

Abstract
Pancreatic cancer is characterized by K-Ras mutations in over 90% of the cases. The mutations make the tumors aggressive and resistant to current therapies resulting in very poor prognoses. Valiant efforts to drug mutant K-Ras and related proteins for the treatment of cancers with Ras mutations have been elusive. The need thus persists for therapies to target and suppress the hyperactive K-Ras mutant proteins to normal levels of activity. Polyisoprenylated cysteinyl amide inhibitors (PCAIs) of polyisoprenylated methylated protein methyl esterase (PMPMEase) were designed to disrupt polyisoprenylated protein metabolism and/or functions. The potential for PCAIs to serve as targeted anticancer agents for pancreatic cancer was evaluated in pancreatic ductal adenocarcinoma (PDAC) cell lines expressing mutant (MIAPaCa-2 and Panc-1) and wild type (BxPC-3) K-Ras proteins. The PCAIs inhibited MIAPaCa-2 and BxPC-3 cell viability and induced apoptosis with EC50 values as low as 1.9 µM. The PCAIs, at 0.5 µM, inhibited MIAPaCa-2 cell migration by 50%, inhibited colony formation and disrupted F-actin filament organization. The PCAIs blocked MIAPaCa-2 cell progression at the G0/G1 phase. These results reveal that the PCAIs disrupt pertinent biological processes that lead to pancreatic cancer progression and thus have the potential to act as targeted effective treatments for pancreatic cancer.
Keywords: PCAIs, PMPMEase, pancreatic cancer, cell migration, cell proliferation, F-actin
Introduction
Although pancreatic cancer is the twelfth most diagnosed cancer, its aggressive nature and lack of effective therapies makes it the fourth most deadly cancer in the United States, and based on current trends, it is expected to become the second leading cause of cancer-related deaths by 2020 [1]. The abysmal five-year survival rates of less than 5% implies that 53,070 new pancreatic cases are projected in 2016 while as much as 41,780 are expected to die from it [2-4]. In the absence of new tools for early diagnosis and novel targeted and more effective therapies, the prognosis for PDAC is certain to remain poor. The poor prognoses associated with pancreatic cancers are linked to resistance to most therapies due mainly to mutations in the tumor suppressor genes p53, p16 and Smad4, and most importantly the Ras proto-oncogene that regulates cell growth, survival and proliferation [5]. The monomeric G-protein family of proteins, which includes Ras, function as molecular switches. They cycle through the activated GTP-bound and the inactivated GDP-bound states following GTP hydrolysis using their intrinsic GTPase activities [6]. This GTPase activity is abrogated in the hyperactive mutant forms of the Ras proteins that drive the vast majority of pancreatic cancer cases [7]. The perpetual growth signaling by the GTP-bound mutant proteins makes the tumors resistant to drugs such as the monoclonal antibodies and tyrosine kinase inhibitors that are directed mainly at such upstream targets as the epidermal growth factor receptor. Targeted disruption of Ras metabolism and/or functional interactions in tumors with mutant Ras is desirous as a counter measure to these deleterious hyperactive effects.
For about three decades, the essential polyisoprenylation pathway modifications have attracted most of the drug discovery and development attempts to drug oncogenic Ras [8]. To this effect, the initial step involving the coupling of the polyisoprenyl moiety to the mercapto group of cysteine catalyzed by polyisoprenyl transferase attracted the most attention, with some polyisoprenylation inhibitors undergoing clinical trials [9]. Inhibitors of Ras converting enzyme-1 [10,11] and polyisoprenylated protein methyl transferase (PPMTase) [12] have also been developed for potential anticancer uses. Some of the polyisoprenylated compounds that were initially developed as PPMTase inhibitors were subsequently demonstrated to dislodge Ras from plasma membranes thereby perturbing its growth signaling activity [8,13-17]. Despite these efforts, cancers with K-Ras mutations continue to be a major burden. A more recent approach has targeted PMPMEase, a partner enzyme of the last and only reversible step of the pathway. This approach takes into account a prior observation that the methylation step may functionally regulate polyisoprenylated protein function [18]. We previously purified and identified PMPMEase as a serine esterase with a hydrophobic active site as was expected from the substrate specificities studies and irreversible inhibition with mechanism-based inactivators [19,20]. More rigorous substrate kinetics studies clearly indicated the preference of the enzyme for polyisoprenylated substrates [21,22]. The results from these studies were employed in the design of targeted mechanism-based irreversible inhibitors that were then used to demonstrate the pertinent role of PMPMEase in regulating cell growth and survival [23]. Other studies using various classes of synthetic and endogenous compounds have been used to demonstrate that PMPMEase inhibition suppresses various cellular events that promote cancer progression [24-26]. That PMPMEase metabolizes and modulates polyisoprenylated protein function has recently been reported by another group [27]. Recently, we reported on the synthesis of the PCAIs with significant apoptotic effects on pancreatic cancer cell lines [28]. The present manuscript expands on these initial findings by demonstrating that the PCAIs inhibit various biological phenomena that promote tumor growth, cell migration and invasion in cell lines that harbor various Ras mutational.
Materials and methods
Materials
Human pancreatic adenocarcinoma BxPC-3, MIAPaCa-2, Panc-1, HPAF II and embryonic kidney HEK-293 cell lines were purchased from ATCC (Manassas, VA). Dulbecco’s minimum essential medium (DMEM), RPMI 1640 medium and Dulbecco PBS buffer (DPBS) were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS), penicillin and streptomycin were purchased from Atlanta Biological (Atlanta, GA). CellTiter-Blue Cell Viability Assay reagents were purchased from Promega (Madison, WI). Calcein AM was from Life Technologies (Waltham, MA). LifeAct-TagRFP plasmid was purchased from Ibidi (Verona, WI).
Cell culture
BxPC-3 cells were grown in RPMI 1640 medium while MIAPaCa-2, Panc-1, HPAF II and HEK-293 cells in DMEM. The growth media were supplemented with 10% heat inactivated FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were cultured in a 5% CO2/95% humidified airflow incubator maintained at 37°C and sub-cultured when 80-90% confluent. Unless otherwise stated, assays were conducted with experimental media containing 5% FBS.
Effect of PCAIs on cell viability
Cells (10,000/well) were plated in 96-well plates containing 100 μL experimental media per well and treated for 48 h with 0-50 μM of each of the PCAIs dissolved in Dulbecco PBS. Control cells were treated with equivalent volumes of Dulbecco PBS. The treatments were repeated after 24 h. To determine the effects on cell viability, 20 μL of 0.02% resazurin reagent prepared in Dulbecco PBS was added to each well at the end of the 48 h treatment. After 2 h incubation, fluorescence was measured at an excitation wavelength of 560 nm and a detection wavelength of 590 nm using the FLx 800 Microplate Fluorescence Reader from BioTek (Winooski, VT). The EC50 values were determined from the non-linear regression plots of the logs of the PCAIs concentrations against cell viability using Graph Pad Prism 5 software (San Diego, CA).
Effect of PCAIs on cell proliferation
Cells (5,000/well) were plated in 96-well plates for 24 h and the viability of cells in 8 of the wells was determined using CellTiter-Blue Cell Viability Assay kit as previously described [24] to be used as the baseline relative fluorescence unit (RFU) value at the onset of the treatments. The cells in the remaining wells were treated for 48 h and the viabilities similarly determined. Cell proliferation was computed as follows: Cell proliferation = 5000 cells × (RFU of treated cells after 48 h - RFU of untreated cells at 0 h)/(RFU of untreated cells at 0 h).
Effect of PCAIs on cell survival and colony formation
MIAPaCa-2 cells (500; 1,000; 20,000 and 50,000/well) were plated in triplicates in 6-well plates for 24 h. The cells were treated with PCAIs at the onset and repeated 24 h after the initial treatment. Thereafter, growth medium was replaced every 3 days until visible colonies were observed (10-14 days). The medium for each well was replaced with 1 mL of 0.07% glutaraldehyde and incubated for 30 min at room temperature to fix the viable cells. Dead cells were rinsed off with distilled water and the wells air-dried for 30 min with the aid of hood vents. Crystal violet solution (0.1%, 1 mL) was added to each well and incubated for 30 min before rinsing several times with distilled water. The plates were air-dried at room temperature for 2 h and photographed. The fluorescent crystal violet dye taken up by the live cells was solubilized by the modified method published by Gillies et al. [29]. Briefly, cells that picked up the dye were distained with 1 mL solution of 0.05 M NaH2PO4 (in a 1:1 mixture of distilled water and ethanol) and quantified at 540 nm in a plate reader (Bio-Tek EL800). Cell survival was calculated as the percentages of the fluorescence values of the treated cells relative to those of the untreated controls. In another experiment, cells were treated as described above and after imaging the plates, colonies containing 50 or more cells were counted using a multifunctional colony counter (e-count, Vernon Hills, IL). The plating efficiency (PE) was calculated as [number of colonies formed/number of cells seeded] × 100 for the untreated wells. The surviving fraction (SF) was calculated as follows: SF = (number of colonies formed after treatment/number of cells seeded) × PE as previously reported [30].
Mechanism of PCAIs-induced cell death
Two methods were used to determine the mode of the observed PCAIs-induced cell death. The first method was the modified acridine orange/ethidium bromide (AO/EB) staining method [31]. Briefly, exponentially growing BxPC-3 or MIAPaCa-2 cells were harvested using trypsin-EDTA and 5,000 cells seeded per well in 8-well Lab-Tech tissue culture chamber slides and incubated for 24 h in experimental medium containing 5% FBS. Thereafter, cells were treated either with PBS (controls) or with 0-2 µM of NSL-BA-056 at the onset and again after 24 h later. After 48 h exposure to the test compound, the cells were stained with AO/EB (10 mg/mL) and images of the cells were captured using an Olympus fluorescent microscope (20 × objective) with a 480/30 nm excitation filter fitted with an Olympus DP70 camera.
In another study, Annexin V, a calcium-dependent phospholipid-binding protein with a high affinity for phosphatidylserine (PS) was used to further assess the mechanism of cell death. Briefly, 5 × 105 cells were seeded into six-well culture plates (Costar Corning, NY) and cultured for 24 h in experimental medium containing 5% FBS. Thereafter, the cells were treated with 0-2 µM NSL-BA-055 and incubated for 48 h. The cells were harvested using trypsin-EDTA, washed and stained with the BD Pharmingen Annexin V-propidium iodide FITC apoptosis reagents according to the manufacturer’s protocol. Briefly, cells were washed twice with Dulbecco PBS maintained at 4°C and re-suspended in 100 µL of binding buffer solution in a 5 mL culture tube. Annexin V and propidium iodide (5 µL each) were added and gently stirred before incubating in the dark at room temperature for 15 min. Binding buffer (400 µL) was added to each tube and the cells analyzed for apoptosis using the Becton Dickinson FACSCalibur flow cytometer (San Jose, CA, USA). For each sample, a total of 10,000 individual events from the gated subpopulation were separately analyzed. CellQuest software was used for acquisition and analysis of the data and the percentage of cells in each phase was determined with ModFit 3.0 software (Verity Software House, Topsham, ME). The percentage of cells in each phase was plotted using the GraphPad Prism 5 as shown in the results.
Effect of the PCAIs on the cell cycle progression
The effect of PCAIs on pancreatic cancer cell progression was investigated using the C6 Accuri flow cytometer (Accuri Cytometers, Ann Arbor, MI). MIAPaCa-2 cells (1 × 106 per well) were seeded overnight in experimental medium containing 5% FBS before treating with 0-10 μM NSL-BA-055 at the onset, followed by a repeat treatment 24 h later. The cells were incubated over 5% CO2 at 37°C. At 48 h from the onset of treatment, the cells were trypsinized, and centrifuged at 2500 rpm for 10 min at room temperature. Each pellet was re-suspended in 1 mL of cold PBS, centrifuged at 2500 rpm for 5 min and the supernatant decanted. Modified Vindelov’s reagent (500 μL from a mixture consisting of 100 mL PBS, 1 mg Ribonuclease A, 7.5 mg propidium Iodide) [32] was added to each tube after which they were gently mixed and incubated overnight in the dark at 4°C. The phase distribution of the cells was analyzed within 24 h using the C6 Accuri flow cytometer. A total of 10,000 events from the gated subpopulation were separately analyzed from each sample. C6 Accuri flow cytometer software was used for the acquisition and analysis of the data. The percentage of cells in each phase was determined in the gated population of singlet cells and plotted using the GraphPad Prism 5.
Effect of PCAIs on pancreatic cancer cell migration and invasion
The wound healing/scratch method was used to determine the effect of PCAIs on MIAPaCa-2 cell motility. The cells (1 × 106/well) were seeded in six-well culture plates in medium containing 10% FBS and placed in 5% CO2/95% humidified airflow incubator maintained at 37°C. The medium was replaced every 48 h and 4 days after, 99% confluent monolayer of cells was observed. The wounds were created on the monolayers by introducing three uniform straight scratches on the surface of each well. Cell debris were washed with Dulbecco PBS and growth medium containing 10% FBS was added. Images of the wounds were taken immediately with an Olympus DP70 microscope. The wounded monolayers of cells were then treated with 0-2 µM of NSL-BA-055. Images were captured and documented at 12 and 24 h time intervals. Prior to capturing the images at the 24 h interval, cells were stained with Calcein AM (for better visualization and demarcation of wounds). The pictures were analyzed by counting the number of cells that migrated into the wounded areas using NIH ImageJ software (http://rsb.info.nih.gov/ij/).
In another experiment, MIAPaCa-2 cells were cultured to about 90% confluent and starved for 12 hours. The Matrigel invasion chambers (inserts) were hydrated per manufacturer’s protocol by placing 0.5 mL of serum-free DMEM (1x) in the top chambers and 0.75 mL in the bottom chambers for at least 2 h in humidified 5% CO2 cell culture incubator maintained at 37°C. After hydration, 50,000 cells in 500 µL were added to the top chamber with or without NSL-BA-055. Growth medium with 10% FBS was added to the bottom chamber of each well and the invasion device incubated for 24 h. The cells were washed with PBS and fixed with 3.7% formaldehyde for 20 minutes. They were then rinsed with PBS and permeabilized with 100% methanol for 20 minutes at room temperature. After rinsing with PBS, the cells were stained with 0.1% crystal violet for 1 h. The invasion chamber inserts were rinsed at least 3 times with Dulbecco PBS and finally in deionized water. Cells at the top of the Matrigel membranes that did not migrate were removed with wetted cotton swaps. The invaded cells at the bottom side of the membranes were observed with an Olympus phase contrast microscope using a 20 × objective and images obtained with an Olympus DP70 camera. The captured images of invaded cells were analyzed using NIH ImageJ software and plotted as the mean numbers of migrated cells versus NSL-BA-055 concentration.
Effect of PCAIs on F-actin filament organization
MIAPaCa-2 cells were transfected with the LifeAct-TagRFP plasmid obtained from Ibidi, according to the manufacturer recommendations. The plasmid harbors a gene that encodes for a protein that fluoresces red upon interaction with F-actin [33]. Briefly, MIAPaCa-2 cells in a 75 cm2 culture flask at more than 90% confluent were transfected with the plasmid using 20 μL of Lipofectamine obtained from Invitrogen (Grand Island, NY) per manufacturer’s recommendation. Transformed cells selected using G418, antibiotic [34] were treated with 0-5 µM of either NSL-BA-055 or NSL-BA-056 for 24 h and the Nikon Eclipse Ti Microscope with time-lapse was used to capture images at 0, 6, 12, 18, and 24 h after treatment. The NIS-Elements software was used to measure the surface areas of at least ten cells per captured image for at least 4 images of each treatment concentration.
Statistical analysis
All results were expressed as the means ± SEM for N = 4. Data were analyzed using one-way analysis of variance (ANOVA). Statistical differences between control and treated groups were determined either by Dunnett’s post-test comparisons or Tukey post-test comparisons. Significance was defined as *P < 0.05; **P < 0.01 and ***P < 0.001. The concentrations that inhibited 50% of the activities (EC50) were obtained from nonlinear regression curves using GraphPad Prism version 5.0 for Windows (San Diego, CA).
Results
PCAIs selectively inhibit human pancreatic cancer cell viability and proliferation
Previous studies on the effects of PCAIs on cell viability indicated that NSL-BA-036, NSL-BA-040, NSL-BA-055 and NSL-BA-056 were very effective against MIAPaCa-2 cells [28]. In the current study, these PCAIs were used to further investigate their effectiveness against a range of biological phenomena that promote cancer progression. First, we investigated whether PCAIs (Figure 1A) preferentially inhibit the viability of different cancer cells of pancreatic origin compared to transformed human embryonic kidney cells. Three human pancreatic cancer cell lines, MIAPaCa-2, Panc-1 and BxPC-3, and human embryonic kidney (HEK-293) cells were used. As shown in Figure 1B, there was a concentration-dependent decrease in viability with increasing concentrations of the PCAIs. The results (Figure 1B and Table 1) show that NSL-BA-055 was the most effective with EC50 values of 2.4, 1.9 and 2.0 µM for MIAPaCa-2, Panc-1 and BxPC-3 cells, respectively. On the contrary, the EC50 value for NSL-BA-055 against HEK-293 was in excess of 50 µM (Figure 1). The EC50 values for the standard pancreatic cancer drugs, gemcitabine and erlotinib, were also in excess of 50 µM (Figure 1B and Table 1). Figure 2 shows the anti-proliferative effect of the PCAIs against MIAPaCa-2 and BxPC-3 cells. Some of the untreated control BxPC-3 and MIAPaCa-2 cells more than tripled in number within 48 h. However, cells treated with PCAIs showed a concentration-dependent reduction in cell proliferation with cell numbers decreasing as the concentration of the PCAIs increased. The concentrations of the PCAIs that inhibited 50% of the growth (GI50 values) were 0.35 µM for BxPC3 cells and 0.40 µM for MIAPaCa-2 cells treated with NSL-BA-055 and 0.60 µM for both BxPC3 and MIAPaCa-2 cells treated with NSL-BA-056 (Table 2). The PCAIs are thus capable of halting pancreatic cell proliferation at low micro- to sub-micromolar concentrations. These results show that PCAIs are several-fold more effective at halting the growth and killing of pancreatic cancer cells than the currently marketed pancreatic cancer drugs. Furthermore, the PCAIs are more selectively toxic to pancreatic cancer cells than the normal embryonic kidney cells.
Figure 1.
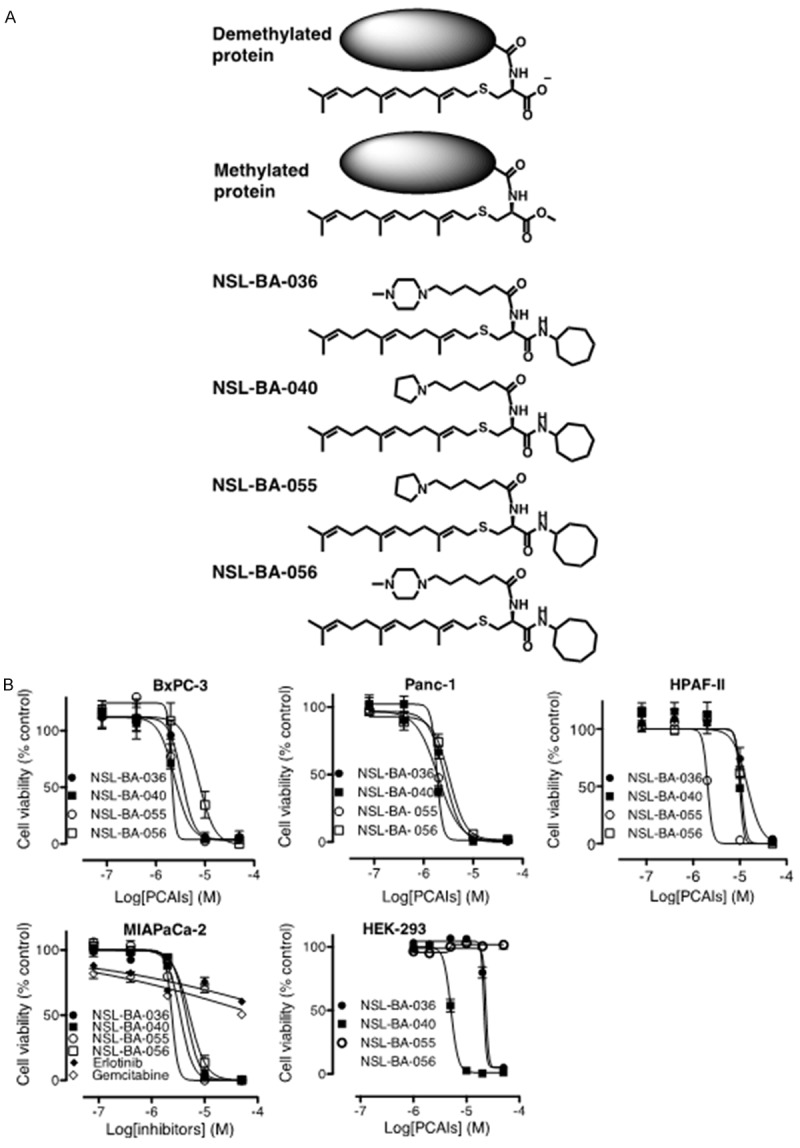
PCAIs inhibit the viabilities of human pancreatic cancer cells. A: Chemical structures of the PCAIs showing their relationship to the polyisoprenyl secondary modifications on proteins. B: Cultured MIAPaCa-2, Panc-1, BxPC-3 and HEK-293 cells were treated for 48 h as described in the methods with the indicated concentrations of PCAIs, erlotinib and gemcitabine. The cell viabilities were evaluated using the resazurin reduction fluorescence assay. The results are the means (± SEM, N = 4) and are representative of four independent determinations.
Table 1.
Relative inhibition of pancreatic cancer and normal cell viabilities by the PCAIs and compared to current therapies
| EC50 (µM) | |||||
|---|---|---|---|---|---|
|
| |||||
| MIAPaCa-2 | Panc-1 | BxPC-3 | HPAF-II | HEK-293 | |
| NSL-BA-036 | 4.4 | 2.6 | 3.3 | 15 | 26 |
| NSL-BA-040 | 3.4 | 1.8 | 2.4 | 9.9 | 8 |
| NSL-BA-055 | 2.4 | 1.9 | 2.0 | 2.1 | > 50 |
| NSL-BA-056 | 5.1 | 3.3 | 7.3 | 11 | 24 |
| Erlotinib | > 50 | ND | ND | > 50 | ND |
| Gemcitabine | > 50 | ND | ND | > 50 | ND |
Figure 2.
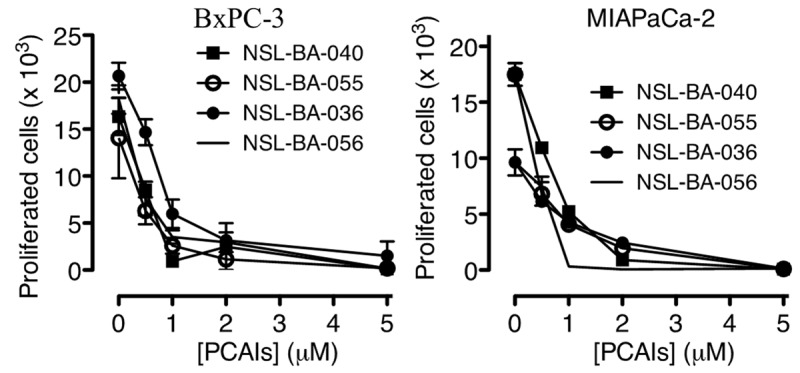
PCAIs inhibit human pancreatic cancer cell proliferation. Cultured MIAPaCa-2 (5,000 cells) and BxPC-3 (5000 cells) cells were treated with the indicated concentrations of PCAIs for 48 h and the cell proliferation was determined as described in the methods. The results are the means (± SEM, N = 4) and are representative of four independent experiments.
Table 2.
Relative growth inhibition of pancreatic cancer cells by the PCAIs
| GI50 (µM) | ||||
|---|---|---|---|---|
|
|
||||
| NSL-BA-036 | NSL-BA-040 | NSL-BA-055 | NSL-BA-056 | |
| MIAPaCa-2 | 0.80 | 0.85 | 0.40 | 0.60 |
| BxPC-3 | 0.75 | 0.90 | 0.35 | 0.56 |
PCAIs inhibit colony formation and cell survival
To investigate the ability of PCAIs at inhibiting human pancreatic cell survival and colony formation, the colony formation and cell survival assays were conducted on MIAPaCa-2 cells. Figure 3 shows the survival fractions for MIAPaCa-2 cells treated with NSL-BA-055 and NSL-BA-056. These reveal a significant decrease in cell colony formation relative to increases in the concentration of both PCAIs (Figure 3). The number of colonies formed relative to the number of cells plated decreased significantly (P < 0.001) when 5,000 cells were plated and treated with 1 µM concentrations of the respective PCAIs compared to the number of colonies formed in the control plated with 500 but untreated cells. The number of surviving colonies approached zero when 50,000 cells were treated with 2 µM of the PCAIs. To validate the clonogenic assay, similar experiments were conducted with NSL-BA-055 and NSL-BA-056. When the cells were stained with crystal violet, washed and the dye dissolved and quantified and the mean optical density (OD) values plotted, the graphs show a concentration-dependent inhibition of viable cells (Figure 3C). It was also noticed that Less than 50% of the treated cells survive at concentrations higher than 1 µM.
Figure 3.
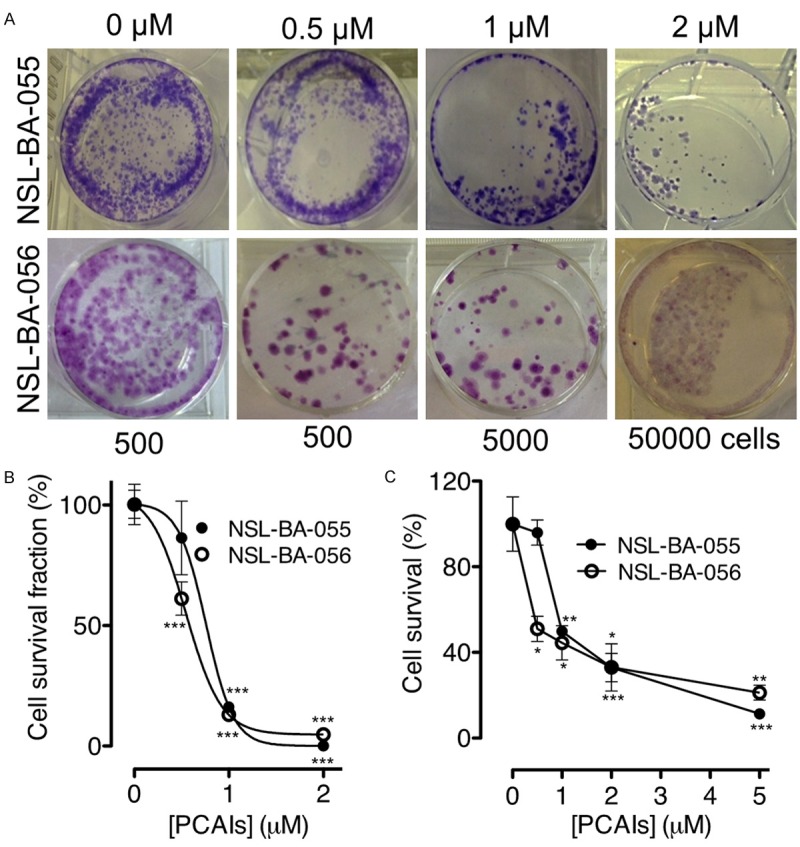
PCAIs inhibit pancreatic cancer colony formation and cell survival. Clonogenic cell survival assays were performed on MIAPaCa-2 cells treated with NSL-BA-055 and NSL-BA-056 (0-5 µM) for 48 h as described in the methods. A: Fluorescent images of the clones of survived cells were captured using an Olympus D70 microscope. B and C: Cell colonies with a minimum of 50 cells were counted and used to derive the cell survival fraction and cell survival plots as described in the methods. The results are the means (± SEM, N = 4) and are representative of four independent experiments. Statistical differences between control and treated groups were determined either by Dunnett’s post-test comparisons. Significance was defined as *P < 0.05; **P < 0.01 and ***P < 0.001.
PCAIs induce apoptosis in pancreatic cancer cells by arresting growth at G0/G1 phase
We further investigated whether PCAIs’ inhibition of pancreatic cancer cell viability, proliferation and survival is due to apoptosis. To achieve this, BxPC-3 and MIAPaCa-2 cells were treated for 48 h with varying concentrations of NSL-BA-055 and NSL-BA-056. The analysis for apoptosis was conducted on BxPC-3 cells using the EB/AO assay. As shown in Figure 4A, 0.5 µM of NSL-BA-056 induced cell death by apoptosis as revealed by the differential uptake of the fluorescent DNA binding dyes (EB/AO). The number of apoptotic cells increased with increasing concentration as depicted by the increasing proportions of orange cell nuclei in the AO/EB overlays (Figure 4A). The Annexin V/propidium iodide and FACS method for determining sub-diploid cell populations [4] was used to analyze the apoptotic effect of the PCAIs on MIAPaCa-2 cells. As shown in Figure 4B, the PCAIs induced apoptosis in MIAPaCa-2 cells in a concentration-dependent manner, with early apoptosis occurring at sub-micro molar concentrations in both assays.
Figure 4.
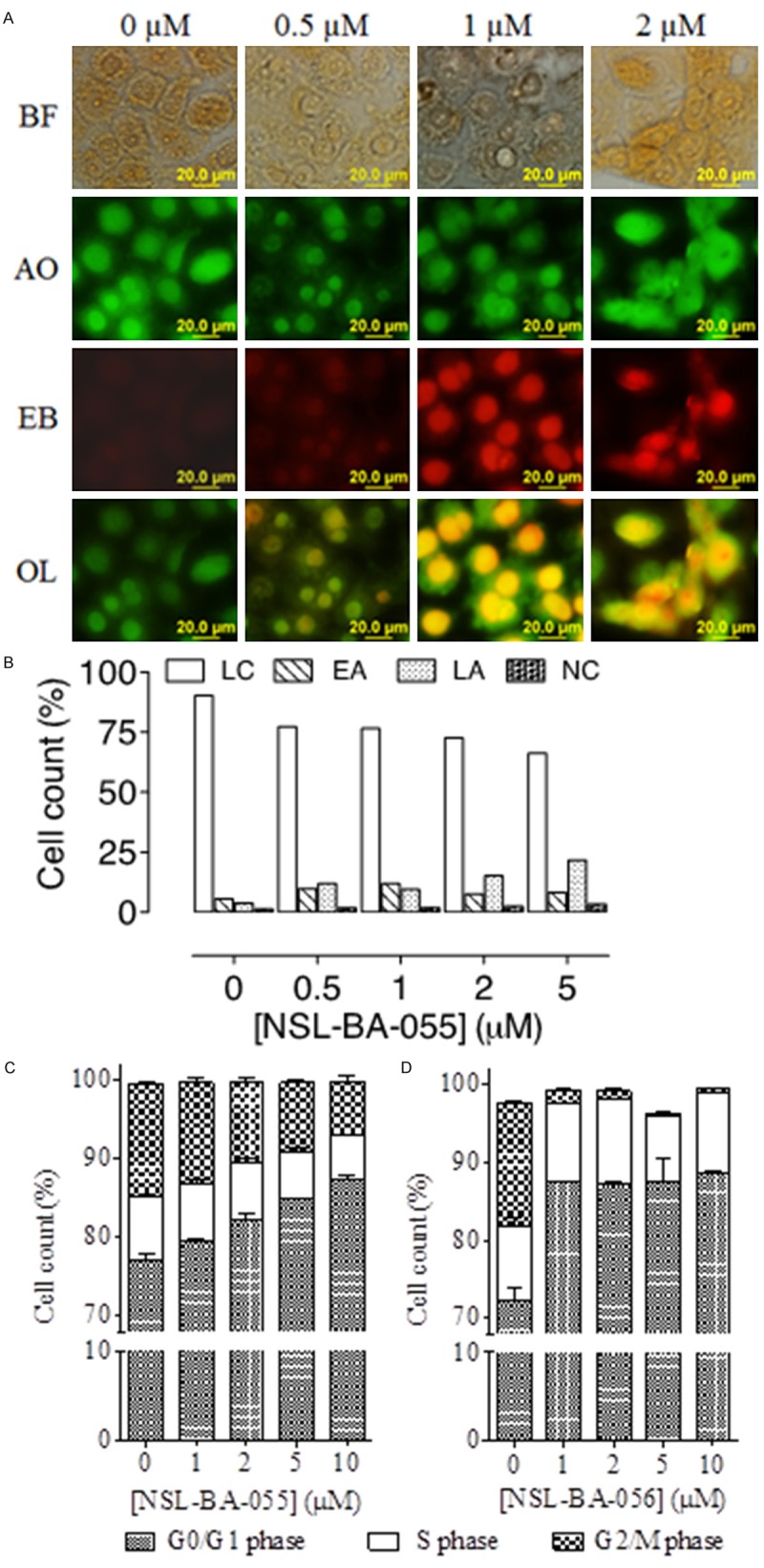
PCAIs induce apoptosis in pancreatic cancer cells by arresting growth at the G0/G1 phase. A: BxPC-3 cells were treated with NSL-BA-056 for 48 h, stained with 10 µg/mL AO/EB and images captured and analyzed as described in the methods. AO stained live cells green and EB stained dying cells with compromised membranes red. BF, Bright Field; AO, Acridine Orange; EB, Ethidium Bromide; OL, overlay of AO and EB images. B and C: MIAPaCa-2 cells were treated with 0-10 µM of either NSL-BA-055 or NSL-BA-056 for 48 h, stained with Vindelov’s reagent as described in the methods and analyzed using a C6 flow cytometer as described in the methods. The relative percent cell counts in each phase of the cell cycle after treatment was plotted. D: MIAPaCa-2cells were treated with NSL-BA-055 for 48 h, stained with FITC-labeled Annexin V and propidium iodide and analyzed as described in the methods. The results are the means (± SEM, N = 4) and are representative of four independent experiments.
To determine whether the effects of PCAIs on the cells involved changes in cell cycle, MIAPaCa-2 cells were treated with the respective PCAIs for 48 h, stained with the Vindelov reagent and analyzed as described in the methods. Figure 4C, 4D reveal a significant concentration-dependent accumulation of cells at the G0/G1 phase coupled with an associated depletion of cells from the G2 phase in the treated MIAPaCa-2 cells when compared to the controls (P < 0.05). The S-phase appeared to be least affected by the PCAIs.
PCAIs inhibit pancreatic cancer cell migration and invasion by disrupting F-actin filaments
The cell migration and invasion methods were used to determine the potential of PCAIs to inhibit the metastatic tendencies of pancreatic cancer cells. In the wound-healing assay, 2 µM of NSL-BA-055 inhibited MIAPaCa-2 cell migration by about 75% (Figure 5A, 5B). Using the Matrigel invasion chambers method, it was further determined that NSL-BA-055 inhibits MIAPaCa-2 cell migration and invasion in a concentration-dependent manner as shown on Figure 5C, 5D. Treatment with sub-micromolar concentrations of NSL-BA-055 resulted in a 4-fold inhibition of the number of cells that migrated and invaded the membrane (Figure 5D). To determine whether the inhibition of cell migration may be linked to effects on actin filament organization, MIAPaCa-2 cells expressing an RFP-conjugate protein that fluoresces red on interacting with F-actin were used. The RFP-tagged protein did not have any significant effects on the relative viability of the cells compared to the non-transfected cells as depicted by the near-identical cell viability EC50 values following treatment with NSL-BA-055 (results not shown). Most importantly, the transfected cells expressing the RFP-tagged protein maintained the morphological characteristics of the non-transfected cells. Since changes in F-actin organization usually translate into alterations in cell morphology [35,36], the morphological characteristics of the transfected cells were used to quantify the effects of the PCAIs on F-actin. As shown in Figure 6A, cells in the treated wells were mostly round in nature in stark contrast to the elongated and more expansive shapes of the untreated cells. Plots of the cell surface area revealed significant decreases with increasing concentrations of the PCAIs (Figure 6B). It was also observed that treatment with 1 to 5 µM of NSL-BA-055 and NSL-BA-056 reduced the average surface areas of the cells by 50 to 80% within 24 h (P < 0.001) compared to the controls (Figure 6B).
Figure 5.
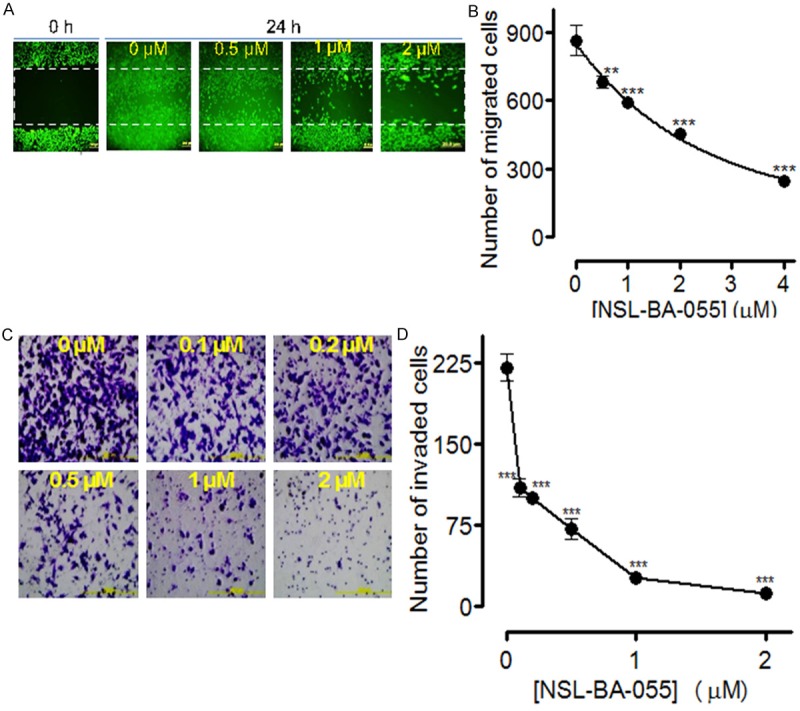
PCAIs inhibit pancreatic cancer cell migration and invasion. A: MIAPaCa-2 cells were incubated in 12-well plates for 24 h, a wound was created with pipette tip, and the cells treated with the indicated concentrations of the NSL-BA-055. After 24 h, the extent of closure was monitored under the microscope and photographed. Representative images of four independent experiments conducted in triplicates are shown. B: MIAPaCa-2 cells were treated with the indicated concentrations of NSL-BA-055. C and D: The cell invasion assay was performed in the Matrigel invasion chambers as described in the methods. All Representative images were 10 × magnification and all data of invaded cells were represented as ± SEM, N = 8-10. Statistical differences between control and treated groups were determined by Dunnett’s post-test comparisons. Significance was defined as *P < 0.05; **P < 0.01 and ***P < 0.001.
Figure 6.
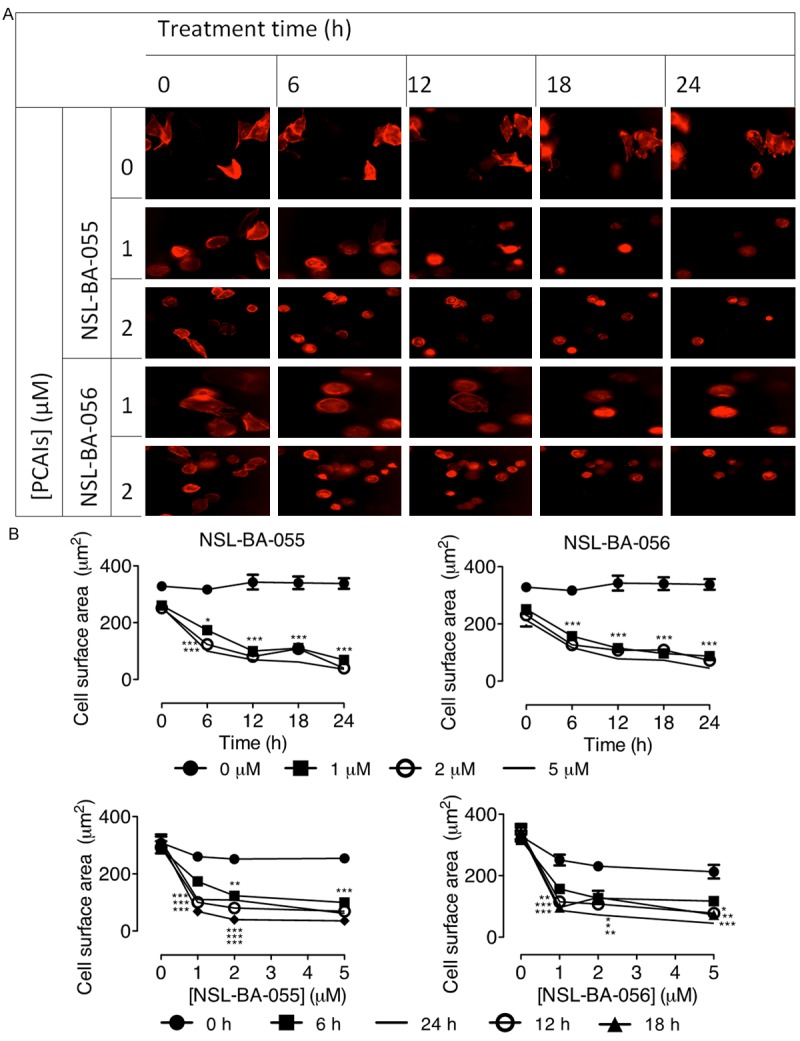
PCAIs alter pancreatic cancer cell morphology by disrupting F-actin filaments. Transfected MIAPaCa-2 cells were treated for 24 h with the indicated concentrations of PCAIs and their effect on F-actin expression was quantified by measuring the cell surface areas using Nikon NIS-Elements software. A: Representative images of treated and untreated control cells were captured at 20 × magnification using Nikon Eclipse Ti Microscope at the indicated time points. B: Cell surface area at various treatment time intervals (upper panels) and concentration after 24 h treatment (lower panels). The results are expressed as the means (± SEM, N = 15-20). Statistical differences between the treated and untreated controls were determined by Tukey post-test comparisons. Significant differences are shown by *P < 0.05; **P < 0.01 and ***P < 0.001.
Discussion
Some of the most effective cancer therapies have been those targeting the molecular alterations driving the disease in the afflicted individuals in what is widely described as personalized medicine. Therefore, the identification of biomarkers has offered opportunities for the development of the usually more effective targeted therapies. Unfortunately this has not been the case for pancreatic cancer in which the agents designed to target the mutated hyperactive Ras that drives tumor progression in the preponderance of cases have not been effective. We recently reported that PMPMEase, an enzyme that has been demonstrated to metabolize polyisoprenylated proteins in cells [27], is overexpressed in various cancer tissues including those from pancreatic cancer [28,37,38]. This implies that PMPMEase may be a useful target for agents that could potentially be useful against pancreatic cancer. The PCAIs, though very poor inhibitors of PMPMEase [28], were far more effective against cancer cell viability in preliminary screening results [28]. The PCAIs, being polyisoprenylated small molecules, may be interfering with the polyisoprenylation-dependent functional interactions of the endogenous polyisoprenylated protein substrates of the enzyme, possibly uncoupling growth signals originating from the cell surface receptors from their intracellular effectors. This has been demonstrated in previous studies with the polyisoprenylated small molecule, farnesylthiosalicylic acid (FTS) in which the effects against cell viability were coupled with dislodging of Ras from its intracellular signaling location on the cytosolic side of the plasma membranes [14,39].
The observation in the current study that the PCAIs disrupt F-actin filaments causing the cells to become more rounded as well as inhibiting cell proliferation and viability by blocking cell cycle progression suggests possible interference with polyisoprenylated protein functions. Interference with Ras interactions is certain to inhibit the growth-stimulatory signaling of even the constitutively active mutant forms found in the majority of pancreatic cancer cases. The observed apoptotic and cell cycle arrest suggest possible disruption of Ras signaling. Also, the recent demonstration that PMPMEase hydrolyzes RhoA methyl esters [40] suggests that PMPMEase overexpression in pancreatic cancer may contribute to its progression. RhoA plays an important role in focal adhesion in which F-actin is organized in the formation filopodia and lamellipodia necessary for cell movement [41,42]. As such, disruption of RhoA interactions would be expected to alter cell shape and suppress the cells’ ability to migrate and invade as observed in this study. In addition to the PCAIs’ ability to curtail cell proliferation and induce apoptosis, disrupting cell migration has the potential to limit metastasis, which is one of the most devastating aspects of cancer biology.
The polyisoprenylation pathway has attracted a lot of research interest due to the role polyisoprenylated Ras, Rho and related proteins play in regulating cell biological activities that are of significance to cancer progression [23,43-45]. Although the Ras mutations destroy their ability to function as molecular switches, the deleterious hyperactive changes that spur the cancerous tendencies can be mitigated if the interactions involving the activated proteins can be mitigated with molecules such as the PCAIs. This is plausible given that the essential nature of the polyisoprenylation modifications to the proteins’ functional interactions have been aptly demonstrated [10,14,17,46,47]. If the PCAIs disrupt the polyisoprenylation-dependent functional interactions, the consequences would be similar to blocking the secondary modifications that are essential to their functions.
Molecular profiling approaches have revealed that the activation of growth signaling intermediates are at the core of molecular events that initiate and/or promote various tumors. For example, the overexpression of growth factors [48,49], growth factor receptors and/or activating mutations of their enzyme domains [50-52] and signaling downstream factors such as Ras [17,53-57] and kinases [58-61] have all been demonstrated to play major roles in promoting various cancers. The fact that Ras and Rho are intracellular and downstream to the growth factors and their receptors implies that the PCAIs may potentially be useful not only for treating cancers in which the monomeric GTPases are hyperactive but may also be effective against those cases that are promoted by growth factors and their receptors.
Acknowledgement
The research reported in this publication was supported by the National Cancer Institute (NCI) and National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Grant SC1CA190505 and by the National Institute On Minority Health and Health Disparities (NIMHD) of the NIH under Award Number G12 MD007582 (previous award NIH/NCRR/RCMI G12 RR03020). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
None.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Fleming JB, Shen GL, Holloway SE, Davis M, Brekken RA. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol Cancer Res. 2005;3:413–423. doi: 10.1158/1541-7786.MCR-04-0206. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, Wingo PA, Howe HL, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975-2001, with a special feature regarding survival. Cancer. 2004;101:3–27. doi: 10.1002/cncr.20288. [DOI] [PubMed] [Google Scholar]
- 4.Howlader N, Ries LA, Mariotto AB, Reichman ME, Ruhl J, Cronin KA. Improved estimates of cancer-specific survival rates from population-based data. J Natl Cancer Inst. 2010;102:1584–1598. doi: 10.1093/jnci/djq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt CM, Powell ES, Yiannoutsos CT, Howard TJ, Wiebke EA, Wiesenauer CA, Baumgardner JA, Cummings OW, Jacobson LE, Broadie TA, Canal DF, Goulet RJ Jr, Curie EA, Cardenes H, Watkins JM, Loehrer PJ, Lillemoe KD, Madura JA. Pancreaticoduodenectomy: a 20-year experience in 516 patients. Arch Surg. 2004;139:718–725. doi: 10.1001/archsurg.139.7.718. discussion 725-717. [DOI] [PubMed] [Google Scholar]
- 6.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 7.Spangler C, Spangler CM, Spoerner M, Schaferling M. Kinetic determination of the GTPase activity of Ras proteins by means of a luminescent terbium complex. Anal Bioanal Chem. 2009;394:989–996. doi: 10.1007/s00216-008-2517-7. [DOI] [PubMed] [Google Scholar]
- 8.Blum R, Cox AD, Kloog Y. Inhibitors of chronically active ras: potential for treatment of human malignancies. Recent Pat Anticancer Drug Discov. 2008;3:31–47. doi: 10.2174/157489208783478702. [DOI] [PubMed] [Google Scholar]
- 9.Adjei AA. Ras signaling pathway proteins as therapeutic targets. Curr Pharm Des. 2001;7:1581–1594. doi: 10.2174/1381612013397258. [DOI] [PubMed] [Google Scholar]
- 10.Elad G, Paz A, Haklai R, Marciano D, Cox A, Kloog Y. Targeting of K-Ras 4B by S-trans,trans-farnesyl thiosalicylic acid. Biochim Biophys Acta. 1999;1452:228–242. doi: 10.1016/s0167-4889(99)00144-5. [DOI] [PubMed] [Google Scholar]
- 11.Hill BT, Perrin D, Kruczynski A. Inhibition of RAS-targeted prenylation: protein farnesyl transferase inhibitors revisited. Crit Rev Oncol Hematol. 2000;33:7–23. doi: 10.1016/s1040-8428(99)00053-0. [DOI] [PubMed] [Google Scholar]
- 12.Casey PJ, Seabra MC. Protein prenyltransferases. J Biol Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg L, Israeli R, Kloog Y. FTS and 2-DG induce pancreatic cancer cell death and tumor shrinkage in mice. Cell Death Dis. 2012;3:e284. doi: 10.1038/cddis.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraitzer A, Kloog Y, Zilberman M. Novel farnesylthiosalicylate (FTS)-eluting composite structures. Eur J Pharm Sci. 2009;37:351–362. doi: 10.1016/j.ejps.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Haklai R, Elad-Sfadia G, Egozi Y, Kloog Y. Orally administered FTS (salirasib) inhibits human pancreatic tumor growth in nude mice. Cancer Chemother Pharmacol. 2008;61:89–96. doi: 10.1007/s00280-007-0451-6. [DOI] [PubMed] [Google Scholar]
- 16.Reif S, Weis B, Aeed H, Gana-Weis M, Zaidel L, Avni Y, Romanelli RG, Pinzani M, Kloog Y, Bruck R. The Ras antagonist, farnesylthiosalicylic acid (FTS), inhibits experimentally-induced liver cirrhosis in rats. J Hepatol. 1999;31:1053–1061. doi: 10.1016/s0168-8278(99)80318-3. [DOI] [PubMed] [Google Scholar]
- 17.Kloog Y, Cox AD. RAS inhibitors: potential for cancer therapeutics. Mol Med Today. 2000;6:398–402. doi: 10.1016/s1357-4310(00)01789-5. [DOI] [PubMed] [Google Scholar]
- 18.Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Gomes AQ, Seabra MC, Young SG. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J Biol Chem. 2001;276:5841–5845. doi: 10.1074/jbc.C000831200. [DOI] [PubMed] [Google Scholar]
- 19.Oboh OT, Lamango NS. Liver prenylated methylated protein methyl esterase is the same enzyme as Sus scrofa carboxylesterase. J Biochem Mol Toxicol. 2008;22:51–62. doi: 10.1002/jbt.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamango NS. Liver prenylated methylated protein methyl esterase is an organophosphate-sensitive enzyme. J Biochem Mol Toxicol. 2005;19:347–357. doi: 10.1002/jbt.20100. [DOI] [PubMed] [Google Scholar]
- 21.Duverna R, Ablordeppey SY, Lamango NS. Biochemical and docking analysis of substrate interactions with polyisoprenylated methylated protein methyl esterase. Curr Cancer Drug Targets. 2010;10:634–648. doi: 10.2174/156800910791859443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamango NS, Duverna R, Zhang W, Ablordeppey SY. Porcine Liver Carboxylesterase Requires Polyisoprenylation for High Affinity Binding to Cysteinyl Substrates. Open Enzym Inhib J. 2009;2:12–27. doi: 10.2174/1874940200902010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilar B, Amissah F, Duverna R, Lamango NS. Polyisoprenylation potentiates the inhibition of polyisoprenylated methylated protein methyl esterase and the cell degenerative effects of sulfonyl fluorides. Curr Cancer Drug Targets. 2011;11:752–762. doi: 10.2174/156800911796191015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amissah F, Taylor S, Duverna R, Ayuk-Takem LT, Lamango NS. Regulation of polyisoprenylated methylated protein methyl esterase by polyunsaturated fatty acids and prostaglandins. Eur J Lipid Sci Technol. 2011;113:1321–1331. doi: 10.1002/ejlt.201100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amissah F, Duverna R, Aguilar BJ, Poku RA, Lamango NS. Polyisoprenylated methylated protein methyl esterase is both sensitive to curcumin and overexpressed in colorectal cancer: implications for chemoprevention and treatment. Biomed Res Int. 2013;2013:416534. doi: 10.1155/2013/416534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayuk-Takem L, Amissah F, Aguilar BJ, Lamango NS. Inhibition of polyisoprenylated methylated protein methyl esterase by synthetic musks induces cell degeneration. Environ Toxicol. 2014;29:466–477. doi: 10.1002/tox.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cushman I, Cushman SM, Potter PM, Casey PJ. Control of RhoA methylation by carboxylesterase I. J Biol Chem. 2013;288:19177–19183. doi: 10.1074/jbc.M113.467407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguilar BJ, Nkembo AT, Duverna R, Poku RA, Amissah F, Ablordeppey SY, Lamango NS. Polyisoprenylated methylated protein methyl esterase: a putative biomarker and therapeutic target for pancreatic cancer. Eur J Med Chem. 2014;81:323–333. doi: 10.1016/j.ejmech.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillies RJ, Didier N, Denton M. Determination of cell number in monolayer cultures. Anal Biochem. 1986;159:109–113. doi: 10.1016/0003-2697(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 30.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 31.Cohen JJ. Apoptosis. Immunol Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- 32.Vindelov LL. Flow microfluorometric analysis of nuclear DNA in cells from solid tumors and cell suspensions. A new method for rapid isolation and straining of nuclei. Virchows Arch B Cell Pathol. 1977;24:227–242. [PubMed] [Google Scholar]
- 33.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorman CM, Lane DP, Watson CJ, Rigby PW. The regulation of gene expression in murine teratocarcinoma cells. Cold Spring Harb Symp Quant Biol. 1985;50:701–706. doi: 10.1101/sqb.1985.050.01.086. [DOI] [PubMed] [Google Scholar]
- 35.Girard PR, Nerem RM. Shear stress modulates endothelial cell morphology and F-actin organization through the regulation of focal adhesion-associated proteins. J Cell Physiol. 1995;163:179–193. doi: 10.1002/jcp.1041630121. [DOI] [PubMed] [Google Scholar]
- 36.Hashemzadeh-Bonehi L, Curtis PS, Morley SJ, Thorpe JR, Pain VM. Overproduction of a conserved domain of fission yeast and mammalian translation initiation factor eIF4G causes aberrant cell morphology and results in disruption of the localization of F-actin and the organization of microtubules. Genes Cells. 2003;8:163–178. doi: 10.1046/j.1365-2443.2003.00623.x. [DOI] [PubMed] [Google Scholar]
- 37.Poku RA, Amissah F, Duverna R, Aguilar BJ, Kiros GE, Lamango NS. Polyisoprenylated methylated protein methyl esterase as a putative drug target for androgen-insensitive prostate cancer. Ecancermedicalscience. 2014;8:459. doi: 10.3332/ecancer.2014.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amissah F, Duverna R, Aguilar BJ, Poku RA, Kiros GE, Lamango NS. Polyisoprenylated methylated protein methyl esterase overexpression and hyperactivity promotes lung cancer progression. Am J Cancer Res. 2014;4:116–134. [PMC free article] [PubMed] [Google Scholar]
- 39.Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer. Methods Enzymol. 2008;439:467–489. doi: 10.1016/S0076-6879(07)00432-6. [DOI] [PubMed] [Google Scholar]
- 40.Cushman I, Casey PJ. RHO methylation matters: a role for isoprenylcysteine carboxylmethyltransferase in cell migration and adhesion. Cell Adh Migr. 2011;5:11–15. doi: 10.4161/cam.5.1.13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mejillano MR, Kojima S, Applewhite DA, Gertler FB, Svitkina TM, Borisy GG. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 42.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 43.Newman P, Kube E, Gerke V, Weber K. Polyisoprenylation of the CAAX motif--an in vitro protein synthesis study. Biochim Biophys Acta. 1991;1080:227–230. doi: 10.1016/0167-4838(91)90006-l. [DOI] [PubMed] [Google Scholar]
- 44.Schaber MD, O’Hara MB, Garsky VM, Mosser SC, Bergstrom JD, Moores SL, Marshall MS, Friedman PA, Dixon RA, Gibbs JB. Polyisoprenylation of Ras in vitro by a farnesyl-protein transferase. J Biol Chem. 1990;265:14701–14704. [PubMed] [Google Scholar]
- 45.Weber K, Plessmann U, Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett. 1989;257:411–414. doi: 10.1016/0014-5793(89)81584-4. [DOI] [PubMed] [Google Scholar]
- 46.Powers S. Protein prenylation: a modification that sticks. Curr Biol. 1991;1:114–116. doi: 10.1016/0960-9822(91)90295-8. [DOI] [PubMed] [Google Scholar]
- 47.Finegold AA, Schafer WR, Rine J, Whiteway M, Tamanoi F. Common modifications of trimeric G proteins and ras protein: involvement of polyisoprenylation. Science. 1990;249:165–169. doi: 10.1126/science.1695391. [DOI] [PubMed] [Google Scholar]
- 48.Al Zobair AA, Al Obeidy BF, Yang L, Yang C, Hui Y, Yu H, Zheng F, Yang G, Xie C, Zhou F, Zhou Y. Concomitant overexpression of EGFR and CXCR4 is associated with worse prognosis in a new molecular subtype of non-small cell lung cancer. Oncol Rep. 2013;29:1524–1532. doi: 10.3892/or.2013.2254. [DOI] [PubMed] [Google Scholar]
- 49.Livasy CA, Reading FC, Moore DT, Boggess JF, Lininger RA. EGFR expression and HER2/neu overexpression/amplification in endometrial carcinosarcoma. Gynecol Oncol. 2006;100:101–106. doi: 10.1016/j.ygyno.2005.07.124. [DOI] [PubMed] [Google Scholar]
- 50.Chen CC, Chiu HH, Yen LC, Chang HJ, Chang MS, Tsai JR, Chen YF, Lin SR. Overexpression of EGFR pathway-related genes in the circulation is highly correlated with EGFR mutations and overexpression in paired cancer tissue from patients with non-small cell lung cancer. Oncol Rep. 2010;23:639–645. [PubMed] [Google Scholar]
- 51.Zhang L, Yang J, Cai J, Song X, Deng J, Huang X, Chen D, Yang M, Wery JP, Li S, Wu A, Li Z, Liu Y, Chen Y, Li Q, Ji J. A subset of gastric cancers with EGFR amplification and overexpression respond to cetuximab therapy. Sci Rep. 2013;3:2992. doi: 10.1038/srep02992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez-Lacaci I, Kannan S, De Santis M, Bianco C, Kim N, Wallace-Jones B, Ebert AD, Wechselberger C, Salomon DS. RAS transformation causes sustained activation of epidermal growth factor receptor and elevation of mitogen-activated protein kinase in human mammary epithelial cells. Int J Cancer. 2000;88:44–52. doi: 10.1002/1097-0215(20001001)88:1<44::aid-ijc7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 53.Chen JC, Zhuang S, Nguyen TH, Boss GR, Pilz RB. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J Biol Chem. 2003;278:2807–2818. doi: 10.1074/jbc.M207943200. [DOI] [PubMed] [Google Scholar]
- 54.Gaengel K, Mlodzik M. Egfr signaling regulates ommatidial rotation and cell motility in the Drosophila eye via MAPK/Pnt signaling and the Ras effector Canoe/AF6. Development. 2003;130:5413–5423. doi: 10.1242/dev.00759. [DOI] [PubMed] [Google Scholar]
- 55.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 56.Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy (Review) Int J Oncol. 2003;22:237–252. [PubMed] [Google Scholar]
- 57.Perkins G, Lievre A, Ramacci C, Meatchi T, de Reynies A, Emile JF, Boige V, Tomasic G, Bachet JB, Bibeau F, Bouche O, Penault-Llorca F, Merlin JL, Laurent-Puig P. Additional value of EGFR downstream signaling phosphoprotein expression to KRAS status for response to anti-EGFR antibodies in colorectal cancer. Int J Cancer. 2010;127:1321–1331. doi: 10.1002/ijc.25152. [DOI] [PubMed] [Google Scholar]
- 58.Gotoh N, Toyoda M, Shibuya M. Tyrosine phosphorylation sites at amino acids 239 and 240 of Shc are involved in epidermal growth factor-induced mitogenic signaling that is distinct from Ras/mitogen-activated protein kinase activation. Mol Cell Biol. 1997;17:1824–1831. doi: 10.1128/mcb.17.4.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joneson T, Fulton JA, Volle DJ, Chaika OV, Bar-Sagi D, Lewis RE. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem. 1998;273:7743–7748. doi: 10.1074/jbc.273.13.7743. [DOI] [PubMed] [Google Scholar]
- 60.Yart A, Laffargue M, Mayeux P, Chretien S, Peres C, Tonks N, Roche S, Payrastre B, Chap H, Raynal P. A critical role for phosphoinositide 3-kinase upstream of Gab1 and SHP2 in the activation of ras and mitogen-activated protein kinases by epidermal growth factor. J Biol Chem. 2001;276:8856–8864. doi: 10.1074/jbc.M006966200. [DOI] [PubMed] [Google Scholar]
- 61.Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]