

Abstract
Increasing evidence suggests that microRNA-101 (miR-101) is involved in the progression of various human cancers, including papillary thyroid carcinoma (PTC). However, the biological functions of miR-101 and underlying molecular mechanisms in PTC remain largely unknown. In this study, we demonstrated that miR-101 underexpression in PTC tissue was associated with lymph node metastasis and poor prognosis of PTC patients. MiR-101 reduced PTC cell proliferation, apoptosis resistance, and invasion. Ubiquitin-specific protease 22 (USP22) was confirmed as a direct target of miR-101. USP22 restoration attenuated the inhibitory effects of miR-101 on PTC malignant traits in vitro. In vivo, miR-101 overexpression or USP22 depletion reduced the tumorigenesis of PTC. Overall, our findings provide new insight into the mechanism of PTC inhibition by miR-101, suggesting the potential of miR-101 as a therapeutic target in PTC patients.
Keywords: MicroRNA-101, papillary thyroid carcinoma, ubiquitin-specific protease 22, tumorigenesis
Introduction
Thyroid cancer is the most common and highly prevalent endocrine malignancy with rapidly increasing incidence over the past few decades [1-3]. Papillary thyroid carcinoma (PTC) is the most frequent type of thyroid tumors. Although the majority of PTC patients have a good prognosis after surgical resection in combination with radioiodine and levothyroxine treatment, metastasis and recurrence usually occur [4]. Many patients have died mainly as a result of insufficient specific diagnostic biomarkers and therapeutic strategies [5]. Thus, the successful prevention and treatment of PTC require a thorough understanding of its biological process and novel diagnostic and prognostic biomarkers.
MicroRNAs (miRNAs) are an abundant class of non-coding RNAs that regulate gene expression at either the post-transcriptional or translational level by interacting with the 3’ untranslated regions (3’-UTRs) of target mRNA [6]. Emerging evidence has suggested that miRNAs play crucial roles in malignant progression [7]. MiR-101 is a tumor suppressor in gastric cancer [8], hepatocellular carcinoma [9], breast cancer [10] and PTC [11-13] by regulating growth, apoptosis, and metastasis. However, the biological function and precise mechanism of miR-101 in PTC remain unclear.
Ubiquitin-specific protease 22 (USP22), a novel human deubiquitinating enzyme gene, is one of a small set of marker genes capable of predicting metastatic potential and therapeutic outcome in many human cancers, including PTC [14-19]. Some previous studies demonstrated that aberrant USP22 expression is associated with poor prognosis of patients with invasive breast cancer, colorectal cancer and PTC [14-16]. USP22 regulates a number of cellular traits, including growth, differentiation, cell cycle progression, transcriptional activation, and signal transduction [20]. Silencing USP22 inhibits proliferation and induces cell cycle arrest in bladder cancer cells [17]. USP22 depletion promotes human brain glioma apoptosis [18]. Targeting USP22 suppresses growth and metastasis of anaplastic thyroid carcinoma (ATC) [19]. However, the clinicopathological significance and biological roles of USP22 in PTC has not yet been elucidated.
In this study, we found that miR-101 expression was lower in PTC tissues and downregulation of miR-101 was inversely associated with lymph node metastasis and prognosis of PTC patients. USP22 was identified as the direct functional target of miR-101. MiR-101 inhibited PTC cell proliferation, apoptosis resistance and invasion, whereas USP22 restoration counteracted the inhibitory effects of miR-101 in vitro. In vivo results were highly consistent with in vitro findings, miR-101 overexpression or USP22 depletion reduced the growth and metastasis of PTC. Collectively, these results suggested miR-101 as a potential target for PTC treatment.
Materials and methods
Ethics statement
The study was approved by the Institutional Review Board of the Tangdu Hospital of the Fourth Military Medical University. Written informed consent was obtained from all participating patients. Animal experiments were approved by the Institutional Committee for Animal Research and performed in accordance with national guidelines for the care and use of laboratory animals.
Clinical tissue specimens
Between 2008 and 2012, tissue specimens were obtained from 120 PTC patients at the Department of General Surgery, Tangdu Hospital, the Fourth Military Medical University. All patients had not received radiation therapy or chemotherapy prior to surgery. Every patient was followed up to 2015 or until death. Tissue samples were classified according to the World Health Organization criteria and stored in liquid nitrogen or -80°C for RNA or protein extraction.
Cell culture and transfection
A benign human thyroid follicular cell line Nthy-ori 3-1 and five human PTC cell lines HTH83, NIM-1, TPC-1, B-CPAP, and K1 were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, which certifies the origin and identity. HEK293T was obtained from American Type Culture Collection (Manassas, VA, USA). HTH83, NIM-1, TPC1 and B-CPAP cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, BRL, Grand Island, USA) containing 10% fetal bovine serum (FBS; HyClone, Victoria, Australia). K1 cell was grown in DMEM/Ham’s F12/MCDB ratio 2:1:1. Nthy-ori 3-1 and HEK293T cells were maintained in Roswell Park Memorial Institute 1640 (Gibco) supplemented with 10% FBS. All cell lines were kept at 37°C and 5% CO2 in a humidified atmosphere.
Establishment of K1-luciferase (luc) cell line
K1 cells were infected with the lentivirus pLV-luc obtained from Inovogen Biotechnology (Delhi, India). Cells were selected with puromycin (Sigma; 200 μg/mL) to generate clones stably expressing luciferase. After 16 days of screening, the K1-luc cell line was established from a single clone.
Plasmid and lentivirus constructs
3’-UTR-wild-type (WT) or 3’-UTR-mutant (MUT) plasmids were prepared by inserting USP22-3’-UTR-WT containing a putative miR-101 binding site or its mutant sequence into the pGL3 control plasmid (Promega, Madison, WI, USA). The pUNOI-hUSP22 (open reading frame) plasmid was purchased from InvivoGen (Hong Kong, China). All constructs were confirmed by DNA sequencing. For lentiviral expression of miR-101, complementary DNA (cDNA) strands corresponding to the pre-miR-101 sequence were synthesized and cloned into the AgeI/EcoRI sites of pGCsi-H1-CMV-GFP (GeneChem, Shanghai, China). HEK293T cells were transfected with the constructs along with pMD2.G and psPAX2 packaging plasmids. At 48 h after transfection, the viral supernatants were collected, centrifuged, filtered, and used to infect K1-luc cells. A short hairpin RNA (shRNA) was designed based on the USP22 sequence. A scrambled shRNA was used as a control. Paired deoxyribonucleotide oligos encoding the shRNAs were synthesized, annealed, and cloned into the EcoRI/NcoI sites of the pLKO.1 vector (Addgene, Cambridge, MA, USA). HEK293T cells were co-transfected with the constructs, pCMV-VSVG, and pCMV-bA.9 plasmids. The viral supernatants were harvested, filtered, and used to infect K1-luc cells. Cells were selected with 5 μg/mL puromycin (Sigma) to generate stable shRNA-expressing clones.
Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was isolated from fresh tissues and cells with TRIzol reagent (Invitrogen, San Diego, CA, USA). cDNA was synthesized by reverse transcriptase (Epicentre, Madison, WI, USA) or with miScript Reverse Transcription Kit (Qiagen, Hilden, Germany) and then amplified with SYBR Premix Ex Taq™ (TaKaRa, Otsu, Shiga, Japan) according to the manufacturer’s instructions. qPCR was performed with an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, White Plains, NY, USA). mRNA and miRNA levels were determined by the 2-ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 as the internal controls. Primer sequences for qPCR were as follows: for USP22, 3’-GCTGCATTCC TGCCTCTA-5’ (forward), 3’-GCTGCATTCCTGCCTCTA-5’ (reverse); for GAPDH, 3’-GGAAATCGTGCGTGACATT-5’ (forward), 3’-CAGGCAGCTCGTAGCTCTT-5’ (reverse); for miR-101, 3’-CTACAGTACTGTGATAACTGAA-5’ (forward), Universal Primer (QIAGEN) (reverse); and for U6, RNU6B_2 miScript Primer (QIAGEN) (forward), Universal Primer (QIAGEN) (reverse).
Dual-luciferase reporter assay
K1 cells were seeded into 24-well plates. When the confluence reached 80%, K1 cells were co-transfected with 100 ng of miR-101 or miR-NC mimics (RiboBio, Guangzhou, China) and pGL3-USP22-3’-UTR-WT or pGL3-USP22-3’-UTR-MUT plasmid using Lipofectamine 2000 (Invitrogen). Luciferase reporter gene assay was conducted with the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Renilla luciferase gene was co-transfected as a control for normalization.
Cell viability assay
Cells were grown on 96-well plates and transfected with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid. 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) assay was performed at 0, 24, 48, 72, and 96 h after transfection. In brief, 0.25 mg/mL of MTT was added to each well and incubated at 37°C for 4 h. The supernatant was decanted, and 0.15 mL of dimethyl sulfoxide (Sigma) was added. The plates were immediately read at 540 nm by using a scanning multi-well spectrometer (Bio-Tek instruments Inc., Winooski, VT, USA).
Colony formation assay
After transfection with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid, K1 cells (1 × 103) were plated on 6-well plates precoated with 1% agar (Sigma). At 14th day post-culture, colonies were fixed in 70% ethanol, stained with 0.5% crystal violet (Sigma), and counted under the microscope (Olympus, Tokyo, Japan). Five random fields were selected for each well to determine the total number of colonies.
5-Ethynyl-20-deoxyuridine (EdU) incorporation assay
Cell proliferation was determined by EdU (RiboBio) staining according to the manufacturer’s instructions. In brief, cells were treated with 20 μM EdU in medium in 24-well plates for 2 h and then fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were subsequently stained with Apollo solution and Hoechst 33342. Six random fields were selected for each well and cells were photographed and counted under an inverted fluorescent microscope (Carl Zeiss, Berlin, Germany).
Migration and invasion assay
Cell migration and invasion assays were performed in Transwell chambers. For migration assays, 5 × 103 cells suspended in medium without serum were added to the upper chamber of Transwell inserts with 8 μm pore (BD Biosciences, Franklin Lakes, NJ, USA). Medium containing 10% FBS was added to the lower chamber as a chemoattractant. For invasion assays, 1 × 104 cells were added to the upper chamber of Tranwells precoated with Matrigel (BD Biosciences). After 24 h of incubation, cells that did not migrate or invade through the pores were carefully removed. The filters were fixed in methanol and stained with 4’,6-diamidino-2-phanylindole (DAPI; Sigma). Five random fields were selected from per chamber and cells were counted under an inverted fluorescent microscope (Carl Zeiss).
Nucleosomal fragmentation assay
After 48 h of transfection with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid, cell apoptosis was quantified by nucleosomal fragmentation (Cell Death Detection ELISA PLUS; Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s protocol. The absorbance values were normalized with reference to control-treated cells to derive the nucleosomal enrichment factor.
Quantitative caspase-3 activity assay
Caspase-3 activity was detected using the Caspase-3/CPP32 Colorimetric Assay Kit (BioVision, Palo Alto, CA, USA) according to the manufacturer’s instructions. After 48 h of transfection with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid, 1 × 106 cells were incubated with 50 μL of chilled lysis buffer on ice for 10 min. The supernatant was collected after centrifugation at 10,000 × g. Protein (150 μg) in a total volume of 50 μL was added to 50 μL of 2 × reaction buffer containing 5 μL of N-Acetyl-Asp-Glu-Val-Asp-pNA substrate (200 μM final concentrations). After 2 h of incubation at 37°C, N-Acetyl-Asp-Glu-Val-Asp-pNA cleavage was monitored by detecting enzyme-catalyzed pNA release at 405 nm with a microplate reader (Bio-Tek instruments Inc.).
Terminal transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay was performed as previously described [10]. Tissue specimens were fixed with 10% formalin overnight, embedded in paraffin, non-serially sectioned (4 μm), and mounted on polylysine-covered slides. After deparaffinization in xylene and rehydration in a graded series of ethanol solutions, the sections were rinsed with PBS and incubated with FITC-labeled TdT nucleotide mix at 37°C for 60 min. The sections were then rinsed twice with PBS and counterstained with 10 mg/mL DAPI. TUNEL-positive cells were imaged by fluorescence microscope (Carl Zeiss), counted, and ultimately expressed as a percentage of total cells.
Western blot analysis
Proteins were extracted from fresh tissues and cells, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes (Millipore, Bedford, MA, USA), and subjected to immunoblot analyses. Western blot was performed with primary antibodies targeting USP22, Rb (from Cell Signaling Technology, Danvers, MA, USA), cyclin D2, E-cadherin (from Abcam, Cambridge, UK), vimentin, snail, Bax, Bcl-2 (all from Abnova, Taiwan, China), cleaved (cl)-caspase-3, caspase-3, and β-actin (all from Sigma), followed by horseradish peroxidase-conjugated secondary antibody (Sigma). Bands were visualized using an enhanced chemiluminescence kit (Santa Cruz, Dallas, TX, USA).
In vivo tumor growth, metastasis, and apoptosis assays
For tumor growth assay, 6-week-old male mice with severe combined immune deficiency (SCID; Institute of Zoology, Chinese Academy of Sciences, Beijing, China) received subcutaneous injections of 1 × 106 K1-luc cells infected with a control lentivirus or a recombinant lentivirus expressing a miR-101 precursor or shUSP22 (n = 6 mice/group). At 1, 3, 5, 7, 9, and 12 weeks after inoculation, tumor volume was monitored and calculated as follows: tumor volume = width2 × length/2. All mice were sacrificed by euthanasia at 12 weeks post-inoculation, and the tumors were removed. For tumor metastasis assay, 6-week-old male SCID mice were injected with 1 × 106 K1-luc cells infected with a control lentivirus or a recombinant lentivirus expressing a miR-101 precursor or shUSP22 through the tail vein (n = 6 mice/group). The mice were monitored for general health status and evidence of morbidity related to the primary tumor or metastasis, and then sacrificed by euthanasia at 8 weeks post-inoculation. Anatomized mice were examined for metastasis in the lung. Lungs with visible tumor colonies were fixed and embedded in paraffin, and three non-sequential sections per animal were obtained. The sections were stained with hematoxylin and eosin (Maixin Biotech.) and analyzed for the presence of metastasis by light microscopy. The total numbers of metastases per lung section were counted and averaged. For apoptosis assays, the tissue sections were stained using TUNEL kits. TUNEL-positive cells were examined under a fluorescent microscope.
Bioluminescence imaging and quantification
Bioluminescence imaging was performed as previously described [10]. Male 6-week-old SCID mice received 1 × 106 K1-luc cells (in 100 μl of PBS) that were infected with a control lentivirus or a recombinant lentivirus expressing a miR-101 precursor or shUSP22 via subcutaneous injection. Tumor growth was measured by in vivo luciferase imaging of the xenografts at 5 weeks after treatment. The in vivo luciferase imaging was performed by intraperitoneal injection of the mice with D-luciferin (Promega) at a dose of 150 mg/kg per mouse. The mice were anesthetized, and images were acquired using the Xenogen IVIS imaging system. The signals in defined regions of interest were quantified as photon flux (photons/s/cm2) using Living Image software (Xenogen Corporation, Berkeley, CA, USA).
Statistical analysis
Data are expressed as means ± SD from three independent experiments and analyzed by Student’s t-test and ANOVA. Survival curves were plotted using the Kaplan-Meier method. Log-rank tests were conducted to assess the statistical significance between cases with high or low levels of miR-101. Correlation between miR-101 expression and lymph node metastasis was analyzed by chi-square test. All P values were two-sided and obtained using SPSS 13.0 software package (SPSS Inc., IL, USA). P < 0.05 was considered statistically significant.
Results
MiR-101 downregulation is inversely associated with prognosis and USP22 expression in PTC
qPCR assay was performed to quantify miR-101 levels in 20 pairs of frozen human PTC specimens and the corresponding adjacent non-cancerous specimens. Figure 1A shows lower miR-101 expression in PTC tissues than in normal tissues. PTC patients with low miR-101 levels had poor survival rate (Figure 1B) and high frequency of lymph node metastasis (Figure 1C) compared to PTC patients with high expression of miR-101. USP22 was highly expressed in PTC tissues measured by qPCR assay (Figure 1D) and USP22 mRNA expression was inversely associated with miR-101 level in PTC specimens (Figure 1E). Western blot analysis showed that the expression tendency of USP22 protein was similar to that of USP22 mRNA (Figure 1F). Compared with Nthy-ori 3-1 cell line, miR-101 expression decreased in the five PTC cell lines HTH83, TPC-1, K1, NIM-1 and B-CPAP, whereas USP22 expression increased (Figure 1G and 1H). The K1 cell line was selected for further study because it had the lowest level of miR-101 and highest level of USP22. These results indicated that miR-101 downregulation is negatively correlated with the prognosis of PTC patients and USP22 expression.
Figure 1.
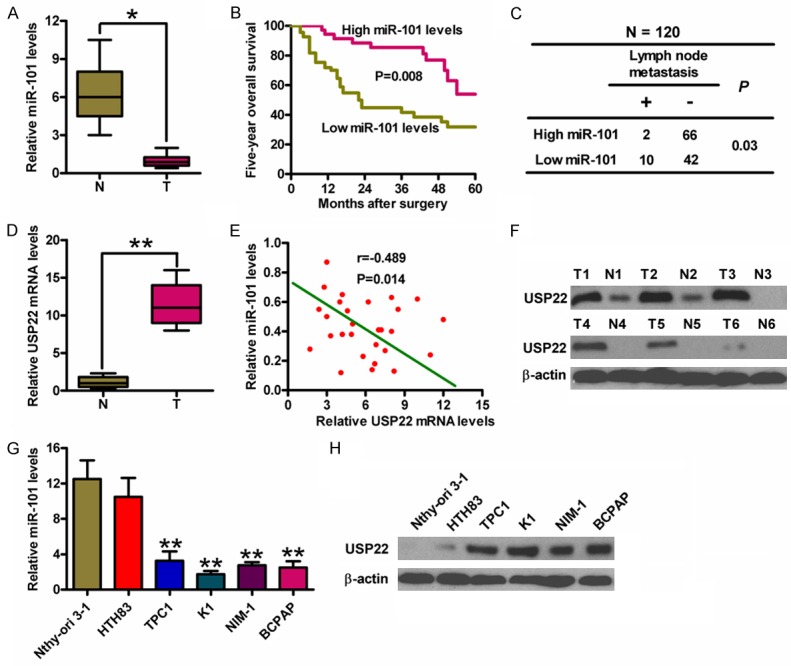
The levels of miR-101 and USP22 in PTC tissues and cell lines and the association between miR-101 expression and clinicopathological characteristics of PTC. (A) qPCR analysis of miR-101 expression in PTC (T) and the adjacent non-cancerous (N) tissues (n = 20). U6 was used as the endogenous control. (B) Kaplan-Meier survival curves of 60 PTC patients divided by miR-101 levels. (C) The association between miR-101 expression and lymph node metastasis (n = 120). (D) qPCR analyses of USP22 mRNA levels in T and N tissues (n = 20). GAPDH was used as the internal control. (E) The inverse association of miR-101 level and USP22 expression in PTC patients. (F) Western blot analysis of USP22 protein expression in T and N tissues. β-actin was used as the internal control. qPCR (G) and Western blot (H) assays were performed to determine miR-101 and USP22 expression in five PTC cell lines (HTH83, TPC-1, K1, NIM-1 and B-CPAP) and a Nthy-ori 3-1 cell line. U6 and β-actin were used as the endogenous controls. All data are shown as means ± SD of three separate experiments. *P < 0.05, **P < 0.01, as compared with N group (A, D); **P < 0.01, as compared with Nthy-ori 3-1 group (G).
MiR-101 inhibits PTC cell proliferation and motility
MiR-NC and miR-101 mimics were transfected into K1 cells to explore biological functions of miR-101 in PTC. MTT assay showed that cell viability was significantly reduced in miR-101-transfected cells than that in miR-NC-transfected cells (Figure 2A). Cells transfected with miR-101 mimics exhibited the decrease in colony formation (Figure 2B). EdU incorporation assay also showed that miR-101 suppressed K1 cell proliferation (Figure 2C). In addition, miR-101 enhanced caspase-3 activity and induced K1 cell apoptosis (Figure 2D and 2E). Subsequently, Transwell assays were performed to evaluate whether miR-101 inhibits PTC cell migration and invasion. As shown in Figure 2F and 2G, miR-101 markedly reduced the migration and invasion of K1 cells. These results demonstrated that miR-101 inhibits the proliferation and motility and induces apoptosis of PTC cells in vitro.
Figure 2.

MiR-101 inhibited proliferation, apoptosis resistance, migration and invasion of PTC cells. K1 cells were transfected with miR-NC or miR-101 mimics. (A) MTT assay was performed to determine cell viability. (B) Representative photographs of the colony formation of K1 cells and related quantitative analysis. (C) EdU staining was performed and the percentages of EdU-positive cells were counted. Scale bar: 10 mm. The caspase-3 activity (D) and enrichment factor (E) were analyzed. Cell migration (F) and invasion (G) were determined using Transwell chambers, and the percentage of migrated and invaded cells was calculated. Scale bar: 10 mm. All data are shown as means ± SD of three separate experiments. *P < 0.05, **P < 0.01, as compared with miR-NC group.
MiR-101 directly targets USP22 in PTC cells
Three algorithms (TargetScan, miRanda, and PicTar) were used to search for the candidate target genes of miR-101 (Figure 3A). A complementary miR-101 sequence was identified in the 3’-UTR of the USP22 mRNA (Figure 3B); therefore, this gene was selected for further analysis. Dual-luciferase reporter assays revealed that miR-101 suppressed luciferase activity of the reporter plasmid with USP22-3’-UTR-WT but had little effect on the activity of a reporter fused to USP22-3’-UTR-MUT in K1 cells (Figure 3C). qPCR and Western blot analyses demonstrated that miR-101 hampered USP22 mRNA and protein expression in K1 cells (Figure 3D and 3E). These results suggested that USP22 is a direct target of miR-101 in PTC cells.
Figure 3.
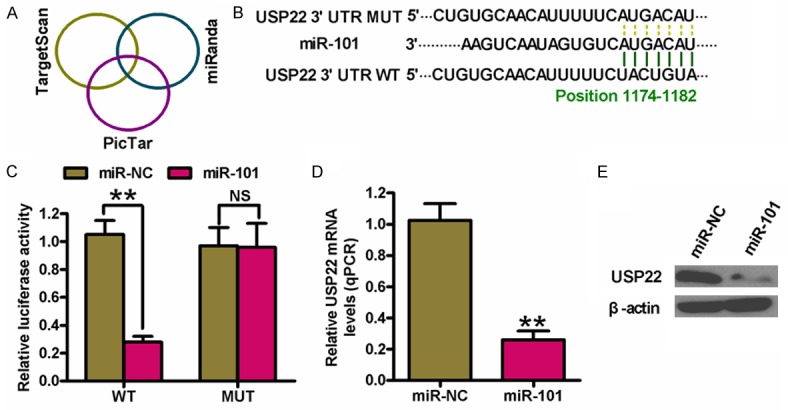
Identification of USP22 as a direct target of miR-101. (A) The putative target genes of miR-101 were predicted using TargetScan, PicTar, and miRanda softwares. (B) The predicted binding sites of miR-101 in the 3’-UTR of USP22. (C) Dual-luciferase reporter assays were performed 24 h after co-transfection of K1 cells with miR-NC or miR-101 mimics and a pGL3 construct containing WT or MUT 3’-UTR of USP22. Data were normalized to those from cells co-transfected with miR-NC and pGL3 plasmid. The mRNA (D) and protein (E) levels of USP22 in K1 cells transfected with miR-NC or miR-101 mimics were measured by qPCR and Western blot assays. GAPDH and β-actin were used as internal controls, respectively. All data are shown as means ± SD of three separate experiments. **P < 0.01, as compared with miR-NC group.
USP22 restoration counteracts the suppressive effects of miR-101 on PTC cell proliferation and apoptosis resistance
We investigated whether USP22 was the functional target of miR-101 in PTC. Figure 4A shows that USP22 overexpression markedly reduced miR-101-mediated inhibition of cell viability. USP22 upregulation counteracted miR-101-decreased colony formation (Figure 4B). EdU incorporation assay also revealed that USP22 restoration reduced miR-101-suppressed K1 cell proliferation (Figure 4C). In addition, USP22 overexpression inhibited miR-101-enhanced caspase-3 activity and nucl-eosomal fragmentation (Figure 4D and 4E). Mechanistically, miR-101 decreased the expressions of cyclin D2 and Bcl-2, whereas increased the levels of Rb, Bax, and cl-caspase-3. Nevertheless, USP22 restoration reversed miR-101-modulated expression of the above mentioned molecules. These results indicated that miR-101 inhibited USP22-elicited PTC cell proliferation and apoptosis resistance.
Figure 4.

USP22 restoration counteracted the suppressive effects of miR-101 on PTC cell proliferation and apoptosis resistance. K1 cells were transfected with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid. (A) Cell viability was measured by MTT assay. (B) The colony number was calculated. (C) The EdU-positive cells were counted to assess cell proliferation. Caspase-3 activity (D) and nucleosomal fragmentation (E) were determined. (F) The levels of USP22, cyclin D2, Rb, Bcl-2, Bax, cl-caspase-3, and caspase-3 were measured by western blot analysis. β-actin was used as endogenous control. All data are shown as means ± SD of three separate experiments. *P < 0.05, as compared with miR-NC group or miR-101 group; #P < 0.05, as compared with miR-101 group.
USP22 overexpression reduces the inhibitory effects of miR-101 on PTC cell migration and invasion
We next investigated whether USP22 is the key mediator of the effects of miR-101 on PTC cell motility. Restored USP22 reduced the miR-101-decreased K1 cell migration (Figure 5A) and invasion (Figure 5B). Epithelial-mesenchymal transition (EMT) is a hallmark of metastatic neoplasms. As shown in Figure 5C, miR-101 upregulated epithelial cell marker E-cadherin, whereas downregulated vimentin and snail, which was characteristic of mesenchymal cells. However, USP22 restoration reversed the above effects of miR-101. These results suggested that miR-101 inhibited PTC cell migration and invasion partly by targeting USP22.
Figure 5.
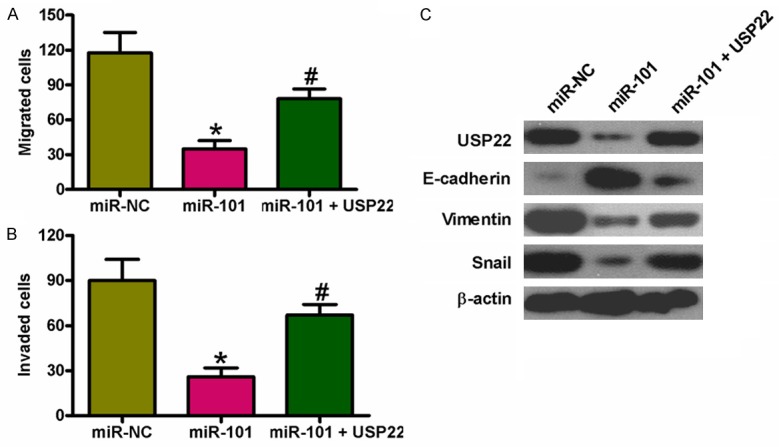
USP22 overexpression reduced the inhibitory effects of miR-101 on PTC cell migration and invasion. K1 cells were transfected with miR-NC, miR-101 mimics, or miR-101 mimics + USP22-expressing plasmid. Transwell assays were performed to detect cell migration (A) and invasion (B), and the number of migrated and invaded cells was calculated. (C) Expression of USP22 and invasion-related proteins containing E-cadherin, vimentin, and snail was measured by western blot analysis. β-actin was used as endogenous control. All data are shown as means ± SD of three separate experiments. *P < 0.05, as compared with miR-NC group; #P < 0.05, as compared with miR-101 group.
MiR-101 overexpression or USP22 depletion suppresses PTC tumorigenesis and metastasis in vivo
Subcutaneous injection of K1-luc cells stably expressing a miR-101 precursor or shUSP22 into SCID mice produced tumors within 1 week. The tumor volumes were measured every 2 weeks, and the mice were sacrificed at 12 weeks after tumor cell implantation. The size and volume of the tumors derived from K1-luc cells stably expressing miR-101 were lower than those of the tumors in the control group (Figure 6A-C). The rate of lung metastasis was also lower in xenograft tumors overexpressing miR-101 than that in tumors derived from control cells (Figure 6D). Furthermore, the number of TUNEL-positive cells was more in tumors overexpressing miR-101 than that in control tumors (Figure 6E). Similar to the effects of miR-101 overexpression, knockdown of USP22 inhibited tumor growth and lung metastasis, and induced PTC cell apoptosis (Figure 6A-E). Molecular analyses of the tumor tissues showed that miR-101 overexpression or USP22 depletion reduced the levels of cyclin D2 and snail, whereas upregulated the expressions of Rb, E-cadherin, and cl-caspase-3 (Figure 6F). These results suggested that the suppressive effects of miR-101 on in vivo PTC tumor growth, lung metastasis, and apoptosis are mediated by USP22 reduction.
Figure 6.

MiR-101 overexpression or USP22 knockdown suppressed tumor growth and metastasis and promoted apoptosis of PTC cells in vivo. SCID mice were injected subcutaneously or via their tail veins with K1-luc cells infected with a control lentivirus (Lenti-pGCsi or Lenti-pLKO.1) or a recombinant lentivirus expressing a miR-101 precursor (Lenti-pGCsi-miR-101) or shUSP22 (Lenti-shUSP22). A. In vivo luciferase imaging of the xenografts at 5 weeks after implanted with K1-luc cells. B. Representative gross photos of tumors after 12 weeks of implantation. C. Tumor volume was measured and calculated every two weeks. D. The numbers of metastatic foci in the lungs of mice from various groups at 8 weeks after tail vein injection. E. TUNEL assay was performed to detect the percentage of apoptotic cells. F. Representative results of western blot analyses of USP22, cyclin D2, Rb, E-cadherin, snail, cl-caspase-3, and caspase-3 in tumor tissues. β-actin was used as endogenous control. All data are shown as means ± SD of three separate experiments. *P < 0.05.
Discussion
PTC patients have died mainly as a result of insufficient specific diagnostic biomarkers and therapeutic strategies [5]. Understanding the novel mechanisms of PTC development and identifying new targets to prevent PTC progression are the main challenges in the improvement of PTC treatment. Previous studies showed that miR-101 inhibits the progression of various types of tumors [8-13]. However, little is known with regards to the regulatory roles of miR-101 in human PTC cells. Therefore, the present study aimed to determine the role of miR-101 in PTC progression and its molecular mechanisms. In this study, we found that miR-101 suppressed tumorigenesis of PTC by targeting USP22. The predominant findings were as follows. First, miR-101 was significantly underexpressed and inversely associated with lymph node metastasis and survival rate of PTC patients. Second, USP22 was confirmed as a direct target of miR-101. Third, miR-101 markedly reduced PTC cell proliferation, motility and apoptosis resistance, which were reversed by USP22 restoration. Lastly, the inhibitory effects of miR-101 on malignant phenotypes of PTC were mimicked by USP22 knockdown in vivo. Overall, these results suggested the potential prognostic role of miR-101 in PTC and indicated that miR-101 was a tumor suppressor of PTC by targeting USP22.
Increasing evidences showed that miR-101 serves a critical function in numerous pathological progresses and may be used as a highly promising diagnostic and/or prognostic marker of cancers [10,11]. In this study, we demonstrated that miR-101 expression was lower in PTC tissues than those in matched non-cancerous tissues, indicating its clinical significance. MiR-101 levels were inversely associated with lymph node metastasis, suggesting that miR-101 may serve a suppressor in PTC. We also evaluated the therapy outcome-predictive power to further confirm the potential clinical utility of miR-101. Kaplan-Meier analysis showed that the five-year overall survival rate of patients with high levels of miR-101 was significantly higher than that of patients with low miR-101 levels. Therefore, miR-101 may be a future diagnostic and/or prognostic marker of PTC patients.
USP22 has been shown to promote the proliferation of human ATC cell 8505C by facilitating cell cycle progression, which was supported by the observed G1 phase arrest and concomitant reduction in the S and G2/M phase when USP22 was depleted [19]. Consistently, we demonstrated that USP22 silencing inhibited the proliferation of human PTC cell K1. Previous studies showed that Rb is a pivotal regulator in the G1 checkpoint of cell cycle [21,22]. A delayed G1-to-S transition is usually accompanied by an enhancement of Rb, which is indeed observed in USP22 knockdown PTC cells in our study, suggesting that USP22 may promote cell proliferation by modulating the Rb/E2F pathway [23]. Cyclin D2 is a very important early G1 phase cell cycle regulator and is essential in regulating Rb function in thyroid carcinogenesis [24]. Previous study also showed that USP22 promotes cell cycle progression by positively regulating the PI3K/Akt pathway [22,23]. Here, we showed that cyclin D2 expression was significantly suppressed upon USP22 silencing in PTC cells. This findings together with previous reports suggested that USP22 promoted cell cycle progression possibly via PI3K/Akt/cyclin D2/Rb pathway in PTC cells.
It has been reported that USP22 silencing induced apoptosis of breast [14], colorectal [15], and anaplastic thyroid [19] cancer cells. Consistently, in the current study, USP22 knockdown markedly induced PTC cell apoptosis, as evidenced by the results of caspase-3 activity, nucleosomal enrichment factor and TUNEL assays. In the attempt to explore the mechanisms by which USP22 silencing leads to PTC cell apoptosis, we found that the proapoptotic molecules Bax and cl-caspase-3 were upregulated in response to USP22 knockdown, consistent with increased activation of caspase-3, whereas anti-apoptotic molecule Bcl-2 were downregulated. Although it remains elusive whether USP22 downregulates Bax or upregulates Bcl-2 via direct transcriptional or posttranslational regulation, further investigations will help to determine whether the well characterized deubiquitinating enzyme activity of USP22 plays a role in the apoptotic machinery of PTC cells.
USP22 is positively associated with invasion and metastasis of multiple types of malignancies [14-19]. EMT is known to be a central mechanism responsible for invasion and metastasis of various cancers, which endows the epithelial cells with mesenchymal-like properties, e.g. increased cell motility, and decreased intercellular adhesion [25,26]. Downregulation of E-cadherin and upregulation of vimentin and snail are hallmarks of cells undergoing EMT [27]. In this study, we showed that USP22 restoration significantly reduced miR-101-elicited decrease in expressions of vimentin and snail and increase in E-cadherin expression in PTC cells. In parallel, we achieved significantly suppressed lung metastasis formation by USP22 knockdown in vivo, which was consistent with the inhibitory effects of miR-101. These data suggest that USP22 serves as a critical regulator of PTC metastasis by promoting EMT.
In conclusion, the results presented here show that miR-101 was downregulated in PTC and was negatively correlated with tumor growth and metastasis. We also demonstrated that miR-101 is an independent prognostic factor in PTC patients and miR-101 directly targets USP22 in PTC cells. The results of in vitro and in vivo studies confirmed that miR-101 reduced the growth and invasion of ATC cells by regulating USP22-mediated expression and activation of a series of pro-tumorigenesis molecules. These results suggested that miR-101 inhibited tumor development and metastasis, and highlight miR-101 as a novel prognostic marker and potential therapeutic target in ATC.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81372859). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Leenhardt L, Grosclaude P, Cherie-Challine L. Increased incidence of thyroid carcinoma in france: a true epidemic or thyroid nodule management effects? Report from the French Thyroid Cancer Committee. Thyroid. 2004;14:1056–1060. doi: 10.1089/thy.2004.14.1056. [DOI] [PubMed] [Google Scholar]
- 2.Prasad NB, Somervell H, Tufano RP, Dackiw AP, Marohn MR, Califano JA, Wang Y, Westra WH, Clark DP, Umbricht CB, Libutti SK, Zeiger MA. Identification of genes differentially expressed in benign versus malignant thyroid tumors. Clin Cancer Res. 2008;14:3327–3337. doi: 10.1158/1078-0432.CCR-07-4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh A, Butuc R, Lopez R. Metastatic papillary thyroid carcinoma with absence of tumor focus in thyroid gland. Am J Case Rep. 2013;14:73–75. doi: 10.12659/AJCR.883834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voutilainen PE, Multanen MM, Leppaniemi AK, Haglund CH, Haapiainen RK, Franssila KO. Prognosis after lymph node recurrence in papillary thyroid carcinoma depends on age. Thyroid. 2001;11:953–957. doi: 10.1089/105072501753211028. [DOI] [PubMed] [Google Scholar]
- 5.Vriens MR, Suh I, Moses W, Kebebew E. Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid. 2009;19:1343–1349. doi: 10.1089/thy.2009.1607. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang HJ, Ruan HJ, He XJ, Ma YY, Jiang XT, Xia YJ, Ye ZY, Tao HQ. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur J Cancer. 2010;46:2295–2303. doi: 10.1016/j.ejca.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 9.Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y, Zhuang SM. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 10.Li JT, Jia LT, Liu NN, Zhu XS, Liu QQ, Wang XL, Yu F, Liu YL, Yang AG, Gao CF. MiRNA-101 inhibits breast cancer growth and metastasis by targeting CX chemokine receptor 7. Oncotarget. 2015;6:30818–30830. doi: 10.18632/oncotarget.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang C, Lu S, Jiang J, Jia X, Dong X, Bu P. Hsa-microRNA-101 suppresses migration and invasion by targeting Rac1 in thyroid cancer cells. Oncol Lett. 2014;8:1815–1821. doi: 10.3892/ol.2014.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin X, Guan H, Li H, Liu L, Liu J, Wei G, Huang Z, Liao Z, Li Y. miR-101 inhibits cell proliferation by targeting Rac1 in papillary thyroid carcinoma. Biomed Rep. 2014;2:122–126. doi: 10.3892/br.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, He M, Hou Y, Liang B, Zhao L, Ma S, Yu Y, Liu X. Expression profiles of microRNAs and their target genes in papillary thyroid carcinoma. Oncol Rep. 2013;29:1415–1420. doi: 10.3892/or.2013.2263. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, Pang D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol. 2011;137:1245–1253. doi: 10.1007/s00432-011-0998-9. [DOI] [PubMed] [Google Scholar]
- 15.Liu YL, Yang YM, Xu H, Dong XS. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J Gastroenterol Hepatol. 2010;25:1800–1805. doi: 10.1111/j.1440-1746.2010.06352.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Li YP, Chen JH, Yuan SF, Wang L, Zhang JL, Yao Q, Li NL, Bian JF, Fan J, Yi J, Ling R. Prognostic significance of USP22 as an oncogene in papillary thyroid carcinoma. Tumour Biol. 2013;34:1635–1639. doi: 10.1007/s13277-013-0696-0. [DOI] [PubMed] [Google Scholar]
- 17.Lv L, Xiao XY, Gu ZH, Zeng FQ, Huang LQ, Jiang GS. Silencing USP22 by asymmetric structure of interfering RNA inhibits proliferation and induces cell cycle arrest in bladder cancer cells. Mol Cell Biochem. 2011;346:11–21. doi: 10.1007/s11010-010-0585-4. [DOI] [PubMed] [Google Scholar]
- 18.Li ZH, Yu Y, Du C, Fu H, Wang J, Tian Y. RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol Lett. 2013;5:1290–1294. doi: 10.3892/ol.2013.1188. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Zhao HD, Tang HL, Liu NN, Zhao YL, Liu QQ, Zhu XS, Jia LT, Gao CF, Yang AG, Li JT. Targeting ubiquitin-specific protease 22 suppresses growth and metastasis of anaplastic thyroid carcinoma. Oncotarget. 2016;7:31191–31203. doi: 10.18632/oncotarget.9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 21.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 22.Fu J, Lv H, Guan H, Ma X, Ji M, He N, Shi B, Hou P. Metallothionein 1G functions as a tumor suppressor in thyroid cancer through modulating the PI3K/Akt signaling pathway. BMC Cancer. 2013;13:462. doi: 10.1186/1471-2407-13-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu YL, Jiang SX, Yang YM, Xu H, Liu JL, Wang XS. USP22 acts as an oncogene by the activation of BMI-1-mediated INK4a/ARF pathway and Akt pathway. Cell Biochem Biophys. 2012;62:229–235. doi: 10.1007/s12013-011-9287-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Guigon CJ, Fan J, Cheng SY, Zhu GZ. Pituitary homeobox 2 (PITX2) promotes thyroid carcinogenesis by activation of cyclin D2. Cell Cycle. 2010;9:1333–1341. doi: 10.4161/cc.9.7.11126. [DOI] [PubMed] [Google Scholar]
- 25.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 26.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 27.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]