

Abstract
Pancreatic cancer (PC) is a lethal disease which is characterized by chemoresistance. Components of the cell cytoskeleton are therapeutic targets in cancer. βIV-tubulin is one such component that has two isotypes—βIVa and βIVb. βIVa and βIVb isotypes only differ in two amino acids at their C-terminus. Studies have implicated βIVa-tubulin or βIVb-tubulin expression with chemoresistance in prostate, breast, ovarian and lung cancer. However, no studies have examined the role of βIV-tubulin in PC or attempted to identify isotype specific roles in regulating cancer cell growth and chemosensitivity. We aimed to determine the role of βIVa- or βIVb-tubulin on PC growth and chemosensitivity. PC cells (MiaPaCa-2, HPAF-II, AsPC1) were treated with siRNA (control, βIVa-tubulin or βIVb-tubulin). The ability of PC cells to form colonies in the presence or absence of chemotherapy was measured by clonogenic assays. Inhibition of βIVa-tubulin in PC cells had no effect chemosensitivity. In contrast, inhibition of βIVb-tubulin in PC cells sensitized to vinca alkaloids (Vincristine, Vinorelbine and Vinblastine), which was accompanied by increased apoptosis and enhanced cell cycle arrest. We show for the first time that βIVb-tubulin, but not βIVa-tubulin, plays a role in regulating vinca alkaloid chemosensitivity in PC cells. The results from this study suggest βIVb-tubulin may be a novel therapeutic target and predictor of vinca alkaloid sensitivity for PC and warrants further investigation.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is a deadly disease, with a five-year survival rate of 6% [1]. It currently ranks as the fourth leading cause of cancer-related deaths [1], and is predicted to become the second leading cause of cancer death in the USA by 2030 due to a lack of improvement in treatments [2]. Currently, the best known treatments for PDA prolong survival by ~8 to 16 weeks [3], [4]. The difficulty in treating PDA stems from its propensity to acquire chemoresistance and metastasize [5], [6]. Thus, new therapeutic approaches are needed to target these processes.
Dysregulation of proteins which comprise the cell cytoskeleton and/or microtubule network have been implicated in chemotherapy drug resistance and aggressive disease in different tumor types [7], [8]. The microtubule network forms part of the cell cytoskeleton and consists of cylindrical assemblies of α- and β-tubulin heterodimers [8], [9]. Microtubules are essential for spindle formation during chromosome alignment and segregation processes of mitosis [8], [10], [11]. Moreover, they play a vital role in the trafficking of molecules within cells. Hence, it is not surprising that microtubules have been subject to extensive research as therapeutic targets for cancer [7].
There are seven distinct β-tubulin isotypes (βI, βII, βIII, βIVa, βIVb, βV and βVI) that are expressed on different genes and have differential tissue and cell specific expression [8], [9]. These β-tubulin isotypes share high amino acid sequence and structural homology and can be distinguished by the last 15 to 20 amino acids of their highly divergent carboxy terminal tail [8], [9]. The importance of β-tubulin is highlighted by the fact that tubulin-binding agents (TBAs) used in the clinic (for example, taxanes and vinca alkaloids) bind to the β-tubulin subunit and at high doses cause stabilization or destabilization of microtubules leading to mitotic arrest and cell death [7], [8], [11], [12]. However, there are drawbacks with the use of TBAs. Namely, they can be associated with side effects such as peripheral neuropathy and neutropenia [11], [13]. Cancer cells may also acquire resistance to TBAs [7]. Interestingly, this can be associated with altered expression of specific β-tubulin isotypes [8], [14], [15], [16], [17], [18].
βIII-tubulin is the most extensively studied β-tubulin isotype in relation to cancer, and its dysregulation has been associated with increased chemoresistance and poor patient outcome in different cancers (reviewed in [8]). Two studies reported that βIII-tubulin was highly expressed in PDA tumors following surgical resection, but absent in normal acinar and pancreatic islets [19], [20]. Recently, our laboratory demonstrated for the first time that silencing βIII-tubulin expression in PDA cells significantly decreased their clonogenic growth and increased chemosensitivity to broad classes of chemotherapy drugs in vitro [20]. Furthermore, stable suppression of βIII-tubulin in PDA cells using shRNA reduced tumor growth and metastases in an orthotopic PDA mouse model [20].
Other β-tubulin isotypes have also been reported to be differentially expressed in cancer cells and play a role in regulating chemotherapy drug sensitivity. For example, βII-tubulin was shown to be present at high levels in breast, lung and head and neck cancers [21], [22], [23]. Moreover, the high βII-tubulin expression in head and neck cancers correlated to a poor survival outcome. A functional role for βII-tubulin in non-small cell lung cancer (NSCLC) cells was reported by Gan et al. [24]. In this study the authors used RNA interference (RNAi) to silence βII-tubulin expression in NSCLC cells in vitro. This led to increased sensitivity to different types of vinca alkaloid drugs. Interestingly, we showed that silencing βII-tubulin in PDA cells had no effect on cell growth or chemosensitivity [20]. This suggests that the β-tubulin isotypes may have unique and distinct biological roles in different tumor types.
βIV-tubulin is another β-tubulin isotype which has been suggested to be involved in regulating chemoresistance [16], [17], [18], [24], [25], [26]. However, only limited studies have explored the role of βIV-tubulin in cancer cells. There are two isotypes of this tubulin: βIVa- and βIVb-tubulin, encoded by the TUBB4 and TUBB2C genes, respectively. In health, βIVa-tubulin is predominantly expressed in the brain, whereas βIVb-tubulin is expressed in the testis, heart and skeletal muscle [27]. These two isotypes only differ by 2 amino acids in their carboxy terminus [28], [29]. βIVa-tubulin levels have been reported to be up-regulated in several cancers (ovarian, lung, prostate) [16], [17], [26] and its expression is increased in taxol-resistant prostate and lung cancer cells. Recently, βIVa-tubulin expression was shown to increase in NSCLC cells when exposed to anchorage-independent growth conditions [30] and correlated with resistance to paclitaxel [30]. βIVb-tubulin is also dysregulated in cancer cells. Shalli et al. [18] reported increased expression of βIVb-tubulin in docetaxel-resistant breast cancer cells when compared to their respective docetaxel-sensitive parent cells. In another study, Gan et al. [24] showed that silencing βIVb-tubulin (by siRNA) in NSCLC cells did not change sensitivity to paclitaxel, but markedly increased sensitivity to vinca alkaloids via increased apoptosis and cell cycle arrest [24]. However, no functional role for either of the two βIV-tubulin isotypes has been established in PDA. Furthermore, there is little information describing the relative biological contribution of each isotype within cancer cells. Using a gene-silencing approach we specifically and potently silenced the expression of βIVa- and βIVb-tubulin in PDA cells and determined their role in regulating cell growth and chemotherapy drug sensitivity. Herein, we report for the first time that βIVb-tubulin appears to be the major βIV-tubulin isotype in PDA cells, and that silencing its expression in vitro: 1) decreases clonogenic anchorage-dependent cell growth and 2) increases sensitivity to vinca alkaloids via an induction in apoptosis.
Materials and Methods
Immunohistochemistry
Human PDA tissue specimens were collected by surgical removal. The use of human PDA tissue sections was approved by the UNSW Human Research Ethics Committee (HCEC# HC14039). Paraffin-embedded tissue sections were stained with βIV-tubulin antibody (1:200) using methods as previously described [20]. 3,3′ diaminobenzidine (DAB) was used as the substrate and sections were counterstained using hematoxylin. The specificity of the primary antibodies was confirmed by including several negative controls: (a) omission of the primary antibody and (b) incubation with isotype control antibodies at the same concentration as the primary antibodies.
Culture and Maintenance of Pancreatic Cancer Cell Lines
PDA cell lines, MiaPaCa-2, HPAF-II and AsPC1 were obtained from the American Type Culture Collection (ATCC). MiaPaCa-2 is a PDA line derived from a primary PDA tumor, while HPAF-II and AsPC1 are PDA lines derived from metastatic sites (HPAF-II and AsPC1 derived from Ascites). All cell lines were validated using short tandem repeat profiling (CellBank Australia). MiaPaCa-2 cells were grown in Dulbecco's Modified Eagle Media (Gibco by Life Technologies Pty Ltd., Mulgrave, VIC, Australia) supplemented with 10% fetal bovine serum (FBS), 2.5% horse serum and 2 mM L-glutamine. HPAF-II cells were grown in Minimal Essential Media (Gibco by Life Technologies Pty Ltd., Mulgrave, VIC, Australia) supplemented with 10% FBS, 2 mM L-glutamine and 1 mM sodium pyruvate. AsPC1 cells were grown in Roswell Park Memorial Institute-1640 medium (Gibco by Life Technologies Pty Ltd., Mulgrave, VIC, Australia) supplemented with 10% FBS and 2 mM L-glutamine. All cancer cell lines were incubated at 37 °C in humidified 5% CO2 atmosphere, and were lifted with 0.25% trypsin/EDTA. Cells were routinely tested for mycoplasma and found to be negative.
siRNA Transfection
Cancer cells were transfected using Lipofectamine® 2000 (Invitrogen by Life Technologies Pty Ltd., Mulgrave, VIC, Australia) with ON-TARGETplus SMARTpool siRNAs targeting βIVb-tubulin, βIVa-tubulin or non-silencing (ns-siRNA) (Millenium Science Pty Ltd., Mulgrave, VIC, Australia). Transfection was performed with 100 nM siRNA at 24 hours post-seeding into a 6-well plate.
Real-Time Quantitative PCR (qPCR)
Total RNA from transfected cells was extracted using the Qiagen RNeasy Plus kit (Qiagen Pty Ltd., Chadstone Centre, VIC, Australia) according to the manufacturer's instructions and stored at −80 °C. Concentration and purity of samples were determined with a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Scoresby, VIC, Australia) by measuring the absorbance ratio at A260/280 and A260/230. 500 ng of total extracted RNA was reverse transcribed using a High Capacity cDNA Reverse Trancription kit (Applied Biosystems by Life Technologies Pty Ltd., Mulgrave, VIC, Australia) into cDNA, and real-time PCR was performed using the Quantitect Power SYBR® Green PCR kit (Qiagen Pty Ltd., Chadstone Centre, VIC, Australia) and ViiA™ 7 Real-Time PCR machine (Life Technologies Pty Ltd., Mulgrave, VIC, Australia). The following primer sequences were used: βIVa-tubulin primer (Cat. No. L-009652-00-0020, Millennium Science Pty Ltd., Mulgrave, VIC, Australia); βIVb-tubulin forward primer, 5′ AAAGAATTCGATGCCACAGCCGAGGAGGA-3′, βIVb-tubulin reverse primer, 5′-AAATCTAGAATGAAAATGCTTTAATGG-3′. β2 microglobulin (β2M) was used as housekeeping gene, and βIVa- and βIVb-tubulin mRNA levels were normalized to β2M mRNA levels as previously described by McCarroll et al. [20].
Western Blot
Whole cell lysates were prepared and western blot analysis was performed as previously described [24], [31], [32]. Antibodies for βIV-tubulin (Abcam cat. ab11315) and GAPDH were purchased from Abcam Ltd. Protein bands were visualized using Amersham Enhanced Chemiluminescent Substrate (ECL) Western Blot Detection Reagent (GE Healthcare Australia Pty Ltd., Rydalmere, NSW, Australia) and an ImageQuant LAS4000 luminometer (GE Healthcare, Rydalmere, NSW, Australia).
Clonogenic Assays
Following transfection (24 h post-transfection), clonogenic assays in the presence or absence of chemotherapy drugs were carried out as previously described [20]. Briefly, cells were seeded into 6-well plates (MiaPaCa-2 = 300cells/well; AsPC1/HPAF-II = 500 cells/well) and incubated with drug 48 h post-transfection, for a total of 72 h. Colonies (>50 cells) were counted after staining with crystal violet.
Immunofluorescence Staining
Immunofluorescence staining was performed as previously described [20]. Chamber slides were incubated with the βIV-tubulin antibody (1:500) at 4 °C overnight in a humidified chamber and then incubated with an AlexaFluor-488 anti-mouse secondary antibody (1:1000, Invitrogen) for 30 min. Alternatively slides were incubated with the α-tubulin antibody (1:500; Sigma Cat. T9026) at 4 °C for 40 min, and then incubated with an AlexaFluor-555 secondary antibody (1:1000) for 30 min at room temperature. The slides were then mounted using undiluted ProLong® Gold Antifade Reagent (Invitrogen) and sealed with a coverslip. Images were captured using a Leica SP5 confocal microscope.
Cell Cycle Assay
Cell cycle assays (48 h post-transfection) were performed as previously described [20].
Apoptosis Assays
Annexin V Assay: PDA cells were treated (48 h post-transfection) with fresh culture medium containing increasing amounts (0.1 nM, 0.3 nM, 0.5 nM) of vincristine for 24 h. Both adherent and floating cells were collected and cell death was measured using the Annexin V-PE/7-AAD-FITC reagent (Guava Nexin reagent, Millipore) as previously described [20]. Samples were acquired on a Guava EasyCyte HT System with InCyte software. Caspase 3/7 activity assay: PDA cells were treated (48 h post-transfection) with fresh culture medium containing increasing amounts (0.1 nM, 0.3 nM, 0.5 nM) of vincristine for 24 h. Caspase 3/7 activity in cell lysates was measured using a luminescent plate reader as previously described [20].
Statistical Analyses
Data were expressed as mean ± SEM and analyzed using GraphPad Prism 6 (GraphPad Software, Inc.). One-way ANOVA (Dunnett's multiple comparison test) or paired t-test were used where appropriate to measure statistical significance.
Results
βIV-Tubulin is Expressed in Human Pancreatic Ductal Adenocarcinoma Cells
Currently, there are no antibodies that can distinguish between βIVa- or βIVb-tubulin due to their high sequence homology, thus we could only detect total βIV-tubulin protein. We first assessed whether βIV-tubulin was present in human pancreatic tumor tissue. Total βIV-tubulin was detected using immunohistochemistry in PDA tissue collected after surgical resection. βIV-tubulin was highly expressed in pancreatic tumor cells (Figure 1, A and B). In addition, we measured βIV-tubulin expression in cell lysates collected from three different PDA cell lines derived from primary (MiaPaCa-2) and metastatic (AsPC1, HPAF-II) sites by Western blot. βIV-tubulin levels were higher in all PDA cell lines (MiaPaCa-2, HPAF-II and AsPC1) relative to normal human pancreatic ductal epithelial (HPDE) cells (cells of origin of PDA; Figure 1C).
Figure 1.
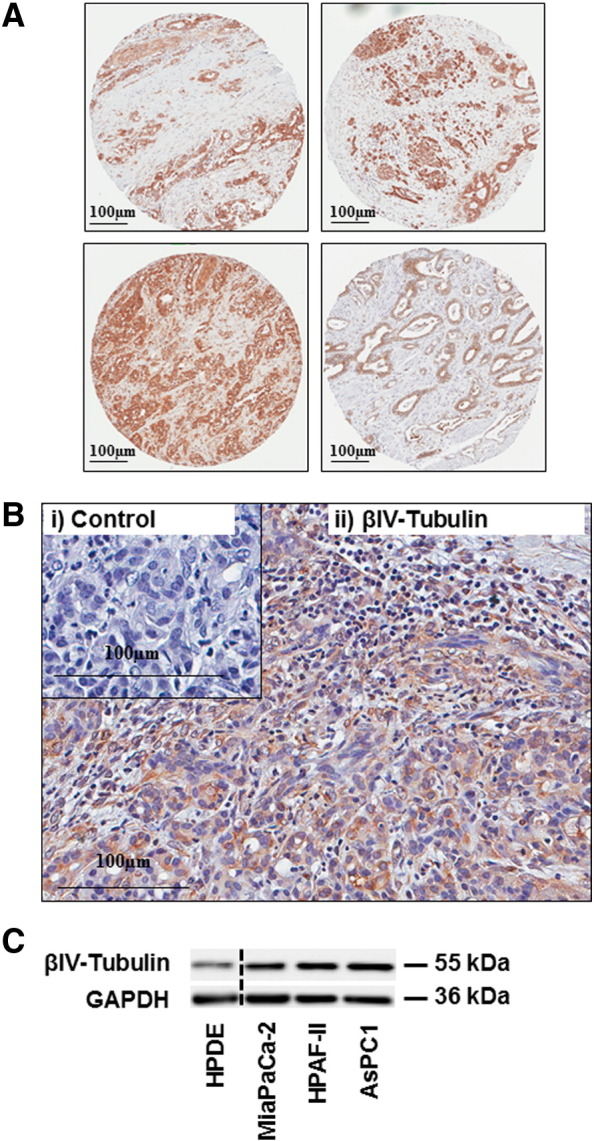
βIV-tubulin expression in PDA patient tissue and PDA cell lines. A) Immunohistochemistry for βIV-tubulin in A) human PDA tissue microarray specimens. B) Magnified field of human PDA tissue stained for βIV-tubulin. Panels show tissue stained with either isotype control antibody (i) or βIV-tubulin antibody (ii). The isotype control was negative and tumor elements had strong immunoreactivity for βIV-tubulin. C) Representative Western blot (n = 3) for βIV-tubulin in protein extracts from pancreatic cancer cell lines and human pancreatic ductal epithelial cells (HPDE). GAPDH was used as a protein loading control.
The Effect of Silencing βIVa- and βIVb-Tubulin Expression in Pancreatic Cancer Cells
To establish if βIVa- or βIVb-tubulin could be silenced in PDA cells, we transfected three independent PDA cell lines (MiaPaCa-2, HPAF-II and AsPC1) with an siRNA pool (four siRNA sequences that bind different regions of the transcript) targeting either βIVa-tubulin, βIVb-tubulin or a non-silencing siRNA control (ns-siRNA). RNA was harvested 72 h post-transfection and gene silencing efficiency was evaluated by qPCR. βIVa-tubulin siRNA treatment resulted in potent inhibition of βIVa-tubulin gene expression in all PDA cell lines compared to ns-siRNA controls (MiaPaCa-2: 98.5 ± 0.2% inhibition; HPAF-II: 71.3 ± 1.97% inhibition; AsPC1: 88.3 ± 2.1% inhibition; Figure 2, A–C). Likewise, treatment of cells with βIVb-tubulin siRNA resulted in a potent reduction in βIVb-tubulin gene expression in all three cell lines compared to ns-siRNA controls (MiaPaCa-2: 76 ± 4.6% inhibition; HPAF-II: 71.3 ± 3.6% inhibition; AsPC1: 83.4 ± 3.9% inhibition; Figure 2, D–F). Importantly, silencing βIVa-tubulin in all PDA cells had no effect on βIVb-tubulin gene levels (Figure 2, A–C), and silencing βIVb-tubulin had no effect on βIVa-tubulin gene expression (Figure 2, D–F). These results demonstrated the high specificity and activity of the siRNA for each gene and provided a valuable tool to delineate the roles of βIVa-tubulin and βIVb-tubulin in PDA cells. To determine the relative contribution of each isotype to the total βIV-tubulin pool, protein was harvested from PDA cells 72 h post-transfection and total βIV-tubulin protein detected by Western blot. Silencing βIVa-tubulin in our PDA cell lines resulted in a negligible reduction in total βIV-tubulin protein (Figure 2, G–I). In contrast, βIVb-tubulin siRNA markedly reduced total βIV-tubulin protein expression in HPAF-II cells (Figure 2H) and completely abolished total βIV-tubulin protein expression in MiaPaCa-2 and AsPC1 cells (Figure 2, G–I). Taken together, these results suggest that βIVb-tubulin comprised the majority of the βIV-tubulin pool in PDA cells.
Figure 2.

βIVa- and βIVb-tubulin silencing in pancreatic cancer cell lines. A-H) Real-time PCR analysis of βIVa- and βIVb-tubulin silencing in MiaPaCa-2 (A and D), HPAF-II (B and E) and AsPC1 (C and F) cells. RNA was harvested from cells 72 h post-transfection with ns-siRNA, βIVa-Tub siRNA or βIVb-tubulin siRNA. βIVa- and βIVb- tubulin mRNA levels were normalized to 18S mRNA. G-I) Western blot analysis of βIVa- and βIVb- tubulin silencing in protein extracts from MiaPaCa-2 (G), HPAF-II (H) and ASPC1 (I) cells. Cell lysates were harvested from cells 72 h after transfection with control siRNA (ns-siRNA), βIVa-tubulin siRNA (βIVa-Tub siRNA) or βIVb-tubulin siRNA (βIVb-Tub siRNA). GAPDH was used as a loading control. Asterisks indicate significance (**P ≤ .01, ***P ≤ .001, ****P ≤ .0001; n ≥ 3).
The Effect of Silencing βIVa- and βIVb-Tubulin on Pancreatic Cancer Cell Clonogenic Growth and Chemosensitivity
Given the importance of β-tubulin isotypes in regulating growth and chemotherapy drug sensitivity in cancer cells, we determined the effect of silencing either βIVa- or βIVb-tubulin expression in PDA cells on their clonogenic growth potential and chemosensitivity to a structurally and functionally diverse range of drugs: Gemcitabine (an anti-metabolite and first-line treatment for PDA), Paclitaxel (a tubulin-stabilizing agent; albumin-bound paclitaxel is currently used in combination with gemcitabine in the clinic) and Vincristine (a tubulin-destabilizing agent). Silencing βIVa-tubulin alone had no effect on the ability of any of the PDA cells to form cell colonies (Figure 3, A–I). Furthermore, with the exception of paclitaxel in AsPC1 cells (Figure 3F), βIVa-tubulin inhibition had no significant effect on chemosensitivity in PDA cells (Figure 3, A–I). In contrast, βIVb-tubulin knockdown alone reduced the ability of the metastases-derived PDA cell lines HPAF-II (Figure 4, B, E and H) and AsPC1 (Figure 4, C, F and I) to form cell colonies. These results suggest that βIVb-tubulin may have a specific role in regulating PDA cell growth. βIVb-tubulin knockdown also had a pronounced effect on PDA chemosensitivity. βIVb-tubulin gene silencing increased the sensitivity of HPAF-II cells to gemcitabine (Figure 4B) and paclitaxel (Figure 4E). AsPC1 cells were also sensitized to paclitaxel following knockdown of βIVb-tubulin (Figure 4F). Notably, silencing βIVb-tubulin in all PDA cells resulted in increased sensitivity to the vinca alkaloid vincristine compared to controls (Figure 4, G–I). Based on these results, we focused on βIVb-tubulin for all further experiments.
Figure 3.

The effect of βIVa-tubulin silencing on pancreatic cancer cell clonogenic cell growth and chemosensitivity. Bars represent the number of MiaPaCa-2, HAPF-II or AsPC1 cell colonies (mean + S.E.M. as a % of mock treatment) that formed from low density seeding following transfection with control siRNA (ns-siRNA), or βIVa-tubulin siRNA (βIVa-Tub siRNA) and 72 h culture in titrations of Gemcitabine (A-C), Taxol (D-F) or Vincristine (G-F). Asterisks indicate significance relative to the 0 nM control for the same siRNA. Hashes indicate significance relative to the ns-siRNA control of the same drug dose (* or #P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001; n ≥ 4).
Figure 4.

The effect of βIVb-tubulin silencing on pancreatic cancer cell clonogenic cell growth and chemosensitivity. Bars represent the number of MiaPaCa-2, HPAF-II or AsPC1 cell colonies (mean + S.E.M. as a % of mock treatment) that formed from low density seeding following transfection with control siRNA (ns-siRNA), or βIVb-tubulin siRNA (βIVb-Tub siRNA) and 72 h culture in titrations of Gemcitabine (A-C), Paclitaxel (D-F) or Vincristine (G-F). Asterisks indicate significance relative to the 0 nM control for the same siRNA. Hashes indicate significance relative to the ns-siRNA control of the same drug dose (* or #P ≤ .05, ** or ##P ≤ .01, *** or ###P ≤ .001, **** or ####P ≤ .0001; n ≥ 3).
Before continuing with our investigation of βIVb-tubulin in PDA cells, we assessed the effect of silencing βIVb-tubulin expression in non-tumorigenic human pancreatic duct epithelial (HPDE; Cells of origin of PDA). We observed that βIVb-tubulin knockdown had no effect on the proliferation of normal HPDE cells (Figure 5). Given βIVb-tubulin knockdown was not toxic in HPDE cells, we did not pursue any further functional analysis in these cells.
Figure 5.
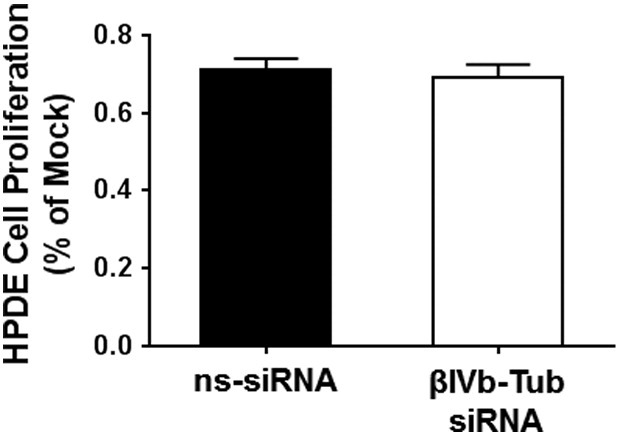
The effect of βIVb-tubulin silencing on human pancreatic ductal epithelial cell growth. Graph demonstrating that knockdown of βIVb-tubulin in human pancreatic ductal epithelial (HPDE) cells does not affect their proliferation (as % of mock transfection). Cells transfected with control siRNA (ns-siRNA) served as controls; n = 3.
Given βIVb-tubulin knockdown sensitized all PDA cell lines to vincristine, we next investigated whether βIVb-tubulin played a broader role in regulating PDA cell sensitivity to vinca alkaloids. Clonogenic assays were repeated using MiaPaCa-2 and HPAF-II cells in the presence of vinblastine or vinorelbine (both FDA approved vinca alkaloids). βIVb-tubulin knockdown significantly sensitized both MiaPaCa-2 and HPAF-II cells to vinblastine and vinorelbine (Figure 6, A–D). Our results suggest that silencing βIVb-tubulin in PDA cells chemosensitizes them to all major vinca alkaloids used in the clinic.
Figure 6.

The effect of βIVb-tubulin silencing on pancreatic cancer cell chemosensitivity to vinblastine and vinorelbine. A-D) Bars represent the number of MiaPaCa-2 (A-B) or HPAFII (C and D) cell colonies (mean + S.E.M. as a % of untreated control siRNA [ns-siRNA] cells) that formed from low density seeding following transfection with ns-siRNA or βIVb-tubulin siRNA (βIVb-Tub siRNA) and 72 h culture in titrations of vinorelbine or vinblastine. Asterisks indicate significance relative to the 0 nM control for the same siRNA. Hashes indicate significance relative to the ns-siRNA control of the same drug dose (** or ##P ≤ .01, *** or ###P ≤ .001, **** or ####P ≤ .0001; n = 3).
The Effect of Silencing βIVb-Tubulin in Pancreatic Cancer Cells on Microtubule Structure and Cell Cycle
To determine if βIVb-tubulin knockdown disrupts the microtubule network in PDA cells, we examined the effect of silencing βIVb-tubulin in MiaPaCa-2 and HPAF-II cells on gross microtubule structure by fluorescence confocal microscopy. Despite potent knockdown of βIV-tubulin protein, no significant effects were observed on the microtubule structure, as shown by staining of α-tubulin, the partner protein of β-tubulin in microtubules (Figure 7, A–B). In addition, no changes in the gene expression levels of total β-tubulin or the closely related β-tubulin isotypes βI, βII or βIII-tubulin were observed in cells treated with βIVb-tubulin siRNA (48 and 72 h post-transfection) (Supplementary Figure 1, A–D). These results demonstrated that there was no significant compensation in other β-tubulin genes when βIVb-tubulin was inhibited.
Figure 7.

The effect of βIVb-tubulin silencing on pancreatic cancer cell morphology and cell cycle. A and B) Confocal microscopy for α-tubulin and βIV-tubulin in MiaPaCa-2 (A) and HPAF-II (B) cells transfected with control siRNA (ns-siRNA) (top panels) or βIVb-tubulin siRNA (βIVb-Tub siRNA; bottom panels). Overlaid fluorescence images are shown in the far right panel of each row. C and D) Cell cycle distribution was analyzed by propidium iodide staining and flow cytometry. Bars represent % of MiaPaCa-2 (C) or HPAFII (D) cells in G0/G1-phase, S-phase, or G2/M-phase (mean ± S.E.M.); n = 3.
We next examined whether the effects of βIVb-tubulin knockdown on PDA clonogenic growth and chemosensitivity were induced via a similar mechanism to TBAs. Treatment of cells with TBAs results in an accumulation of cells in G2/M phase (preparation for cell division/mitosis) of the cell cycle and a reduction of cells in G1 phase (preparation for replication) and S phase (replication). We first measured cell cycle distribution in PDA cells transfected with either βIVb-tubulin siRNA or ns-siRNA, using propidium iodide staining and flow cytometry. In contrast to TBAs, silencing βIVb-tubulin alone in MiaPaCa-2 cells did not significantly alter their cell cycle distribution relative to ns-siRNA controls (Figure 7, C and D). Given we observed chemosensitization to vincristine, but not paclitaxel, in all three lines, we repeated the experiment using MiaPaCa-2 cells in the presence/absence of vincristine (tubulin destabilizer) and paclitaxel (tubulin stabilizer). Consistent with their roles as tubulin binding agents, both paclitaxel and vincristine caused cell cycle arrest in mitosis (observed as an increase in G2/M phase cell population; Figure 8). βIVb-tubulin knockdown had no effect on paclitaxel-induced cell cycle changes, relative to ns-siRNA controls (Figure 8A). However, βIVb-tubulin knockdown significantly enhanced the arrest of cells in mitosis induced by vincristine (observed as an additional increase in G2/M phase cell population; Figure 8B).
Figure 8.

The effect of βIVb-tubulin silencing on pancreatic cancer cell cycle in the presence of paclitaxel or vincristine. Cell cycle distribution was analyzed by propidium iodide staining and flow cytometry. Bars represent % of MiaPaCa-2 cells in G1-phase, S-phase, or G2/M-phase (mean + S.E.M.). 72 h post-transfection with either ns-siRNA or βIII-Tub siRNA cells were incubated for eight hours with (A) paclitaxel or (B) vincristine. Asterisks indicate significance relative to the 0 nM control for the same siRNA. Hashes indicate significance relative to the ns-siRNA control of the same drug dose (** or ##P ≤ .01, *** or ###P ≤ .001, **** or ####P ≤ .0001; n = 3).
βIVb-tubulin knockdown in pancreatic cancer cells induces apoptosis in the presence of drug
Finally, we investigated whether βIVb-tubulin knockdown was exerting its effects on chemosensitivity through the induction of apoptosis. MiaPaCa-2 cells were transfected with βIVb-tubulin or control siRNA (ns-siRNA) and treatments commenced 48 hours post-transfection. Cells were cultured in the presence of increasing concentrations of vincristine (0.1–0.5 nM) for a total of 24 h. Apoptosis was examined by measuring caspase 3/7 activity and Annexin V/7AAD staining and flow cytometry. PDA cells treated with control (ns-siRNA) siRNA showed only a slight increase in caspase 3/7 activity or annexin V/7ADD staining when treated with highest concentration of vincristine (0.5 nM) (Figure 9, A and B). In contrast, cells treated with βIVb-tubulin siRNA showed a small increase in caspase 3/7 activity in the absence of drug (Figure 8A). Addition of vincristine (0.3-0.5 nM) enhanced the increase in caspase 3/7 (Figure 9A). These results also correlated with a significant increase in annexin V/7ADD staining (Figure 9B). These data indicate that the chemosensitisation of PDA cells with suppressed βIVb-tubulin is mediated via an induction of apoptosis.
Figure 9.

The effect of βIVb-tubulin silencing in pancreatic cancer cells on apoptosis. MiaPaCa-2 cells were transfected with control siRNA (ns-siRNA) or βIVb-tubulin siRNA (βIVb-Tub siRNA). 48 h later, cells were cultured in vincristine (0.1 nM, 0.3 nM, 0.5 nM) for a further 24 h. Apoptosis was then measured using a caspase 3/7 activity assay (A) or Annexin V and 7AAD staining and flow cytometry (B). Asterisks indicate significance relative to the 0 nM control for the same siRNA. Hashes indicate significance relative to the ns-siRNA control of the same drug dose. Asterisks indicate significance (* or #P < .05, **P < .01, *** or ### P ≤ .001, **** or ####P < .0001; n ≥ 3).
Discussion and Conclusions
Dysregulated expression of β-tubulins in cancer cells is associated with increased tumor aggressiveness and resistance to chemotherapy agents in different cancers [7], [8]. However, this is the first study to describe the functional role of βIV-tubulin in pancreatic cancer cells. Moreover, no study has attempted to delineate the individual roles of βIVa-tubulin and βIVb-tubulin within the same cancer cell type. Herein, we report for the first time novel roles for βIVb-tubulin in regulating: (1) PDA cell anchorage-dependent growth; and (2) sensitivity to chemotherapeutic drugs. Importantly, we demonstrate that βIVb-tubulin has functionally distinct roles in PDA cells when compared to βIVa-tubulin, and is a potential therapeutic target which may sensitize PDA cells to vinca alkaloids.
A major limitation to understanding the functional role of βIV-tubulin in cancer biology has been a lack of specific antibodies and inhibitors to distinguish between the βIVa- and βIVb-tubulin isotypes. To assess the role of both βIV-tubulins in PDA cells we used an RNAi approach which was designed to silence either βIVa-tubulin or βIVb-tubulin expression with high selectivity and specificity. Knockdown of βIVa-tubulin using siRNA did not affect βIVb-tubulin gene expression in PDA cells. Similarly, knockdown of βIVb-tubulin had no effect on βIVa-tubulin gene expression. These data demonstrated potent knockdown of each isotype at the gene level, and confirmed that the siRNAs used in this study were specific. We also showed that when βIVa-tubulin was silenced using siRNA there was very little change in the protein expression of total βIV-tubulin in PDA cells. In contrast, when βIVb-tubulin was silenced using siRNA, total βIV-tubulin protein expression was significantly decreased or in the case of AsPC1 and MiaPaCa-2 cells completely abolished. This suggests that βIVb-tubulin is the major isotype in the total βIV-tubulin pool in PDA cells. These results are in accordance with a study [27] which measured the gene expression of all seven β-tubulin isotypes in a panel of different tumor and non-tumor cells. βIVb-tubulin was found to be expressed at high levels in a large number of different tumor cells, while βIVa-tubulin expression was low.
Silencing βIVa-tubulin expression in PDA cells (primary and metastases derived) had no effect on the clonogenic cell growth in the absence of chemotherapy drugs. In addition, silencing βIVa-tubulin expression in PDA cells, broadly speaking, had no effect on chemosensitivity. The only exception to this was a small increase in sensitivity to paclitaxel at one drug concentration in AsPC1 cells. These results are not surprising given that βIVa-tubulin appears to make up very little of the total βIV-tubulin pool in PDA cells. To date, there are few reported studies examining βIV-tubulin in cancer [16], [17], [18], [24], [25], [26], [33]. Only four studies have correlated increased βIVa-tubulin expression with increased chemoresistance (to estramustine and paclitaxel) in prostate, ovarian and lung cancer cells [16], [17], [26], [30]. This study is the first to explore the effect of silencing βIVa-tubulin expression in any cancer.
Similarly, few studies have explored the role of βIVb-tubulin in cancer cells [18], [24], [25], [33]. Notably, we demonstrated that βIVb-tubulin knockdown has significant effects on PDA cell growth and chemosensitivity. For example, knockdown of βIVb-tubulin in PDA cells significantly decreased clonogenicity in two out of three PDA cells. These results are in contrast to Gan et al. [24] which reported no effect on non-small cell lung cancer (NSCLC) cell growth following silencing of βIVb-tubulin in the absence of chemotherapeutics. Therefore, it is likely that the different β-tubulin isotypes play different roles in different cell types. Interestingly, unlike TBAs, the decrease in PDA cell growth following knockdown of βIVb-tubulin was not associated with any changes in cell cycle progression. Studies examining the mechanism of action for βIVb-tubulin regulation on PDA tumorigenicity are currently under investigation in our laboratory.
βIVb-tubulin knockdown had marked effects on PDA cell chemosensitivity. Firstly, silencing βIVb-tubulin expression increased the sensitivity of HPAF-II cells to gemcitabine and paclitaxel. AsPC1 cells were also shown to have increased sensitivity to paclitaxel following βIVb-tubulin knockdown, while no effect was observed in MiaPaCa-2 cells. These cell-line specific effects indicate that βIVb-tubulin may play a broad role in modulating chemoresistance in specific subsets of PDA cells. In contrast, βIVb-tubulin knockdown sensitized all PDA cell lines to vincristine. Further investigation revealed increased sensitivity to other vinca alkaloids such as vinblastine and vinorelbine following knockdown of βIVb-tubulin. Both drugs are widely used to treat different solid and hematologic tumors. Importantly, in the past vinca alkaloids have been reported to have minimal activity against pancreatic cancer [34]. Our study provides new information on PDA cell resistance to vinca alkaloids demonstrating that βIVb-tubulin plays an important role in regulating sensitivity to these types of drugs. Together, these data provide new opportunities to develop inhibitors against βIVb-tubulin, or to select patients which express low levels of βIV-tubulin which may allow for re-purposing of vinca alkaloids for the treatment of PDA.
Importantly, silencing βIVb-tubulin in non-tumorigenic human HPDE cells had no effect on their viability, suggesting that our observed anti-proliferative effects were specific to cancer cells. This is in contrast to TBAs, which can arrest mitosis in any cell by stabilizing/destabilizing all β-tubulin isotypes [8]. As a result, TBAs are dose-limited by off-target toxicity, especially peripheral neuropathy and neutropenia [35]. Despite this, TBAs are still effectively used in the clinic and off-target toxicity is well managed [36]. For example, vincristine has limited myelotoxicity but some neurotoxicity associated with clinical sensory neuropathy, occasionally requiring dose reduction but rarely cessation.
While βIVb-tubulin is also expressed in normal tissues (testis, heart and skeletal muscle), it is unlikely that silencing a single β-tubulin isotype in these cells would have the same off-target toxicity as TBAs (which bind to all β-tubulin isotypes). In fact, the ability of βIVb-tubulin inhibition to sensitize PDA cells to vinca alkaloids also means that vinca alkaloid dosing could potentially be reduced in combination with βIVb-tubulin inhibition. We have also made significant advances in siRNA delivery technology [37], which not only overcomes the lack of specific inhibitors against βIVb-tubulin, but can also target therapy to PDA tumors. To highlight the therapeutic potential of targeting a single β-tubulin isotype up-regulated in PDA, we have previously administered therapeutic doses of βIII-tubulin siRNA with nanoparticle which effectively inhibited βIII-tubulin protein levels in orthotopic pancreatic tumors without any off-target toxicity in other organs in mice [37].
A question that remains to be answered is how βIVb-tubulin exerts its effect on PDA cell growth and chemosensitivity? It is established that TBAs, which bind to all β-tubulins predominantly exert their anti-cancer effect by inducing potent cell cycle arrest which coincides with alterations to the structure of the microtubule network and eventual cell death. Our data showed that knockdown of βIVb-tubulin alone did not affect any stages of the cell cycle, nor did it disrupt the cytoskeleton in PDA cells. However, βIVb-tubulin knockdown in PDA cells enhanced vincristine-induced mitotic arrest, but not paclitaxel-induced mitotic arrest. We also found that silencing βIVb-tubulin in PDA cells also enhanced apoptosis induced by vincristine treatment. Taken together, the results suggest that βIVb-tubulin knockdown enhances the ability of vinca alkaloids to arrest mitosis and consequently induce apoptosis.
As mentioned above, our most pronounced effects on PDA cell chemosensitivity were observed in the presence of vinca alkaloids. These class of drugs inhibit cell division by destabilizing microtubules. Vinca alkaloid resistance in cancer cells is often associated with overexpression of ABC drug efflux pumps P-glycoprotein and MRP1, under the control of AKT signaling [38], [39], [40], [41]. It is possible that βIVb-tubulin may contribute to this signaling through its unique C-terminus tail. β-tubulin isotypes differ in the last 15–20 amino acids of their carboxy-terminal tail [27], [42]. Importantly, this region is subject to a diverse range of post-translational modifications [43], [44]. These modifications have the potential to influence vital protein–protein interactions with the cell cytoskeleton/microtubule network [45], [46]. For example, post-translational modifications have been identified for βIII-tubulin in ovarian cancer cells when exposed to stress including hypoxia and glucose-starvation [47], [48]. These modifications allowed βIII-tubulin to form direct protein–protein interactions with key signaling proteins which are involved in promoting cell survival and metastases. In support of this concept, a study by Miller et al. [25] identified a novel post-translational modification in βIVb-tubulin in liver tumors isolated from an orthotopic hepatic cancer mouse model. The post-translational modification was increased during the growth and development of liver cancer and was present in metastases in lungs of mice. Therefore, it is possible that post-translational modifications in βIVb-tubulin may allow for critical protein–protein interactions to occur in PDA cells, which in turn, activate signaling pathways that promote cell survival when the cells are exposed to chemical stress such as chemotherapy drugs.
Another mode of action for βIVb-tubulin in regulating chemosensitivity in PDA cells which should be considered is regulation of microtubule dynamics in the presence of chemotherapy drugs. This highly dynamic process involves the lengthening and shorting of microtubules. This is most critical during cell division and TBAs interfere with this process. For example, Gan et al. [32] demonstrated that NSCLC cells with knockdown of βIII-tubulin had increased suppression of microtubule dynamics when in the presence of low concentrations of vincristine. This correlated to increased cell death. Therefore, it is possible that a similar scenario also occurs in PDA cells with βIVb-tubulin knockdown.
Collectively, this work provides valuable novel insight into the functional roles of βIVa- and βIVb-tubulin in PDA cells. βIVb-tubulin appears to be the major β-tubulin isotype making up total βIV-tubulin in PDA cells, and plays an important role in modulating PDA cell growth and sensitivity to vinca alkaloids. These results provide a rationale for further investigation of sensitisation of PDAs to this class of agents, in an in vivo setting.
The following is the supplementary data related to this article.
The effect of βIVb-tubulin silencing on β-tubulin mRNA expression levels. Graphs showing that knockdown of βIVb-tubulin in MiaPaCa-2 PDA cells does not alter the gene expression levels of total β-tubulin, βI-tubulin, βII-tubulin, βIII-tubulin 48 or 72 h post-transfection. Cells treated with control siRNA (ns-siRNA) served as controls; n = 3.
Footnotes
This work was supported by grants from the National Health and Medical Research Council (NHMRC; P A. Phillips, J McCarroll, M Kavallaris and D Goldstein; APP1024895), Cancer Council New South Wales (P A. Phillips, J McCarroll, M Kavallaris and D Goldstein), Cure Cancer Australia Foundation Grant (P A. Phillips), Cancer Institute NSW Fellowship (J McCarroll, G Sharbeen), NHMRC CDF Fellowship (P A. Phillips, APP1024896), and NHMRC Senior Research Fellowship (Maria Kavallaris, APP1058299). M Kavallaris is funded by the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036) and an NHMRC Program Grant (APP1091261). Biospecimens were provided by the Australian Pancreatic Cancer Genome Initiative (APGI) which is made possible by an Avner Pancreatic Cancer Foundation grant. We would also like to acknowledge Mr Gino Iori for his valuable consumer involvement in this project.
Conflict of interest disclosure: The authors declare no competing financial interest.
References
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardiere C. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein D., El-Maraghi R.H., Hammel P., Heinemann V., Kunzmann V., Sastre J., Scheithauer W., Siena S., Tabernero J., Teixeira L. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst. 2015;107(2) doi: 10.1093/jnci/dju413. http://dx.doi.org/10.1093/jnci/dju413 [pii: dju413] [DOI] [PubMed] [Google Scholar]
- 5.Wang Z., Li Y., Ahmad A., Banerjee S., Azmi A.S., Kong D., Sarkar F.H. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8(1):27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 6.Zalatnai A., Molnar J. Review. Molecular background of chemoresistance in pancreatic cancer. In Vivo. 2007;21(2):339–347. [PubMed] [Google Scholar]
- 7.Katsetos C.D., Draber P. Tubulins as therapeutic targets in cancer: From bench to bedside. Curr Pharm Des. 2012;18(19):2778–2792. doi: 10.2174/138161212800626193. [DOI] [PubMed] [Google Scholar]
- 8.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 9.Verdier-Pinard P., Pasquier E., Xiao H., Burd B., Villard C., Lafitte D., Miller L.M., Angeletti R.H., Horwitz S.B., Braguer D. Tubulin proteomics: towards breaking the code. Anal Biochem. 2009;384(2):197–206. doi: 10.1016/j.ab.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goncalves A., Braguer D., Kamath K., Martello L., Briand C., Horwitz S., Wilson L., Jordan M. Resistance to taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci U S A. 2001;98(20):11737–11742. doi: 10.1073/pnas.191388598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordan M.A., Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 12.Rieder C.L., Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Deve Cell. 2004;7(5):637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M., Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat. 2007;10(4):162–181. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Banerjee A. Increased levels of tyrosinated alpha-, beta (III)-, and beta (IV)-tubulin isotypes in paclitaxel-resistant MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2002;293(1):598–601. doi: 10.1016/S0006-291X(02)00269-3. [DOI] [PubMed] [Google Scholar]
- 15.Karki R., Mariani M., Andreoli M., He S., Scambia G., Shahabi S., Ferlini C. βIII-Tubulin: biomarker of taxane resistance or drug target? Expert Opin Ther Targets. 2013;17(4):461–472. doi: 10.1517/14728222.2013.766170. [DOI] [PubMed] [Google Scholar]
- 16.Kavallaris M., Kuo D., Burkhart C.A., Regl D.L., Norris M.D., Haber M., Horwitz S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest. 1997;100(5):1282–1293. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranganathan S., Benetatos C., Colarusso P., Dexter D., Hudes G. Altered beta-tubulin isotype expression in paclitaxel-resistant human prostate carcinoma cells. Br J Cancer. 1998;77(4):562–566. doi: 10.1038/bjc.1998.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalli K., Brown I., Heys S.D., Schofield A.C. Alterations of β-tubulin isotypes in breast cancer cells resistant to docetaxel. FASEB J. 2005;19(10):1299–1301. doi: 10.1096/fj.04-3178fje. [DOI] [PubMed] [Google Scholar]
- 19.Lee K.M., Cao D., Itami A., Pour P.M., Hruban R.H., Maitra A., Ouellette M.M. Class III beta-tubulin, a marker of resistance to paclitaxel, is overexpressed in pancreatic ductal adenocarcinoma and intraepithelial neoplasia. Histopathology. 2007;51(4):539–546. doi: 10.1111/j.1365-2559.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 20.McCarroll J.A., Sharbeen G., Liu J., Youkhana J., Goldstein D., McCarthy N., Limbri L.F., Dischl D., Ceyhan G.O., Erkan M. betaIII-tubulin: a novel mediator of chemoresistance and metastases in pancreatic cancer. Oncotarget. 2015;6(4):2235–2249. doi: 10.18632/oncotarget.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cucchiarelli V., Hiser L., Smith H., Frankfurter A., Spano A., Correia J.J., Lobert beta-tubulin isotype classes II and V expression patterns in nonsmall cell lung carcinomas. Cell Motil Cytoskeleton. 2008;65(8):675–685. doi: 10.1002/cm.20297. [DOI] [PubMed] [Google Scholar]
- 22.Cullen K.J., Schumaker L., Nikitakis N., Goloubeva O., Tan M., Sarlis N.J., Haddad R.I., Posner M.R. beta-Tubulin-II expression strongly predicts outcome in patients receiving induction chemotherapy for locally advanced squamous carcinoma of the head and neck: a companion analysis of the TAX 324 trial. J Clin Oncol. 2009;27(36):6222–6228. doi: 10.1200/JCO.2009.23.0953. [DOI] [PubMed] [Google Scholar]
- 23.Dozier J.H., Hiser L., Davis J.A., Thomas N.S., Tucci M.A., Benghuzzi H.A., Frankfurter A., Correia J.J., Lobert beta class II tubulin predominates in normal and tumor breast tissues. Breast Cancer Res. 2003;5(5):R157–R169. doi: 10.1186/bcr631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan P.P., Kavallaris M. Tubulin-targeted drug action: Functional significance of class II and class IVb β-tubulin in Vinca alkaloid sensitivity. Cancer Res. 2008;68(23):9817–9824. doi: 10.1158/0008-5472.CAN-08-1501. [DOI] [PubMed] [Google Scholar]
- 25.Miller L.M., Menthena A., Chatterjee C., Verdier-Pinard P., Novikoff P.M., Horwitz S.B., Angeletti R.H. Increased levels of a unique post-translationally modified βIVb-tubulin isotype in liver cancer. Biochemistry. 2008;47(28):7572–7582. doi: 10.1021/bi8005225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ranganathan S., Dexter D.W., Benetatos C.A., Chapman A.E., Tew K.D., Hudes G.R. Increase of βIII-and βIVa-tubulin isotypes in human prostate carcinoma cells as a result of estramustine resistance. Cancer Res. 1996;56(11):2584–2589. [PubMed] [Google Scholar]
- 27.Leandro-García L.J., Leskelä S., Landa I., Montero-Conde C., López-Jiménez E., Letón R., Cascón A., Robledo M., Rodríguez-Antona C. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton. 2010;67(4):214–223. doi: 10.1002/cm.20436. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee A., Roach M., Trcka P., Luduena R. Preparation of a monoclonal antibody specific for the class IV isotype of beta-tubulin. Purification and assembly of alpha beta II, alpha beta III, and alpha beta IV tubulin dimers from bovine brain. J Biol Chem. 1992;267(8):5625–5630. [PubMed] [Google Scholar]
- 29.Ludueña R., Banerjee A. The isotypes of tubulin. In: Fojo T., editor. The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Humana Press; 2008. pp. 123–175. [Google Scholar]
- 30.Atjanasuppat K., Lirdprapamongkol K., Jantaree P., Svasti J. Non-adherent culture induces paclitaxel resistance in H460 lung cancer cells via ERK-mediated up-regulation of betaIVa-tubulin. Biochem Biophys Res Commun. 2015;466(3):493–498. doi: 10.1016/j.bbrc.2015.09.057. [DOI] [PubMed] [Google Scholar]
- 31.Gan P.P., Pasquier E., Kavallaris M. Class III β-tubulin mediates sensitivity to chemotherapeutic drugs in non–small cell lung cancer. Cancer Res. 2007;67(19):9356–9363. doi: 10.1158/0008-5472.CAN-07-0509. [DOI] [PubMed] [Google Scholar]
- 32.Gan P.P., McCarroll J.A., Po'uha S.T., Kamath K., Jordan M.A., Kavallaris M. Microtubule dynamics, mitotic arrest, and apoptosis: drug-induced differential effects of betaIII-tubulin. Mol Cancer Ther. 2010;9(5):1339–1348. doi: 10.1158/1535-7163.MCT-09-0679. [DOI] [PubMed] [Google Scholar]
- 33.Gan P.P., McCarroll J.A., Byrne F.L., Garner J., Kavallaris M. Specific beta-Tubulin Isotypes Can Functionally Enhance or Diminish Epothilone B Sensitivity in Non-Small Cell Lung Cancer Cells. PLoS One. 2011;6(6) doi: 10.1371/journal.pone.0021717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conroy T. Activity of vinorelbine in gastrointestinal cancers. Crit Rev Oncol Hematol. 2002;42(2):173–178. doi: 10.1016/s1040-8428(01)00180-9. [DOI] [PubMed] [Google Scholar]
- 35.Dumontet C., Jordan M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8(8):2086–2095. doi: 10.1158/1535-7163.MCT-09-0366. [DOI] [PubMed] [Google Scholar]
- 37.Teo J., McCarroll J.A., Boyer C., Youkhana J., Sagnella S.M., Duong H.T., Liu J., Sharbeen G., Goldstein D., Davis T.P. A Rationally Optimized Nanoparticle System for the Delivery of RNA Interference Therapeutics into Pancreatic Tumors in Vivo. Biomacromolecules. 2016;17(7):2337–2351. doi: 10.1021/acs.biomac.6b00185. [DOI] [PubMed] [Google Scholar]
- 38.Akan I., Akan S., Akca H., Savas B., Ozben T. Multidrug resistance-associated protein 1 (MRP1) mediated vincristine resistance: effects of N-acetylcysteine and Buthionine sulfoximine. Cancer Cell Int. 2005;5(1):22. doi: 10.1186/1475-2867-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barancik M., Bohacova V., Sedlak J., Sulova Z., Breier A. LY294,002, a specific inhibitor of PI3K/Akt kinase pathway, antagonizes P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci. 2006;29(5):426–434. doi: 10.1016/j.ejps.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 40.van Zanden J.J., de Mul A., Wortelboer H.M., Usta M., van Bladeren P.J., Rietjens I.M., Cnubben N.H. Reversal of in vitro cellular MRP1 and MRP2 mediated vincristine resistance by the flavonoid myricetin. Biochem Pharmacol. 2005;69(11):1657–1665. doi: 10.1016/j.bcp.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Zhang H., Li B., Bai S.W., Wang H.J. Constitutively active Akt contributes to vincristine resistance in human retinoblastoma cells. Cancer Invest. 2010;28(2):156–165. doi: 10.3109/07357900903179641. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan K.F., Cleveland D.W. Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proc Natl Acad Sci U S A. 1986;83(12):4327–4331. doi: 10.1073/pnas.83.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janke C., Bulinski J.C. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12(12):773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- 44.Luchko T., Torin Huzil J., Stepanova M., Tuszynski J. Conformational analysis of the carboxy-terminal tails of human β-tubulin isotypes. Biophys J. 2008;94(6):1971–1982. doi: 10.1529/biophysj.107.115113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huzil J.T., Chen K., Kurgan L., Tuszynski J.A. The roles of beta-tubulin mutations and isotype expression in acquired drug resistance. Cancer Inform. 2007;3:159–181. [PMC free article] [PubMed] [Google Scholar]
- 46.Panda D., Miller H.P., Banerjee A., Luduena R.F., Wilson L. Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proc Natl Acad Sci U S A. 1994;91(24):11358–11362. doi: 10.1073/pnas.91.24.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cicchillitti L., Di Michele M., Urbani A., Ferlini C., Donati M.B., Scambia G., Rotilio D. Comparative proteomic analysis of paclitaxel sensitive A2780 epithelial ovarian cancer cell line and its resistant counterpart A2780TC1 by 2D-DIGE: The role of ERp57. J Proteome Res. 2009;8(4):1902–1912. doi: 10.1021/pr800856b. [DOI] [PubMed] [Google Scholar]
- 48.Raspaglio G., De Maria I., Filippetti F., Martinelli E., Zannoni G.F., Prislei S., Ferrandina G., Shahabi S., Scambia G., Ferlini C. HuR regulates β-tubulin isotype expression in ovarian cancer. Cancer Res. 2010;70(14):5891–5900. doi: 10.1158/0008-5472.CAN-09-4656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of βIVb-tubulin silencing on β-tubulin mRNA expression levels. Graphs showing that knockdown of βIVb-tubulin in MiaPaCa-2 PDA cells does not alter the gene expression levels of total β-tubulin, βI-tubulin, βII-tubulin, βIII-tubulin 48 or 72 h post-transfection. Cells treated with control siRNA (ns-siRNA) served as controls; n = 3.