

Abstract
The signaling networks that drive the aging process, associated functional deterioration, and pathologies has captured the scientific community's attention for decades. While many theories exist to explain the aging process, the production of reactive oxygen species (ROS) provides a signaling link between engagement of cellular senescence and several age-associated pathologies. Cellular senescence has evolved to restrict tumor progression but the accompanying senescence-associated secretory phenotype (SASP) promotes pathogenic pathways. Here, we review known biological theories of aging and how ROS mechanistically control senescence and the aging process. We also describe the redox-regulated signaling networks controlling the SASP and its important role in driving age-related diseases. Finally, we discuss progress in designing therapeutic strategies that manipulate the cellular redox environment to restrict age-associated pathology.
Keywords: Oxidative stress, Aging, Senescence, Senescence-associated secretory phenotype (SASP), Reactive oxygen species (ROS)
Graphical abstract
Highlights
-
•
Cellular aging corresponds to a cumulative failure of repair mechanisms, that mitigate damage from oxidants.
-
•
Age-associated redox imbalance drives pathological pathways.
-
•
The precise redox triggers that control the senescence-associated secretory phenotype are yet to be elucidated.
-
•
ROS-sensitive signaling networks may be potential therapeutic targets in restricting age-related pathologies.
1. Biological theories of aging
Why do we age? The answer to this perennial question has encouraged the scientific community to pursue hypotheses that can delineate the mechanisms behind the human body's functional decline with age. However, despite medical advances there remain challenges with respect to delineating controls that regulate the span of human life and health. Elucidation of these control mechanisms may lead to better-targeted interventions for the gamut of diseases propagated by age. Ultimately, amelioration of age-associated pathologies and comorbidities would limit age-related frailty and improve the well-being of our aging population.
Biological aging is the progressive manifestation of accumulated cellular damage with age, and is determined by environmental or genetic factors [1], [2], [3], [4]. The process of aging is complex and can be explained by a number of complimentary theories. The wear and tear theory, proposed by August Weismann in 1882, compares organisms to machines, suggesting that cells ‘wear out’ with time and accumulate damage through excessive consumption of fat, sugar, or exposure to UV radiation, leading to declines in functional efficiency of organs. However, this theory disregards the ability of cells to repair damage. The stochastic theory which is a by-product of the wear and tear theory, postulates that aging is due to the accumulation of damage in cells resulting from failure of wound repairing mechanisms, infections, UV radiation, or environmental stress [5]. Evolutionary theories of aging support the concept that specific genes implicated in longevity are involved in the stability and maintenance of somatic cells, and are susceptible to mutations with age. Medawar's mutation accumulation theory links aging to natural selection, hypothesizing that there is no selection pressure on the aging population, as there is no known evolutionary mechanism to eliminate a population of mutations that cause deleterious effects only in aged species [6]. This suggests that detrimental late-acting alleles may accumulate with age and exert adverse effects on survival. The antagonistic pleiotropy model suggests that a pleiotropic gene that selectively increases fitness and survival during the early stages of life can still induce adverse late-acting effects in the population [7]. In the context of cellular aging, this could apply to replicative senescence that suppresses tumor formation early in life, but may promote cancer in later stages. The disposable soma theory, put forth as an extension of the antagonistic pleiotropy model, highlights the balance between somatic maintenance and repair versus reproduction and suggests that with age extensive maintenance would be necessary as cells can accumulate multiple lesions in parallel [8], [9]. The rate of living theory puts across the idea that organisms that metabolize oxygen more rapidly have a higher energy expenditure and typically have shorter lifespans that can be extended by caloric restriction [10], [11], [12], [13]. According to the cross-linking (or glycosylation) theory of aging, cross-linked proteins or sugar moieties can bind to DNA causing replicative damage or an age-related decline in protein turnover that can be linked to the loss of functional proteins, further promoting age-associated pathologies [14].
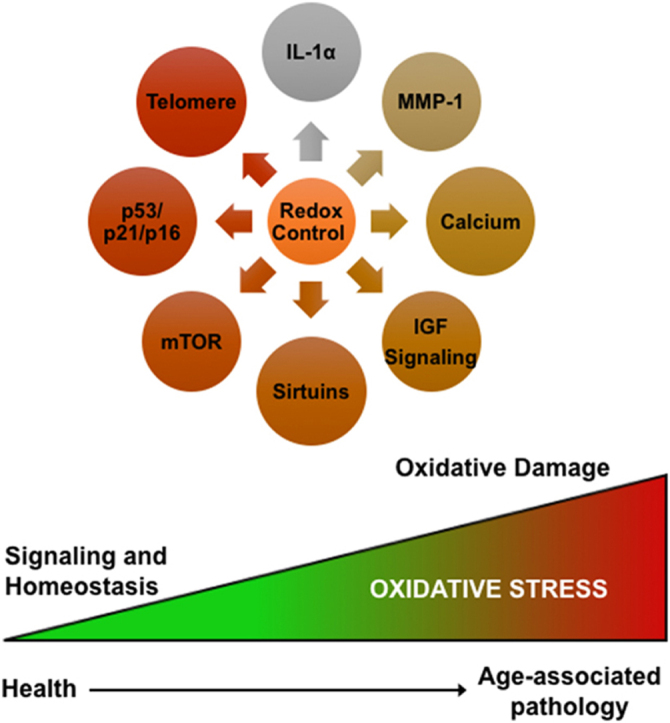
While all of the aforementioned theories have been examined and laboriously tested, the mitochondrial free radical theory of aging, later termed the oxidative stress theory (OST), is currently one of the most popular correlative theories of the aging process. OST explains aging at the molecular level and results from failure to maintain oxidative defenses, mitochondrial integrity, proteostasis, barrier structures, DNA repair, telomeres, immune function, metabolic regulation, and regenerative capacity (Fig. 1). More than half a century ago, Gerschman introduced that oxygen free radicals, commonly considered to be too reactive to exist in biological systems, are generated in situ in response to radiation and oxygen poisoning and are responsible for the associated toxicities [15], [16]. Later, Denham Harman proposed that hydroxyl and hydroperoxyl radicals generated endogenously from oxygen-consuming metabolic processes play crucial roles in the aging process [17]. This hypothesis coalesced from a number of observations: (i) indications that free radicals are produced in vivo; (ii) free radicals cause pervasive damage to macromolecules in vitro; and (iii) the deleterious effects of exposure to irradiation and hyperoxia are alleviated by antioxidant administration. Free radicals are highly reactive atoms or molecules with one or more unpaired electron(s) in their outermost shell and can be formed when oxygen interacts with certain molecules. They can be formed endogenously by natural biological processes, or generated upon exposure to external stimuli. Reactive oxygen species (ROS) collectively describes a number of reactive molecules, such as superoxide (O2•-), hydroxyl radical (OH•), hydroperoxyl radical (HO2•), nitric oxide (NO•), nitrogen dioxide (NO2•), and peroxyl (ROO•), produced as by-products during the mitochondrial electron transport of aerobic respiration or by oxidoreductase enzymes. Also, hydrogen peroxide (H2O2), ozone (O3), singlet oxygen (1O2), hypochlorous acid (HOCl), nitrous acid (HNO2), peroxynitrite (ONOO–), dinitrogen trioxide (N2O3), and lipid peroxide (LOOH), are oxidants that can easily lead to free radical reactions in cellular systems [18]. Expanding knowledge of the mechanisms of ROS generation, intracellular sources, antioxidant defenses, and the chemistry of ROS-induced oxidative damage, was critical to improve understanding of the homeostasis between pro-oxidants and anti-oxidants, thereby evolving the free radical theory into the OST of aging [19], [20]. The “structural damage-based” hypothesis driving this theory is that age-associated functional losses are due to the accrual of oxidative damage to macromolecules such as lipids, DNA, and proteins by free radicals, and that the progressive oxidant/anti-oxidant imbalance leads to the consequent disruption of redox-regulated signaling mechanisms. Beckman and Ames divided this hypothesis into “strong” and “weak” versions: The strong version proposed that oxidative damage determines life span, while the weak version correlated oxidative damage to age-associated pathologies [21].
Fig. 1.

Schematic representation of the oxidative stress theory of aging. The key features of this hypothesis are that increase in oxidants and concomitant failure of antioxidant mechanisms cause structural damage to macromolecules which accumulates with age leading to corresponding decline or loss in function.
Although widely accepted, the OST has been severely refuted in recent years; most criticisms try to discredit the theory pointing at failure to increase longevity by increasing antioxidants and the production of fewer free radicals in certain long-living species [22], [23], [24], [25], [26]. Recently, a critical idea has emerged that ROS at non-toxic levels function as signaling molecules that induce protective defenses against age-dependent damage, and that peroxisomes may be implicated in the regulation of aging [27]. Currently, the two major traits correlating organismal aging and longevity to the OST of aging are considered to focus primarily on the species-specific low mitochondrial ROS generation rates at complex I of the electron transport chain (ETC) in long-lived animals, and low levels of fatty acid unsaturation on cellular and mitochondrial membranes [28], [29], [30].
2. Cellular senescence
Cellular senescence, also termed replicative senescence, was first proposed four decades ago [31], [32] and has since been proven to be a significant tumor constraining mechanism that halts the proliferation of primary mammalian cells after a finite number of population doublings [33], [34], [35], [36], [37] allowing the organism to antagonize the potentially detrimental effects of uncontrolled growth. Proliferation is checked by persistently arresting cell growth at the G1- [38], [39], [40], [41], [42], G2- [43], [44], [45], G2/M- [46] or S-phase of the cell cycle leading to failure of DNA replication. So far, senescence-associated growth arrest has been shown to depend functionally on the tumor- suppressor pathways controlled by p16INK4a and pRB (retinoblastoma protein), as well as by p53. Progressive telomere attrition due to persistent DNA damage responses [47] has been attributed to be one of the clocking mechanisms that keeps track of the number of times a cell divides, providing an appropriate trigger for the onset of telomere-induced senescence through the p53 pathway [34], [48], [49], [50], [51], [52]. ROS can induce a senescent growth arrest in vitro, by triggering the DNA damage response (DDR) pathway, with ATM or ATR kinases blocking progression of the cell cycle through stabilization of p53 and transcriptional activation of the cyclin-dependent kinase (CDK) inhibitor p21 [53]. However, in response to persistent DNA damage, p16INK4a is activated via p38-MAPK-mediated mitochondrial dysfunction and ROS production. Nuclear lamin B1 downregulation triggers modifications in chromatin methylation and induces senescence progression with the formation of highly condensed regions of chromatin called senescence-associated heterochromatin foci [54]. These foci are enriched in chromatin modifications that associate with genes required for maintenance of senescence-associated growth arrest. Senescence can also be induced by cell-type specific stress independent of telomere shortening, resulting in increased expression of the tumor suppressor p16, in which case it is termed stress-induced senescence [42], [55], [56], [57]. This includes chronic signaling by anti-proliferative cytokines that increase cellular oxidative stress and potentiate senescence [58], [59], [60]. Oncogene-induced senescence (OIS), characterized by aberrant DNA replication and the associated DNA damage response, is affected by activated oncogenes or the loss of tumor-suppressor genes in healthy cells [45], [61], [62], [63], [64] and upregulation of the CDK inhibitors p15INK4b, p16INK4a, and p21CIP1 [65]. Therapy-induced senescence (TIS) has been described in tumor cell lines in response to induction with selective preoperative neoadjuvant chemotherapeutics or radiation agents, and is considered crucial to mitigate toxicity-related side effects, thereby presenting a novel therapeutic target for cancer [66], [67].
3. Senescence-Associated Secretory Phenotype (SASP)
Cellular senescence is a significant tumor constraining mechanism that halts cellular proliferation in response to damage that occurs during replication. However, senescent cells acquire a deleterious, irreversible senescence-associated secretory phenotype (SASP) involving secretion of soluble factors (interleukins, chemokines, and growth factors), degradative enzymes like matrix metalloproteases (MMPs), and insoluble proteins/extracellular matrix (ECM) components [33], [68] that can alter tissue microenvironments and affect neighboring epithelial cells in a paracrine fashion to promote tumor progression [65], [69], [70]. Also termed the senescence-messaging secretome (SMS) [71], these factors promote shedding of membrane-associated proteins, degradation of signaling molecules, and/or ECM processing, thereby disrupting normal tissue framework and function [72]. Morphologically, senescence is characterized by cellular enlargement and flattening with an accompanying increase in cell volume, increased size of organelles including Golgi, lysosomes and nucleus, appearance of vacuoles in the cytoplasm and ER and an increase in the number of cytoplasmic microfilaments.
The senescent phenotype has been long associated with driving organismal aging, and has recently been linked to development and tissue repair. The SASP is primarily a persistent DDR coupled as a positive feedback loop with the senescent stimuli ROS, prompting telomere-dependent or –independent signaling mechanisms to facilitate growth arrest. The non-cell-autonomous activities mediated by SASP serve to deplete stem and progenitor cells and demonstrate that the heterogeneous functional networks of senescence are formed by multiple effector pathways involving highly dynamic signal amplification [73]. The SASP can also be uncoupled from the senescence-associated cell-cycle arrest, as it exemplifies a perpetual state of damage in comparison to growth arrest. SASP has been characterized by analysis and conservation of factors between human and mouse cells cultured under physiological oxygen conditions [74] and its occurrence has been seen in vivo in mice and humans [75], [76] and amongst diverse proliferative cell types (fibroblasts, epithelial cells, endothelial cells, astrocytes, etc.) [76], [77], [78].
The SASP components that modulate neighboring cells can lead to the activation of signaling cascades involved in several disease processes. It is sufficient for a given tissue to be comprised of less than 20% of senescent cells to exert SASP-mediated systemic effects [79]. The gamut of downstream effects initiated by SASP include tumorigenesis (paracrine), immunomodulation (paracrine), senescence amplification (paracrine and autocrine), transformation of the tissue microenvironment (paracrine), and perturbation of stem cell niche [80]. Continued exposure to senescent cells has been shown to potentially promote senescence in healthy bystander fibroblasts via gap junction-mediated cell-cell contacts and processes implicating ROS [81]. While a deluge of evidence points out to the role of ROS in inducing senescence, Lawless et al. employed an empirical stochastic step model of replicative senescence to suggest that increased mitochondrial ROS generation in senescent cells is a repercussion of SASP rather than the reverse [82].
The proteins of the SASP component are broadly conserved, although differences exist among cell types [76]. The contribution of non-protein factors such as nucleotides, bradykines, prostenoids, or ceramides to SASP's effects is also relatively unexplored. The composition of the SMS may vary dynamically with time after the initiation of senescence and partly depends on the senescence-inducing mechanism. This ‘late-induced’ senescence phenomenon propagates by forming cytoplasmic chromatin fragments with DNA lesions that eventually undergo lysosome-mediated proteolysis causing overall histone loss, thereby illustrating perpetual genomic and epigenomic remodeling in senescence [76].
3.1. Beneficial effects of SASP
The SASP is fundamentally a response to genomic damage that allows damaged cells to communicate their compromised state to neighboring cellsand stimulates tissue repair and/or regeneration after insult by attracting immune cells and inducing local inflammation. However, it has been indicated that senescence is not consistently followed by immune cell infiltration and inflammation [83] as observed in highly persistent senescent cells in human melanocytic nevi. The SASP also includes several chemokines and cytokines that activate immune cells to specifically target and eventually clear senescent cells from tissues [84], [85]. SASP factors such as IL-6, IL-8, protease inhibitor plasminogen activator inhibitor-1 (PAI-1), and pleiotropic protein insulin-like growth factor binding protein-7 (IGFBP-7) are also implicated in tumor suppressive growth arrest of cells [75], [86], [87]. The SASP-produced MMPs also limit fibrosis during wound healing [88] or following liver injury [89], [90]. Additionally, developmentally programmed and transiently-regulated senescence is established to be vital in promoting tissue remodeling, embryogenesis and patterning [91], [92], [93]. Finally, the deterioration of the immune system with age coupled with the kinetics and efficiency of senescent-cell clearance determines the switch of senescence from a temporal programmed process to a persistent stochastic response. The persistent stochastic response involves a multitude of concurrent signals that are paramount to the progression of chronic age-related pathologies.
3.2. SASP in disease
Cellular senescence and consequently SASP factors are involved in several acute and chronic pathological processes (Fig. 2). In a murine model, this association was supported by the delayed onset of age-related diseases when senescent cells were genetically ablated [94]. Considering that the SASP components differ in character from cytokines to growth factors and proteases the effects they can convey individually or more importantly collaboratively in different tissues may vary as well. Byun et al. have proposed that senescent cells have a definitive role in age-associated pathologies including their ability to modulate young cells through 1) release of inflammatory SASP factors i.e. IL-1; 2) secretion of autocrine or paracrine factors that modulate the activity of other senescent cells or disease-prone young cells (EMT activation by IL-6 and IL-8); 3) expression of SASP receptors (IL-17); and 4) release of anti-inflammatory molecules such as IL-10 [95]. The SASP can promote distinct disease pathologies by perpetuating a pro-inflammatory state and promoting cell transdifferentiation. For example, various cardiovascular risk factors including obesity, hypertension, diabetes and atherosclerosis that compromise metabolism are associated with an inflammatory state and increased cellular senescence and SASP [96]. Vascular calcification, another risk factor for cardiovascular disease, is linked to a SASP-driven osteoblastic transdifferentiation of senescent smooth muscles cells [97], while atherogenesis has been shown to be in part mediated by the pro-inflammatory IL-1α [98]. Obese individuals with type-2 diabetes show increased levels of SASP factors IL-6, IL-8, and the chemokine MCP-1 [96]. Moreover, both IL-6 and IL-1β are independent predictors of diabetes [96]. In many prevalent neurodegenerative conditions including Alzheimer's Disease, brain tissue biopsies are present with increased levels of p16INK4a- positive astrocytes, MMP-1 and IL-6 [99], [100]. Other prevalent pathologies which share several damaging SASP profiles including IL-6, IL-8, GM-CSF, MMP-1 [101] are chronic obstructive pulmonary disease (COPD), biliary cirrhosis and cholangitis [102], and osteoarthritis [103].
Fig. 2.

Schematic representation of diseases propagated by age. Cellular senescence and the associated secretory phenotype have been implicated in initiation and progress of several acute and chronic pathological maladies driven by age-associated inflammation (inflammaging) and oxidative stress-mediated damage. AMD – Age-related macular degeneration; COPD – Chronic obstructive pulmonary disease).
Although cellular senescence can serve as a tumorigenic restrictive mechanism, it can also promote tumor growth and tumor invasiveness, through SASP-mediated regulatory effects [104], [105]. A known pathway for SASP-promoted cancer metastasis is the induction of epithelial to mesenchymal transition (EMT); a process that can have both beneficial wound healing effects, and pathological fibrotic and invasiveness-promoting ones [106]. EMT is also a crucial process that can elicit fibrosis in Crohn's disease and graft loss after kidney transplantation [107], [108]. Moreover, decreased renal function due to both acute and chronic kidney injury has also been associated with cellular senescence and SASP [109], [110].
Though the SASP signature of specific pathological processes still remains to be elucidated, great attention is increasingly being paid to the effects of both biological and stress-induced senescence in different tissues. Both the beneficial and deleterious effects of SASP suggest a possible use of the implicated factors as therapeutic targets for disease control. In addition, the oxidative stress-mediated regulation of biological aging and age-associated pathologies has become a promising target for therapeutic strategies against age-associated maladies. Here, we review the recent progress in understanding the role of the oxidative stress and the cellular redox environment in senescence, with a focus on redox-control of senescence-regulatory factors. The effects of oxidative stress on cellular senescence at a cellular and nuclear level are depicted in Fig. 3, Fig. 4 respectively (Fig. 3, Fig. 4).
Fig. 3.

Schematic representation of the redox-control of pro-aging mechanisms. Oxidative stress-mediated regulation of several factors and mechanistic pathways plays a critical role in senescence regulation. Several of these components can interact with each other to induce effects of the senescent phenotype. AP‐1 – Activator protein 1; Ca2+– Calcium; FOXO – Forkhead box O; IGF-1 – Insulin-like growth factor-1; IL – Interleukin; MMP – Matrix metalloproteinase; mTORC – Mammalian target of rapamycin complex; NF-κB – Nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K – Phosphoinositide 3-kinase; Pot-1 – Protection of telomeres protein 1; pRb – Retinoblastoma protein; ROS – Reactive oxygen species; SIRT – Sirtuin; SOD2 – Superoxide dismutase 2, mitochondrial; Trf – Telomeric repeat binding factor.
Fig. 4.

Schematic representation of the redox-control of pro-aging mechanisms in the nucleus. Oxidative stress-mediated regulation of nuclear proteins can happen via expression of factors induced by DNA damage-mediated responses or at the transcriptional/translational level via modulation of protein activity, thereby allowing for precise control over senescence effector mechanisms. AP-1 – Activator protein 1; FOXO – Forkhead box O; IL – Interleukin; NF-κB – Nuclear factor kappa-light-chain-enhancer of activated B cells; Pot-1 – Protection of telomeres protein 1; pRb – Retinoblastoma protein; SIRT – Sirtuin; Trf – Telomeric repeat binding factor.
4. Redox Control of SASP and Age-Related Pathologies
4.1. Mammalian target of rapamycin pathway
The mammalian target of rapamycin (mTOR) protein is a highly conserved large protein kinase of the phosphatidylinosital 3 kinase-related kinase (PI3KK) family, and an intracellular target of rapamycin, a pharmacological immunosuppressant approved by FDA (Food and Drug Administration, USA) in therapies against various types of cancers [111], [112]. The conserved TOR complexes, mTORC1 and mTORC2, can be differentiated by their distinct associated proteins, Raptor and Rictor, respectively. mTOR complexes play crucial roles in mediating nutrient signaling, autophagy and growth regulation (mTORC1) and also regulate cell survival and spatial organization of the cytoskeleton (mTOR2) [113], [114].
Inhibition of mTORC1 by rapamycin may both counter damage-inducing senescence mechanisms and enhance repair pathways, and as a consequence, result in extended lifespan in model organisms [115]. Over the past decade, the impact of mTOR signaling on cellular aging has gained traction with several studies performed in multiple cellular systems that argue for a pivotal role of mTOR in the senescent program [112], [116]; however, little is known with respect to how cellular redox state contributes to mTOR regulatory function in senescence.
Cellular stresses, such as hypoxia, or low energy status, which can be sensed by the AMP-sensitive kinase (AMPK) inhibit mTOR signaling [111]. Conversely, elevated mTOR activity in response to excess nutrients leads to increased oxidative stress by enhancing mitochondrial oxygen consumption. Cysteine oxidation enhances phosphorylation of S6K, which is a direct readout of mTORC1 activation [117]. Conversely, treatment with reducing agents effectively suppresses mTORC1 activation via inhibition of S6K phosphorylation. Furthermore, oxidants destabilize the mTOR-Raptor interaction but not the mTOR-Rictor interaction suggesting that a redox-sensitive mechanism may underlie amino acid-dependent mTORC1, but not mTORC2 regulation [118]. These studies also indicate that the phosphorylation of tuberous sclerosis complex (TSC2) tumor suppressor protein regulates Rheb GTPase activity which plays an essential role in mTORC1 regulation in response to cellular redox potential. GRp58 (also known as ERp57), a widely expressed protein disulfide isomerase regulating protein-protein interactions through a redox-based mechanism acts via its two thioredoxin-like domains and has shown to be an mTOR-interacting protein [119]. These findings suggest that a redox-sensitive mechanism regulates the activity of mTORC1, and the interaction between Raptor and proteins with redox-sensing abilities are potentially involved in the assembly and regulation of mTOR complexes. In contrast, Liu et al. demonstrated that mTOR regulation under hypoxic conditions is independent of cellular redox change [120] as H2O2-scavenging by catalase failed to attenuate the hypoxic hypo-phosphorylation of p70S6K and 4EBP1. More recently, regulation of the mTORC2 complex formation and stability has been demonstrated to be redox sensitive [121], as knockdown of p22phox inhibits NADPH-dependent superoxide generation and decreases downstream Rictor-associated mTORC2 complex activity without affecting Raptor-associated mTORC1 complex. This likely occurs through redox-control of Rictor expression and its subsequent interaction with mTORC2. mTORC1 also regulates mRNA translation and protein synthesis under conditions that favor growth, and there is evidence to suggest that regulation of mRNA translation can modulate longevity in several model organisms [122]. The cellular redox status may likely play a role in global reduction in mRNA translation through TOR signaling and would be advantageous to the aging process allowing for better maintenance of protein homeostasis. Taken together, these observations indicate that approaches that exploit the redox-sensitive nature of both mTOR complexes might prove useful in the design of therapeutics that target age-associated disorders.
4.2. IL-1alpha
Pro-inflammatory members of the interleukin-1 (IL-1) family of cytokines (IL-1α and β) are important mediators of the host defense response to infection, but can also aggravate inflammation that is central to the progression of many disease processes [123]. IL-1α, one of the upstream regulators of SASP, is a dual-function cytokine which is rapidly expressed not only upon stimulation but is present constitutively in healthy cells. It is synthesized as a precursor protein that is subsequently cleaved into two functional fragments that have distinct subcellular localizations by proteases such as calpain [124], CTL/NK-granzyme-B, mast cell chymase, or neutrophil elastase [125]. However, IL-1α cleavage by calpain is rare and only serves to increase the potency of the mature form of the protein in activating pro-inflammatory mediators of SASP.
Basal and tumor necrosis factor (TNF)-induced expression of IL-1α has been shown to be regulated by superoxide via modulation of mitochondrial superoxide dismutase (SOD2) that in turn influences the cellular redox homeostasis [126]. Their findings indicate a critical role for a superoxide-reactive intermediate of mitochondrial origin in regulating IL-1α mRNA stability both constitutively and in response to TNF. The IL‐1α proximal promoter contains binding sites for the transcription factor AP-1 [127], that has previously been established to be activated by an H2O2-dependent mechanism [128], again suggesting the redox-dependency of IL-1α induction. In exploration of the mechanistic drivers of IL-1α, redox-engineered fibrosarcoma cells were used to demonstrate that the cellular redox state mediates IL-1α expression and processing through the Ca2+ dependent protease calpain and alterations in calcium (Ca2+) homeostasis [129]. Steady state levels of intracellular hydrogen peroxide (H2O2) significantly promoted IL-1α expression and nuclear translocation, where it serves as a pro-inflammatory mediator by increasing NF-κB activity, thereby further delineating the role of IL-1α in ROS-mediated inflammation. Similarly, in serially cultured senescent human fetal lung fibroblasts, the redox-dependent processing of IL-1α by calpain has been shown to be critical to the regulation of SASP [70]. Senescence-associated upregulation of the mature form of IL-1α promoted metastatic progression of neighboring healthy epithelial cells, triggering an epithelial-mesenchymal transition and was reversed by antioxidant treatment, implying a redox-driven control mechanism. More recently, Laberge et al. have shown that mTOR drives the SASP and that treatment with the mTOR inhibitor rapamycin partly suppresses the secretory phenotype and tumor-promoting ability via translational inhibition of IL-1α [130]. Despite its role in the regulation and amplification of SASP, there exist few studies that describe aberrant activation mechanisms for IL-1α, thus understanding these mechanistic controls is necessary to allow successful therapeutic intervention to reduce the deleterious effects of the normal aging process.
4.3. Matrix metalloproteinase regulation
Matrix metalloproteinases (MMPs) are a group of endopeptidases with important roles in wound healing, tissue remodeling and cellular growth [131], [132]. Abnormally high MMP expression is associated with age-related and chronic diseases such as cancer, Alzheimer's, atherosclerosis, osteoarthritis, and lung emphysema [132]. The mitochondrial redox control of MMPs has been previously reported, where ROS are shown as key regulators of MMPs including MMP-1, −2, −7, and −9, [133], [134], [135] while also being upregulated by high levels of MMP-2 [136]. In particular, the association between MMP-1 and ROS and the significance of these interactions in degenerative disease etiology is well established. MMP-1, cleaves the structural connective tissue components collagen type I, II and II, and is detected only at low levels under physiological conditions, but can be abnormally high in several age-associated pathologies [132], [137]. The increased expression of MMP-1 can be both age- and ROS-dependent [128], [138], [132]. ROS and age associated MMP-1 expression involves the recruitment of the primary transcription factors Ets-1/AP-1 (c-Jun/c-Fos) to the distal promoter region of MMP-1, along with the differential recruitment of chromatin-modifying proteins P/CAF and HDAC2. These coordinated events mediated by ROS are responsible for induction of an active MMP-1 promoter complex. Multiple studies also show that while oxidants such as H2O2, and antioxidant inhibitors such as aminotriazole can enhance MMP expression [139], treatment with antioxidant agents such as catalase, Vitamin A, Vitamin E, trans retinoic acid, resveratrol, and N-acetyl cysteine can block or decrease MMP expression in different cell lines [140], [141], [142], [143], [144], [145], [146]. More recently, ROS-activated MMPs in the wall of cerebral vessels has also been shown to be exacerbated with age in a mouse model that recapitulates cerebromicrovascular alterations present in elderly humans [147], adding to the growing body of literature supporting age-associated redox-driven MMP induction. This redox sensitive regulation of MMPs provides a definitive rationale for the use of antioxidant-based therapies in the treatment of degenerative disorders associated with aberrant matrix destruction propagated by age.
4.4. IGF Signaling and FOXO regulation
The insulin/insulin-like growth factor-1 (IGF-1) signal transduction pathway controls various proliferative, survival and metabolic signaling networks. IGF-1 in association with its downstream effectors was the first evolutionarily conserved longevity regulatory network identified from yeast to humans [148], [149]. Impaired insulin/IGF-1 signaling lowers the oxidative burden and the associated oxidative damage in cells which contributes to increased longevity [150], [151], [152].
Forkhead box O (FOXO) transcription factor family members (FOXO1, FOXO3a, FOXO4, and FOXO6 in mammals; DAF-16 in C. elegans; DFOXO in D. melanogaster) are evolutionarily conserved AKT substrates that regulate genes involved in cell cycle, apoptosis, DDR, metabolism, oxidative stress defense mechanisms, and aging [153]. The activity of FOXO proteins is primarily regulated by signaling in response to IGF receptor engagement in a redox-dependent fashion [154]. FOXO family members also confer protection from oxidative stress through transcription of antioxidant genes, SOD2, catalase, peroxiredoxin 3, and members of the sestrin family [155], [156], [157]. The ROS-induced formation of cysteine-thiol disulfide-dependent complexes of FOXO with the p300/CBP (CREB-binding protein) acetyltransferase has also been implicated in the modulation of biological activity of FOXO4 [158]. Overall, FOXOs serve as stress sensors and appear to influence both pro- (inducing senescence and apoptosis) and anti-aging (ROS scavenging) mechanisms.
Senescent endothelial, epithelial, and fibroblast cells demonstrate high expression levels of IGF-binding proteins (IGFBPs) including IGFBP-2, −3, −4, −5, and −6 [72], [159], [160], [161], and their regulating proteins (IGFBP-rPs) IGFBP-rP1, and 2 [162], [163]. IGFBP-rP1 promotes senescence in part through p21 and independent of p53 [164]. Recently, IGFBP7, a potential tumor suppressor, has been identified as a marker of oncogenic BRAF-induced senescence, via negative feedback loops [87], [165]. The initial response to the insulin receptor activation is the NADPH oxidase-dependent generation of H2O2 [166] that plays both positive and negative autoregulatory roles in the regulation of insulin/IGF-1 signaling. The endogenous production of H2O2 is implicated in the autophosphorylation of the insulin receptor β-subunit (IRβ) tyrosine motif [167], [168]·H2O2-dependent priming of IRβ and associated proteins, coupled with reversible oxidative inhibition of protein tyrosine phosphatases, enables IRβ phosphorylation and sustains insulin/IGF-1 signaling.
The insulin/IGF-1 pathway turns from a growth-promoting regulator in early stages and into a senescence-inducing pathway during the later stages of life [169], [170]. A detailed characterization of the regulatory role of ROS in insulin/IGF-1-mediated signaling at all stages in life is crucial to design therapeutics targeting age-related disorders.
4.5. Calcium (Ca2+)
Calcium is a ubiquitous secondary signaling molecule regulating a disparate range of physiological process, including muscle contraction, gene transcription, and cell proliferation [171]. Comprehensive reviews by Ureshino et al., and Farfariello et al. summarize the Ca2+ signaling networks that may mediate senescence via multiple regulatory pathways including proliferation and cell cycle arrest, apoptosis, autophagy, alterations in membrane potential, altered telomerase activity, and SASP [172], [173]. The role of Ca2+ as a key signaling intermediate involved in SASP regulation has only recently emerged [70].
Ca2+ signaling is central to driving morphological changes that are the hallmarks of senescence. Senescence-associated increase in cell volume is normally countered by regulatory volume decrease (RVD) which preserves the structural integrity of the cell [174]. Senescence-associated cell volume changes lead to membrane stretching or osmotic swelling-based activation of TRP vanilloid 4 (TRPV4) or TRP melastatin 7 (TRPM7) channels that mediate Ca2+ influx from the extracellular space, subsequently activating RVD counter-regulatory signals [175], [176]. Mitochondrial Ca2+ importers maintain homeostatic regulation of the electron transport and increased Ca2+ influx decreases overall ATP production, elevating cytosolic NADH, decreasing sirtuin activity leading to senescence [177]. Similarily, recent work by Wiel et al., identified two calcium channels, inositol 1,4,5-trisphosphate receptor, type 2 (ITPR2) localized in the ER and the mitochondrial calcium uniporter (MCU), as novel regulators of senescence program by increasing Ca2+ uptake [178].
In aged human fetal lung fibroblasts, senescence-associated increases in basal intracellular Ca2+ is implicated in downstream activation of the Ca2+-dependent protease calpain, and the subsequent regulation of IL-1α [70]. Treatment of senescent fibroblasts with BAPTA-AM, a Ca2+chelator or calpain inhibition reduces the senescence-associated expression of IL-8 and IL-6, respectively. These observations suggest there exists both a redox and Ca2+-dependent process in the regulation of the prominent SASP factors IL-6 and IL-8. During OIS and replicative senescence, ITPR2 triggers Ca2+ release from the endoplasmic reticulum (ER), with subsequent mitochondrial Ca2+ accumulation via MCU channel, leading to reduced mitochondrial membrane potential, ROS accumulation, and senescence. Conversely, intracellular Ca2+ reservoirs in the mitochondria are released in response to stress, and facilitate activation of cAMP-responsive element binding protein 1 (CREB) that upregulates p21CIP1/WAF1 expression, thereby inhibiting cell proliferation [179]. Sharov et al. have shown that age-dependent loss in approximately 2.8 mol of cysteine /mol of sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA) as a result of oxidation, may in part be responsible for the age-dependent decline in SERCA activity by about 40% [180]. In a similar model employing young (5 month old) and aged (21 month old) transgenic mice, myocyte‐specific catalase overexpression limited oxidative modification of SERCA and preserved SR Ca2+, SERCA activity and myocyte relaxation [181]. The ability of catalase overexpression to preserve SERCA activity strongly suggests that the post-translational modifications of SERCA in the senescent heart are H2O2-dependent. Though the role of Ca2+ dysregulation in physiological and organ-specific aging processes has been explored, further work is needed to decipher the connection between Ca2+ dynamics and oxidative damage-induced senescence.
4.6. Sirtuins
Silent information regulator (Sir2) genes and their functional orthologs are conserved from bacteria to humans and constitute a family of NAD+-dependent protein histone deacetylases (HDACs) called sirtuins (SIRT). Initially described as gene silencers, sirtuins are implicated in a wide variety of physiological processes including apoptosis, lipid metabolism, cellular response to stress, mitochondrial biogenesis, fatty acid oxidation, insulin production, and inflammation. SIRT regulates the lifespan-extending effects of caloric restriction improved glucose tolerance, inhibit of degenerative disorders, improve endothelial function, promote regression of atherosclerotic plaques and prevent cancer [182]. The sirtuins class III family is comprised of seven human sirtuins (SIRT1-7) that exhibit deacetylation, demyristoylase, ADP-ribosylation, lipoamidase, demalonylation, desuccinylation, or deglutarylation activity depending on the substrate. SIRT 1–7 are localized in various sub-cellular compartments in mammalian cells: SIRT1, 6, and 7 are localized mainly in the nucleus [183], [184], [185], SIRT2 in the cytoplasm [186], SIRT3, 4, and 5 are active in the mitochondria [187]. As sirtuin activity requires NAD+ as a cofactor, cellular redox status-based NAD+/NADH ratio can further affect sirtuins [188] at the transcriptional or post-transcriptional level of activity [189], [190]. Recent work has established a role for some sirtuins in cellular senescence.
SIRT1 is the most widely studied and well-characterized sirtuin family member and is involved in the regulation of DNA repair and apoptosis, mitochondrial biogenesis, glucose and insulin homeostasis, and cell stress responses. However, our knowledge pertaining to the molecular redox control of SIRT1 is limited. A pro-oxidative shift in the cellular environment has been shown to induce SIRT1 expression in neural progenitors both in vitro and in vivo [191]. Additionally, the SIRT1 promoter is directly trans-activated by hypoxia-inducible factor (HIF) transcription factors in response to a hypoxic environment [192]. SIRT1-mediated protection against oxidative stress-induced cellular damage involves induction of SOD2 expression via deacetylation and modulation of FOXOs, p53, p21, and proteins involved in DNA damage and repair in distinct cell types [193], [194], [195]. This fits well with the observation that oxidative stress promotes acetylation of p53 by removing the inhibitory effect of SIRT1 on p53, activating the downstream target p21, and inducing premature senescence followed by pro-tumorigenic IL-6 overexpression [196]. A similar trend is observed in the interaction of SIRT1 with RelA/p65 protein in the NF-κB complex which deacetylates lysine 310 and potentiates the transactivation capacity of the NF-κB complex [197]. Exposure to cigarette smoke oxidatively decreases SIRT1 levels and concomitantly increases NF-κB-dependent release of pro-inflammatory mediators in MonoMac6 cells and in mouse lungs [198], [199]. The role of SIRT1-FOXO3 axis in cellular adaptation to hypoxia-associated oxidative stress has also been elucidated by Kume et al. who demonstrate that hypoxia inhibits SIRT1 activity and prevents nuclear translocation of FOXO3, subsequent autophagy, and cell cycle arrest leading to an increase in mitochondrial oxidative damage in kidneys of aged mice [200]. Interestingly, there are ambiguities in the effect of SIRT1 on FOXO activity with variations according to gene or function; SIRT increases transcription of genes involved in cell cycle arrest and resistance to oxidative stress and represses genes promoting FOXO-dependent cell death [201]. Similar to the studies described above, ionizing radiation-induced ROS also promotes cellular senescence in articular chondrocytes by negative post-translational regulation of SIRT1 through the ROS-dependent activation of p38 [202]. In contrast, SIRT1 has been implicated in promoting oxidative stress through IRS-2/Ras/ERK signaling downstream of insulin/IGF-1 receptors [203]. There also is a strong link between the insulin/IGF-1 longevity pathway and SIRT1 activity in senescence. The IGF-1/SIRT1/p53 interplay involves prolonged IGF-1 treatment which inhibits SIRT1 deacetylase activity, resulting in increased p53 acetylation, stabilization, and activation leading to premature cellular senescence [204]. Ectopic expression of SIRT1 effectively abrogates IGF-1-induced cellular senescence, thereby linking IGF-1-SIRT1-p53 to cellular senescence. Recently, SIRT1 has also been shown to dissociate chromatin structure and epigenetically repress the expression of SASP factors IL-6 and IL-8 [205]. However, oxidative stress or DNA damage-mediated can dissociate SIRT1 from the promoter regions of IL-6 and IL-8 increasing their levels systemically and further expanding senescent cell populations. The RNA-binding protein HuR has been shown to stabilize SIRT1 mRNA by binding its 3′ untranslated region in a manner that is inhibited by redox-dependent post-translational modifications. Overall, alterations in SIRT1 activity in response to oxidative stress appear to promote genomic instability, enhance inflammation and senescence and offers an additional target for therapeutics that limit the process of senescence.
SIRT3 acts as a tumor suppressor gene, owing to its deacetylation role which enhances the activity of two significant targets, SOD2 and isocitrate dehydrogenase 2 (IDH2), the latter of which generates NADPH for the glutathione pathway of ROS detoxification [206], [207], [208]. SIRT3 is also essential for the caloric restriction-mediated mitigation of oxidative damage by regulating the glutathione antioxidant system, thereby regulating senescence-associated pathologies propagated by oxidative stress [209]. Lack of SIRT3 leads to activation of Akt signaling suggesting a potential role in increasing cellular ROS levels and as a driver of senescence, by affecting mitochondrial function and ATP generation, ROS detoxification, and the mitochondrial unfolded protein response (UPR). Both SIRT3 transcript and protein levels are induced by the generation of mitochondrial ROS [210]. In mice fed with high fat diets for a prolonged 12-week period, evidence of hepatic redox stress, and an associated reduction in SIRT3 transcript and protein levels has been established [211]. SIRT3 is involved in cancer progression, neurodegenerative and cardiovascular disease as well as metabolic syndromes [212], and is likely a critical participant in many age-related disease processes.
SIRT6 is another key member of the sirtuin family with varied functions in DNA damage repair, maintenance of genomic stability, inflammation, tumor suppression and participates in longevity regulation [213]. The promise of employing targeting strategies against SIRT6 for limiting age-associated disease processes by impeding cellular senescence and delaying premature aging [214] or through apoptosis engagement has been established [215]. Recently, Pan et al. demonstrated that SIRT6 deficiency in human adult stem cells promotes premature and accelerated cellular senescence and is accompanied by elevated ROS, disrupted redox homeostasis, and increased sensitivity to oxidative stress. SIRT6 complexes with and transcriptionally coactivates nuclear factor erythroid 2-related factor 2 (NRF2), a critical regulator of antioxidant responses, thereby modulating stem cell redox homeostasis, providing a unique target for constraining this senescence-associated oxidative-stress based stem cell attrition [216]. The best-known actions of SIRT6 as a tumor-suppressing protein include its inhibitory effects on c-Jun by blocking IGF-Akt signaling, and Myc, while suppression of chronic inflammation is achieved by repressing TNF-α and NF-κB activity [217], thus enabling varying levels of senescence enforcement.
4.7. Telomere maintenance
The ends of linear eukaryotic chromosomes are protected from damage by the inclusion of tandemly repeated DNA sequences and specialized proteins, called telomeres. In mammalian species, the telomeric sequence TTAGGG extends in a 5’ to 3’ direction with a T-loop structure at the end comprised of a G-rich 3’ overhang and associated proteins that offers telomeric protection. The repeats and the bound proteins form a complex structure called sheltrin, that camouflage chromosomal ends from being recognized as damaged subsequently preventing DNA end-joining, recombination, and repair processes. Over the course of cell division, chromosomal attrition occurs, as the 5’ to 3’ nature of the DNA replication machinery results in incomplete replication of the lagging parent strand. In eukaryotes, this is resolved by the DNA polymerase enzyme telomerase, which adds telomeric repeat sequences to the ends of chromosomes de novo and compensates for their erosion. Telomere-associated proteins may also localize to other sub-nuclear or sub-cellular sites, and interact with other associated proteins to regulate telomere length [218].
Senescence is usually triggered when the terminal restriction fragment of telomeres shorten from an average length of 15–20 kb to 4–7 kb, by contributing to a persistent DDR that propagates and maintains senescence-associated proliferation arrest, with dysfunctional telomeres, and damaged nuclear foci termed DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) [219]. While telomere attrition has mostly been attributed to senescence induction, certain studies suggest that DNA damage, and the ensuing damage to telomere structure may be sufficient to trigger senescence independent of telomere shortening [220], [221]. Also, the complex molecular T-loop structure at the tips of telomeres that are modulated differently in different cell populations, may also actively determine the cellular proliferative capacity, independent of telomere length.
Comprehensive evidence establishes the theory that progressive telomere loss contributes to replicative cellular senescence and is an indicator of age-related disease. Telomere maintenance is regulated by genetic factors and is additively determined by telomerase activity, cell turnover rate, and stress-inducing non-genetic environmental elements. The G-rich composition of telomeres renders them highly susceptible to oxidative damage with the formation of oxidative damage lesions 8-oxo-2'-deoxyguanosine (8-oxo dG) [47], [222] which can induce telomere uncapping in a cell cycle-dependent manner by titrating out essential factors such as protection of telomeres protein 1 (POT1). POT1 protects against cellular senescence by promoting telomere elongation, 3′ overhang protection and inhibits chromosomal aberrations [223]. Telomeric DNA is defective in the repair of oxidant-induced single-strand breaks, therefore telomere shortening resulting from unrepaired or residual oxidative damage determines the occurrence of mutations, and unrepaired nucleotides that interfere with the replication fork [53]. This persistent stress-induced telomeric damage is characteristically independent of telomerase activity and telomere length. The repair deficiency in telomeres can be attributed to the binding of telomeric repeat binding factor 2 (TRF2) that makes the strand breaks inaccessible to DNA repair enzymes [224], inhibits ATM kinase phosphorylation [225], and interacts with polymerase β [226], thereby negatively impacting DNA repair. This also suggests that telomere-driven senescence occurs as a potential tumor-suppression mechanism in response to accumulating genomic damage, and not after a set number of cellular divisions as clocked by telomeres. Mechanistically, telomeric foci contain multiple DDR factors signaling through ATM to p53, up-regulating p21 and causing cell cycle arrest at the G1 phase. Telomere- and DNA damage-independent up-regulation of p16 was also observed, suggesting that distinctive senescence effector pathways are not mutually exclusive and can develop in parallel, resulting in a mixed cell population, or an individual cell responding to collective signals [42].
Recently, analysis of 3D chromosomal architecture using combined biochemical, cytological, and computational techniques has revealed significant decreases in volume, and increases in chromosome compaction during cellular senescence [227]. This unique senescent chromosomal signature is attributed to an increase in short-range chromatin contacts and genome-wide contraction of chromosome arms. It is hypothesized that the structural 3D organization of chromosomes may regulate senescence-associated cell cycle arrest. Mathematical models have been employed to quantitatively predict the role of telomere shortening in senescence. One such model assessed rates of telomere loss occurring from oxidative damage and activity of C-strand specific exonuclease, and the possible interactions between these two mechanisms [228]. The model predicts that under lower oxidative burden the end-replication problem and C-strand processing are major contributors to telomere loss; however under normoxic or hyperoxic conditions, it is predicted that single strand breaks induce shortening. Another model incorporates a shortening factor and autonomous telomere loss and assumes that senescence incidence is proportional to telomere length [229]. A simple negative feedback regulation model of telomere reduction is proposed, suggesting that telomere erosion is dependent on telomere length.
Ectopic expression of telomerase alone [230], or in combination with p16INK4A/pRb inactivation [231] can also prevent telomeric loss-dependent senescence in primary fibroblasts and extend replicative lifespan. Likewise, studies suggests that loss of telomeres correlates with depletion of antioxidant enzyme levels and decreases in oxidative burden by antioxidant treatment might limit the telomere shortening rates and delay cellular senescence. However, a critical review and meta-analysis on human data performed by Simons reveals no causal involvement of telomere length in the aging process, suggesting that thorough investigation of the mechanistic in vivo role of telomeres is crucial to assess their function in senescence [232].
4.8. Tumor suppressors p53, p21, p16
The G1 phase of the cell cycle is the main checkpoint at which DDR pathways activate cellular senescence as a protective mechanism to induce cell cycle arrest. [38], [233]. Both the p16INK4a/pRB and p53/p21 tumor suppressor pathways induce cell growth arrest through senescence activation [234], [235]. Both p16 and p53 display compensatory interactions [236], [237] and both have a role in senescence initiation and maintenance [79], [238], [239].
p21 and p16 are cyclin-dependent kinase inhibitors involved in preventing cell cycle progression by preventing pRB phosphorylation [37], [240]. In general terms, DNA damage activates DDR, a p53/21-dependent response that is originally transient but which can become persistent if the damage is severe enough, and which can also be reversed through the p16/pRB pathways [219], [241]. After DDR activation, the p53/p21 pathway deactivates the cyclin E-CDK2 complex responsible for pRB phosphorylation [242], while the p16/pRB pathway inhibits pRB phosphorylation by deactivation of kinase CDK4/6 [239]. Preventing pRB phosphorylation allows pRB to retain its association with the transcription factor E2F1, preventing the expression of E2F1 target genes that are necessary for the cell transition from G1 to the S phase [239].
p53 has been shown to both promote and inhibit cellular senescence [243], [244] partly through its up and down regulation of ROS, and its effects on mTOR. Also, a positive feedback loop between p53 and ROS has been shown to promote senescence in human fibroblasts – a loop that can be counteracted by the cell cycle mediator cullin-4B (CUL4B) via p53 ubiquitination in stressed cells [245]. Moreover, ROS promotes p53 acetylation and induction of premature senescence by inhibition of the deacetylase SIRT1 in fibroblasts [246]. In epithelial cancer cells, the transcription factor FOXp3 accelerates p53-mediated senescence while also increasing ROS levels and p21 expression, demonstrating the intricate link between ROS and these pathways [247]. It has also been suggested that ROS can induce senescence through activation of the p16INK4a/pRB pathway by activating p38 MAPK and extracellular regulated protein kinase (Erk) [248], although in extrinsically induced senescence models, the same pathway was engaged [249].
While there is evidence of the regulatory role of ROS in the p16INK4a/pRB and p53/p21 pathways that lead to senescence and certain pathologies, much remains to be elucidated to fully understand the implication of oxidative stress as both an effector and affected agent in cellular senescence.
5. Summary
The complexity of the aging process and the drive to comprehend its molecular underpinnings, steers focus towards approaches to improve organismal healthspan and lifespan. Despite the transition from cell-culture curiosity to a potential regulator of cancer and aging, cellular senescence remains enigmatic and continues to raise a variety of complex of questions. Oxidative stress plays a major role in several pathways that lead to cellular senescence and the associated secretory phenotype. Although initially intended to be a protective response to oncogenic insult, cellular senescence induced by DNA-damaging agents and ROS, can potentially elicit harmful effects and can drive many age-related diseases. While the ROS-driven SASP has proven to be beneficial in certain conditions, it is also associated with perpetuating a pathological inflammatory state. The ability to genetically ablate senescent cells has clearly established a causal role for senescence in many degenerative disease processes, and while there is much unknown with respect to the role of oxidative stress in such processes, the evidence compiled here suggests that precise ROS-mitigation may limit senescence and senescence-associated pathologies. However, translation of these findings into relevant human interventions is at present restricted due to our incomplete understanding of both the basic molecular biology of senescent cells in vivo and the etiology of senescence-associated pathologies. While the long-standing notions of the free radical theory of aging postulating a causal role of ROS in the aging process (Harman, 1956) seem to fit with several studies, there are a number of observations that suggest that the cellular effects of ROS, with regard to inducing senescence, do not unequivocally translate into organismal aging. Therefore, efforts should be made to escalate research directed at resolving the basic biology of cellular aging and the redox-control of these mechanisms to generate integrated therapeutic strategies to improve human health and lifespan.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Kirkwood T.B.L. Understanding the odd science of aging. Cell. 2005;120(4):437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 2.Ljubuncic P., Reznick A.Z. The evolutionary theories of aging revisited - a mini-review. Gerontology. 2009;55(2):205–216. doi: 10.1159/000200772. [DOI] [PubMed] [Google Scholar]
- 3.Jin K. Modern biological theories of aging. Aging Dis. 2010;1(2):72–74. [PMC free article] [PubMed] [Google Scholar]
- 4.Lipsky M.S., King M. Vol. 61. Elsevier; 2015. pp. 460–466. (Biological Theories of Aging. Disease-a-Month). [DOI] [PubMed] [Google Scholar]
- 5.Peng C., Wang X., Chen J., Jiao R., Wang L., Li Y.M. Biology of ageing and role of dietary antioxidants. Biomed. Res Int. 2014;2014:831841. doi: 10.1155/2014/831841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medawar P.B. An unsolved problem of biology. Evol. Health Dis. 1952:24. [Google Scholar]
- 7.Williams G.C. Pleiotropy, natural selection, and the evolution of senescence. Sci. Aging Knowl. Environ. 2001;2001(1):cp13. [Google Scholar]
- 8.Kirkwood T.B.L. R. Holiday FRS. The evolution of aging and longevity. Proc. R. Soc. L B Biol. Sci. 1979;205:531–546. doi: 10.1098/rspb.1979.0083. [DOI] [PubMed] [Google Scholar]
- 9.Kirkwood T.B.L., Austad S.N. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 10.Brys K., Vanfleteren J.R., Braeckman B.P. Testing the rate-of-living/oxidative damage theory of aging in the nematode model Caenorhabditis elegans. Exp. Gerontol. 2007;42(9):845–851. doi: 10.1016/j.exger.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Colman R.J., Anderson R.M., Johnson S.C., Kastman E.K., Kosmatka K.J., Beasley T.M. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barzilai N., Huffman D.M., Muzumdar R.H., Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61(6):1315–1322. doi: 10.2337/db11-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finkel T. The metabolic regulation of aging. Nat. Med. 2015;21(12):1416–1423. doi: 10.1038/nm.3998. [DOI] [PubMed] [Google Scholar]
- 14.Bjorksten J., Tenhu H. The crosslinking theory of aging - added evidence. Exp. Gerontol. 1990;25(2):91–95. doi: 10.1016/0531-5565(90)90039-5. [DOI] [PubMed] [Google Scholar]
- 15.Gerschman R., Gilbert D.L., Nye S.W., Dwyer P., Fenn W.O. Oxygen poisoning and x-irradiation: a mechanism in common. Science. 1954;119(3097):623–626. doi: 10.1126/science.119.3097.623. [DOI] [PubMed] [Google Scholar]
- 16.Gerschman R., Gilbert D.L., Nye S.W., Fenn W.O. Influence of x-irradiation on oxygen poisoning in mice. Proc. Soc. Exp. Biol. Med. 1954;86(1):27–29. doi: 10.3181/00379727-86-21002. [DOI] [PubMed] [Google Scholar]
- 17.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 18.Genestra M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal. 2007;19(9):1807–1819. doi: 10.1016/j.cellsig.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Sohal R.S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sohal R.S., Orr W.C. Vol. 52. Elsevier Inc; 2012. pp. 539–555. (The redox stress hypothesis of aging. Free Radic Biol Med). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckman K.B., Ames B.N. The free radical theory of aging matures. Physiol. Rev. 1998;78(2):547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 22.Muller F.L., Lustgarten M.S., Jang Y., Richardson A., Van Remmen H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007;43(4):477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 23.Sanz A., Stefanatos R.K.A. The mitochondrial free radical theory of aging: a critical view. Curr. Aging Sci. 2008;1(1):10–21. doi: 10.2174/1874609810801010010. [DOI] [PubMed] [Google Scholar]
- 24.Pérez V.I., Bokov A., Remmen H., Mele J., Van Remmen H., Mele J. Is the Oxidative Stress Theory of Aging Dead? Biochim Biophys. Acta. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lapointe J., Hekimi S. When a theory of aging ages badly. Cell Mol. Life Sci. 2010;67(1):1–8. doi: 10.1007/s00018-009-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rugarli E., Trifunovic A. Vol. 1847. Elsevier B.V; 2015. pp. 1345–1346. (Is Mitochondrial Free Radical Theory of Aging Getting Old? Biochim Biophys Acta - Bioenerg). [DOI] [PubMed] [Google Scholar]
- 27.Liu Y., Long J., Liu J. Mitochondrial free radical theory of aging: who moved my premise? Geriatr. Gerontol. Int. 2014;14(4):740–749. doi: 10.1111/ggi.12296. [DOI] [PubMed] [Google Scholar]
- 28.Barja G. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013;19(12):1420–1445. doi: 10.1089/ars.2012.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barja G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014:1–27. doi: 10.1016/B978-0-12-394625-6.00001-5. (In) [DOI] [PubMed] [Google Scholar]
- 30.Jones D.P. Vol. 5. Elsevier; 2015. pp. 71–79. (Redox theory of aging. Redox Biol). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 32.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 33.Campisi J. Cancer, aging and cellular senescence. Vivo (Brooklyn) 2000;14(1):183–188. [PubMed] [Google Scholar]
- 34.Campisi J., Kim S.H., Lim C.S., Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp. Gerontol. 2001;36(10):1619–1637. doi: 10.1016/s0531-5565(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 35.Wright W.E., Shay J.W. Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr. Opin. Genet Dev. 2001;11(1):98–103. doi: 10.1016/s0959-437x(00)00163-5. [DOI] [PubMed] [Google Scholar]
- 36.Dimri G.P. What has senescence got to do with cancer? Cancer Cell. 2005;7(6):505–512. doi: 10.1016/j.ccr.2005.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collado M., Blasco M. a., Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130(2):223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Afshari C. a., Vojta P.J., Annab L. a., Futreal P. a., Willard T.B., Barrett J.C. Investigation of the role of G1/S cell cycle mediators in cellular senescence. Exp. Cell Res. 1993:231–237. doi: 10.1006/excr.1993.1306. [DOI] [PubMed] [Google Scholar]
- 39.Di A., Steven L., Kris P.L., Wahl G.M. DNA damage triggers a prolonged p53- dependent G arrest ana long-term induction of Cipl in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- 40.Ogryzko V.V., Hirai T.H., Russanova V.R., Barbie D. a., Howard B.H. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol. Cell Biol. 1996;16(9):5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a) Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 42.Herbig U., Jobling W.A., Chen B.P.C., Chen D.J., Sedivy J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell. 2004;14(4):501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J., Woods D., McMahon M., Bishop J.M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12(19):2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olsen C.L., Gardie B., Yaswen P., Stampfer M.R. Raf-1-induced growth arrest in human mammary epithelial cells is p16-independent and is overcome in immortal cells during conversion. Oncogene. 2002;21(41):6328–6339. doi: 10.1038/sj.onc.1205780. [DOI] [PubMed] [Google Scholar]
- 45.Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 46.Wada T., Joza N., Cheng H.M., Sasaki T., Kozieradzki I., Bachmaier K. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat. Cell Biol. 2004;6(3):215–226. doi: 10.1038/ncb1098. [DOI] [PubMed] [Google Scholar]
- 47.Hewitt G., Jurk D., Marques F.D.M., Correia-Melo C., Hardy T., Gackowska A. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. Nat. Publ. Group, a Div. Macmillan Publ. Ltd. All Rights Reserve. 2012;28(3):708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harley C.B., Futcher A.B., Greider C.W., Calvin H.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 49.Campisi J. The biology of replicative senescence. Eur. J. Cancer Part A. 1997;33(5):703–709. doi: 10.1016/S0959-8049(96)00058-5. [DOI] [PubMed] [Google Scholar]
- 50.Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 51.Lundberg A.S., Hahn W.C., Gupta P., Weinberg R. a. Genes involved in senescence and immortalization. Curr. Opin. Cell Biol. 2000;12(6):705–709. doi: 10.1016/s0955-0674(00)00155-1. [DOI] [PubMed] [Google Scholar]
- 52.Shawi M., Autexier C. Telomerase, senescence and ageing. Mech. Ageing Dev. 2008;129(1–2):3–10. doi: 10.1016/j.mad.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 53.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27(7):339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 54.Zhang R., Chen W., Adams P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 2007;27(6):2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huschtscha L.I., Noble J.R., Neumann A.A., Moy E.L., Barry P., Melki J.R. Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res. 1998;58(16):3508–3512. [PubMed] [Google Scholar]
- 56.Itahana K., Zou Y., Itahana Y., Martinez J.-L., Beausejour C., Jacobs J.J.L. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell Biol. 2003;23(1):389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fridlyanskaya I., Alekseenko L., Nikolsky N. Senescence as a general cellular response to stress: a mini-review. Exp. Gerontol. 2015;72:124–128. doi: 10.1016/j.exger.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 58.Vijayachandra K., Lee J., Glick A.B. Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 2003;63(13):3447–3452. [PubMed] [Google Scholar]
- 59.Zhang H., Cohen S.N. Smurf2 up-regulation activates telomere-dependent senescence. Genes Dev. 2004;18(24):3028–3040. doi: 10.1101/gad.1253004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moiseeva O., Mallette F.A., Mukhopadhyay U.K., Moores A., Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell. 2006;17(4):1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Courtois-Cox S., Jones S.L., Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27(20):2801–2809. doi: 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- 62.Gorgoulis V.G., Halazonetis T.D. Oncogene-induced senescence: the bright and dark side of the response. Curr. Opin. Cell Biol. 2010;22(6):816–827. doi: 10.1016/j.ceb.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 63.Hills S.A., Diffley J.F.X. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014 19;24(10):R435–R444. doi: 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 64.Aird K.M., Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015;356(2 Pt A):204–210. doi: 10.1016/j.canlet.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Ewald J.A., Desotelle J.A., Wilding G., Jarrard D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010;102(20):1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramello M.C., Tosello Boari J., Canale F.P., Mena H.A., Negrotto S., Gastman B. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014;5:e1507. doi: 10.1038/cddis.2014.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young A.R.J., Narita M. SASP reflects senescence. EMBO Rep. 2009;10(3):228–230. doi: 10.1038/embor.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krtolica A., Parrinello S., Lockett S., Desprez P.Y., Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. USA. 2001;98(21):12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCarthy D. a., Clark R.R., Bartling T.R., Trebak M., Melendez J.A. Redox-control of the senescence regulator Interleukin-1 α and the secretory phenotype. J. Biol. Chem. 2013;288(45):32149–32159. doi: 10.1074/jbc.M113.493841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuilman T., Peeper D.S. Senescence-messaging secretome: sms-ing cellular stress. Nat. Rev. Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 72.Coppé J.-P., Desprez P.-Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young A.R.J., Narita M., Narita M. Cell senescence as both a dynamic and a static phenotype. Methods Mol. Biol. 2013;965:1–13. doi: 10.1007/978-1-62703-239-1_1. [DOI] [PubMed] [Google Scholar]
- 74.Coppé J.-P., Patil C.K., Rodier F., Krtolica A., Beauséjour C.M., Parrinello S. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. In: Blagosklonny M.V., editor. Vol. 5. 2010. p. e9188. (PLoS One). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acosta J.C., O’Loghlen A., Banito A., Guijarro M.V., Augert A., Raguz S. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 76.Coppé J.-P., Patil C.K., Rodier F., Sun Y., Muñoz D.P., Goldstein J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. Public Libr. Sci. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Salminen A., Ojala J., Kaarniranta K., Haapasalo A., Hiltunen M., Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011;34(1):3–11. doi: 10.1111/j.1460-9568.2011.07738.x. [DOI] [PubMed] [Google Scholar]
- 78.Erusalimsky J.D., Kurz D.J. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005;40(8–9):634–642. doi: 10.1016/j.exger.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 79.Campisi J., d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8(9):729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 80.Hoare M., Narita M. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. Nat. Publ. Group. 2013;15(8):887–889. doi: 10.1038/ncb2811. [DOI] [PubMed] [Google Scholar]
- 81.Nelson G., Wordsworth J., Wang C., Jurk D., Lawless C., Martin-Ruiz C. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11(2):345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lawless C., Jurk D., Gillespie C.S., Shanley D., Saretzki G., von Zglinicki T. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS One. 2012;7(2):e32117. doi: 10.1371/journal.pone.0032117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benz G., Hölzel D., Schmoeckel C. Inflammatory cellular infiltrates in melanocytic nevi. Am. J. Derm. 1991;13(6):538–542. doi: 10.1097/00000372-199113060-00003. [DOI] [PubMed] [Google Scholar]
- 84.Xue W., Zender L., Miething C., Dickins R.A., Hernando E., Krizhanovsky V. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007 8;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krizhanovsky V., Yon M., Dickins R.A., Hearn S., Simon J., Miething C. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuilman T., Michaloglou C., Vredeveld L.C.W.W., Douma S., van Doorn R., Desmet C.J. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 87.Wajapeyee N., Serra R.W., Zhu X., Mahalingam M., Green M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Demaria M., Ohtani N., Youssef S.A., Rodier F., Toussaint W., Mitchell J.R. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell. 2014;31(6):722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jun J.-I., Lau L.F. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2010;2(9):627–631. doi: 10.18632/aging.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jun J.-I., Lau L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. Nat. Publ. Group. 2010;12(7):676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Muñoz-Espín D., Cañamero M., Maraver A., Gómez-López G., Contreras J., Murillo-Cuesta S. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 92.Storer M., Mas A., Robert-Moreno A., Pecoraro M., Ortells M.C., Di Giacomo V. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–1130. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 93.Meuter A., Rogmann L.-M., Winterhoff B.J., Tchkonia T., Kirkland J.L., Morbeck D.E. Markers of cellular senescence are elevated in murine blastocysts cultured in vitro: molecular consequences of culture in atmospheric oxygen. J. Assist Reprod. Genet. 2014;31(10):1259–1267. doi: 10.1007/s10815-014-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baker D.J., Wijshake T., Tchkonia T., LeBrasseur N.K., Childs B.G., van de Sluis B. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nat. Nat. Publ. Group, a Div. Macmillan Publ. Ltd. All Rights Reserve. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Byun H., Lee Y., Kim J. From cell senescence to age-related diseases: differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015;48(10):549–558. doi: 10.5483/BMBRep.2015.48.10.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palmer A.K., Tchkonia T., LeBrasseur N.K., Chini E.N., Xu M., Kirkland J.L. Cellular senescence in Type 2 diabetes: a therapeutic opportunity. Diabetes. 2015;64(7):2289–2298. doi: 10.2337/db14-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burton D.G.A., Matsubara H., Ikeda K. Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp. Gerontol. 2010;45(11):819–824. doi: 10.1016/j.exger.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 98.Freigang S., Ampenberger F., Weiss A., Kanneganti T.-D., Iwakura Y., Hersberger M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. Nat. Publ. Group, a Div. Macmillan Publ. Ltd. All Rights Reserve. 2013;14(10):1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 99.Bhat R., Crowe E.P., Bitto A., Moh M., Katsetos C.D., Garcia F.U. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7(9):e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chinta S.J., Woods G., Rane A., Demaria M., Campisi J., Andersen J.K. Cellular senescence and the aging brain. Exp. Gerontol. 2015;68:3–7. doi: 10.1016/j.exger.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar M., Seeger W., Voswinckel R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. Am. Thorac. Soc. 2014;51(3):323–333. doi: 10.1165/rcmb.2013-0382PS. [DOI] [PubMed] [Google Scholar]
- 102.Meng L., Quezada M., Levine P., Han Y., McDaniel K., Zhou T. Functional role of cellular senescence in biliary injury. Am. J. Pathol. 2015;185(3):602–609. doi: 10.1016/j.ajpath.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benderdour M., Martel-Pelletier J., Pelletier J.-P., Kapoor M., Zunzunegui M.-V., Fahmi H. Vol. 8. Bentham Science Publishers; 2015. pp. 147–157. (Cellular Aging, Senescence and Autophagy Processes in Osteoarthritis. Curr. Aging Sci.). [DOI] [PubMed] [Google Scholar]
- 104.Campisi J., Andersen J.K., Kapahi P., Melov S. Vol. 21. Elsevier Ltd; 2011. pp. 354–359. (Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Coppé J.-P., Desprez P.-Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Laberge R.-M., Awad P., Campisi J., Desprez P.-Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5(1):39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bedi S., Vidyasagar A., Djamali A. Epithelial-to-mesenchymal transition and chronic allograft tubulointerstitial fibrosis. Transpl. Rev. 2008;22(1):1–5. doi: 10.1016/j.trre.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scharl M., Huber N., Lang S., Fürst A., Jehle E., Rogler G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn's disease associated intestinal fibrosis. Clin. Transl. Med. 2015;4:1. doi: 10.1186/s40169-015-0046-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Canaud G., Bonventre J.V. Vol. 30. Oxford University Press; 2015. pp. 575–583. (Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial Transplant). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ferenbach D.A., Bonventre J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. Nat. Publ. Group, a Div. Macmillan Publ. Ltd. All Rights Reserve. 2015;11(5):264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 112.Xu S., Cai Y., Wei Y. mTOR signaling from cellular senescence to organismal. Aging Aging Dis. 2014;5(4):263–273. doi: 10.14336/AD.2014.0500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 114.Zoncu R., Efeyan A., Sabatini D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chung K.W., Kim D.H., Park M.H., Choi Y.J., Kim N.D., Lee J. Recent advances in calorie restriction research on aging. Exp. Gerontol. 2013;48(10):1049–1053. doi: 10.1016/j.exger.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 116.Dulic V. Senescence regulation by mTOR. Methods Mol. Biol. 2013:15–35. doi: 10.1007/978-1-62703-239-1_2. (Clifton, N.J.) [DOI] [PubMed] [Google Scholar]
- 117.Sarbassov D.D., Sabatini D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005;280(47):39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- 118.Yoshida S., Hong S., Suzuki T., Nada S., Mannan A.M., Wang J. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem. American Society for. Biochem. Mol. Biol. 2011 16;286(37):32651–32660. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ramírez-Rangel I., Bracho-Valdés I., Vázquez-Macías A., Carretero-Ortega J., Reyes-Cruz G., Vázquez-Prado J. Vol. 31. American Society for Microbiology (ASM); 2011. pp. 1657–1671. (Regulation of mTORC1 complex assembly and signaling by GRp58/ERp57. Mol Cell Biol). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu L., Wise D.R., Diehl J.A., Simon M.C. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol Chem. Am. Soc. Biochem. Mol. Biol. 2008;283(45):31153–31162. doi: 10.1074/jbc.M805056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nayak B.K., Feliers D., Sudarshan S., Friedrichs W.E., Day R.T., New D.D. Stabilization of HIF-2α through redox regulation of mTORC2 activation and initiation of mRNA translation. Oncogene. NIH Public Access. 2013;32(26):3147–3155. doi: 10.1038/onc.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kaeberlein M, Kennedy BK. Hot topics in aging research: Protein translation and TOR signaling, 2010. Aging Cell. 2011 Apr;10(2):185–190. [DOI] [PMC free article] [PubMed]
- 123.Rider P., Carmi Y., Voronov E., Apte R.N. Interleukin-1α. Semin. Immunol. 2013;25(6):430–438. doi: 10.1016/j.smim.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 124.Kobayashi Y., Yamamoto K., Saido T., Kawasaki H., Oppenheim J.J., Matsushima K. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc. Natl. Acad. Sci. USA. 1990;87(14):5548–5552. doi: 10.1073/pnas.87.14.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Afonina I.S., Tynan G. a., Logue S.E., Cullen S.P., Bots M., Lüthi A.U. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell. 2011;44(2):265–278. doi: 10.1016/j.molcel.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Melendez J.A., Davies K.J. Manganese superoxide dismutase modulates interleukin-1alpha levels in HT-1080 fibrosarcoma cells. J. Biol. Chem. 1996;271(31):18898–18903. doi: 10.1074/jbc.271.31.18898. [DOI] [PubMed] [Google Scholar]
- 127.Moerman-Herzog A.M., Barger S.W. A polymorphism in the upstream regulatory region of the interleukin-1α gene confers differential binding by transcription factors of the AP-1 family. Life Sci. 2012;90(25–26):975–979. doi: 10.1016/j.lfs.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dasgupta J., Kar S., Liu R., Joseph J., Kalyanaraman B., Remington S.J. Reactive oxygen species control senescence-associated matrix metalloproteinase-1 through c-Jun-N-terminal kinase. J. Cell Physiol. 2010;225(1):52–62. doi: 10.1002/jcp.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McCarthy D. a., Ranganathan A., Subbaram S., Flaherty N.L., Patel N., Trebak M. Redox-control of the alarmin, Interleukin-1α. Redox Biol. 2013;1(1):218–225. doi: 10.1016/j.redox.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Laberge R.-M., Sun Y., Orjalo A.V., Patil C.K., Freund A., Zhou L. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. Nat. Publ. Group. 2015;17(8):1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pardo A., Selman M. MMP-1: the elder of the family. Int J. Biochem Cell Biol. 2005;37(2):283–288. doi: 10.1016/j.biocel.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 132.Kar S., Subbaram S., Carrico P.M., Melendez J.A. Vol. 174. Elsevier B.V; 2010. pp. 299–306. (Redox-control of matrix metalloproteinase-1: a critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lu Y., Wahl L.M. Oxidative stress augments the production of matrix metalloproteinase-1, cyclooxygenase-2, and prostaglandin E2 through enhancement of NF- κB activity in lipopolysaccharide-activated human primary monocytes. J. Immunol. 2005;175:5423–5429. doi: 10.4049/jimmunol.175.8.5423. [DOI] [PubMed] [Google Scholar]
- 134.Nelson K.K., Melendez J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004;37(6):768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 135.Touyz R.M. Mitochondrial redox control of matrix metalloproteinase signaling in resistance arteries. Arterioscler Thromb. Vasc. Biol. 2006;26(4):685–688. doi: 10.1161/01.ATV.0000216428.90962.60. [DOI] [PubMed] [Google Scholar]
- 136.Prado A., Pernomian L., Azevedo A., Castro M., Bendhack L., Tanus-Santos J. Matrix metalloproteinase 2 increases ROS in aortic tissue (1153.10) FASEB J. 2014 (Apr;28(1_Supplement)(:1153.10-) [Google Scholar]
- 137.Brkic M., Balusu S., Libert C., Vandenbroucke R.E. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflamm. 2015;2015:620581. doi: 10.1155/2015/620581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fisher G.J., Quan T., Purohit T., Shao Y., Cho M.K., He T. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009;174(1):101–114. doi: 10.2353/ajpath.2009.080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wenk J., Brenneisen P., Wlaschek M., Poswig A., Briviba K., Oberley T.D. Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. J. Biol. Chem. 1999;274(36):25869–25876. doi: 10.1074/jbc.274.36.25869. [DOI] [PubMed] [Google Scholar]
- 140.Zaw KK, Yokoyama Y, Abe M, Ishikawa O. Catalase restores the altered mRNA expression of collagen and matrix metalloproteinases by dermal fibroblasts exposed to reactive oxygen species. Eur J Dermatol. Jan;16(4):375–379. [PubMed]
- 141.Orbe J., Rodríguez J.A., Arias R., Belzunce M., Nespereira B., Pérez-Ilzarbe M. Antioxidant vitamins increase the collagen content and reduce MMP-1 in a porcine model of atherosclerosis: implications for plaque stabilization. Atherosclerosis. 2003;167(1):45–53. doi: 10.1016/s0021-9150(02)00392-1. [DOI] [PubMed] [Google Scholar]
- 142.Fisher G.J., Talwar H.S., Lin J., Voorhees J.J. Molecular mechanisms of photoaging in human skin in vivo and their prevention by all-trans retinoic acid. Photochem. Photobiol. 1999;69(2):154–157. doi: 10.1562/0031-8655(1999)069<0154:mmopih>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 143.Lee B., Moon S.-K. Resveratrol inhibits TNF-alpha-induced proliferation and matrix metalloproteinase expression in human vascular smooth muscle cells. J. Nutr. 2005;135(12):2767–2773. doi: 10.1093/jn/135.12.2767. [DOI] [PubMed] [Google Scholar]
- 144.Hozumi A., Nishimura Y., Nishiuma T., Kotani Y., Yokoyama M. Induction of MMP-9 in normal human bronchial epithelial cells by TNF-alpha via NF-kappa B-mediated pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281(6):L1444–L1452. doi: 10.1152/ajplung.2001.281.6.L1444. [DOI] [PubMed] [Google Scholar]
- 145.Ranganathan A.C., Nelson K.K., Rodriguez A.M., Kim K.H., Tower G.B., Rutter J.L. Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. J. Biol. Chem. 2001;276(17):14264–14270. doi: 10.1074/jbc.M100199200. [DOI] [PubMed] [Google Scholar]
- 146.Warner R.L., Bless N.M., Lewis C.S., Younkin E., Beltran L., Guo R. Time-dependent inhibition of immune complex-induced lung injury by catalase: relationship to alterations in macrophage and neutrophil matrix metalloproteinase elaboration. Free Radic. Biol. Med. 2000;29(1):8–16. doi: 10.1016/s0891-5849(00)00282-3. [DOI] [PubMed] [Google Scholar]
- 147.Toth P., Tarantini S., Springo Z., Tucsek Z., Gautam T., Giles C.B. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. Wiley-Black. 2015;14(3):400–408. doi: 10.1111/acel.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barbieri M., Bonafè M., Franceschi C., Paolisso G. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003;285(5):E1064–E1071. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- 149.Kenyon C. The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos Trans. R. Soc. Lond. B Biol. Sci. 2011;366(1561):9–16. doi: 10.1098/rstb.2010.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Murakami S., Salmon A., Miller R.A. Multiplex stress resistance in cells from long-lived dwarf mice. FASEB J. 2003;17(11):1565–1566. doi: 10.1096/fj.02-1092fje. [DOI] [PubMed] [Google Scholar]
- 151.Richardson A., Liu F., Adamo M.L., Van Remmen H., Nelson J.F. The role of insulin and insulin-like growth factor-I in mammalian ageing. Best. Pr. Res Clin. Endocrinol. Metab. 2004;18(3):393–406. doi: 10.1016/j.beem.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 152.van der Horst A., Burgering B.M.T. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007;8(6):440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 153.Martins R., Lithgow G.J., Link W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2015:8. doi: 10.1111/acel.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vurusaner B., Poli G., Basaga H. Vol. 52. Elsevier Inc; 2012. pp. 7–18. (Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med.). [DOI] [PubMed] [Google Scholar]
- 155.GJPL Kops, Dansen T.B., Polderman P.E., Saarloos I., KWA Wirtz, Coffer P.J. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 156.Chiribau C.B., Cheng L., Cucoranu I.C., Yu Y.-S., Clempus R.E., Sorescu D. FOXO3A regulates peroxiredoxin III expression in human cardiac fibroblasts. J. Biol. Chem. 2008;283(13):8211–8217. doi: 10.1074/jbc.M710610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Honda Y., Tanaka M., Honda S. Redox regulation, gene expression and longevity. Geriatr. Gerontol. Int. 2010;10(Suppl 1):S59–S69. doi: 10.1111/j.1447-0594.2010.00591.x. [DOI] [PubMed] [Google Scholar]
- 158.Dansen T.B., Smits L.M.M., Van Triest M.H., De Keizer P.L.J., Van Leenen D., Koerkamp M.G. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009;5(9):664–672. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 159.Kim K.S., Kim M.-S., Seu Y.B., Chung H.Y., Kim J.H., Kim J.-R. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell. 2007;6(4):535–545. doi: 10.1111/j.1474-9726.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- 160.Kim K.S., Seu Y.B., Baek S.-H., Kim M.J., Kim K.J., Kim J.H. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol. Biol. Cell. 2007;18(11):4543–4552. doi: 10.1091/mbc.E07-03-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Severino V., Alessio N., Farina A., Sandomenico A., Cipollaro M., Peluso G. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013;4:e911. doi: 10.1038/cddis.2013.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.López-Bermejo A., Buckway C.K., Devi G.R., Hwa V., Plymate S.R., Oh Y. Characterization of insulin-like growth factor-binding protein-related proteins (IGFBP-rPs) 1, 2, and 3 in human prostate epithelial cells: potential roles for IGFBP-rP1 and 2 in senescence of the prostatic epithelium. Endocrinology. 2000;141(11):4072–4080. doi: 10.1210/endo.141.11.7783. [DOI] [PubMed] [Google Scholar]
- 163.Kim K.-H., Park G.-T., Lim Y.-B., Rue S.-W., Jung J.-C., Sonn J.-K. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-beta-mediated signaling pathway. Biochem Biophys. Res Commun. 2004;318(4):819–825. doi: 10.1016/j.bbrc.2004.04.108. [DOI] [PubMed] [Google Scholar]
- 164.Zuo S., Liu C., Wang J., Wang F., Xu W., Cui S. IGFBP-rP1 induces p21 expression through a p53-independent pathway, leading to cellular senescence of MCF-7 breast cancer cells. J. Cancer Res Clin. Oncol. 2012;138(6):1045–1055. doi: 10.1007/s00432-012-1153-y. [DOI] [PubMed] [Google Scholar]
- 165.Courtois-Cox S., Genther Williams S.M., Reczek E.E., Johnson B.W., McGillicuddy L.T., Johannessen C.M. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10(6):459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dröge W. Oxidative aging and insulin receptor signaling. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2005;60(11):1378–1385. doi: 10.1093/gerona/60.11.1378. [DOI] [PubMed] [Google Scholar]
- 167.Mahadev K., Zilbering A., Zhu L., Goldstein B.J. Insulin-stimulated Hydrogen Peroxide Reversibly Inhibits Protein-tyrosine Phosphatase 1B in Vivo and Enhances the Early Insulin Action Cascade. J. Biol. Chem. 2001;276(24):21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 168.Goldstein B.J., Mahadev K., Wu X., Zhu L., Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid. Redox Signal. 2005;7(7–8):1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rincon M., Muzumdar R., Atzmon G., Barzilai N. The paradox of the insulin/IGF-1 signaling pathway in longevity. Mech. Ageing Dev. 2004;125(6):397–403. doi: 10.1016/j.mad.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 170.Salminen A., Kaarniranta K. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-kappaB signaling. Cell Signal. 2010;22(4):573–577. doi: 10.1016/j.cellsig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 171.Berridge M., Lipp P., Bootman M. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 172.Ureshino R.P., Rocha K.K., Lopes G.S., Bincoletto C., Smaili S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014;21(1):123–137. doi: 10.1089/ars.2013.5777. [DOI] [PubMed] [Google Scholar]
- 173.Farfariello V., Iamshanova O., Germain E., Fliniaux I., Prevarskaya N. Vol. 1853. Elsevier B.V; 2014. pp. 1974–1979. (Calcium homeostasis in cancer: A focus on senescence. Biochim. Biophys. Acta - Mol. Cell Res). [DOI] [PubMed] [Google Scholar]
- 174.Lang F., Busch G.L., Ritter M., Völkl H., Waldegger S., Gulbins E. Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 1998;78(1):247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- 175.Becker D., Blase C., Bereiter-Hahn J., Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J. Cell Sci. 2005;118(Pt 11):2435–2440. doi: 10.1242/jcs.02372. [DOI] [PubMed] [Google Scholar]
- 176.Numata T., Shimizu T., Okada Y. Vol. 19. Karger Publishers; 2007. pp. 1–8. (Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol. Biochem.). [DOI] [PubMed] [Google Scholar]
- 177.Marcu R., Wiczer B.M., Neeley C.K., Hawkins B.J. Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD+/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell Biol. 2014;34(15):2890–2902. doi: 10.1128/MCB.00068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wiel C., Lallet-Daher H., Gitenay D., Gras B., Le Calvé B., Augert A. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014;5:3792–3793. doi: 10.1038/ncomms4792. [DOI] [PubMed] [Google Scholar]
- 179.Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C. Vol. 21. 2002. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation; pp. 53–63. (EMBO J). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sharov V.S., Dremina E.S., Galeva N.A., Williams T.D., Schöneich C. Vol. 394. Biochem J. Portland Press Ltd; 2006. p. 605. (Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC–electrospray-tandem MS: selective protein oxidation during biological aging). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Qin F., Siwik D.A., Lancel S., Zhang J., Kuster G.M., Luptak I. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J. Am. Heart Assoc. Wiley-Black. 2013;2(4):e000184. doi: 10.1161/JAHA.113.000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Blander G., Guarente L. The Sir2 family of protein deacetylases. Annu Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 183.Vaziri H., Dessain S.K., Eaton E.N., Imai S.-I., Frye R.A., Pandita T.K. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell. 2001;107(2):149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 184.Mahlknecht U., Ho A.D., Voelter-Mahlknecht S. Chromosomal organization and fluorescence in situ hybridization of the human Sirtuin 6 gene. Int J. Oncol. 2006;28(2):447–456. [PubMed] [Google Scholar]
- 185.Ford E., Voit R., Liszt G., Magin C., Grummt I., Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006;20(9):1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell. 2003;11(2):437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 187.Michan S., Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. Portland Press Ltd. 2007;404(1):1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lin S.-J., Ford E., Haigis M., Liszt G., Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18(1):12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nat. Nat. Publ. Group. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 190.Zhang Q., Wang S.-Y., Fleuriel C., Leprince D., Rocheleau J.V., Piston D.W. Metabolic regulation of SIRT1 transcription via a HIC1: ctbp corepressor complex. Proc. Natl. Acad. Sci. USA. 2007;104(3):829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 191.Prozorovski T., Schulze-Topphoff U., Glumm R., Baumgart J., Schröter F., Ninnemann O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. Nat. Publ. Group. 2008;10(4):385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- 192.Chen R., Dioum E.M., Hogg R.T., Gerard R.D., Garcia J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011;286(16):13869–13878. doi: 10.1074/jbc.M110.175414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kume S., Haneda M., Kanasaki K., Sugimoto T., Araki S., Isono M. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006;40(12):2175–2182. doi: 10.1016/j.freeradbiomed.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 194.Furukawa A., Tada-Oikawa S., Kawanishi S., Oikawa S. H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell Physiol. Biochem. 2007;20(1–4):45–54. doi: 10.1159/000104152. [DOI] [PubMed] [Google Scholar]
- 195.Tanno M., Kuno A., Yano T., Miura T., Hisahara S., Ishikawa S. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J. Biol. Chem. 2010;285(11):8375–8382. doi: 10.1074/jbc.M109.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Volonte D., Zou H., Bartholomew J.N., Liu Z., Morel P.A., Galbiati F. Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6) J. Biol. Chem. 2015;290(7):4202–4214. doi: 10.1074/jbc.M114.598268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yeung F., Hoberg J.E., Ramsey C.S., Keller M.D., Jones D.R., Frye R.A. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. EMBO Press. 2004;23(12):2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yang S.-R., Wright J., Bauter M., Seweryniak K., Kode A., Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;292(2):L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 199.Rajendrasozhan S., Yang S.-R., Kinnula V.L., Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008;177(8):861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kume S., Uzu T., Horiike K., Chin-Kanasaki M., Isshiki K., Araki S.-I. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. Am. Soc. Clin. Investig. 2010;120(4):1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 202.Hong E.-H., Lee S.-J., Kim J.-H.J.-I.J.-S., Lee K.-H., Um H.-D., Kim J.-H.J.-I.J.-S. Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J. Biol. Chem. Am. Soc. Biochem. Mol. Biol. 2010;285(2):1283–1295. doi: 10.1074/jbc.M109.058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Li Y., Xu W., McBurney M.W., Longo V.D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8(1):38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tran D., Bergholz J., Zhang H., He H., Wang Y., Zhang Y. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell. 2014;13(4):669–678. doi: 10.1111/acel.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hayakawa T., Iwai M., Aoki S., Takimoto K., Maruyama M., Maruyama W. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One. 2015;10(1):e0116480. doi: 10.1371/journal.pone.0116480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Qiu X., Brown K., Hirschey M.D., Verdin E., Chen D. Vol. 12. Elsevier Inc; 2010. pp. 662–667. (Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab.). [DOI] [PubMed] [Google Scholar]
- 207.Tao R., Coleman M.C., Pennington J.D., Ozden O., Park S.H., Jiang H. Vol. 40. Elsevier Inc; 2010. pp. 893–904. (Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Mol Cell). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bell E.L., Emerling B.M., Ricoult S.J.H., Guarente L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene. Macmillan Publ. Ltd. 2011;30(26):2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Someya S., Yu W., Hallows W.C., Xu J., Vann J.M., Leeuwenburgh C. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143(5):802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chen Y., Zhang J., Lin Y., Lei Q., Guan K.-L., Zhao S. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12(6):534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bao J., Scott I., Lu Z., Pang L., Dimond C.C., Sack M.N. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 2011;49(7):1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.McDonnell E., Peterson B.S., Bomze H.M., Hirschey M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 2015;26(9):486–492. doi: 10.1016/j.tem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liao C.-Y., Kennedy B.K. SIRT6, oxidative stress, and aging. Cell Res. Nature Publishing Group; 2016. pp. 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ghosh S., Zhou Z. SIRTain regulators of premature senescence and accelerated aging. Protein Cell. 2015;6(5):322–333. doi: 10.1007/s13238-015-0149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Meter M., Van, Mao Z., Gorbunova V., Seluanov A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle Taylor Fr. 2011:15. doi: 10.4161/cc.10.18.17435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pan H., Guan D., Liu X., Li J., Wang L., Wu J. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Nat. Publ. Gr. Nat. Publ. Group. 2016;26(2):190–205. doi: 10.1038/cr.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wątroba M., Szukiewicz D. The role of sirtuins in aging and age-related diseases. Adv. Med Sci. 2015;61(1):52–62. doi: 10.1016/j.advms.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 218.Blackburn E.H., Epel E.S., Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350(6265):1193–1198. doi: 10.1126/science.aab3389. [DOI] [PubMed] [Google Scholar]
- 219.Rodier F., Muñoz D.P., Teachenor R., Chu V., Le O., Bhaumik D. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011;124(Pt 1):68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Toussaint O., Medrano E.E., von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000;35(8):927–945. doi: 10.1016/s0531-5565(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 221.Chen Q.M., Prowse K.R., Tu V.C., Purdom S., Linskens M.H. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp. Cell Res. 2001;265(2):294–303. doi: 10.1006/excr.2001.5182. [DOI] [PubMed] [Google Scholar]
- 222.Kawanishi S., Oikawa S. Mechanism of telomere shortening by oxidative stress. Ann. N. Y Acad. Sci. 2004;1019:278–284. doi: 10.1196/annals.1297.047. [DOI] [PubMed] [Google Scholar]
- 223.Tsolou A., Passos J.F., Nelson G., Arai Y., Zglinicki T. von. ssDNA fragments induce cell senescence by telomere uncapping. Exp. Gerontol. 2008;43(10):892–899. doi: 10.1016/j.exger.2008.08.043. [DOI] [PubMed] [Google Scholar]
- 224.Richter T., Saretzki G., Nelson G., Melcher M., Olijslagers S., von Zglinicki T. TRF2 overexpression diminishes repair of telomeric single-strand breaks and accelerates telomere shortening in human fibroblasts. Mech. Ageing Dev. 2007;128(4):340–345. doi: 10.1016/j.mad.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 225.Karlseder J., Hoke K., Mirzoeva O.K., Bakkenist C., Kastan M.B., Petrini J.H.J. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004;2(8):E240. doi: 10.1371/journal.pbio.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fotiadou P., Henegariu O., Sweasy J.B. DNA polymerase beta interacts with TRF2 and induces telomere dysfunction in a murine mammary cell line. Cancer Res. 2004;64(11):3830–3837. doi: 10.1158/0008-5472.CAN-04-0136. [DOI] [PubMed] [Google Scholar]
- 227.Criscione S.W., De Cecco M., Siranosian B., Zhang Y., Kreiling J.A., Sedicy J.M. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016;2(2):1–12. doi: 10.1126/sciadv.1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Proctor C.J., Kirkwood T.B.L. Modelling telomere shortening and the role of oxidative stress. Mech. Ageing Dev. 2002;123(4):351–363. doi: 10.1016/s0047-6374(01)00380-3. [DOI] [PubMed] [Google Scholar]
- 229.Op Den Buijs J., Van Den Bosch P.P.J., Musters M.W.J.M., Van Riel N. a.W. Mathematical modeling confirms the length-dependency of telomere shortening. Mech. Ageing Dev. 2004;125(6):437–444. doi: 10.1016/j.mad.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 230.Mendelsohn A.R., Larrick J.W. Ectopic expression of telomerase safely increases health span and life span. Rejuvenation Res. 2012;15(4):435–438. doi: 10.1089/rej.2012.1359. [DOI] [PubMed] [Google Scholar]
- 231.Kiyono T., Foster S.A., Koop J.I., McDougall J.K., Galloway D.A., Klingelhutz A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396(6706):84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 232.Simons M.J.P. Vol. 24. Elsevier B.V; 2015. pp. 6–11. (Questioning causal involvement of telomeres in aging. Ageing Res Rev). [DOI] [PubMed] [Google Scholar]
- 233.Mombach J.C.M., Bugs C.A., Chaouiya C. Modelling the onset of senescence at the G1/S cell cycle checkpoint. BMC Genom. BioMed. Cent. 2014:15. doi: 10.1186/1471-2164-15-S7-S7. (Suppl 7(7)(:S7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chen J., Huang X., Halicka D., Brodsky S., Avram A., Eskander J. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 2006;290(4):H1575–H1586. doi: 10.1152/ajpheart.00364.2005. [DOI] [PubMed] [Google Scholar]
- 235.Chandler H., Peters G. Stressing the cell cycle in senescence and aging. Curr. Opin. Cell Biol. 2013;25(6):765–771. doi: 10.1016/j.ceb.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 236.Takeuchi S., Takahashi A., Motoi N., Yoshimoto S., Tajima T., Yamakoshi K. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010;70(22):9381–9390. doi: 10.1158/0008-5472.CAN-10-0801. [DOI] [PubMed] [Google Scholar]
- 237.Yamakoshi K., Takahashi A., Hirota F., Nakayama R., Ishimaru N., Kubo Y. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J. Cell Biol. 2009;186(3):393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Adams P.D. Vol. 36. Elsevier Inc; 2009. pp. 2–14. (Healing and Hurting: Molecular Mechanisms, Functions, and Pathologies of Cellular Senescence. Mol Cell). [DOI] [PubMed] [Google Scholar]
- 239.Rayess H., Wang M.B., Srivatsan E.S. Cellular senescence and tumor suppressor gene p16. Int J. Cancer. 2012;130(8):1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Campisi J. Aging, cellular senescence, and cancer. Annu Rev. Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rodier F., Coppé J.-P., Patil C.K., Hoeijmakers W.A.M., Muñoz D.P., Raza S.R. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009;11(8):973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Muñoz-Espín D., Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. Nat. Publ. Group. 2014;15(7):482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 243.Demidenko Z.N., Korotchkina L.G., Gudkov A.V., Blagosklonny M.V. Paradoxical suppression of cellular senescence by p53. Proc. Natl. Acad. Sci. USA. 2010;107(21):9660–9664. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Vigneron A., Vousden K.H. p53, ROS and senescence in the control of aging. Aging (Albany NY) 2010;2(8):471–474. doi: 10.18632/aging.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wei Z., Guo H., Liu Z., Zhang X., Liu Q., Qian Y. CUL4B impedes stress-induced cellular senescence by dampening a p53-reactive oxygen species positive feedback loop. Free Radic. Biol. Med. 2015;79:1–13. doi: 10.1016/j.freeradbiomed.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 246.Volonte D., Zou H., Bartholomew J.N., Liu Z., Morel P.A., Galbiati F. Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6) J. Biol. Chem. Am. Soc. Biochem. Mol. Biol. 2015;290(7):4202–4214. doi: 10.1074/jbc.M114.598268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kim J.-E., Shin J.-S., Moon J.-H., Hong S.-W., Jung D.-J., Kim J.H. Vol. 30. Nature Publishing Group; 2016. pp. 1–12. (Foxp3 is a key downstream regulator of p53-mediated cellular senescence. Oncogene). [DOI] [PubMed] [Google Scholar]
- 248.Probin V., Wang Y., Zhou D. Busulfan-induced senescence is dependent on ROS production upstream of the MAPK pathway. Free Radic. Biol. Med. NIH Public Access. 2007;42(12):1858–1865. doi: 10.1016/j.freeradbiomed.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhang Y., Zhang Y., Zhong C., Xiao F., Barceloux D.G., Hayflick L. Vol. 4. Nature Publishing Group; 2016. p. 34578. (Cr(VI) induces premature senescence through ROS-mediated p53 pathway in L-02 hepatocytes. Sci. Rep.). [DOI] [PMC free article] [PubMed] [Google Scholar]