000GP CEU+TSI” panels from the IMPUTE website used as reference for imputation. This reference panel contained HapMap 3 data (from release #2, Feb 2009) and 1,000 Genomes Project data from Pilot 1 genotypes (released in August 2009). Finally, the imputed genetic dataset was filtered, excluding SNPs with MAF<0.01, HWE P<1E-06, or >2% missing genotypes, and excluding subjects with >5% missing genotypes. After genetic quality control, the dataset included 735,843 SNPs in 431 PD cases and 427 sibling controls [2]. Further sub-setting to sib-pairs with complete covariate data, including pesticide exposure data as well as age, sex and smoking, resulted in a dataset of 364 case-control sib- pairs that were included in the gene-environment interaction analysis.
2.4. Statistical Analysis
All genetic analyses were performed using conditional logistic regression to account for the case-control matching (i.e. sibling pairs). We first performed a genome-wide scan for SNP-pesticide interaction effects on PD risk. Conditional logistic regression was used to fit models with the binary pesticide exposure variable and the SNP genotype coded as the minor allele count (0,1,2 for observed SNPs or the imputed “dosage”), with and without the interaction term between these two variables. The covariates age (at time of enrollment), gender and smoking ever/never (smoking at least 100 cigarettes in life) were included in all models. Principal components derived from genome-wide SNP data were not included as covariates, as the discordant-sib pair data protects the analysis from spurious associations arising from population stratification. A one degree-of-freedom (1df) likelihood ratio test of the interaction effect was performed by comparing the models with and without the interaction term. While 735,843 SNPs were analyzed, multiple testing correction for 735,843 SNPs would be highly conservative, due to the extensive linkage disequilibrium (LD) between SNPs. In particular, among the total analyzed SNPs, 149,817 were genotyped, and the remaining SNPs were imputed based on their LD with the observed SNPs and are thus not independent. Using the software simpleM with a window size of 5000 SNPs [13], we estimated that approximately 54,000 independent SNPs were included in the analysis, suggesting a significance threshold of 9.3E-07 for the SNP-level tests.
We then performed gene-pesticide interaction tests at the gene level, to identify genes that might show evidence of excess interaction with pesticide exposure when all available SNPs in/near the gene are considered. To evaluate evidence for GE interactions at the gene level, we applied Principal Component (PC) Analysis to the SNP data, and then tested for PC-pesticide interactions using conditional logistic regression. PC analysis is a dimension reduction technique that has been shown to be a powerful approach for gene-level association tests [14]; here we adapted the approach of Gauderman et al. to study GE interactions in a sample of discordant siblings. SNPs were first assigned to genes if they mapped within 20 kb upstream or downstream of a gene. By this mapping rule, a total of 454,993 SNPs were mapped to 23,765 genes (with the number of SNPs mapping to a gene ranging from 1 to 1,204, median=11, interquartile range=25). We then considered two ways of evaluating GE interactions at the gene-level. First, we used the first PC for each gene (PC1), which captures the most genetic variation across that gene, to represent the genetic variation in the gene, and tested the effect of the interaction between PC1 and pesticide use on risk of PD; i.e. we performed a 1df test of the PC1*pesticide interaction term based on conditional logistic regression models. We also considered the first k PCs for each gene that in total explain at least 80 % of the variance in the genotypes for that gene, performed k PC-pesticide interaction tests for the gene, and then combined the k P-values using Fisher’s p-value combination method [15] providing a global test of PC-pesticide interaction. We refer to these two approaches as the “PC1” gene-level test and the “All-PC” gene-level test. We note that alternative approaches could have been used, for instance the All-PC test could have been performed using a joint likelihood ratio test of all the PC-pesticide interaction terms. However, this test poses challenges for large genes, where many PCs are required to represent the genetic variation in the gene, leading to a very high degree-of-freedom test. We therefore selected the approach described above based on combining individual PC-pesticide interaction p-values using Fisher’s methods. Post-hoc sensitivity analyses demonstrated that for the top gene-level results presented in the paper, these two alternative approaches provided very similar results. For the gene-level analyses, the number of tests is equal to the total number of genes analyzed; thus, because 23,765 genes were analyzed, the threshold for genome-wide significance was 0.05/23765 = 2.1e-06. The statistical packages SAS (version 9.2; SAS Institute Inc., Cary, NC) and R (version 2.13; www.cran.r-project.org) were used for all analyses.
Power was estimated using the Quanto software package (http://hydra.usc.edu/gxe) [16]. Assuming a marginal environmental effect (relative risk) of 1.2 and a marginal log-additive genetic relative risk of 1.1, the sample of 364 discordant sib-pairs provides 74% power to detect an interaction effect (Rge) of 5 for a SNP with a minor allele frequency (MAF) of 0.2. Under the same scenario, but a SNP with MAF = 0.4, the power to detect an interaction with Rge= 5 would be 95%.
3. Results
A summary of the subject characteristics is shown in Table 1.
Table 1.
Subject Characteristics
| Control (N=364) |
Case (N=364) |
Total (N=728) |
p value* | |
|---|---|---|---|---|
| Age at study: mean (sd) | 64.8 (11.0) | 65.4 (10.6) | 65.1 (10.8) | 0.08 |
| Years of school: mean(sd) | 13.8 (2.9) | 13.8 (3.1) | 13.8 (3.0) | 0.75 |
| Gender | <.0001 | |||
| Female | 191 (52.5%) | 145 (39.8%) | 336 (46.2%) | |
| Male | 173 (47.5%) | 219 (60.2%) | 392 (53.8%) | |
| Smoking (ever/never) | 0.0037 | |||
| No | 185 (50.8%) | 219 (60.2%) | 404 (55.5%) | |
| Yes | 179 (49.2%) | 145 (39.8%) | 324 (44.5%) | |
| Farm or garden pesticides (ever/never) | 0.16 | |||
| No | 249 (68.4%) | 233 (64.0%) | 482 (66.2%) | |
| Yes | 115 (31.6%) | 131 (36.0%) | 246 (33.8%) |
P-value from univariate conditional logistic regression models
A Manhattan plot of the P-values for the SNP-pesticide interaction analysis (at the SNP level) is shown in Figure 1, and the top 10 SNP association results are listed in Table 2. A QQ plot of the p-values is shown in Supplemental Figure S1. None of the individual SNP P-values are significant after correction for multiple testing. The top SNP-pesticide interaction signal, rs67383717 (P=2.4E-06) maps to LINC00476 near the ERCC6L2 gene on chromosome 9. A SNP in LD with this SNP (rs591486 r2=0.78 in the 1000 Genomes CEU population), which maps to intron 2 of ERCC6L2, had the second smallest interaction P-value in our analysis (P=2.7E-06). Other top results included SNPs in or near the genes ADAM29, PTPRF, and BACH2. SNP effect sizes in the pesticide exposure strata (unexposed and exposed) for the top 10 interaction SNPs are shown in Table 3. These results indicate that for the BACH2 SNPs, the SNP has no effect on risk of PD in unexposed individuals, but the minor allele is associated with increased risk in exposed individuals. For the ERCC6L2 and ADAM29 SNPs, the odds ratios indicate that the minor allele is associated with increased risk in unexposed, but is protective in exposed individuals. The opposite pattern is revealed for PTPRF, where the minor allele appears to be protective in unexposed, but is associated with increased risk in those exposed to pesticides.
Figure 1.
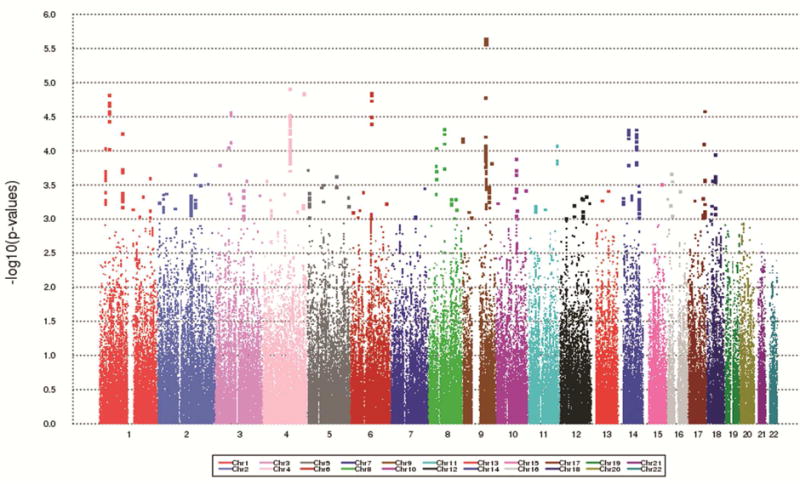
Manhattan plot of p-values from genome-wide SNP-pesticide interaction analysis.
Table 2.
Results of gene-by-pesticide interaction analyses at the SNP level: top 10 SNP-pesticide interaction association signals.
| SNP | Chr | Position (build 36) | Gene | Major Allele | Minor Allele | MAF | OR ratio | P value |
|---|---|---|---|---|---|---|---|---|
| rs67383717 | 9 | 97666369 | LINC00476, upstream of ERCC6L2 | C | A | 0.469 | 0.29 | 2.4E-06 |
| rs591486 | 9 | 97689867 | ERCC6L2 | A | G | 0.467 | 0.30 | 2.7E-06 |
| rs6851004 | 4 | 116951675 | PGAM4P2, KRT18P21 | T | A | 0.246 | 4.06 | 1.3E-05 |
| X6.90946408 | 6 | 90946408 | BACH2 | C | T | 0.089 | 9.19 | 1.4E-05 |
| X6.90949107 | 6 | 90949107 | BACH2 | A | G | 0.089 | 9.18 | 1.4E-05 |
| rs10013344 | 4 | 176146286 | ADAM29 | C | T | 0.178 | 0.22 | 1.5E-05 |
| rs10005382 | 4 | 176156562 | ADAM29 | C | G | 0.179 | 0.23 | 1.5E-05 |
| X6.90956325 | 6 | 90956325 | BACH2 | A | G | 0.089 | 9.11 | 1.5E-05 |
| rs539096 | 1 | 43845007 | PTPRF | G | A | 0.202 | 3.66 | 1.5E-05 |
| rs6919587 | 6 | 90959852 | BACH2 | T | A | 0.088 | 9.06 | 1.6E-05 |
MAF = minor allele frequency; OR ratio = ratio of odds ratios for allele effect in pesticide exposed vs. pesticide unexposed strata
Table 3.
Top SNP-pesticide interactions. Allelic effects in the pesticide exposed and unexposed strata for the top interacting SNPs from Table 2.
| SNP | Minor allele OR (95% CI) in unexposed | Minor allele OR (95% CI) in exposed |
|---|---|---|
| rs67383717 | 1.56 (1.09, 2.24) | 0.46 (0.28,0.75) |
| rs591486 | 1.59 (1.11, 2.27) | 0.47 (0.29,0.77) |
| rs6851004 | 0.69 (0.45, 1.05) | 2.80 (1.48,5.29) |
| X6.90946408 | 0.83 (0.43, 1.60) | 7.63 (2.29,25.46) |
| X6.90949107 | 0.83 (0.43, 1.60) | 7.66 (2.30,25.56) |
| rs10013344 | 1.92 (1.11, 3.32) | 0.43 (0.22,0.83) |
| rs10005382 | 1.90 (1.11, 3.25) | 0.43 (0.22,0.83) |
| X6.90956325 | 0.84 (0.44, 1.61) | 7.63 (2.29,25.38) |
| rs539096 | 0.55 (0.36, 0.84) | 2.01 (1.14,3.53) |
| rs6919587 | 0.84 (0.44, 1.61) | 7.61 (2.29,25.27) |
Top results of the gene-level GE analyses are presented in Supplemental Table S1. The QQ plots for the two genome-wide gene-level GE analyses are shown in Supplemental Figure S2, demonstrating that the expected distribution of P-values was obtained with both methods, which indicates that both methods attained correct type 1 error rates. In both analyses, no results reached the genome-wide significance level of P<2.1E-06. Top ranking genes in the gene-level analyses included two genes that were also identified in the SNP-level tests: ERCC6L2 (P=4.7E-05 with PC1, P=7.3E-05 with All-PC) and PTPRF (P=1.2E-04 with All-PC).
Supplemental Table S2 shows the results from the All-PC gene-level analysis for several candidate genes that have previously been reported to interact with pesticides to alter risk of PD. No evidence for interaction with pesticide exposure was observed for any of these candidate genes.
4. DISCUSSION
We performed a genome-wide association analysis for GE interaction effects at the gene level as well as the SNP level after correcting for multiple testing. We found no statistically significant interactions between genes and pesticide exposure for the risk of developing PD at the genome-wide level, despite the fact that both SNP-level and gene-level analyses were performed. The lack of significant GE interaction effects at the SNP level is consistent with findings from other genome-wide association studies of GE interactions, which have demonstrated that current studies tend to be underpowered to detect SNP-environment interactions at the genome-wide significant level. Although, in the present study, there were no statistically significant interactions between genes and pesticide exposure after stringent Bonferroni correction, the current genome-wide scan for SNP- and gene-pesticide interactions identified several biologically plausible candidates for further investigation.
The top SNP-pesticide interaction results with biological plausibility included SNPs in ERCC6L2, PTPRF, and BACH2. Gene-level tests provided further marginal evidence of effects on PD risk via pesticide interactions for ERCC6L2 and PTPRF. ERCC6L2 has been implicated in DNA repair and mitochondrial function [17], providing a potential link to PD given the substantial evidence that mitochondrial dysfunction plays a central role in the pathophysiology of this disease [18]. In an in vitro experiment, ERCC6L2 had characteristics of an early DNA damage-response protein that traffics to the mitochondria and the nucleus in a reactive oxygen species-dependent fashion [17]. Most pesticides preferentially inhibit the mitochondrial electron transport chain, disrupting oxidative phosphorylation [19]. Therefore, there may be joint effects of pesticide exposure and SNPs in ERCC6L2 that impair mitochondrial function and increase the risk of PD, which deserves further investigation.
While not significantly associated with PD in prior GWAS, meta-analysis of prior studies [3] included in the PDGene database (www.pdgene.org) provides additional evidence of association of PD with SNPs in PTPRF (P<10−4 for 8 SNPs in the PTPRF region, and P<0.05 for 164 out of the total 444 SNPs analyzed in this locus including SNP rs539096 implicated in our study) [2]. PTPRF acts as a protein-tyrosine phosphatase that negatively regulates the insulin signaling pathway. Recent evidence indicates that PD and type 2 diabetes, both age-related chronic diseases, share remarkably similar dysregulated pathways [20]. Pesticides have a role as endocrine-disrupting chemicals in the development of type 2 diabetes. Therefore, there may be interactions between PTPRF SNPs and pesticide exposure in PD risk, which warrant further investigation.
Of the prior gene-pesticide interactions reported in PD, none were detected at a genome-wide or nominal significance level. It is, however, important to note that our results for the previously-reported candidate genes (Supplemental Table S1) are based on the gene-level, rather than SNP-level analyses, as most of the specific functional SNPs in these genes are not represented in our SNP data. This limits our findings, as the SNPs that were genotyped in these genes may not adequately tag the known functional variants and therefore the gene-level analyses are not a direct replication of the earlier results.
This study has several limitations, including the retrospective collection of pesticide exposure data and the use of proxy informants in a small number of subjects. Also, because the analyzed genetic data was obtained as part of the first published GWAS of PD, after quality control the genetic data included only 149,817 SNPs, which after imputation provided 735,843 SNPs for analysis. This is low genomic coverage as compared to modern genome-wide genotyping followed by imputation. Furthermore, the sample size analyzed in this study is small, particularly for investigating GE interactions at the genome-wide level. However, the discordant sib-pair study design that was used generally provides greater power to detect GE interactions than the population based case-control design, and thus smaller case-sib samples are required to achieve similar power to detect GE interactions [21]. Moreover, powerful statistical methods based on gene-level rather than SNP-level tests of GE interaction effects were applied, providing suggestive evidence of GE effects that warrant further investigation. While the gene-level analyses have the advantage of potentially increasing the power of association analysis, they also have limitations. In particular, interpretation of the results is not as straightforward as in standard SNP-environment interaction analyses; thus, while a test may provide statistical evidence that the effect of variation in a gene on risk of disease is modified by the environment, the effect sizes are not readily interpretable. Moreover, statistical methods for analysis of interactions at the gene-level are still under development, and the optimal approach for this type of analysis has not yet been established.
Given the limitations of the present study, in order to conclusively assess the role of the SNPs and genes nominated here in PD susceptibility, validation studies in large independent samples with high-density genotyping are warranted. To this end, important next steps to assess the role of GE interactions in disease risk will involve data harmonization efforts across multi-centric, carefully recruited and well-characterized datasets.
Supplementary Material
HIGHLIGHTS.
Environmental and genetic risk factors contribute to the development of PD.
There have been few studies of gene and pesticide exposure (PE) interactions in PD.
We present a genome-wide gene-environment interaction analysis of PE and risk of PD.
Several top results may implicate genes with biological plausibility for PD risk.
Further investigation of the role of these genes in PD risk is warranted.
Acknowledgments
This work was supported by funding from the National Institutes of Health grant 2R01ES10751 to DMM, and a Mayo Clinic Division of Biomedical Statistics and Informatics Meritorious Award to JMB. The authors wish to thank Vimal Patel, PhD, Medical & Scientific Writer at NorthShore Neurological Institute for assistance with the preparation of this manuscript.
CONFLICTS OF INTEREST
D.M. Maraganore has received philanthropic support from the Auxiliary of NorthShore University HealthSystem; serves on the editorial board of Parkinsonism and Related Disorders; is an author on 2 pending patents: (1) method to treat Parkinson’s disease and (2) method to predict Parkinson’s disease; and receives research support from the Agency for Health Care Research and Quality (1R01HS024057-01). R. Frigerio’s spouse has received philanthropic support from the Auxiliary of NorthShore University HealthSystem; serves on the editorial board of Parkinsonism and Related Disorders; is an author on 2 pending patents: (1) method to treat Parkinson’s disease and (2) method to predict Parkinson’s disease; and receives research support from the Agency for Health Care Research and Quality (1R01HS024057-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gao HM, Hong JS. Gene-environment interactions: key to unraveling the mystery of Parkinson’s disease. Progress in neurobiology. 2011;94:1–19. doi: 10.1016/j.pneurobio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, Destefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, Maraganore DM, Martin ER, Martinez M, Myers RH, Nalls MA, Pankratz N, Payami H, Satake W, Scott WK, Sharma M, Singleton AB, Stefansson K, Toda T, Tung JY, Vance J, Wood NW, Zabetian CP, Young P, Tanzi RE, Khoury MJ, Zipp F, Lehrach H, Ioannidis JP, Bertram L. Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson’s Disease Genetics: The PDGene Database. PLoS genetics. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. Pesticide exposure and risk for Parkinson’s disease. Annals of neurology. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- 5.Ritz BR, Paul KC, Bronstein JM. Of Pesticides and Men: a California Story of Genes and Environment in Parkinson’s Disease. Curr Environ Health Rep. 2016;3:40–52. doi: 10.1007/s40572-016-0083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet. 2005;6:287–298. doi: 10.1038/nrg1578. [DOI] [PubMed] [Google Scholar]
- 7.Winham SJ, Biernacka JM. Gene-environment interactions in genome-wide association studies: current approaches and new directions. Journal of child psychology and psychiatry, and allied disciplines. 2013;54:1120–1134. doi: 10.1111/jcpp.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214–1220. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- 10.Frigerio R, Sanft KR, Grossardt BR, Peterson BJ, Elbaz A, Bower JH, Ahlskog JE, de Andrade M, Maraganore DM, Rocca WA. Chemical exposures and Parkinson’s disease: a population-based case-control study. Mov Disord. 2006;21:1688–1692. doi: 10.1002/mds.21009. [DOI] [PubMed] [Google Scholar]
- 11.Brighina L, Frigerio R, Schneider NK, Lesnick TG, de Andrade M, Cunningham JM, Farrer MJ, Lincoln SJ, Checkoway H, Rocca WA, Maraganore DM. Alpha-synuclein, pesticides, and Parkinson disease: a case-control study. Neurology. 2008;70:1461–1469. doi: 10.1212/01.wnl.0000304049.31377.f2. [DOI] [PubMed] [Google Scholar]
- 12.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X, Becker LC, Becker DM, Starmer JD, Province MA. Avoiding the high Bonferroni penalty in genome-wide association studies. Genetic epidemiology. 2010;34:100–105. doi: 10.1002/gepi.20430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gauderman WJ, Murcray C, Gilliland F, Conti DV. Testing association between disease and multiple SNPs in a candidate gene. Genetic epidemiology. 2007;31:383–395. doi: 10.1002/gepi.20219. [DOI] [PubMed] [Google Scholar]
- 15.Fisher RA. Statistical Methods for Research Workers. London: Oliver and Boyd; 1932. [Google Scholar]
- 16.Gauderman WJ, Morrison JM. QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006 http://hydra.usc.edu/gxe.
- 17.Tummala H, Kirwan M, Walne AJ, Hossain U, Jackson N, Pondarre C, Plagnol V, Vulliamy T, Dokal I. ERCC6L2 mutations link a distinct bone-marrow-failure syndrome to DNA repair and mitochondrial function. Am J Hum Genet. 2014;94:246–256. doi: 10.1016/j.ajhg.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 19.Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem. 2007;100:1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santiago JA, Potashkin JA. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol Med. 2013;19:176–186. doi: 10.1016/j.molmed.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Gauderman WJ. Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med. 2002;21:35–50. doi: 10.1002/sim.973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.