

Abstract
The aim of this study was to evaluate the played by oxidative stress in the apoptotic response in different brain areas of rats chronically treated with supra‐physiological doses of nandrolone decanoate (ND). Immunohistochemical study and Western blot analysis were performed to evaluate cells' apoptosis and to measure the effects of expression of specific mediators, such as NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells), Bcl‐2 (B‐cell lymphoma 2), SMAC/DIABLO (second mitochondria‐derived activator of caspases/direct IAP‐binding protein with low PI) and VMAT2 (vesicular monoamine transporter 2) on apoptosis. The results of the present study indicate that a long‐term administration of ND promotes oxidative injury in rat brain specific areas. A link between oxidative stress and NF‐κB signalling pathways is supported by our results. In addition to high levels of oxidative stress, we consistently observed a strong immunopositivity to NF‐κB. It has been argued that one of the pathways leading to the activation of NF‐κB could be under reactive oxygen species (ROS)‐mediated control. In fact, growing evidence suggests that although in limited doses, endogenous ROS may play an activating role in NF‐κB signalling, while above a certain threshold, they may negatively impact upon this signalling. However, a mutual crosstalk between ROS and NF‐κB exists and recent studies have shown that ROS activity is subject to negative feedback regulation by NF‐κB, and that this negative regulation of ROS is the means through which NF‐κB counters programmed cells.
Keywords: nandrolone decanoate, oxidative stress, neurotoxicity, apoptosis
Introduction
Anabolic androgenic steroids (AASs) are a group of synthetic compounds obtained by selective chemical manipulations of the 19‐carbon testosterone molecule that affect the pharmacokinetics as well as the ratio of the anabolic/androgenic effect 1. Misuse of AASs by athletes is widely acknowledged, and worldwide non‐medical use is increasing in adolescents and adults, typically in individuals seeking physical strength, enhanced appearance and performance 2, 3, 4, 5, 6, 7, 8. AASs can be legally prescribed to treat conditions resulting from steroid hormone deficiency, such as delayed puberty and hypogonadism, as well as other diseases, such as bone marrow failure syndromes, bone mineralization and some muscle‐wasting disorders 9.
Although the potential effects on the nervous system have not been well defined, a wide range of physical and psychiatric adverse effects has been described in the literature 10, 11. Early behavioural effects include increased confidence, energy and motivation accompanied by irritability and agitation 12, 13, whereas prolonged use is usually associated with loss of inhibition and impulsive and markedly aggressive behaviour 13 by significantly modifying both serotonergic and noradrenergic neurotransmission 14. Currently, it is not yet fully clarified whether AASs are toxic to neurons and whether their abuse is a risk factor for chronic neurodegenerative disorders, although growing evidence supports a neurodegenerative potential for AASs 15, 16. The neurodegenerative effects of long‐term AASs abuse seem to be a phenomenon that has not yet been taken into consideration, probably because of the fact that most of the AAS users are still under the age of 50 and even if they might have incurred in neurotoxic effects, they are still too young to exhibit gross cognitive or motor deficits 17, even though neuronal loss has been observed on human AASs abusers 18.
Although the origin of AASs neurodegeneration might be multi‐factorial, oxidative stress could play a critical role. In fact, oxidative stress has been involved in many neurodegenerative human diseases, such as Alzheimer's disease, Parkinson's disease (PD), Huntington's disease, amyotrophic lateral sclerosis and HIV‐associated neurocognitive disorder 19, 20, 21, 22, 23, and the potential effects of disrupting the redox signalling of AASs is evident and this kind of toxicity occurs in numerous organs and systems 24. In addition, recent animal studies have shown that increased neuronal susceptibility to apoptotic stimuli could explain the neurotoxic effects of AASs 25. Long‐term administration of certain AASs leads to behavioural changes in the central nervous system in rodents 26, 27, 28, 29, 30, 31, 32, which may underlie some of the behavioural changes that are observed in AASs abusers 33.
As there is growing evidence of the potential role of oxidative stress and apoptosis for AASs‐mediated neurotoxicity, the aim of this study was to evaluate the role played by oxidative stress in the apoptotic response in different brain areas of rats chronically treated with supra‐physiological doses of nandrolone decanoate (ND), one of the most frequently abused AASs.
Materials and methods
The experiments were performed on 40 adult male Wistar rats (Wistar, Charles River, Lecco, Italy) weighing 200–250 g (10 weeks old). All experimental procedures were in compliance with the EEC Directive (86/609/EEC) on the protection of animals used for experimental and other scientific purposes, and were approved by the Ethical Committee for the Use of Laboratory Animals of the University of Siena. All efforts were made to minimize animal suffering and to reduce the number of animals used.
At the beginning of the experiments there was no statistically significant difference in animals' bodyweight within the group (P > 0.20), as well as between the groups (P > 0.30). All animals were housed in four per cage (55 × 35 × 30 cm), under standard conditions (23 ± 2°C, 50–60% relative humidity, 12 hr/12 hr light/dark cycle with lights on at 08:00, and with free access to food and water). The animals were randomly divided into two groups A: ND treated group and B: control group (submitted to vehicle injection; peanut oil with 10% of benzoic alcohol). Steroid and vehicle were administered by a single intramuscular injection twice a week for 8 weeks. The rats of group A (20 animals) received 3.75 mg ND/kg/week (1.875 mg/kg twice per week). The rats of group B (20 animals) received vehicle, twice a week. One week after the last injection, the rats were killed by decapitation, and blood was immediately collected. The brains were excised and were placed dorsal side up in an ice‐chilled rat brain matrix (World Precision Instruments, Inc., Aston, Stevenage, UK) with slits spaced at 1 mm. Using an ice‐chilled razor blade, the target regions were dissected according to the atlas of Paxinos and Watson. In each case, samples from (pre) frontal cortex (PFC), striatum (S), hippocampus (Hipp) and cerebellum (Cer) were taken. A portion of each sample was immediately frozen in liquid nitrogen and stored at −80°C. The remaining samples were fixed in 10% buffered formalin for 48 hrs.
Biochemical analysis
Malondialdehyde assessment
The extent of lipid peroxidation, a marker of oxidative stress, in rat brain areas was estimated using malondialdehyde (MDA) level calculation. Samples of brain areas were homogenized in a 0.04 M K+‐phosphate buffer (pH 7.4) containing 0.01% butyl hydroxytoluene (1:5 w/v, 0°C) to prevent the artificial oxidation of polyunsaturated free fatty acids during the assay. This homogenate was deproteinized with acetonitrile (1:1) and then centrifuged at 3000 × g for 15 min. The supernatants were used for MDA‐analysis after pre‐column derivatization with 2,4‐dinitrophenylhydrazine. The MDA‐hydrazone was quantified by isocratic reversed‐phase high‐performance liquid cromatography (HPLC) method with UV detection as described by Shara et al. 34.
Histopathological study
Paraffin‐embedded brain tissue specimens were sectioned at 4 μm and stained with haematoxylin and eosin. In addition, an immunohistochemical investigation was performed with antibodies anti‐NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells), Bcl‐2 (B‐cell lymphoma 2), SMAC/DIABLO (second mitochondria‐derived activator of caspases/direct IAP‐binding protein with low PI), VMAT‐2 (vesicular monoamine transporter 2) and apoptosis with TUNEL assay. We used 3‐μm‐thick paraffin sections mounted on slides covered with 3, amminopropyl‐triethoxysilane (Fluka, Buchs, Switzerland). A pre‐treatment was necessary to facilitate antigen retrieval and to increase membrane permeability to the antibodies: for NF‐κB (Santa Cruz Biotechnology, Santa Cruz, CA, USA), boiling 0.25 M ethylenediaminetetraacetic acid buffer; for Bcl‐2 (Millipore–Upstate, Temecula, CA, USA), SMAC/DIABLO (Millipore–Chemicon) boiling in 0.1 M citric acid buffer, and for antibody anti‐VMAT2 (Chemicon), 5 min. proteolytic enzyme at 20°C (Dako, Copenhagen, Denmark). For TUNEL assay (Millipore–Chemicon), we used TdT enzyme: the sections were immerged in proteinase K (20 μg/ml of TRIS) for 15 min. at 20°C. The primary antibody was applied in a 1:50 ratio for NF‐κB and Bcl‐2. The incubation of the primary antibody was for 120 min. at 20°C. For TUNEL assay the sections were covered with the TdT enzyme, diluted in a ratio of 30% in reaction buffer (Apotag Plus Peroxidase In Situ Apoptosis Detection Kit; Chemicon) and incubated for 60 min. at 38°C. The detection system utilized was the LSAB+ kit (Dako), a refined avidin–biotin technique in which a biotinylated secondary antibody reacts with several peroxidase‐conjugated streptavidin molecules. The positive reaction was visualized with 3,3‐diaminobenzidine peroxidation, according to standard methods. Then, the sections were counterstained with Mayer's Haematoxylin, dehydrated, cover‐slipped and observed under a Leica DM4000B optical microscope (Leica, Cambridge, UK). The samples were also examined under a confocal microscope, and a three‐dimensional reconstruction was performed (True Confocal Scanner; Leica TCS SPE). For semiquantitative analysis, slides were scored in a blinded manner by two observers. Staining pattern within each sample was assessed semiquantitatively in the scale 0–5 as follows: −: no immunoreactivity (0%); +: mild immunopositivity in scattered cells (10%); ++: immunopositivity in up to one‐third of cells (33%); +++: immunopositivity in up to one half of cells (50%) and ++++: strong immunopositivity in the majority or in all cells (100%).
Western blot analysis
Western blot analysis was performed. Approximately, 100 mg of frozen brain tissue was dissected and immediately transferred to the RIPA buffer with a protease inhibitor cocktail, and homogenized on ice, utilizing the homogenizer Silent Crusher. The homogenate was centrifuged (12,000 × g for 10 min. at 4°C). The supernatant was collected, estimated by Qubit Fluorometer (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), and boiled for 5 min., at 95°C. Brain total protein extracts (approximately 40 μg/lane) were run on 4–15% SDS PAGE at 80 V for about 2.5 hrs. For Western blot analysis, proteins from SDS gels were electrophoretically transferred to nitrocellulose membranes in mini trans blot apparatus (1 hr at 250 mA). Non‐specific binding was blocked by incubating membranes in Western blocker solution for 1 hr at room temperature. The membranes were incubated with primary antibodies anti‐diluted in Western blocker solution, in a 1:400 ratio overnight at 4°C. Blots were washed with PBS (Tween‐20) and then incubated for 1 hr at room temperature with HRP (horseradish peroxidase)‐conjugated secondary antibodies diluted in Western blocker solution, in a 1:2000 ratio. Membranes were washed with PBS/Tween‐20, and the immune reaction was developed in IMMUNOSTAR Kit Western C (Bio‐Rad Laboratories, Segrate, Milan, Italy) and then visualized by chemiluminescent detection methods. The light was then detected by a photographic film. The image was analysed by Versadoc (Bio‐Rad Laboratories), which detected the chemiluminescent blots of protein staining.
Statistical analysis
Values are presented as mean S.D. The unpaired two‐way Student's t‐test was used to compare the results obtained for ND treated rat group with the control group. P < 0.05 was accepted as indicative of a significant difference between the two groups.
Results
MDA evaluation
The MDA levels were used as a lipoperoxidation index and evidence of oxidative damage. The results obtained showed a strong and significant increase in MDA concentrations (Table 1) in all brain areas examined respect to controls: + 347% PFC, + 669% S, + 446% Hipp, + 86% Cer (Fig. 1).
Table 1.
MDA (nmol/mg tissue) in rat brain areas after administration of nandrolone decanoate 1.875 mg/kg twice per week by intramuscular injection for 8 weeks (each value is the mean ± S.D. of three animals)
| Cont PFC | Nan PFC | Cont Str | Nan Str | Cont Hipp | Nan Hipp | Cont Cer | Nan Cer | |
|---|---|---|---|---|---|---|---|---|
| Mean | 3.5 | 15.5 | 2.61 | 20.1 | 2.7 | 14.6 | 5.8 | 10.8 |
| S.D. | 1.13 | 4.38 | 0.98 | 5.69 | 1.29 | 5.21 | 2.98 | 3.7 |
PFC and Str P < 0.01 versus Cont; Hipp P < 0.02 versus Cont; Cer P < 0.05 versus Cont.
Cont: Control; Nan: nandrolone; PFC: frontal cotex; Str: striatum; Hipp: hippocampus; Cer: cerebellum.
Figure 1.
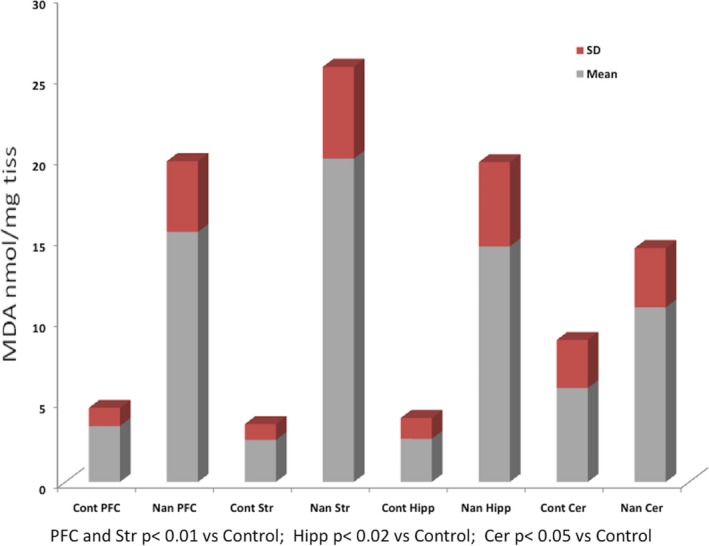
MDA (nmol/mg tissue) in rat brain areas after administration of nandrolone decanoate 1.875 mg/kg twice per week by intramuscular injection for 8 weeks (each value is the mean ± S.D. of three animals). Cont: Control; PFC: frontal cotex; Str: striatum; Hipp: hippocampus; Cer: cerebellum.
Histopathological results
The microscopic evaluation of the sections stained with haematoxylin and eosin revealed in the treated group: red neurons, nuclear shrinkage and perivascular haemorrhages.
The immunohistochemical study of the samples, for each antibody revealed the immunohistochemical findings and gradation of the immunohistochemical reaction were described with an ordinal scale and the median value was reported (Table 2).
Table 2.
Responses NF‐κB, Bcl‐2, VMAT2 and apoptosis with TUNEL assay in brain specimens
| Cont PFC | Nan PFC | Cont Str | Nan Str | Cont Hipp | Nan Hipp | Cont Cer | Nan Cer | Statistical value Nan versus Cont | |
|---|---|---|---|---|---|---|---|---|---|
| Anti‐NF‐κB | + | +++ | + | +++ | + | +++ | + | + | *** |
| Anti‐Bcl‐2 | + | +++ | + | +++ | + | +++ | + | + | *** |
| TUNEL assay | + | +++ | + | +++ | + | +++ | + | + | *** |
| Anti‐VMAT2 | +++ | + | +++ | + | +++ | + | + | + | *** |
| SMAC/DIABLO | + | +++ | + | +++ | + | +++ | + | + | *** |
NS: P > 0.05; *: P < 0.05; **: P < 0.01; ***: P < 0.001. Intensity of immunopositivity was assessed semiquantitatively in the scale 0–5 as follows: −: no immunoreactivity (0%); +: mild immunopositivity in scattered cells (10%); ++: immunopositivity in up to one‐third of cells (33%); +++: immunopositivity in up to two‐third of cells (70%) and ++++: strong immunopositivity in the majority or all cells (100%). In cases of divergent scoring, a third observer decided the final category.
Cont: Control; Nan: nandrolone; PFC: frontal cortex; Str: striatum; Hipp: hippocampus; Cer: cerebellum.
*, **, *** is the value of P. It is an international standard.
+, ++, +++, ++++ is the value of semiquantitative analysis (see the text)
NF‐κB
Anti‐NF‐κB provided strong neuronal positive reaction in brain samples of the treated rats compared to the control group, particularly in PFC, S and Hipp samples. Cerebellum areas had a weaker positivity (Fig. 2).
Figure 2.
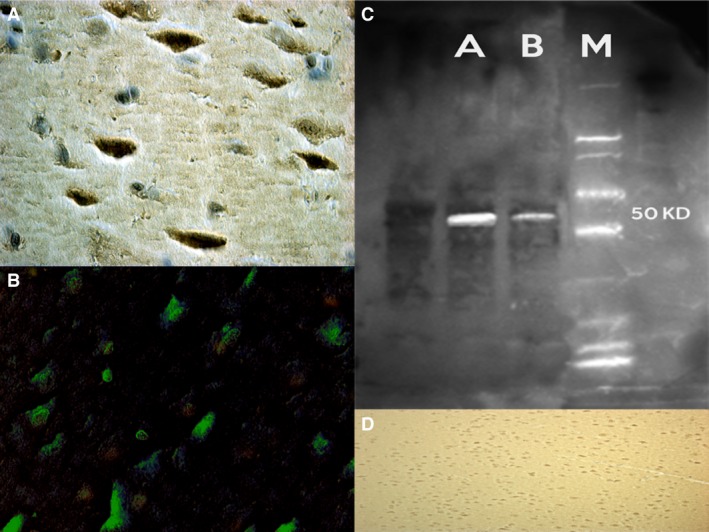
Strong and uniform NF‐kB neuronal positivity was found in the frontal cortex (A), striatum and hippocampus (B), of the nandrolone group. Western blot analysis detects the chemiluminescent blots of NF‐kB (C). (D) control group with negative results.
Bcl‐2
We found a strong positive reaction to the Bcl‐2 in ND group compared to control rats. In detail, our findings revealed that PFC, S and Hipp samples had a stronger positive reaction, whereas a weaker positive reaction was detected in cerebellum (Fig. 3).
Figure 3.
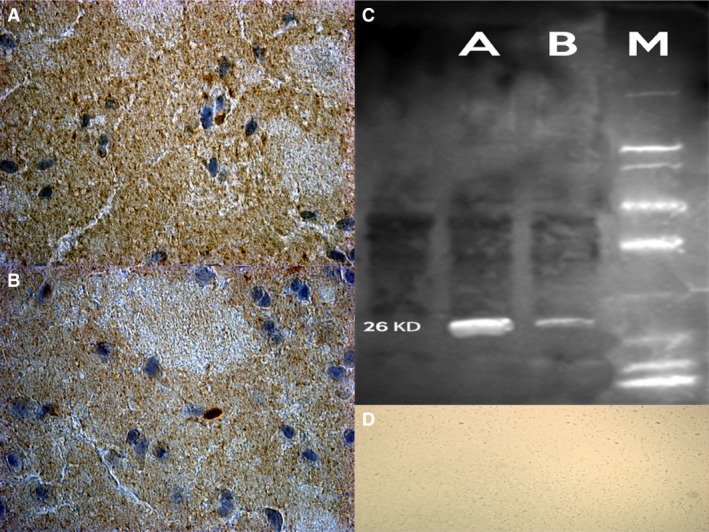
Confocal laser scanning microscopy showed markedly Bcl‐2 positive cytoplasmic reaction (in brown) on the striatum (A) and hippocampus (in brown) (B) in rats after nandrolone treatment. (C) Western blot analysis detects the chemiluminescent blots of Bcl‐2 in the treated group. (D) control group with negative results.
SMAC/DIABLO
A strong localization on the dendrites and neuronal cell body positivity located in PFC, S and Hipp areas was revealed for treated rats, compared to control group (Fig. 4).
Figure 4.
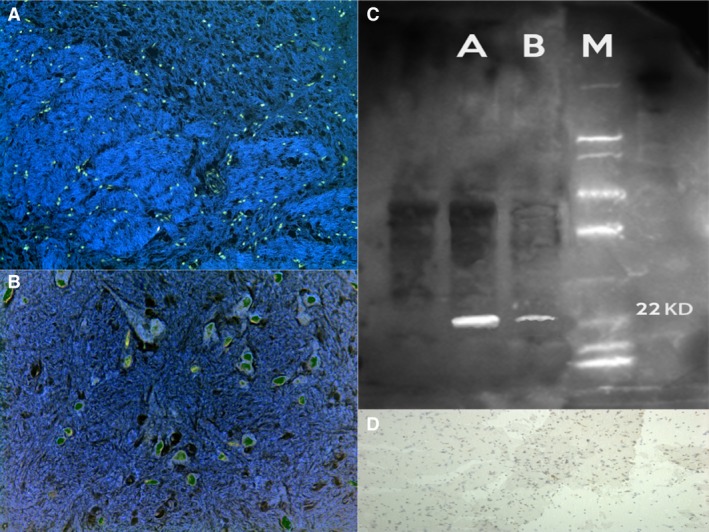
A strong SMAC/DIABLO localization on the dendrites and neuronal cell body positivity located in the frontal cortex (A), striatum (B) and hippocampus areas was revealed for treated rats. (C) Chemiluminescent blots of SMAC/DIABLO in the treated group. (D) Control group with negative results.
VMAT‐2
Anti‐VMAT‐2 immunopositivity was significantly weaker on the dendrites and neuronal cell bodies of treated rats compared to controls. In particular the most significant difference was found in S and Hipp samples (Fig. 5).
Figure 5.
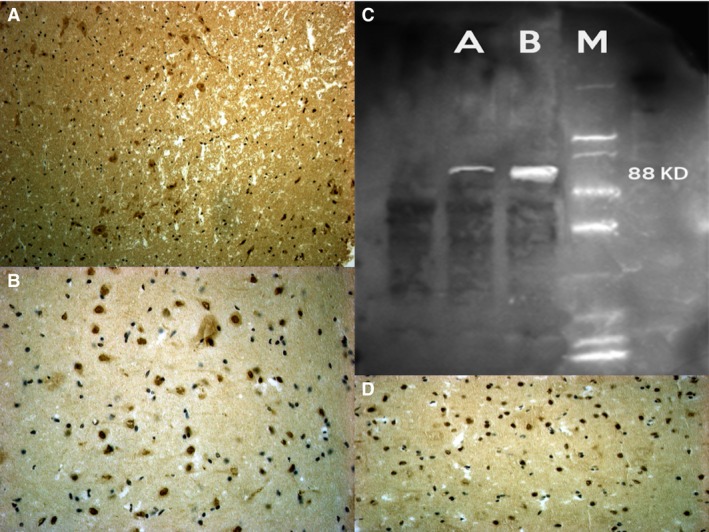
VMAT‐2 weaker reactions on the dendrites and neuronal cell bodies of treated rats compared to controls (D). In particular the most significant difference was found in striatum (A) and hippocampus (B) samples. (C) Western blot analysis detects the chemiluminescent blots of VMAT2 in the treated group.
TUNEL
The immunohistochemical study revealed an intensive positive result to TUNEL assay. The number of TUNEL positive cells that showed the typical morphological features of apoptosis (chromatin condensation, cytoplasmatic blebbing and apoptotic bodies) significantly increased in PFC, S and Hipp when compared with the control group. The neuronal nuclei labelled by TUNEL assay showed an intense, widespread, positive reaction in the treated group, especially in PFC, S and Hipp samples. Spotted positive nuclei were observed in cerebellum samples and in the control group (Fig. 6).
Figure 6.
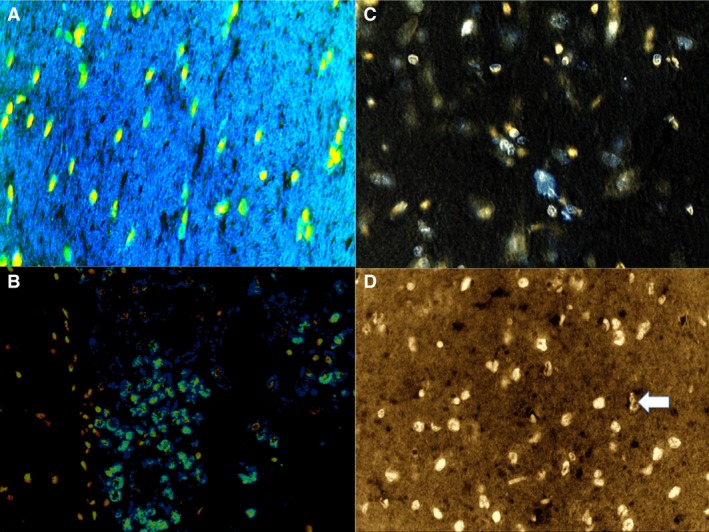
The neuronal nuclei labelled by TUNEL assay showed an intense, widespread, positive reaction in the treated group, especially in the frontal cortex: confocal laser scanning microscopy shows a marked positive nuclear reaction (in green apoptotic bodies) (A), and hippocampus (B) samples. Spotted positive nuclei were observed in cerebellum samples (in bleu) (C) and in the control group (arrow) (D).
Western blot analysis
Furthermore, the induction of expression levels was quantified by Western blot analysis. NF‐κB revealed an intense and massive positive reaction in ND group. Western blot analysis detected the chemiluminescent blots of NF‐κB, Bcl‐2 and SMAC/DIABLO in the treated group; a weak reaction was observed in the treated group for VMAT‐2.
Furthermore, the induction of these protein expression levels was quantified. The results were as follows: NF‐κB/β‐actin 0.60, Bcl‐2/β‐actin 0.60 and SMAC/DIABLO/β‐actin 0.50 for nandrolone‐treated group A; VMAT‐2/β‐actin 0.20 for the same ND group, matching perfectly with the immunohistochemistry results (Figs 2, 3, 4, 5, 6).
Control groups did not show any immunoreactivity for the studied markers, as well as Western blot analysis (reactions were not present), except for VMAT‐2. Our results are summarized in Table 2.
Discussion
In this study, we have investigated the hypothesis that high, chronically administered doses of ND could induce deleterious effects in the brains of rats through a strict mutual crosstalk among apoptotic pathways activation, neuronal degeneration and oxidative stress unbalance. The results of the present study indicate that a long‐term administration of ND, an AAS, promotes oxidative injury in rat brain specific areas. The main metabolites are 3α‐hydroxy‐5β‐estran‐17‐one (3‐norandrosterone) and 3α‐hydroxy‐5β‐estran‐17‐one (2‐noretiocholanolone) 35. The substitution of a methyl group to the carbon atom at position 19 by a hydrogen atom in the testosterone molecule changes considerably the ratio between anabolic and androgenic activities, increasing the concentration of the former compound 35. The genotoxic activity of steroids is also because of an indirect process that takes place in the redox cycle, as well as in the production of oxygen reactive types 36, 37. Thus, the metabolic activation of testosterone derivatives leads to the formation of free radicals and consequently to the induction of oxidative stress. The lipid peroxidation observed in all brain areas tested, as an index of neuronal oxidative injury, is the evidence of this effect. The possible consequence on behaviour, learning, memory and cognitive abilities must be considered.
Our data revealed a strong increase in apoptotic death in brain specimens of treated rats when compared to the control group. Not surprisingly, we found that apoptotic death as indicated by the number of TUNEL+ cells was mostly exacerbated in brain areas of treated rats where the greatest densities of androgen receptors (ARs) were found, namely the hippocampus and deep layers of cerebral cortex where ARs were also localized 38, 39, 40, 41. As expected fewer TUNEL‐positive apoptotic cells were observed in cerebellum samples (P < 0.05).
The mechanisms of the neuropathological effects of AASs have not yet been completely clarified and are still largely unexplored; however, evidence has shown the recurrence of increased neuronal susceptibility to apoptotic stimuli as a source of the neurodegenerative and neurotoxic potential of these compounds 1. It is well known that AASs can exert apoptotic stimuli in various tissues and organs 42, 43, and growing evidence is emerging that apoptotic mechanisms are also partly involved in AASs induced neurotoxicity. Anabolic androgenic steroids mechanisms are similar to the other steroid hormones. In particular they exert their effects by binding to ARs at cellular level, translocating to binding sites on chromatin, promoting gene transcription, stimulating the production of mRNA and ultimately increasing protein synthesis 42. This classic genomic model for steroid hormone action presumes that steroid hormones can freely cross the plasma membrane, enter the cytoplasm, and bind to and activate specific intracellular steroid receptor proteins 44. An apoptotic effect of high dosages of AASs acting on an AR‐mediated genomic pathway has been experimentally demonstrated in dopaminergic neurons (N 27 cells) expressing ARs 45. In this experimental model, androgens enter the cell, bind to the classical intracellular ARs and induce oxidative stress leading to mitochondrial dysfunction. Release of cytochrome c from the mitochondria activates the apoptotic caspase cascade. This effect has been abrogated by the AR antagonist flutamide 25, 45.
In addition to the classical intracellular AR via, AASs can exert an apoptotic effect also through a non‐genomic pathway, involving the rapid rise of intracellular calcium concentration ([Ca2+]i) 46. The rapidity of the calcium modulation response (from seconds to minutes) leads us to presume that the androgen must bind to some sort of receptor at the surface of the cell to achieve this result 44. Interestingly, not all cell types that demonstrate a rapid androgen response express the classic nuclear ARs or are blocked by ARs antagonists. Therefore, it is not yet known whether the receptor located at the cell surface is the classic intracellular AR coupled to other signal transduction machinery located in the membrane or a unique protein, capable of binding androgens and initiating signal transduction cascade 46. Effects of AASs on intracellular Ca2+ represent a classic ‘non‐genomic’ effect; Ca2+ oscillations are a key point in neuronal apoptosis 47, 48. High, supra‐pharmacological doses of testosterone for relatively short periods initiate an apoptotic programme in neuroblastoma cells through a rapid overactivation of intracellular Ca2+ signalling pathways 49. This rapid effect of testosterone on intracellular Ca2+ signalling in neurons occurs in the absence of ARs 33, 49. The apoptotic role of AASs is further supported by the study of Tugyan et al. 50 who, in an animal model (rats), demonstrated that ND caused a significant increase in apoptotic cells and a significant decrease in neuronal counting in the parietal cortex, prefrontal cortex and hippocampal regions of the brain. Neuronal death was induced in the cortical neuronal cultures obtained from rats using high doses of nandrolone. The glial component is important in AASs‐induced neurotoxicity: when ND was administered to mixed (neuronal and glial cells) cortical cell cultures, low doses of the drug were enough to initiate the apoptotic death programme. Similarly in cultures of pure neurons, this toxic effect was inhibited by ARs antagonist flutamide 25. Conclusively, ND appears to be more potent in neurotoxicity when the glial component is present in cell cultures, suggesting that androgen‐induced brain inflammation through the induction of NF‐κB 51 could synergize with androgen in reducing neuronal viability 25. These observations are consistent with our findings of NF‐κB immunoreactivity that showed a strong positive reaction in brain samples of the treated rats compared to control group, particularly in PFC, S and Hypp samples. The NF‐κB family is a family of transcription factors that are central, co‐ordinating regulators of immunity, inflammation, development, growth and cell survival. In non‐stimulated cells, most of the NF‐κB complexes lie latent in the cells' cytoplasm interacting with IkB family inhibitory proteins. A great number of stimuli, including pro‐inflammatory cytokines, bacterial products and stress, can activate NF‐κB from these inactive cytosolic pools. When adequate stimuli occur, the IkBs proteins are quickly phosphorylated by the activating IκB kinase complex. Phosphorylation of inhibitory IκB proteins initiates their ubiquitination and subsequent proteosomal degradation, followed by the release and nuclear translocation of active NF‐κB dimers to regulate expression of target genes, among which are the encoding numerous cytokines, adhesion molecules, growth factors, immune receptors and prosurvival anti‐apoptotic proteins 52, 53, 54, 55, 56, 57, 58, 59, 60. The NF‐κB system is widely expressed in the central nervous system (CNS). Damage‐associated molecular patterns, pathogen‐associated molecular patterns, cytokines, chemokines, neurotransmitters, neurotrophic factors and neurotoxins are known to stimulate NF‐κB activation in the CNS 61, and the IKK/NF‐κB signalling system is thought to be critically involved in the pathogenesis of various neurological diseases 56, 62, 63. One of the earliest recognized unconventional functions of the apoptotic apparatus is represented by the death‐receptor‐mediated activation of NFκB‐regulated inflammation 64. NF‐κB is actually regarded as the matchmaker between apoptosis and inflammation 65. Ligand‐bound death receptors, in particular TNFR1, can potentially trigger a wide range of cellular responses ranging from cell death, because of extrinsic apoptosis or regulated necrosis, to NF‐κB activation. Depending on the cell type and specific context, NF‐κB can transactivate genes with anti‐apoptotic functions, such as BCL‐2, or leading to the production of pro‐inflammatory mediators including tumour necrosis factor‐α and interferon‐γ 58. Many other components of the extrinsic apoptotic pathway, such as some caspases, are also involved in the inflammatory response. Nevertheless, the exact role of the IKK/NF‐κB system in CNS pathology is not yet fully understood, it is argued that because of its pro‐inflammatory function, NF‐κB activation is able to trigger neuronal dysfunction, ageing and cell death, thereby increasing the severity of many CNS diseases 62, 66, 67, 68. Although this aspect has not yet been investigated, it is tempting to speculate that the neurotoxic effect of high doses of AAS can be mediated also by an inflammatory response through the pro‐inflammatory activity of some components of the apoptotic machinery. A link between oxidative stress and NF‐κB signalling pathways is supported by our results. In addition to high levels of oxidative stress, we consistently observed a strong immunopositivity to NF‐κB. It has been argued that one of the pathways leading to the activation of NF‐κB could be under reactive oxygen species (ROS)‐mediated control. In fact, growing evidence suggests that, although in limited doses, endogenous ROS may play an activating role in NF‐κB signalling, above a certain threshold, they may negatively impact upon this signalling 96. Reactive oxygen species are thought to have an inhibitory effect on NF‐κB activity. However, a mutual crosstalk between ROS and NF‐κB exists and recent studies have shown that ROS activity is subject to negative feedback regulation by NF‐κB, and that this negative regulation of ROS is the means through which NF‐κB counters programmed cell 69.
Bcl2 family members regulate the mitochondrial pathway of apoptosis. They are either pro apoptotic (Bak or Bax) or anti‐apoptotic (Bcl2 or Bcl XL), both of which are essential for apoptosis driven by the mitochondrial pathway. These proteins play a role in the permeabilization of the mitochondrial outer membrane on receiving apoptotic signals. Permeabilization leads to the release of cytochrome c, formation of apoptosome complex, activation of caspases, thus triggering morphological changes like membrane blebbing and nuclear fragmentation. Cell survival or apoptosis relies on the delicate balance between the up‐ and down‐regulation of Bcl2 and Bax 70, 71. Bax up‐regulation leads to enhanced susceptibility to apoptosis; on the contrary, Bcl2 up‐regulation leads to neuronal survival 72.
Our findings that chronic exposure to ND can impact VMAT‐2 levels are of interest. In particular, exposure to ND induced a significant (P < 0.001) decrease in VMAT‐2 immunoreactivity as assessed in tissue samples prepared from rat brain. VMAT‐2 is an important regulator of intra‐neuronal monoamine concentrations and disposition. It has been shown to be responsible for sequestrating cytoplasmatic neurotransmitters such as dopamine (DA) within synaptic vesicles. Under physiological conditions, DA is largely confined to synaptic vesicles where it is protected from metabolic breakdown. Sequestration of DA into vesicles provides a protective environment against the intracellular production of ROS. In the cytoplasm, free DA can in fact give rise to the formation of cytotoxic free radicals. Oxidative metabolites of DA may conjugate with α‐synuclein to form an adduct of DA–α‐synuclein, which may stabilize the toxic form of α‐synuclein through a covalent bound to DA quinone 73, while also promoting selective neurotoxicity 74. Normally, the concentration of cytoplasmic DA is kept at a minimum by continuous pumping activity of VMAT‐2 75. Cytosolic DA increases levels of DA‐generated oxy radicals ultimately resulting in degeneration of DAergic neurons. Moving from the study of Cubells et al. 76, it has been argued that the redistribution of DA from a smaller environment inside synaptic vesicles to oxidizing environments outside vesicles favoured the formation of ROS within the DA neurons which contribute to DA loss 77. Therefore, we can say that a change in DA storage and release machinery is associated with DA neurons loss, probably because of a caspase‐independent ROS‐mediated apoptotic pathway 78.
VMAT‐2 is currently considered a marker of dopaminergic neurons integrity with neuroprotective function. Recently, its role in neurodegenerative disorders, such as PD, has been unravelled 79, 80, thus focusing on the fact that VMAT2 defects may be an early abnormality promoting mechanisms leading to nigrostriatal DA neuron death in PD. Studies have indicated that several exogenous substances influence VMAT‐2 81. In particular, psycho‐stimulants, both the releasers (i.e. amphetamine analogous) and uptake blockers (i.e. cocaine‐like drugs) interfere with the activity and sub‐cellular distribution of monoamine transporters (VMAT‐2 and DAT – dopamine transporter), and this mechanism is likely to be related to the neurotoxicity shown by these substances 81. Several investigators have assessed the impact of cocaine on cytoplasmic vesicles, wherein it was determined that cocaine administration increases DA transport into this cytoplasmic vesicular fraction 82. This effect was attributed to a redistribution of VMAT‐2 and associated vesicles from synaptosomal membranes into the cytoplasm 83, thus elucidating the mechanisms whereby cocaine alters DA signalling. Psycho‐stimulants like methamphetamine which act as releasers of DA by disrupting vesicular pH gradients and allowing vesicular DA to redistribute into the cytoplasm 75, 81, have been demonstrated to decrease striatal VMAT‐2 ligand binding 81. Administration of several other agents causing DA release decrease VMAT‐2 activity and/or immunoreactivity in a similar manner; these include a single administration of AMPH 81 and repeated injections of MDMA 84. These drugs can potentiate the oxidative mechanism of DA. VMAT‐2 is able to take up methamphetamine in monoaminergic vesicles, inducing the release of DA to the cytosol which is important for methamphetamine neurotoxicity. The role of cytosolic DA in methamphetamine neurotoxicity has been supported by the fact that the inhibition of DA synthesis protects against methamphetamine neurotoxicity while the inhibition of VMAT‐2 and monoamine oxidase exacerbate methamphetamine neurotoxicity 85.
On the basis of the data presented here and on our findings showing a significant decrease in VMAT 2 immunoreactivity in ND treated rats compared to control rats, we infer that ND chronically administered could induce alterations in the VMAT machinery and alter the VMAT‐2‐mediated DA uptake into monoaminergic vesicles, which is known to be an important neuroprotective mechanism in dopaminergic neurons.
Given previous experimental findings of ROS involvement in pathways leading to the activation of programmed cell death 86, 87, in our experiment, oxidative stress involvement was evaluated in different brain areas where apoptosis was detected and quantified. Our results clearly show that supra‐physiological doses of ND administered chronically are able to disrupt redox metabolism in the brain, characterizing an oxidative stress state in all the studied cerebral areas. The low significant statistical difference of oxidative stress marker (MDA) in cerebellum specimens of treated rats compared to control group may likely reflect the high percentage of cerebellar granule cells that have been demonstrated less vulnerable to oxidative stress‐induced cell death, via a mechanism involving an up‐regulation of the cellular antioxidant defence 88.
Conclusions
Thus, these findings support the idea that oxidative stress plays a pivotal role in AASs‐induced neurotoxiticy. ROS represent a serious hazard for cells because they are powerful oxidizing molecules able to damage proteins, lipids and DNA 89, 90. Reactive oxygen species act as second messengers in various biological responses, among which the induction of programmed cell death is of paramount importance in our understanding of many common diseases and degenerative conditions 91. Holmes et al. 92 investigated the effects of androgens under conditions of oxidative stress to determine whether androgens play a neuroprotective or neurotoxic role in DA neuronal functions. They found that androgens, alone, increased mitochondrial function via a calcium‐dependent mechanism. Androgen pre‐treatment protected cells from oxidative stress‐induced cell death. However, treatment with androgens after the oxidative insult increased cell death, and these effects were, in part, mediated by calcium influx into the mitochondria and the negative effects of androgens were not blocked by either androgen or oestrogen receptor antagonists 93. A membrane‐associated AR was thought to be implicated. The results of this study suggest that androgens are neuroprotective when oxidative stress levels are minimal, but when oxidative stress levels are elevated, androgens exacerbate oxidative stress damage 92. Similar results were reported by Cunningham et al. 94 who demonstrated that testosterone appears to have negative consequences on brain function under conditions of elevated oxidative stress. In a pre‐existing oxidative stress environment, androgens can further exacerbate oxidative stress damage 95, 96. A possible mechanism for androgen‐induced neuroprotection is preconditioning because androgens can moderately increase oxidative stress and apoptosis 25. These results suggest that the level of oxidative stress determines whether androgens play a positive or negative role in neuronal function 91, and it is argued that oxidative stress defines the neuroprotective and neurotoxic properties of androgens, thus acting as a molecular switch for androgen actions 94 (Fig. 7).
Figure 7.
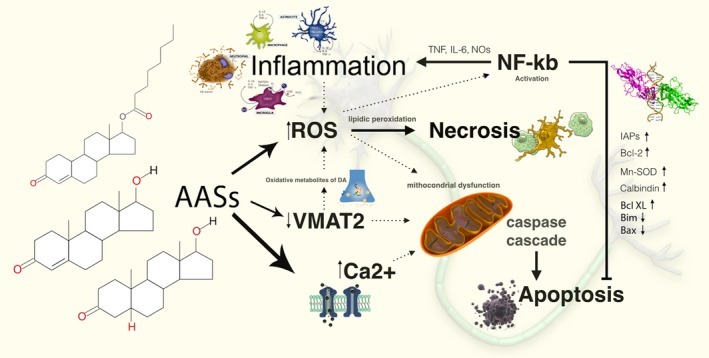
Mechanisms of the neuropathological effects of AASs: evidence has shown the recurrence of increased neuronal susceptibility to apoptotic stimuli as a source of the neurodegenerative and neurotoxic potential of these compounds. ROSs represent a serious hazard for cells, because they are powerful oxidizing molecules able to damage proteins, lipids and DNA. ROSs act as second messengers in various biological responses, among which the induction of programmed cell death is of paramount importance in our understanding of many common diseases and degenerative conditions. Growing evidence suggests that endogenous ROS may play an activating role in NF‐kB signalling, and above a certain threshold, they may negatively impact upon this signalling. ROS are thought to have an inhibitory effect on NF‐kB activity.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
Author contribution
ET wrote the paper; MN performed the research; DC performed the research; SC contributed essential reagents or tools; PF performed the research; LM analysed the data; FPB analysed the data; CP analysed the data; IR performed the research; VF designed the research study and wrote the paper.
References
- 1. Scaccianoce S, Caruso A, Miele J, et al Potential neurodegenerative effect of anabolic androgenic steroid abuse. J Biol Regul Homeost Agents. 2013; 27: 107–14. [PubMed] [Google Scholar]
- 2. Buckley WE, Yesalis CE, Friedl KE, et al Estimated prevalence of anabolic steroid use among male high school seniors. JAMA. 1988; 260: 3441–5. [PubMed] [Google Scholar]
- 3. Pope HG Jr, Katz DL. Affective and psychotic symptoms associated with anabolic steroid use. Am J Psychiatry. 1988; 27: 487–90. [DOI] [PubMed] [Google Scholar]
- 4. Lukas SE. CNS effects and abuse liability of anabolic‐androgenic steroids. Annu Rev Pharmacol Toxicol. 1996; 36: 333–57. [DOI] [PubMed] [Google Scholar]
- 5. Yesalis CE. Epidemiology and patterns of anabolic‐androgenic steroid use. Psychiatric Annals. 1992; 22: 7–8. [Google Scholar]
- 6. Yesalis CE, Barsukiewicz CK, Kopstein AN, et al Trends in anabolic‐androgenic steroid use among adolescents. Arch Pediatr Adolesc Med. 1997; 151: 1197–206. [DOI] [PubMed] [Google Scholar]
- 7. Yesalis CE, Kennedy NJ, Kopstein AN, et al Anabolic‐androgenic steroid use in the United States. JAMA. 1993; 270: 1217–21. [PubMed] [Google Scholar]
- 8. Frati P, Kyriakou C, Del Rio A, et al Smart drugs and synthetic androgens for cognitive and physical enhancement: revolving doors of cosmetic neurology. Curr Neuropharmacol. 2015; 13: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shahidi NT. A review of the chemistry, biological action, and clinical applications of anabolic‐androgenic steroids. Clin Ther. 2001; 23: 1355–90. [DOI] [PubMed] [Google Scholar]
- 10. Brower KJ. Anabolic steroids. Psychiatr Clin North Am. 1993; 16: 97–103. [PubMed] [Google Scholar]
- 11. Clark AS, Fast AS. Comparison of the effects of 17 alpha‐methyltestosterone, methandrostenolone, and nandrolone decanoate on the sexual behavior of castrated male rats. Behav Neurosci. 1996; 110: 1478–86. [DOI] [PubMed] [Google Scholar]
- 12. Midgley SJ, Heather N, Davies JB. Levels of aggression among a group of anabolic‐androgenic steroid users. Med Sci Law. 2001; 41: 309–14. [DOI] [PubMed] [Google Scholar]
- 13. Ambar G, Chiavegatto S. Anabolic‐androgenic steroid treatment induces behavioral disinhibition and downregulation of serotonin receptor messenger RNA in the prefrontal cortex and amygdala of male mice. Genes Brain Behav. 2009; 8: 161–73. [DOI] [PubMed] [Google Scholar]
- 14. Rainer Q, Speziali S, Rubino T, et al Chronic nandrolone decanoate exposure during adolescence affects emotional behavior and monoaminergic neurotransmission in adulthood. Neuropharmacology. 2014; 83: 79–88. [DOI] [PubMed] [Google Scholar]
- 15. Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006; 97: 1634–58. [DOI] [PubMed] [Google Scholar]
- 16. Orlando R, Caruso A, Molinaro G, et al Nanomolar concentrations of anabolic‐androgenic steroids amplify excitotoxic neuronal death in mixed mouse cortical cultures. Brain Res. 2007; 1165: 21–9. [DOI] [PubMed] [Google Scholar]
- 17. Kanayama G, Hudson JI, Pope HG. Illicit anabolic‐androgenic steroid use. Horm Behav. 2010; 58: 111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petersson A, Garle M, Holmgren P, et al Toxicological findings and manner of death in autopsied users of anabolic androgenic steroids. Drug Alcohol Depend. 2006; 81: 241–9. [DOI] [PubMed] [Google Scholar]
- 19. Heikkila R, Cohen G. Inhibition of biogenic amine uptake by hydrogen peroxide: a mechanism for toxic effects of 6‐hydroxydopamine. Science. 1971; 172: 1257–8. [DOI] [PubMed] [Google Scholar]
- 20. Przedborski S, Kostic V, Jackson‐Lewis V, et al Transgenic mice with increased Cu/Zn‐superoxide dismutase activity are resistant to N‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced neurotoxicity. J Neurosci. 1992; 12: 1658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oishi T, Hasegawa E, Murai Y. Sulfhydryl drugs reduce neurotoxicity of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) in the mouse. J Neural Transm Park Dis Dement Sect. 1993; 6: 45–52. [DOI] [PubMed] [Google Scholar]
- 22. Kumar R, Agarwal AK, Seth PK. Free radical‐generated neurotoxicity of 6‐hydroxydopamine. J Neurochem. 1995; 64: 1703–7. [DOI] [PubMed] [Google Scholar]
- 23. Nakamura T, Cho DH, Lipton SA. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol. 2012; 238: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frankenfeld SP, Oliveira LP, Ortenzi VH, et al The anabolic androgenic steroid nandrolone decanoate disrupts redox homeostasis in liver, heart and kidney of male Wistar rats. PLoS ONE. 2014; 9:e102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caraci F, Pistara V, Corsaro A, et al Neurotoxic properties of the anabolic androgenic steroids nandrolone and methandrostenolone in primary neuronal cultures. J Neurosci Res. 2011; 89: 592–600. [DOI] [PubMed] [Google Scholar]
- 26. Thiblin I, Finn A, Ross SB, et al Increased dopaminergic and 5‐hydroxytryptaminergic activities in male rat brain following long‐term treatment with anabolic androgenic steroids. Br J Pharmacol. 1999; 126: 1301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clark AS, Henderson LP. Behavioral and physiological responses to anabolic‐androgenic steroids. Neurosci Biobehav Rev. 2003; 27: 413–36. [DOI] [PubMed] [Google Scholar]
- 28. Kindlundh AM, Rahman S, Lindblom J, et al Increased dopamine transporter density in the male rat brain following chronic nandrolone decanoate administration. Neurosci Lett. 2004; 356: 131–4. [DOI] [PubMed] [Google Scholar]
- 29. Kurling S, Kankaanpaa A, Ellermaa S, et al The effect of sub‐chronic nandrolone decanoate treatment on dopaminergic and serotonergic neuronal systems in the brains of rats. Brain Res. 2005; 1044: 67–75. [DOI] [PubMed] [Google Scholar]
- 30. Henderson LP, Penatti CA, Jones BL, et al Anabolic androgenic steroids and forebrain GABAergic transmission. Neuroscience. 2006; 138: 793–9. [DOI] [PubMed] [Google Scholar]
- 31. Rossbach UL, Steensland P, Nyberg F, et al Nandrolone‐induced hippocampal phosphorylation of NMDA receptor subunits and ERKs. Biochem Biophys Res Commun. 2007; 357: 1028–33. [DOI] [PubMed] [Google Scholar]
- 32. Penatti CA, Costine BA, Porter DM, et al Effects of chronic exposure to an anabolic androgenic steroid cocktail on alpha5‐receptor‐mediated GABAergic transmission and neural signaling in the forebrain of female mice. Neuroscience. 2009; 161: 526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kanayama G, Hudson JI, Pope HG. Long‐term psychiatric and medical consequences of anabolic‐androgenic steroid abuse: a looming public health concern? Drug Alcohol Depend. 2008; 98: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shara MA, Dickson PH, Bagchi D, et al Excretion of formaldehyde, malondialdehyde, acetaldehyde and acetone in the urine of rats in response to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, paraquat, endrin and carbon tetrachloride. J Chromatogr. 1992; 576: 221–33. [DOI] [PubMed] [Google Scholar]
- 35. Lippi G, Franchini M, Banfi G. Biochemistry and phisiology of anabolic androgenic steroids doping. Mini Rev Med Chem. 2011; 11: 362–73. [DOI] [PubMed] [Google Scholar]
- 36. Fischer WH, Keiwan A, Schmitt E, et al Increased formation of micronuclei after hormonal stimulation of cell proliferation in human breast cancer cells. Mutagenesis. 2001; 16: 209–12. [DOI] [PubMed] [Google Scholar]
- 37. do Carmo CA, Goncalves AL, Salvadori DM, et al Nandrolone androgenic hormone presents genotoxic effects in different cells of mice. J Appl Toxicol. 2012; 32: 810–4. [DOI] [PubMed] [Google Scholar]
- 38. Roselli CE, Abdelgadir SE, Ronnekleiv OK, et al Anatomic distribution and regulation of aromatase gene expression in the rat brain. Biol Reprod. 1998; 58: 79–87. [DOI] [PubMed] [Google Scholar]
- 39. Roselli CE, Horton LE, Resko JA. Distribution and regulation of aromatase activity in the rat hypothalamus and limbic system. Endocrinology. 1985; 117: 2471–7. [DOI] [PubMed] [Google Scholar]
- 40. Simerly RB, Chang C, Muramatsu M, et al Distribution of androgen and estrogen receptor mRNA‐containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990; 294: 76–95. [DOI] [PubMed] [Google Scholar]
- 41. Bingaman EW, Baeckman LM, Yracheta JM, et al Localization of androgen receptor within peptidergic neurons of the rat forebrain. Brain Res Bull. 1994; 35: 379–82. [DOI] [PubMed] [Google Scholar]
- 42. Maravelias C, Dona A, Stefanidou M, et al Adverse effects of anabolic steroids in athletes. A constant threat. Toxicol Lett. 2005; 158: 167–75. [DOI] [PubMed] [Google Scholar]
- 43. Frati P, Busardò FP, Cipolloni L, et al Anabolic androgenic steroid (AAS) related deaths: autoptic, histopathological and toxicological findings. Curr Neuropharmacol. 2015; 13: 146–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foradori CD, Weiser MJ, Handa RJ. Non‐genomic actions of androgens. Front Neuroendocrinol. 2008; 29: 169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cunningham RL, Giuffrida A, Roberts JL. Androgens induce dopaminergic neurotoxicity via caspase‐3‐dependent activation of protein kinase Cdelta. Endocrinology. 2009; 150: 5539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Foradori CD, Werner SB, Sandau US, et al Activation of the androgen receptor alters the intracellular calcium response to glutamate in primary hippocampal neurons and modulates sarco/endoplasmic reticulum calcium ATPase 2 transcription. Neuroscience. 2007; 149: 155–64. [DOI] [PubMed] [Google Scholar]
- 47. Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol. 2010; 2: a005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vicencio JM, Estrada M, Galvis D, et al Anabolic androgenic steroids and intracellular calcium signaling: a mini review on mechanisms and physiological implications. Mini Rev Med Chem. 2011; 11: 390–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Estrada M, Varshney A, Ehrlich BE. Elevated testosterone induces apoptosis in neuronal cells. J Biol Chem. 2006; 281: 25492–501. [DOI] [PubMed] [Google Scholar]
- 50. Tugyan K, Ozbal S, Cilaker S, et al Neuroprotective effect of erythropoietin on nandrolone decanoate‐induced brain injury in rats. Neurosci Lett. 2013; 533: 28–33. [DOI] [PubMed] [Google Scholar]
- 51. Gonzales RJ, Duckles SP, Krause DN. Dihydrotestosterone stimulates cerebrovascular inflammation through NFkappaB, modulating contractile function. J Cereb Blood Flow Metab. 2009; 29: 244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grossmann M, O'Reilly LA, Gugasyan R, et al The anti‐apoptotic activities of Rel and RelA required during B‐cell maturation involve the regulation of Bcl‐2 expression. EMBO J. 2000; 19: 6351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karin M, Lin A. NF‐kappaB at the crossroads of life and death. Nat Immunol. 2002; 3: 221–7. [DOI] [PubMed] [Google Scholar]
- 54. Hayakawa M, Miyashita H, Sakamoto I, et al Evidence that reactive oxygen species do not mediate NF‐kappaB activation. EMBO J. 2003; 22: 3356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kucharczak J, Simmons MJ, Fan Y, et al To be, or not to be: NF‐kappaB is the answer–role of Rel/NF‐kappaB in the regulation of apoptosis. Oncogene. 2003; 22: 8961–82. [DOI] [PubMed] [Google Scholar]
- 56. Ghosh S, Hayden MS. New regulators of NF‐kappaB in inflammation. Nat Rev Immunol. 2008; 8: 837–48. [DOI] [PubMed] [Google Scholar]
- 57. Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell. 2008; 132: 344–62. [DOI] [PubMed] [Google Scholar]
- 58. Perkins ND. Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol. 2008; 8: 49–62. [DOI] [PubMed] [Google Scholar]
- 59. Claudio E, Brown K, Siebenlist U. NF‐kappaB guides the survival and differentiation of developing lymphocytes. Cell Death Differ. 2006; 13: 697–701. [DOI] [PubMed] [Google Scholar]
- 60. Tasyriq M, Najmuldeen IA, In LL, et al 7alpha‐hydroxy‐beta‐sitosterol from Chisocheton tomentosus induces apoptosis via dysregulation of cellular Bax/Bcl‐2 ratio and cell cycle arrest by downregulating ERK1/2 activation. Evid Based Complement Alternat Med. 2012; 765316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maqbool A, Lattke M, Wirth T, et al Sustained, neuron‐specific IKK/NF‐kappaB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol Neurodegener. 2013; 8: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Memet S. NF‐kappaB functions in the nervous system: from development to disease. Biochem Pharmacol. 2006; 72: 1180–95. [DOI] [PubMed] [Google Scholar]
- 63. Gupta SC, Sundaram C, Reuter S, et al Inhibiting NF‐kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010; 1799: 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ben‐Neriah Y, Karin M. Inflammation meets cancer, with NF‐kappaB as the matchmaker. Nat Immunol. 2011; 12: 715–23. [DOI] [PubMed] [Google Scholar]
- 65. Galluzzi L, Kepp O, Trojel‐Hansen C, et al Non‐apoptotic functions of apoptosis‐regulatory proteins. EMBO Rep. 2011; 13: 322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van Loo G, De Lorenzi R, Schmidt H, et al Inhibition of transcription factor NF‐kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. 2006; 7: 954–61. [DOI] [PubMed] [Google Scholar]
- 67. Lattke M, Magnutzki A, Walther P, et al Nuclear factor kappaB activation impairs ependymal ciliogenesis and links neuroinflammation to hydrocephalus formation. J Neurosci. 2012; 32: 11511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang G, Li J, Purkayastha S, et al Hypothalamic programming of systemic ageing involving IKK‐beta, NF‐kappaB and GnRH. Nature. 2013; 497: 211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bubici C, Papa S, Dean K, et al Mutual cross‐talk between reactive oxygen species and nuclear factor‐kappa B: molecular basis and biological significance. Oncogene. 2006; 25: 6731–48. [DOI] [PubMed] [Google Scholar]
- 70. Lu G, Kwong WH, Li Q, et al bcl2, bax, and nestin in the brains of patients with neurodegeneration and those of normal aging. J Mol Neurosci. 2005; 27: 167–74. [DOI] [PubMed] [Google Scholar]
- 71. Youle RY, Strasser A. The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9: 47–59. [DOI] [PubMed] [Google Scholar]
- 72. Liu D, Lu C, Wan R, et al Activation of mitochondrial ATP‐dependent potassium channels protects neurons against ischemia‐induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J Cereb Blood Flow Metab. 2002; 22: 431–43. [DOI] [PubMed] [Google Scholar]
- 73. Conway KA, Rochet JC, Bieganski RM, et al Kinetic stabilization of the alpha‐synuclein protofibril by a dopamine‐alpha‐synuclein adduct. Science. 2001; 294: 1346–9. [DOI] [PubMed] [Google Scholar]
- 74. Xu J, Kao SY, Lee FJ, et al Dopamine‐dependent neurotoxicity of alpha‐synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002; 8: 600–6. [DOI] [PubMed] [Google Scholar]
- 75. Riddle EL, Fleckenstein AE, Hanson GR. Role of monoamine transporters in mediating psychostimulant effects. AAPS J. 2005; 7: E847–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cubells JF, Rayport S, Rajendran G, et al Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine‐dependent intracellular oxidative stress. J Neurosci. 1994; 14: 2260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fleckenstein AE, Volz TJ, Hanson GR. Psychostimulant‐induced alterations in vesicular monoamine transporter‐2 function: neurotoxic and therapeutic implications. Neuropharmacology. 2009; 56: 133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Samms WC, Perera RP, Wimalasena DS, et al Perturbation of dopamine metabolism by 3‐amino‐2‐(4′‐halophenyl) propenes leads to increased oxidative stress and apoptotic SH‐SY5Y cell death. Mol Pharmacol. 2007; 72: 744–52. [DOI] [PubMed] [Google Scholar]
- 79. Pifl C, Rajput A, Reither H, et al Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci. 2014; 34: 8210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wu TH, Lu YN, Chuang CL, et al Loss of vesicular dopamine release precedes tauopathy in degenerative dopaminergic neurons in a Drosophila model expressing human tau. Acta Neuropathol. 2013; 125: 711–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Riddle EL, Hanson GR, Fleckenstein AE. Therapeutic doses of amphetamine and methylphenidate selectively redistribute the vesicular monoamine transporter‐2. Eur J Pharmacol. 2007; 571: 25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter‐2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther. 2001; 296: 762–7. [PubMed] [Google Scholar]
- 83. Farnsworth SJ, Volz TJ, Hanson GR, et al Cocaine alters vesicular dopamine sequestration and potassium‐stimulated dopamine release: the role of D2 receptor activation. J Pharmacol Exp Ther. 2009; 328: 807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hansen JP, Riddle EL, Sandoval V, et al Methylenedioxymethamphetamine decreases plasmalemmal and vesicular dopamine transport: mechanisms and implications for neurotoxicity. J Pharmacol Exp Ther. 2002; 300: 1093–100. [DOI] [PubMed] [Google Scholar]
- 85. Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009; 60: 379–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Curtin JF, Donovan M, Cotter TG. Regulation and measurement of oxidative stress in apoptosis. J Immunol Methods. 2002; 265: 49–72. [DOI] [PubMed] [Google Scholar]
- 87. Matsuzawa A, Ichijo H. Stress‐responsive protein kinases in redox‐regulated apoptosis signaling. Antioxid Redox Signal. 2005; 7: 472–81. [DOI] [PubMed] [Google Scholar]
- 88. Ahlbom E, Prins GS, Ceccatelli S. Testosterone protects cerebellar granule cells from oxidative stress‐induced cell death through a receptor mediated mechanism. Brain Res. 2001; 892: 255–62. [DOI] [PubMed] [Google Scholar]
- 89. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007; 87: 245–313. [DOI] [PubMed] [Google Scholar]
- 90. Aguirre J, Lambeth JD. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic Biol Med. 2010; 49: 1342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jones DP. Radical‐free biology of oxidative stress. Am J Physiol Cell Physiol. 2008; 295: C849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Holmes S, Abbassi B, Su C, et al Oxidative stress defines the neuroprotective or neurotoxic properties of androgens in immortalized female rat dopaminergic neuronal cells. Endocrinology. 2013; 154: 4281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Handa RJ, Pak TR, Kudwa AE, et al An alternate pathway for androgen regulation of brain function: activation of estrogen receptor beta by the metabolite of dihydrotestosterone, 5alpha‐androstane‐3beta,17beta‐diol. Horm Behav. 2008; 53: 741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cunningham RL, Singh M, O'Bryant SE, et al Oxidative stress, testosterone, and cognition among Caucasian and Mexican‐American men with and without Alzheimer's disease. J Alzheimers Dis. 2014; 40: 563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cunningham RL, Lumia AR, McGinnis MY. Androgenic anabolic steroid exposure during adolescence: ramifications for brain development and behavior. Horm Behav. 2013; 64: 350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Riezzo I, Turillazzi E, Bello S, et al Chronic nandrolone administration promotes oxidative stress, induction of pro‐inflammatory cytokine and TNF‐α mediated apoptosis in the kidneys of CD1 treated mice. Toxicol Appl Pharmacol. 2014; 280: 97–106. [DOI] [PubMed] [Google Scholar]