

Abstract
The human metastasis associated lung adenocarcinoma transcript 1 (MALAT1) is a long non-coding RNA associated with metastasis, and is a favorable prognostic factor for lung cancer. Recent studies have shown that MALAT1 plays an important role in many malignancies. However, little is known about the role of MALAT1 in glioma. In this study, we determined the expression of MALAT1 and explored its prognostic value in glioma. Further, we investigated the regulatory mechanism of MALAT1 in glioma progression. Our results showed that the expression of MALAT1 was significantly decreased in glioma specimens than in noncancerous brain tissues. In addition, MALAT1 expression was significantly correlated with tumor size, WHO grade and Karnofsky Performance Status (KPS), and was an independent prognostic factor for survival of glioma patients. The gain- and loss-of-function experiments revealed miR-155 down-regulation by MALAT1, resulting in reciprocal effects. Further, MALAT1 suppresses cell viability by down-regulating miR-155. FBXW7 mRNA was identified as a direct target of miR-155 in glioma. The miR-155-induced tumorigenesis is mediated through FBXW7 function. Finally, we found that MALAT1 positively regulated FBXW7 expression, which was responsible for glioma progression mediated by MALAT1-miR-155 pathway. In conclusion, our data demonstrated that MALAT1 may be a novel prognostic biomarker and therapeutic target in glioma. Restoration of MALAT1 levels represents a novel therapeutic strategy against glioma.
Keywords: MALAT1, miR-155, FBXW7, glioma, cell viability
Introduction
Glioma is a common intracranial tumor, accounting for more than 50% of all primary brain tumors [1]. Despite successful surgical resection and chemotherapy, the prognosis of glioma patients remains poor [2]. The molecular mechanisms underlying the etiology of the disease are largely unknown [3]. Therefore, it is essential to identify the critical carcinogenic pathways and identify new and effective diagnostic and prognostic targets for successful management of this devastating disease.
Recently, long non-coding RNAs (lncRNAs) have emerged as new gene regulators and prognostic markers in several cancers including glioma. LncRNAs mediate several biological processes including epigenetic regulation, nuclear import, cell cycle control, nuclear and cytoplasmic trafficking, imprinting, cell differentiation, alternative splicing, RNA decay, transcription and translation [4,5]. Thus, lncRNAs have emerged as new players in cancer research. Several studies have shown that specific lncRNAs function as oncogenes, tumor suppressor genes or both, depending on the conditions [6].
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is an abundantly expressed nuclear lncRNA measuring approximately 8000 nucleotides in length. It was first identified by Ji et al as the lncRNA associated with metastasis and survival in non-small cell lung cancer [7]. Aberrant expression of MALAT1 was also observed in a broad range of human malignant tumors including cervical cancer, breast cancer, bladder cancer and hepatocellular cancer [8-11]. However, the role of MALAT1 in glioma is largely unknown. One study showed that MALAT1 was associated with malignant status and poor prognosis in glioma [12]. However, Han et al reported that it downregulated MMP2 and inactivated ERK/MAPK signaling [13], suggesting the need for additional investigation into the role of MALAT1 as a tumor suppressor in glioma cells.
The role of microRNAs (miRNAs) in gene regulation and cell function has been elucidated in numerous cancers. MiRNAs regulate gene expression primarily via interaction with the 3’UTRs of target mRNAs, resulting in mRNA decay or translational repression. MicroRNA-155 (miR-155) located on chromosome 21q21.3, is known as a B cell integration cluster (BIC) [14]. MiR-155 potentially targets the tumor suppressor genes, SOCS1 and APC, and the kinase WEE1, which blocks the activity of Cdc2 and prevents entry into mitosis [15,16]. However, the specific role of miR-155 in glioma prognosis is largely unknown. The F-box and WD repeat domain containing 7 (FBXW7) protein encodes a substrate adaptor for an E3 SCF ubiquitin ligase complex and negatively regulates the abundance of different oncoproteins [17]. Reduced expression or loss of function of FBXW 7 has been frequently reported in a variety of human cancers, with an overall mutation frequency of 6% [18].
In this study, we investigated the potential role of MALAT1 in glioma progression and further identified the direct targets correlated with the malignant phenotype of glioma. Our results showed that a low MALAT1 expression was frequently associated with concurrent high miR-155 expression in glioma patients. Furthermore, MALAT1 suppressed glioma cell viability by down-regulating miR-155 and activating FBXW7 signaling.
Materials and methods
Clinical samples
Forty-five serum samples, 66 tissues and paired adjacent noncancerous tissues from primary glioma patients were collected at the Cangzhou Central Hospital (China) between 2008 and 2011. Meanwhile, serum samples from 30 healthy volunteers were also collected in this study. All the patients were pathologically confirmed and the tissues were collected immediately during the surgery, and stored at -80°C to prevent RNA loss. They were classified according to the WHO criteria and staged according to the tumor-node-metastasis (TNM) classification. Written informed consent was obtained from all the patients according to the guidelines approved by the Ethics Committee of the Cangzhou Central Hospital.
Cell culture
Human glioma cell lines (U87 and SHG139) were purchased from the Shanghai Institute of Life Sciences Cell Resource Center (Shanghai, China). All the cell lines were cultured in DMEM medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Biowest, France). All the cell cultures were incubated at 37°C and 5% CO2.
RNA oligoribonucleotides and cell transfection
The small interfering RNAs (siRNAs) that specifically target human MALAT1, miR-155 and FBXW7 mRNA were designated as siMALAT1, anti-miR-155 and siFBXW7, respectively. The MALAT1 overexpression plasmid (pMALAT1) was purchased from Addgene. The coding sequence of FBXW7 was amplified and cloned into a PCDNA3.1 vector, and was designated as pFBXW7. The lentivirus vector containing MALAT1 shRNA plasmid (Lv-ShMALAT1) and pre-miR-155 plasmid (Lv-miR-155) was amplified and cloned (Genechem Corporation, Shanghai, China). The negative control duplex (NC) containing both miRNA mimics and siRNA, as well as the single standard negative control RNA for miRNA inhibitors (anti-NC), was not homologous to any human genome sequences. All the RNA oligoribonucleotides were purchased from RiboBio (Guangzhou, China). The transfection of RNA oligoribonucleotides and plasmid was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Quantitative real-time PCR (RT-qPCR)
Total RNA was isolated from glioma specimens or glioma cell lines using TRIzol reagent (Invitrogen). The amounts of miRNAs were quantified in duplicate by quantitative reverse transcription polymerase chain reaction (RT-PCR) using the human TaqMan MicroRNA Assay Kits (Applied Biosystems, Foster City, CA, USA). U6 was used as the internal control for miRNA detection. For mRNA detection, the cDNA was synthesized from 200 ng of extracted total RNA using the PrimeScript RT reagent Kit (Takara Bio Company, Shiga, Japan) and amplified by RT-qPCR using Light Cycler 480 SYBR Green I Master (Roche, Germany) and GAPDH as the control gene. The 2-ΔΔCt method was used to determine the relative quantification of gene expression levels. The premier sequences were as follows: MALAT1 (Forward): GGGTGTTTACGTAGACCAGAACC, (Reverse): CTTCCAAAAGCCTTCTGCCTTAG; FBXW7 (Forward): GGGAGCACTTTGCTGAAATC, (Reverse): CAGCAGCCACTTCTTGAAAC; GAPDH (Forward): GCACCGTCAAGGCTGAGAAC, (Reverse): ATGGTGGTGAAGACGCCAGT.
Cell cycle analysis
After transfection, cells were washed in PBS and fixed in 70% ethanol at 4°C for 2 h. DNA staining was carried out with 10 mg propidium iodide/mL PBS and 2.5 mg Ag DNase-free RNase (Roche Diagnostics)/mL PBS for at least 30 min before flow cytometry using a Coulter EPICS XL flow cytometer (Beckman Coulter, Inc., Fullerton, CA). Cell cycle profiles were generated using flow cytometry with Modifit software (BD Biosciences).
Cell viability assay
Cell viability was quantified using the Cell Counting Kit-8 (Sigma). Briefly, 100 μL of cells from the different transfection groups were seeded on a 96-well plate at a concentration of 2000 cells per well and incubated at 37°C. At 72 h, the optical density was measured at 450 nm using a microtiter plate reader, and the rate of cell survival was expressed as the absorbance. The results represent the mean of three replicates under the same conditions.
Colony formation assay
The transfected glioma cells were placed in a fresh six-well plate and maintained in 1640 medium containing 10% FBS. After 14 days, cells were fixed with methanol and stained with 0.1% crystal violet. Visible colonies were manually counted.
Dual-luciferase reporter assay
The full-length FBXW7 3’-UTR was amplified by PCR and cloned downstream of the firefly luciferase gene in the pGL3 vector (Promega, USA). The vector was named wild-type 3’UTR. The GeneTailor Site-Directed Mutagenesis System (Invitrogen, USA) was used to perform site-directed mutagenesis of the miR-155 binding site in FBXW7 3’-UTR, to obtain a mutant 3’-UTR. These cells were transfected with reporter plasmids and transferred to 96-well plates. After incubating the cells for 48 h, the cells were harvested and assayed using the dual-luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer’s instructions. Each experiment was performed in triplicate.
Western blot and antibodies
Cells were lysed in Laemmli-buffer containing 10% β-mercaptoethanol (Sigma-Aldrich). Equal amounts of cells were resolved by 10% SDS-PAGE. After blotting on a nitrocellulose membrane (Schleicher&Schuell, Dassel, Germany) the membrane was blocked with 3% nonfat dry milk (Biorad) in TBS-T buffer. The membrane was incubated with rabbit anti-human FBXW7 antibody (1:1000; Novas), rabbit anti-human Ki-67 antibody (1:1000, Santa Cruz, CA, USA) and rabbit anti-β-actin (1:2000, Santa Cruz). Finally, the membrane was washed with TBS-T and incubated with a secondary antibody, which was a goat-anti-rabbit antibody conjugated with horseradish peroxidase and diluted 1:5000 (Biorad). The amount of detected protein was visualized by enhanced chemiluminescence (Amersham Biosciences, Freiburg, Germany) and autoradiography followed by densitometry using the ImageJ software (NIH, Bethesda, USA).
Statistical analysis
Statistical analyses were performed with GraphPad Prism 4.0 (GraphPad Software, La Jolla, California, USA). The differences between the two groups were analyzed using the Mann-Whitney U-test. Correlation analyses were carried out using Spearman’s rank correlation method. A log-rank test was used to analyze the statistical differences in survival based on Kaplan-Meier curves. Cox proportional-hazard regression analysis was performed to calculate HR and 95% CI for each covariable. All the differences were regarded as statistically significant when P<0.05.
Results
MALAT1 down-regulation in glioma specimens correlated with better survival
RT-qPCR was used to detect MALAT1 expression levels in cell lines and clinical samples, normalized to GAPDH. The results showed that glioma stem cell lines of U87, SHG44 and SHG139 expressed lower levels of MALAT1 than their parental lines. Further, the expression of MALAT1 was significantly decreased in 66 glioma tissues compared with paired noncancerous tissues (Figure 1B). Similarly, the serum MALAT1 expression was also suppressed in glioma patients compared with healthy individuals (Figure 1C). Moreover, the glioma tissues in 60.6% (40 of 66) of cases showed at least a two-fold lower expression of MALAT1 compared with noncancerous tissues (Figure 1D). We further determined the association between MALAT1 and clinicopathological characters. As shown in Table 1, MALAT1 was significantly correlated with tumor size, WHO grade and Karnofsky Performance Status (KPS). No significant correlations were observed between MALAT1 expression and other clinicopathological factors including gender and age in 66 primary glioma tissues.
Figure 1.

MALAT1 is down-regulated in glioma specimens and related to overall survival. A: The expression level of MALAT1 in seven glioma cell lines was detected by using RT-qPCR method. B: RT-qPCR was used to detect the expression of MALAT1 in 66 primary glioma tissues and noncancerous tissues. C: RT-qPCR was used to determine the expression of MALAT1 in 45 glioma serum samples and 30 healthy individuals. D: MALAT1 expression level was analyzed by using RT-qPCR in 66 primary glioma tissues and expressed as log2 fold change (glioma/normal), and the log2 fold changes were presented as follows: <1, underexpression (40 cases); >1, overexpression (8 cases); the remainder were defined as unchanged (18 cases). E: Kaplan-Meier curves for overall survival were drawn according to MALAT1 expression in 66 primary glioma tissues and were analyzed by using log-rank test. Error bars represent SEM. *P<0.05, **P<0.01.
Table 1.
Clinical characteristics of 66 patients and the expression of MALAT1 in glioma tissues
| Factors | Case | MALAT1 Median (range) | P |
|---|---|---|---|
| Gender | 0.766 | ||
| Male | 36 | 3.14 (0.21-6.59) | |
| Female | 30 | 3.21 (0.57-6.86) | |
| Age (years) | 0.425 | ||
| <60 | 32 | 3.08 (0.43-6.32) | |
| ≥60 | 34 | 3.36 (0.46-5.43) | |
| Tumor size | 0.006 | ||
| <5 cm | 35 | 3.97 (1.24-6.79) | |
| ≥5 cm | 31 | 2.46 (0.36-5.04) | |
| WHO grade | <0.001 | ||
| I | 12 | 2.03 (0.21-5.65) | |
| III | 9 | 2.45 (0.68-6.10) | |
| III | 19 | 3.58 (1.77-6.69) | |
| IV | 26 | 4.39 (2.63-6.86) | |
| KPS | 0.024 | ||
| <90 | 47 | 2.77 (0.57-5.43) | |
| ≥90 | 19 | 3.58 (1.35-6.86) |
KPS: Karnofsky Performance Status.
Furthermore, Kaplan-Meier survival analysis was conducted to investigate the prognostic value of MALAT1 in glioma patients. A median value of MALAT1 (3.17) in 66 primary glioma tissues was used to classify these patients into high and low groups. Patients with high MALAT1 expression manifested longer overall survival compared with those with low MALAT1 (Figure 1E). Moreover, Cox regression mutivariate analysis was used to determine whether MALAT1 expression was an independent predictor of overall survival in glioma patients. The results showed that low MALAT1 was significantly associated with poor survival independent of other clinical covariates (Table 2). Collectively, these results suggest that MALAT1 may function as a tumor-suppressor and served as an independent prognostic factor in glioma.
Table 2.
Multivariate Cox proportional hazards regression model analysis for overall survival in glioma patients
| Factors | Multivariate analysis | ||
|---|---|---|---|
|
| |||
| RR | 95% CI | P | |
| Gender | 0.999 | 0.473-2.012 | 0.998 |
| Age | 1.953 | 0.847-4.318 | 0.094 |
| Tumor size | 1.892 | 1.036-3.012 | 0.036 |
| WHO grade | 3.648 | 1.321-8.937 | 0.011 |
| KPS | 2.351 | 0.731-4.984 | 0.176 |
| MALAT1 expression | 2.796 | 1.157-4.735 | 0.027 |
KPS: Karnofsky Performance Status.
MALAT1 negatively regulates miR-155 expression
Our results showed that MALAT1 was poorly expressed in glioma cells and specimens, and therefore, the underlying function of MALAT1 in glioma was further investigated. Using miRcode (http://www.mircode.org/mircode), a computer algorithm to identify miRNA target genes including lncRNAs (31 205), we found that miR-155 contained two enriched MALAT1 binding sites (Figure 2A), which suggested a potential interaction between MALAT1 and miR-155. The miR-155 expression was detected using RT-qPCR, and was sufficiently up-regulated in primary glioma tissues compared with primary noncancerous tissues (Figure 2B). Further, the glioma cell lines also showed elevated miR-155 expression compared with the normal human astrocyte 1800 cell line (Figure 2C). The Spearman test indicated a significant negative correlation between MALAT1 and miR-155 expression in primary glioma tissues (Figure 2D). To further validate the negative regulation of MALAT1 on miR-155, U87 and SHG139 were used to demonstrate the effect of overexpression and knock-down of MALAT1 on miR-155 expression. RT-qPCR was used to detect the expression of MALAT1 after transfection of pMALAT1 in both glioma cell lines. As shown in Figure 2E, the U87 and SHG139 cells exhibited a significantly increased MALAT1 expression: 3.65-fold (P<0.01) and 4.40-fold (P<0.01), respectively, compared with the control cells (Figure 2E). Similarly, MALAT1 expression levels in U87 and SHG139 cells were decreased by 66.0% (P<0.01) and 64.9% (P<0.01) after transfection with siMALAT1, respectively (Figure 2F). After knockdown of MALAT1, a dramatically enhanced miR-155 expression was observed in U87 and SHG139 cells (Figure 2G). However, miR-155 was down-regulated in the above cell lines showing MALAT1 overexpression (Figure 2H). Similarly, we also interrupted miR-155 expression with anti-miR-155 and miR-155 mimics (Figure 2I and 2J). MALAT1 was also negatively regulated by miR-155 (Figure 2K and 2L). Thus, we conclude that the interaction between MALAT1 and miR-155 has reciprocal effects.
Figure 2.

MALAT1 negatively regulates miR-155 expression in glioma cells. (A) Representation of the miR-155 binding site in MALAT1 based on miRcode (http://www.mircode.org/mircode/). (B) The expression level of miR-155 in 66 primary glioma tissues and noncancerous tissues was detected by using RT-qPCR. (C) The miR-155 expression level was significantly higher in glioma cell lines when compared with that in the normal human astrocyte 1800 cell line. (D) FBXW7 mRNA is decreased following forced expression of miR-155 in primary glioma tissues. (E) The expression levels of MALAT1 in the U87 and SHG139 cells transfected with MALAT1 overexpression vector (pMALAT1) were increased by 3.65-folds and 4.40-folds, respectively, compared with the control cells. (F) The expression levels of MALAT1 in the U87 and SHG139 cells transfected with MALAT1-siRNA (siMALAT1) were decreased by 66.0% and 64.9%, respectively, compared with the control cells. (G) After knockdown of MALAT1, a dramatically enhanced miR-155 expression was observed in U87 and SHG139 cells. (H) MiR-155 expression level was down-regulated in U87 and SHG139 cells with MALAT1 overexpression. (I, J) MiR-155 expression level was dramatically suppressed by anti-miR-155 (I) and overexpressed by miR-155 mimics (J) in U87 and SHG139 cells. (K, L) MALAT1 expression level was up-regulated by anti-miR-155 (K) and down-regulated by miR-155 mimics (L) in U87 and SHG139 cells. Error bars represent SEM. *P<0.05, **P<0.01, ***P<0.001.
MALAT1 inhibits cell viability by down-regulating miR-155 expression in vitro
Following transfection with siMALAT1 or p-MALAT1, the cell viability was evaluated. The results showed that siMALAT1 enhanced cell viability while p-MALAT1 suppressed cell survival in both the glioma cell lines (Figure 3A). Besides, siMALAT1 also promoted colony formation by U87 cells (Figure 3B). We investigated the effect of miR-155 on glioma cell viability. As shown in Figure 3C, miR-155 induced cell proliferation and viability while anti-miR-155 suppressed cell viability. Next, results of cell cycle analysis indicated that both p-MALAT1 and anti-miR-155 decreased the percentage of S-G2 cells and increased the percentage of G0/G1 cells in U87 cells (Figure 3D).
Figure 3.

MALAT1 inhibits cell viability through down-regulating miR-155 expression. (A) CCK8 assay was performed to evaluate the glioma cell growth, and the relative cell viability was determined after transfection for 72 h. siMALAT1 promoted cell growth while p-MALAT1 suppressed cell growth in U87 and SHG139 cells. (B) siMALAT1 also promoted colony formation capacity of U87 cells compared with siNC. (C) miR-155 induced a proliferative effect on cell viability while anti-miR-155 suppressed cell viability. (D) Both p-MALAT1 and anti-miR-155 decreased the percentage of S-G2 cells and increased the percentage of G0/G1 cells in U87 cells. (E, F) MiR-155/anti-miR-155 was transfected into the p-MALAT1/siMALAT1 infected U87 and SHG139 cells, respectively. MiR-155 significantly rescued the growth inhibition induced by MALAT1 overexpression (E), while suppression of miR-155 significantly abrogated proliferative effect induced by MALAT1 knockdown (F). (G) Ki-67 protein was detected by Western blot in U87 cells, and miR-155 dramatically rescued the pMALAT1-induced suppression of Ki-67 expression. Error bars represent SEM. *P<0.05, **P<0.01.
To investigate the antagonistic effect of MALAT1 and miR-155 on cell viability, miR-155 was transfected into p-MALAT1-infected U87 and SHG139 cells. As shown in Figure 3E, miR-155 rescued the growth inhibition induced by MALAT1 overexpression. In addition, suppression of miR-155 significantly abrogated the proliferative effect triggered by MALAT1 knockdown (Figure 3F). Furthermore, the cell proliferation marker Ki-67 was detected by Western blot. MiR-155 dramatically rescued the pMALAT1-induced suppression of Ki-67 expression in both cell lines (Figure 3G). These results suggest that MALAT1 promotes glioma progression via miR-155 regulation.
FBXW7 is identified as a direct target of miR-155 in glioma cells
To elucidate the underlying regulatory mechanism of miR155 in glioma formation and development, we investigated the gene targets. FBXW7 mRNA was predicted as the functional target of miR-155 using several bioinformatics programs including Targetscan and miRnada (Figure 4A). RT-qPCR was used to detect the FBXW7 mRNA expression in primary glioma tissues. A significant negative correlation was found between FBXW7 and miR-155 expression in 66 glioma tissues (r = -0.7459, P<0.001, Figure 4B). Using the gain- and loss-of-function assay, we found that miR-155 knockdown significantly rescued both FBXW7 mRNA and protein expression in glioma cells (Figure 4C and 4D).
Figure 4.
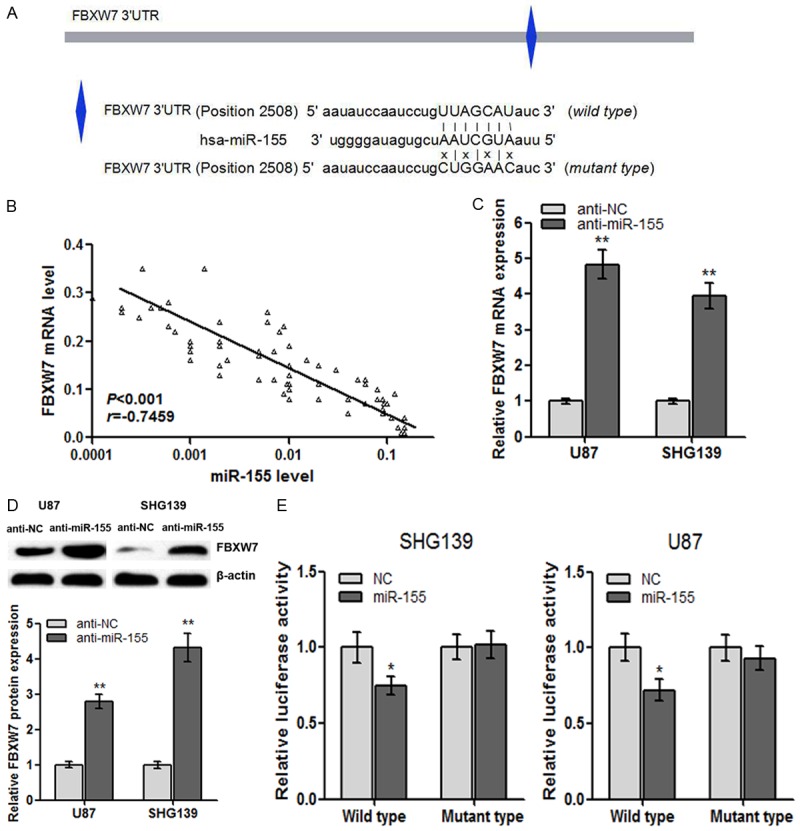
FBXW7 is identified as a direct target of miR-155 in glioma cells. A: Illustration of the the putative predicted miR-155 binding sites in the FBXW7 3’UTR region. B: FBXW7 mRNA is decreased following forced expression of miR-155 in primary glioma tissues. C: FBXW7 mRNA was up-regulated in glioma cell lines transfected with anti-miR-155. D: Western blots showing that anti-miR-155 significantly increased FBXW7 protein expression in U87 and SHG139 cells. E: MiR-155 targets the wild-type but not the mutant 3’UTR of FBXW7 in U87 and SHG139 cells. Error bars represent SEM. *P<0.05, **P<0.01.
The luciferase reporter assay was performed to explore the direct interaction between miR-155 and FBXW7 in glioma. Wild-type and mutant-type luciferase reporter plasmids were constructed as described in Methods. We found that miR-155 significantly inhibited the luciferase activity compared with the negative control miRNA (Figure 4E), suggesting that miR-155 interacted directly with the 3’-UTR of FBXW7 mRNA. In addition, miR-155 failed to inhibit the luciferase activity of the reporter vector containing mutant 3’-UTR of FBXW7 in the miR-155-binding site (Figure 4E). Based on these results, we conclude that miR-155 specifically suppresses FBXW7 protein synthesis in glioma cells.
FBXW7 mediates miR-155-induced tumorigenesis in glioma cells
Based on the direct interaction between miR-155 and FBXW7 expression, we further investigated the functional regulation of miR-155 by FBXW7. U87 cells were transfected with pFBXW7 or empty plasmid. FBXW7 mRNA and protein expression was significantly up-regulated in cells transfected with pFBXW7 compared with empty controls (Figure 5A and 5B). Subsequently, a CCK-8 assay was performed to investigate the effect of FBXW7 on the viability of glioma cells in vitro. As shown in Figure 5C, the FBXW7 overexpression significantly abrogated the cell proliferation capacity of U87 cells. Moreover, the enhanced cell viability induced by miR-155 was adequately suppressed after FBXW7 plasmid transfection (Figure 5D).
Figure 5.

FBXW7 mediates miR-155-induced tumorigenesis in glioma cells. (A, B) FBXW7 mRAN (A) and protein expression levels (B) were significantly up-regulated in cells transfected with pFBXW7 compared with empty controls. (C) FBXW7 overexpression significantly decreased the viability abilities of U87 cells. (D) pFBXW7 significantly abrogated the enhanced cell viability induced by miR-155 in U87 cells. (E, F) FBXW7 mRAN (E) and protein expression levels (F) were silenced in cells transfected with siFBXW7 compared with siNC. (G) FBXW7 knockdown significantly promoted the SHG139 cell proliferation. (H) Anti-miR-155 suppressed the cell viability, and this suppression was significantly reversed by FBXW7 knockdown. Error bars represent SEM. *P<0.05, **P<0.01, ***P<0.001.
However, the expression of FBXW7 in SHG139 cells was transfected by siFBXW7, and the FBXW7 mRNA and protein expression was validated (Figure 5E and 5F). The treated cells were evaluated for viability using a CCK8 assay. Inhibition of FBXW7 expression strongly enhanced cell viability when compared with nonspecific siRNA treatments (Figure 5G). Furthermore, the suppression of cell viability by miR-155 inhibition was significantly reversed by FBXW7 knockdown (Figure 5H). In summary, these results suggest that inhibition of FBXW7 deregulated the cell growth induced by miR-155 in glioma cells.
MALAT1 suppresses cell viability by down-regulating miR-155 and promoting FBXW7 expression
Our results demonstrated that MALAT1 inhibits cell viability by down-regulating miR-155, and the miR-155-induced cell proliferation was inhibited by functionally targeting FBXW7 in glioma cells. Therefore, we wondered whether the cell proliferation mediated by MALAT1 occurred via suppression of miR-155 and promotion of FBXW7. The pMALAT1 was transfected into U87 and SHG139 cells, and the FBXW7 mRNA and protein expression was determined. As shown in Figure 6A and 6B, both the mRNA and protein expression of FBXW7 was significantly increased by pMALAT1 when compared with the pVector. Similarly, FBXW7 mRNA and protein expression was sufficiently down-regulated after MALAT1 was knocked down (Figure 6C and 6D). Next, the cell function assay showed that the enhanced cell viability induced by siMALAT1 was abrogated by FBXW7 overexpression in U87 and SHG139 cells (Figure 6E). However, the FBXW7 knockdown significantly rescued the cell viability suppressed by MALAT1 up-regulation in both cell lines (Figure 6F). Overall, we concluded that MALAT1 inhibited cell viability by suppressing miR-155 and promoting FBXW7 expression.
Figure 6.

MALAT1 suppresses cell viability through down-regulating miR-155 and promoting FBXW7 expression. (A, B) pMALAT1 was transfected into U87 and SHG139 cells, and the FBXW7 mRNA (A) and protein expression level (B) was significantly increased by pMALAT1 when compared with the pVector. (C, D) siMALAT1 was transfected into U87 and SHG139 cells, and the FBXW7 mRNA (C) and protein expression level (D) was sufficiently down-regulated after MALAT1 knockdown. (E) MALAT1 knockdown promoted glioma cell viability, and this enhanced cell viability was abrogated by FBXW7 overexpression. (F) MALAT1 overexpression suppressed glioma cell growth, and this suppression was significantly rescued by FBXW7 knockdown. Error bars represent SEM. *P<0.05, **P<0.01.
Discussion
Gene expression is a complex cellular process that is tightly regulated at several levels to ensure appropriate gene dosage. It is dysregulated in human malignancies, leading to overexpression of tumor-promoting genes, and down-regulation of tumor suppressor genes. It is known that glioma is the most common intracranial tumor due to its high proliferation and defective apoptosis. Thus, it is critical to detect and determine the potential cellular processes that are tightly regulated by specific tumor-promoting genes. The aim of this study was to investigate the clinical significance of MALAT1 expression in human glioma and further investigate the potential regulatory mechanism underlying MALAT1 in glioma progression. Our study showed that the expression of MALAT1 was significantly lower in glioma specimens than in noncancerous brain tissues. Further, MALAT1 expression was significantly correlated with tumor size, WHO grade and KPS and was an independent prognostic factor of overall survival in glioma patients. Furthermore, we demonstrated a novel regulatory pathway of MALAT1 tumor-suppressive function in glioma. MALAT1 suppresses cell viability by down-regulating miR-155 and promoting FBXW7 expression. Thus, MALAT1 represents a potential prognostic and therapeutic target in glioma patients.
MALAT1 is a lncRNA overexpressed in numerous human cancers, such as non-small cell lung cancer [19], hepatocellular carcinoma [20], bladder cancer [8], and glioma [21]. MALAT1 expression was positively correlated with Gleason score, the level of prostate specific antigen (PSA), tumor stage and castration resistance in PCa. Gutschner et al showed that MALAT1-deficient lung cancer cells show impaired migration and formed fewer tumors in mice [22]. Wilusz et al found that MALAT1 acted as a precursor for the production of small RNAs and identified a highly conserved small RNA [23]. However, its functional role in tumorigenesis was apparently linked to cell growth. Ma et al demonstrated that MALAT1 associated with the malignant status and poor prognosis in glioma [12]. However, the underlying mechanism was unclear. Han et al demonstrated the tumor-suppressive function of MALAT1 in glioma cells via down-regulation of MMP2 and inactivation of ERK/MAPK signaling [13]. Consistent with Han et al, our study showed that MALAT1 was down-regulated in glioma and the high expression of MALAT1 was correlated with improved survival in glioma patients suggesting the key role of regulatory pathways.
We first investigated the interaction between MALAT1 with other non-coding RNAs such as miRNA. As a newly described regulatory mechanism, lncRNA influenced post-transcriptional regulation and interfered with miRNA pathways by competing for shared miRNA response elements [24]. In specific cases, lncRNAs contain miRNA response elements (MRE) and act as a natural miRNA sponge to reduce the binding of endogenous miRNAs to target genes. Thus lncRNA may modulate de-repression of miRNA target gene expression [25,26]. Liu et al have shown that lncRNA HOTAIR acts as a competing endogenous RNA (ceRNA) to regulate the expression of human epithelial growth factor receptor 2 (HER2) by competing for miR-331-3p [27]. In our study, we found that miR-155 was sufficiently up-regulated in glioma cells and specimens. By using miRcode, we demonstrated a reciprocal repression of MALAT1 and miR-155, which may represent a new regulatory mechanism of the two non-coding RNAs, consistent with recent studies [28]. Moreover, the gain- and loss-of-function study showed that MALAT1 inhibited cell growth while miR-155 promoted cell growth. Most importantly, the miR-155 rescued the growth inhibition induced by MALAT1 overexpression, while anti-miR-155 significantly abrogated the proliferative effect induced by MALAT1 knockdown. These results suggest that MALAT1 promoteed glioma progression by suppressing miR-155.
We subsequently investigated the underlying regulatory role of miR-155. It is widely accepted that microRNAs play an essential role in the posttranscriptional regulation of gene expression. Previous studies indicated that inhibition of miR-155 induced up-regulation of SOCS1 expression and subsequent inhibition of STAT3 in various malignant events [29]. Li found that miR-155 suppressed proliferation and induced apoptosis by up-regulating BACH1 in renal cancer cells [30]. In our study, the miR-155 target was predicted to explore the mechanism underlying the function of MALAT1 in glioma. Our results established FBXW7 as a direct target of miR-155 using the luciferase assays. FBXW7 is a component of the complex of SKP1, CUL1 and F-box-protein (SCF)-type ubiquitin ligases that target several oncoproteins for ubiquitination and degradation by the proteasome [31]. Several observations indicate that FBXW7 mediated cancer cell growth and tumorigenesis. FBXW7 acts as a tumor suppressor in the degradation of substrates with oncogenic activity frequently overexpressed in breast cancer such as Cyclin E, c-Myc and AURKA [32-34]. Our results showed that FBXW7 significantly suppressed glioma cell viability, and the cell viability suppressed by miR-155 inhibition was significantly reversed by FBXW7 knockdown. These results suggest that FBXW7 mediated the effect of miR-155 on cell viability.
We already found that MALAT1 suppressed cell viability by down-regulating miR-155, and FBXW7 was responsible for the miR-155-induced cell growth. We investigated if MALAT1 suppressed cell viability by down-regulating miR-155 and promoting FBXW7. Cell function assays showed that the enhanced cell viability induced by siMALAT1-1 was abrogated by FBXW7 overexpression. However, FBXW7 knockdown significantly rescued the cell viability suppressed by MALAT1 up-regulation. Overall, we concluded that MALAT1 inhibited cell viability by suppressing miR-155 and promoting FBXW7 expression.
In conclusion, our data demonstrated for the first time that MALAT1 was an independent prognostic factor for the survival of glioma patients. Furthermore, we determined that MALAT1 suppressed glioma cell proliferation via down-regulation of miR-155 and activation of FBXW7 function. Thus, MALAT1 may represent a novel prognostic biomarker and therapeutic target in glioma. Restoration of MALAT1 levels may be a novel therapeutic strategy against glioma.
Acknowledgements
This study was supported by the National Science Foundation of China (No. 81000612).
Disclosure of conflict of interest
None.
References
- 1.Taylor LP. Diagnosis, treatment, and prognosis of glioma: five new things. Neurology. 2010;75:S28–32. doi: 10.1212/WNL.0b013e3181fb3661. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Liu F, Xu Q, Wang X. Analysis of the expression profile of Dickkopf-1 gene in human glioma and the association with tumor malignancy. J Exp Clin Cancer Res. 2010;29:138. doi: 10.1186/1756-9966-29-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu X, Xi L, Zeng J, Yao Q. A functional +61G/A polymorphism in epidermal growth factor is associated with glioma risk among Asians. PLoS One. 2012;7:e41470. doi: 10.1371/journal.pone.0041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339:159–166. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhou S, Wang J, Zhang Z. An emerging understanding of long noncoding RNAs in kidney cancer. J Cancer Res Clin Oncol. 2014;140:1989–1995. doi: 10.1007/s00432-014-1699-y. [DOI] [PubMed] [Google Scholar]
- 7.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying L, Chen Q, Wang Y, Zhou Z, Huang Y, Qiu F. Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol Biosyst. 2012;8:2289–2294. doi: 10.1039/c2mb25070e. [DOI] [PubMed] [Google Scholar]
- 9.Praz V, Jagannathan V, Bucher P. CleanEx: a database of heterogeneous gene expression data based on a consistent gene nomenclature. Nucleic Acids Res. 2004;32:D542–547. doi: 10.1093/nar/gkh107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ, Taft RJ, Rizzi E, Askarian-Amiri M, Bonnal RJ, Callari M, Mignone F, Pesole G, Bertalot G, Bernardi LR, Albertini A, Lee C, Mattick JS, Zucchi I, De Bellis G. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, Strom S, Demetris AJ, Nalesnik M, Yu YP, Ranganathan S, Michalopoulos GK. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006;44:1012–1024. doi: 10.1002/hep.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma KX, Wang HJ, Li XR, Li T, Su G, Yang P, Wu JW. Long noncoding RNA MALAT1 associates with the malignant status and poor prognosis in glioma. Tumour Biol. 2015;36:3355–3359. doi: 10.1007/s13277-014-2969-7. [DOI] [PubMed] [Google Scholar]
- 13.Han Y, Wu Z, Wu T, Huang Y, Cheng Z, Li X, Sun T, Xie X, Zhou Y, Du Z. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016;7:e2123. doi: 10.1038/cddis.2015.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z, Ma T, Huang C, Hu T, Li J. The pivotal role of microRNA-155 in the control of cancer. J Cell Physiol. 2014;229:545–550. doi: 10.1002/jcp.24492. [DOI] [PubMed] [Google Scholar]
- 15.Qin W, Ren Q, Liu T, Huang Y, Wang J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013;587:1434–1439. doi: 10.1016/j.febslet.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 16.Liu Q, Chen J, Wang J, Amos C, Killary AM, Sen S, Wei C, Frazier ML. Putative tumor suppressor gene SEL1L was downregulated by aberrantly upregulated hsa-mir-155 in human pancreatic ductal adenocarcinoma. Mol Carcinog. 2014;53:711–721. doi: 10.1002/mc.22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Zhang J, Zhou L, Sun W, Zheng ZG, Lu P, Gao Y, Yang XS, Zhang ZC, Tao KS, Dou KF. Fbxw7 regulates hepatocellular carcinoma migration and invasion via Notch1 signaling pathway. Int J Oncol. 2015;47:231–243. doi: 10.3892/ijo.2015.2981. [DOI] [PubMed] [Google Scholar]
- 18.Roversi G, Picinelli C, Bestetti I, Crippa M, Perotti D, Ciceri S, Saccheri F, Collini P, Poliani PL, Catania S, Peissel B, Pagni F, Russo S, Peterlongo P, Manoukian S, Finelli P. Constitutional de novo deletion of the FBXW7 gene in a patient with focal segmental glomerulosclerosis and multiple primitive tumors. Sci Rep. 2015;5:15454. doi: 10.1038/srep15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Muller-Tidow C. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 20.Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26:851–858. doi: 10.1038/sj.onc.1209846. [DOI] [PubMed] [Google Scholar]
- 21.Xu C, Yang M, Tian J, Wang X, Li Z. MALAT-1: a long non-coding RNA and its important 3’ end functional motif in colorectal cancer metastasis. Int J Oncol. 2011;39:169–175. doi: 10.3892/ijo.2011.1007. [DOI] [PubMed] [Google Scholar]
- 22.Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, Zornig M, MacLeod AR, Spector DL, Diederichs S. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilusz JE, Freier SM, Spector DL. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008;135:919–932. doi: 10.1016/j.cell.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, Kong R, Xia R, Lu KH, Li JH, De W, Wang KM, Wang ZX. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92. doi: 10.1186/1476-4598-13-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma MZ, Li CX, Zhang Y, Weng MZ, Zhang MD, Qin YY, Gong W, Quan ZW. Long non-coding RNA HOTAIR, a c-Myc activated driver of malignancy, negatively regulates miRNA-130a in gallbladder cancer. Mol Cancer. 2014;13:156. doi: 10.1186/1476-4598-13-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huffaker TB, O’Connell RM. miR-155-SOCS1 as a Functional Axis: Satisfying the Burden of Proof. Immunity. 2015;43:3–4. doi: 10.1016/j.immuni.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Chen T, Zhong Z, Wang Y, Li Y, Zhao X. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol Med Rep. 2012;5:949–954. doi: 10.3892/mmr.2012.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gasca J, Flores ML, Giraldez S, Ruiz-Borrego M, Tortolero M, Romero F, Japon MA, Saez C. Loss of FBXW7 and accumulation of MCL1 and PLK1 promote paclitaxel resistance in breast cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.10481. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singhi AD, Cimino-Mathews A, Jenkins RB, Lan F, Fink SR, Nassar H, Vang R, Fetting JH, Hicks J, Sukumar S, De Marzo AM, Argani P. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol. 2012;25:378–387. doi: 10.1038/modpathol.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Chen Y, Olopade OI. MYC and Breast Cancer. Genes Cancer. 2010;1:629–640. doi: 10.1177/1947601910378691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaye A, Sahin A, Hao Q, Hunt K, Keyomarsi K, Bedrosian I. Cyclin E deregulation is an early event in the development of breast cancer. Breast Cancer Res Treat. 2009;115:651–659. doi: 10.1007/s10549-008-0266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]